
Stable electrically conductive, highly flame-retardant foam composites generated from reduced graphene oxide and silicone resin coatings†
Qian
Wu‡
a,
Chun
Liu‡
a,
Longcheng
Tang
 a,
Yue
Yan
a,
Huayu
Qiu
ab,
Yongbing
Pei
*ab,
Michael J.
Sailor
ac and
Lianbin
Wu
*ab
a,
Yue
Yan
a,
Huayu
Qiu
ab,
Yongbing
Pei
*ab,
Michael J.
Sailor
ac and
Lianbin
Wu
*ab
aKey Laboratory of Organosilicon Chemistry and Materials Technology of Ministry of Education, Hangzhou Normal University, Hangzhou, 311121, P. R. China. E-mail: wulianbin@hznu.edu.cn; peiyongbing@hznu.edu.cn
bCollaborative Innovation Center of Zhejiang Province for Manufacturing of Fluorine Silicon Fine Chemicals and Materials, Hangzhou Normal University, Hangzhou, 311121, P. R. China
cDepartment of Chemistry and Biochemistry, University of California, La Jolla, San Diego, California 92093, USA
First published on 15th October 2020
Abstract
To acheive flexible polyurethane (PU) foam composites with stable electrical conductivity and high flame retardancy involved first coating of graphene oxide (GO) onto PU foam surfaces and then chemically reducing the GO with hydrazine to form reduced GO (RGO). The RGO-coated PU foam is then dipped into a solution containing silicone resin (SiR) and silica nano-particles and cured. The resulting composites (PU-RGO–SiR) show superior flame retardancy, thermal stability and mechanical stability relative to the PU starting materials or PU coated with either RGO or SiR alone. The electrical conductivity of the PU-RGO–SiR composites (as high as 118 S m−1 at room temperature) could almost be retained but with small loss of 9.5% of the original value after 150 cyclic compression. When the samples were subjected to a temperature range from −50 to 400 °C, the electrical conductivity could remain constant at −50 °C, 25 °C, 100 °C, 200 °C, and even at 300 °C and 400 °C; the electrical-conductivity exhibited mild vibration but the vibration range was not beyond 5.6%. Flame retardancy tests show that the limiting oxygen index (LOI) increases from 14.7% for the pure foam to 31.5% for PU-RGO–SiR, and the PU-RGO–SiR composites exhibit a 65% reduction in the peak heat release rate (pHRR) and a 30% reduction in total smoke release (TSR). Thus, stable electrically conductive and highly flame-retardant foam composites have potential applications even in a variety of harsh conditions like high temperature, flame, organic solvents, and external compression.
1. Introduction
Polymeric foams such as flexible foam have been widely used in electrical power, building, and transportation industries due to their excellent elastic, acoustic, and thermal insulation properties.1,2 These polymeric foams are suitable candidates for developing foam composites with multi-functional performance like electrical-conductivity,3 thermal insulation,4,5etc. Amongst these foam composites, electrically conductive foam composites have attracted considerable attention and presented promising applications in various areas such as electronics,6 fire-alarm sensors,7 electromagnetic shielding materials8,9 and so on. Generally, electrically conductive foam composites comprise foam and conductive fillers. Polyurethane (PU) foam is one of the ideal starting polymer substrates for preparing conductive polymer composites.10 The incorporation of conductive fillers includes constructing an interconnected conductive network, as well as the assembly of two-dimensional conductive fillers onto three-dimensional foam structures.11 For electrically conductive foam composites used in various environments, their electrical-conductivity is highly desired to be stable to work normally. When they are directly exposed to harsh conditions ranging from high temperatures to organic solvents, the foam composites might work abnormally, because the effects of temperature, humidity or solvent might fail to keep them stable and even result in loss of electrical conductivity. Thereby, it is of great significance to develop foam composites with stable electrical conductivity, which is a crucial issue to enlarge their application.Actually, most of these polymer foam composites are made of combustible materials. For example, PU foam is easily ignited by a small fire source (flame, cigarettes, etc.), and it can combust completely in several seconds, accompanied by substantial quantities of toxic fumes, molten products, and a high rate of heat release. Amongst the methods used to reduce the flammability of these polymers, the combination of both nano-materials and conventional flame retardants is commonly used.12–18 Layer-by-layer (LbL) deposition is popular to use multilayer coatings due to its low cost, feasibility to control the composition at a nanoscale, and simplicity.19,20 Many kinds of inorganic nano-particles can be deposited including carbon nano-tubes,21,22 graphene oxide (GO),23,24 carbon black,25,26 metal compounds,26–28 clays,29–31 and silicates.32 In addition, organic materials like polydopamine (PDA)33 and combinations of organic/inorganic materials (clay/chitosan,31,34 alginate/clay,35 vermiculite/boehmite,36 and graphene/alginate37) have been deposited by the LbL method as combustion inhibitors.
In particular, GO is composed of two-dimensional sheets of sp2 hybridized carbon atoms, and it has been shown that it can provide flame-retardancy when serving as coatings on PU by the LBL method. This can be ascribed to the intrinsic high specific surface area38 and the physical barrier, which can delay thermal decomposition, reduce heat evolution, and block gas diffusion during combustion. Meanwhile, GO can be used with other organic/inorganic materials to prepare hybrid flame-retardant coating on PU foam for further improvement of flame-retardancy.23,24 Furthermore, considering there are many functional groups such as carboxyl, hydroxyl, epoxy and vinyl distributed on the basal planes or the edges of GO,39,40 many combustion inhibitors such as poly(piperazine spirocyclic pentaerythritol bisphosphonate) (PPSPB),41 2-(diphenylphosphino) ethyltriethoxy silane (DPPES),42 and polyphosphamide (PPA)43 can be attached onto GO nano-sheets to prepare flame-retardant foam.44 Similar to the physical barrier effect of GO, reduced graphene oxide (RGO) could be individually45–47 or simultaneously used with graphitic carbon nitride,48 layered double hydroxides,49 and polydopamine50 to develop flame-retardant materials. For instance, Pan et al. made a comparative study on the flame retardancy of only GO and RGO assembled PU foam by the LBL method, and the results showed that RGO coated PU foams exhibited higher thermal stability than GO coated PU foams in a temperature range from 430 °C to 600 °C.47 Furthermore, Qiu et al. used PDA–RGO complex dispersion to prepare flame-retardant PU foam via dip-coating by the LBL method, and the testing results showed that a synergistic flame-retardant effect was observed when using PDA and RGO together.50
Due to the higher bonding energy of the Si–O bond (460.5 kJ mol−1) relative to the C–C bond (304.0 kJ mol−1) and the C–O bond (358.0 kJ mol−1), the inherent strength of the siloxane (Si–O–Si) bond in silicone resin, silicone rubber, polyhedral oligomeric silsesquioxanes (POSS) and other silicone derivatives was used to reduce the flammability of combustible polymers.51,52 Under combustion conditions, silicones display slow burning rates and comparatively low heat release rates (HRRs). They do not shed molten flaming masses, displaying minimal toxic emissions and low yields of carbon monoxide. Thus silicones have been developed as environmentally friendly flame retardants.53 Prior work by Cai et al. incorporated hyperbranched polysiloxanes into epoxy to improve the thermal stability and flame retardancy of epoxy resin.54 In our previous work, we coated PU foams with silicone resin and found that higher silicon content in the resin resulted in better flame retardancy.55 We also previously reported hierarchical multi-layered coatings on PU foam created by LbL growth of silicone resin and GO and these materials displayed better flame retardancy.7 In this prior work, we found that the thermal decomposition of silicones into the silica crust can act as “barrier-wall” to reduce the amount of combustible gas in the volatile phase, delay the onset of decomposition products, slow the external heat flux, and reduce polymer melt dripping upon combustion.
Previous studies had revealed that RGO and silicone derivatives like POSS and silicon resin are environmentally friendly flame retardants to improve the flame retardancy of composites. As we know, RGO is a kind of electrically conductive nano-material and quite suitable for preparing electrically conductive composites. To the best of our best knowledge, little attention has been focused and seldom research has been done to prepare electrically conductive composites with superior flame retardancy, but which is highly desired to make the electrically conductive composites work normally under harsh conditions like high temperature. Thus, how to obtain electrically conductive composites together with excellent thermal stability or flame retardancy remains an open challenge. Herein, in this work, we prepared electrically conductive polymer composites by coating reduced graphene oxide (RGO). In order to improve the stability of electrical conductivity, as well as to reduce the flammability of the foam composites, we incorporated silica nano-particles into the silicone resin solution prior to dip-coating the RGO coated foam composites. In this way, it would not only provide more pronounced thermal stability and better flame retardancy, but also lead to stable electrical conductivity under a variety of harsh conditions like high temperature, flame attack, organic solvents, water and external compression.
2. Experimental
2.1 Materials
Graphite (600 mesh, 99 wt%) was purchased from Chengdu organic chemistry Co., Ltd. Methyltrimethoxysilane ((CH3)Si(CH3O)3, 98%), diethoxydimethylsilane ((CH3)2Si(C2H5O)2, 98%), dimethoxymethylphenylsilane (CH3C6H5Si(CH3O)2, 98%), hexamethyldisilazane ((CH3)6NHSi, 98%), phenyltrimethoxysilane (C6H5Si(CH3O)3, 98%), hydrazine hydrate (N2H4·H2O, 80 vol%), and double amino-terminated polyetheramine (D230) crosslinking agent were purchased from Sinopharm Chemical Reagent Co., Ltd. Concentrated sulfuric acid (H2SO4, ≥95 wt%), potassium peroxydisulfate (K2S2O8, 98 wt%), hydrochloric acid (HCl, 35 wt%), phosphorus pentoxide (P2O5, 98 wt%), hydrogen peroxide (H2O2, 30 vol%), potassium permanganate (KMnO4, 98%) and others reagents were supplied by Sinopharm Chemical Reagent Co., Ltd (China). PU foam with a density of ∼0.025g cm−3 was provided from Zhejiang Hangzhou Guangsheng Foam Plastic Company. Hydrophilic silica nano-particles (mean diameters 500 nm and 50 nm) were obtained from Macklin Co., Ltd.2.2 Preparation of GO
GO was synthesized via the oxidation of graphite flakes using a modified Hummers’ method.56,57 First, 12 g P2O5, 12 g K2S2O8 and 75 mL of H2SO4 were transferred into a 250 mL round bottom flask and the mixture was stirred until the solids dissolved completely. Graphite (15 g) was then added slowly to the solution. The mixture was heated to 90 °C in an oil bath and maintained at that temperature for 4 h, and then removed from the oil bath and allowed to cool to room temperature. It was then slowly added to a large amount (∼2000 mL) of deionized (DI) water and left overnight. Next, it was filtered and the solids were washed with copious amounts of DI water to remove the residual acid and reactants. The contents of the filter were then dried at 80 °C in a vacuum oven for 12 h. The solid (5 g) was then added to concentrated H2SO4 (115 mL) in a 500 mL round-bottom flask equipped with a mechanical stirrer and immersed in an ice-water bath. Solid KMnO4 was then slowly added to the suspension and care was taken to ensure that the temperature of the mixture was maintained below 10 °C. After the KMnO4 addition was complete, the mixture was heated to 35 °C and maintained at that temperature for 2 h. Ice-cold DI water (800 mL) was slowly added into the mixture, followed by the dropwise addition of 10 mL of 30% aqueous H2O2. The bright yellow mixture was washed with 1 M aqueous hydrochloric acid to remove the metal ions, and the mixture was then dialyzed with DI water. The recovered solid was freeze dried to obtain the GO product.2.3 Preparation of silicone resin
Phenyltrimethoxysilane (100 g), diethoxydimethylsilane (160 g), methyltrimethoxysilane (30 g), dimethoxymethylphenylsilane (64 g), alcohol (250 g) and hydrochloric acid solution (60 mL, 0.65 M HCl) were loaded into a 2000 mL, three-necked, round-bottom flask fitted with a thermometer, a stirrer, and a reflux condenser. The mixture was heated to 65–70 °C (reaction scheme is given in Fig. S1, ESI†). DI water (120 g) was added slowly using a pressure-compensated titration funnel over a period of 1.5–2 h, under constant stirring. After the addition was complete, the mixture was stirred for an additional 4 h. A solution of 0.02 M NaHCO3 was then added to the mixture to neutralize the acid. Once the solution attained a pH of 7, the mixture was filtered and the filtrate was heated to 100 °C under vacuum to remove the solvent. The silicone resin (SiR) was obtained after rapid cooling and characterized by 1H NMR, 29Si NMR, and FTIR (Fig. S2a, b and S3, ESI†).2.4 Preparation of polyurethane-reduced graphene oxide (PU-RGO) foams and polyurethane-silicone resin (PU-SiR)
GO (1.5 g) was dispersed in 750 mL of ethanol and ultrasonicated for 10 min. A cleaned PU foam sample (50 mm length × 50 mm width × 10 mm height) was fully immersed into the above mixture and subjected to vacuum for 15 min. The foam was removed from the liquid mixture and centrifuged at 350 rpm for 5 min. This process was repeated until the GO weight percent was 30 wt% compared to that of the pure PU foam (the correlation between the GO mass and the dip-coating cycle is given in Fig. S4a, ESI†). The PU-GO foam was then placed in a hydrothermal reactor with 1 mL of hydrazine hydrate. The reactor was transferred into a vacuum oven at 90 °C for 2 h and the resultant foam was designated as PU-RGO in this work.Silica nano-particles (15 g, D500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D50 = 2:1 wt/wt, where D500 represents the mean diameter of 500 nm and D50 represents the mean diameter of 50 nm), DI water (0.5 g) and hexamethyldisilazane (HMDS, 100 mL) were added to a 250 mL three-neck, round-bottom flask equipped with a mechanical stirrer, and then the mixture was heated to 100 °C for 4 h. The mixture was then filtered, washed with absolute ethanol, and dried at 100 °C for 12 h under vacuum to obtain hydrophobic silica nano-particles. The two possible transformation mechanisms from hydrophilic to hydrophobic were reported in the previous literature.58 Considering that the HMDS amount was excessive, the hydrophilic hydroxyl group would be completely consumed and the silicyl groups were grafted onto the silica nano-particles, endowing the silica nano-particles with a hydrophobic behavior. Hydrophobic nano-silica (9 g), silicone resin (21 g, prepared as described above), ethanol (50 g) and double amino-terminated polyetheramine D230 (0.6 g) were mixed with a high-speed mechanical mixer (97043-858, VWR International) for 30 min at 2000 rpm in a 100 mL round-bottom flask. The mixture was then subjected to ultrasonication (WB2000-M, WIGGENS) for 15 min and referred to as SiR solution. Subsequently, the PU foam was fully immersed into the above mixture for 15 min under vacuum, and then the sample was centrifuged at 600 rpm for 5 min. The above immersion/centrifugation process was repeated until the weight of the silica nano-particles and silicone resin was approximately 300 wt% of the PU foam weight (typically 4 cycles were required). Finally, the sample was cured at 90 °C for ∼4 h. The resulting coating consisting of silicone resin, hydrophobic silica nano-particles and the polyetheramine (D230) crosslinking agent was designated as PU-SiR.
:D50 = 2:1 wt/wt, where D500 represents the mean diameter of 500 nm and D50 represents the mean diameter of 50 nm), DI water (0.5 g) and hexamethyldisilazane (HMDS, 100 mL) were added to a 250 mL three-neck, round-bottom flask equipped with a mechanical stirrer, and then the mixture was heated to 100 °C for 4 h. The mixture was then filtered, washed with absolute ethanol, and dried at 100 °C for 12 h under vacuum to obtain hydrophobic silica nano-particles. The two possible transformation mechanisms from hydrophilic to hydrophobic were reported in the previous literature.58 Considering that the HMDS amount was excessive, the hydrophilic hydroxyl group would be completely consumed and the silicyl groups were grafted onto the silica nano-particles, endowing the silica nano-particles with a hydrophobic behavior. Hydrophobic nano-silica (9 g), silicone resin (21 g, prepared as described above), ethanol (50 g) and double amino-terminated polyetheramine D230 (0.6 g) were mixed with a high-speed mechanical mixer (97043-858, VWR International) for 30 min at 2000 rpm in a 100 mL round-bottom flask. The mixture was then subjected to ultrasonication (WB2000-M, WIGGENS) for 15 min and referred to as SiR solution. Subsequently, the PU foam was fully immersed into the above mixture for 15 min under vacuum, and then the sample was centrifuged at 600 rpm for 5 min. The above immersion/centrifugation process was repeated until the weight of the silica nano-particles and silicone resin was approximately 300 wt% of the PU foam weight (typically 4 cycles were required). Finally, the sample was cured at 90 °C for ∼4 h. The resulting coating consisting of silicone resin, hydrophobic silica nano-particles and the polyetheramine (D230) crosslinking agent was designated as PU-SiR.
2.5 Preparation of PU-RGO–SiR
The as-prepared PU-RGO foams were fully immersed into the SiR solution under vacuum for 15 min, and subsequently the foams were removed and centrifuged at 600 rpm for 5 min. The above immersion/centrifugation process was repeated until the weight of the SiR coating was approximately 300 wt% compared to the PU foam weight (typically 4 cycles were required). Finally, the sample was cured at 90 °C for ∼4 h. The resulting foam was designated as PU-RGO–SiR in this work. The whole preparation process of the PU-RGO–SiR foam is illustrated in Fig. 1.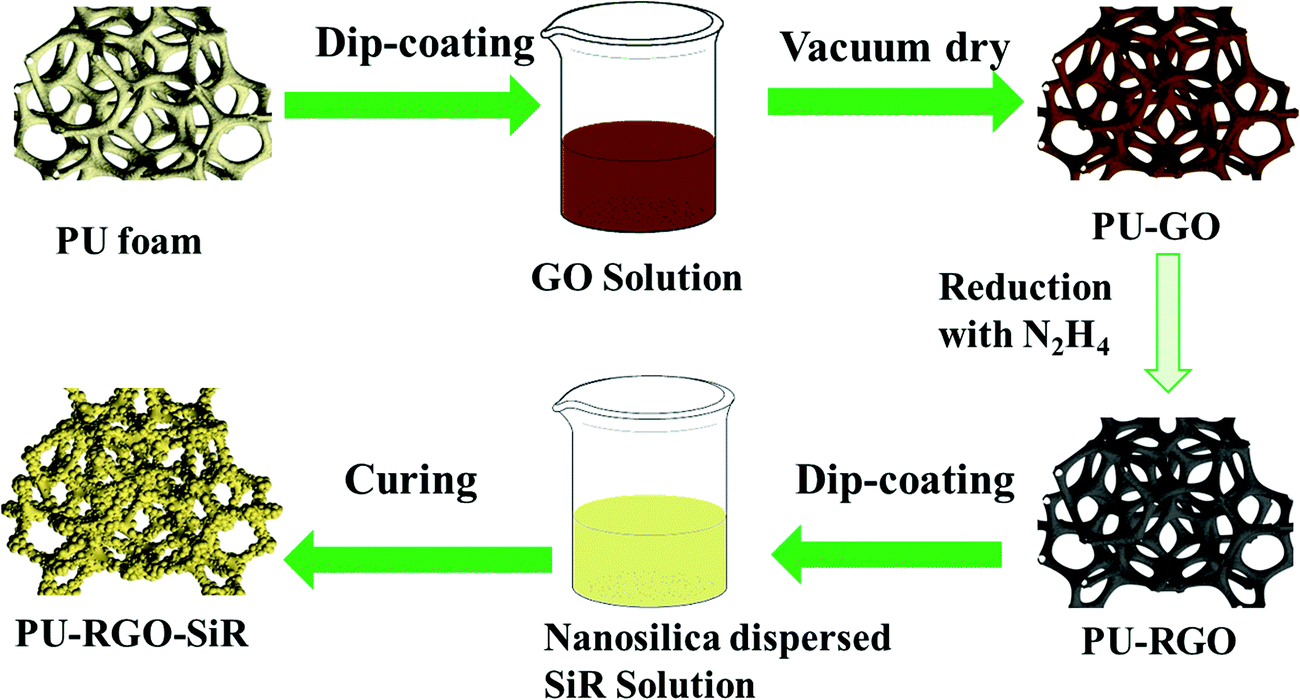 | ||
| Fig. 1 Schematically depicting the process to prepare PU-RGO–SiR foam generated from reduced graphene oxide (RGO) and nanosilica dispersed SiR solution. | ||
2.6 Material characterization
Raman spectra were recorded in the range 1000 to 2000 cm−1 using a SENTERRA Micro Raman spectrometer (Bruker Instruments, Germany) employing a 633 nm He–Ne laser. The morphologies and structures of PU, PU-RGO, PU-RGO–SiR and PU-RGO–SiR (after being subjected to combustion conditions) were examined by scanning electron microscopy (SEM, HITACHI S-4800 and Zeiss Sigma500). XRD measurements on the PU-GO and PU-RGO samples were performed using a D/Max 2550 V X-ray diffractometer (Rigaku, Japan) using graphite-monochromatized Cu-Kα X-rays. The diffraction patterns were recorded in the 2θ range from 5 to 40° at a wavelength of λ = 0.154 nm and at a scan rate of 5° min−1. Fourier-transform infrared (FTIR) spectra were recorded using a Nicolet 7000 FTIR spectrometer (Nicolet Instrument Company, USA) in the frequency range 400 to 4000 cm−1. Thermogravimetric analysis (TGA) was performed with a TA Instruments Q500 instrument, and the analyses were conducted in a nitrogen atmosphere at a heating rate of 10 °C min−1 between room temperature and 800 °C. Compression tests were carried out at room temperature using an Lloyd LS100Plus, Universal Testing Machine at a crosshead speed of 1.0 mm min−1. Typically, the PU-RGO–SiR sample dimension for compression testing was 25.0 mm length × 25.0 mm width × 10.0 mm thickness. Electrical conductivity tests were performed at 25 ± 1% and 50 ± 2% relative humidity, employing an ESCORT 3146A multimeter with a two-electrode method; the electrical conductivity (σ) was calculated as follows: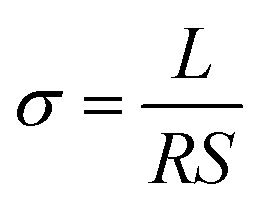 | (1) |
Flammability properties were evaluated using a vertical burning test. Before testing, the specimens were conditioned at 25 ± 1 °C and 50 ± 2% relative humidity for 72 h. The typical specimen dimension was 125 ± 5 mm length × 13 ± 0.5 mm width × 10 ± 0.5 mm thickness, and the long axis of each specimen was positioned approximately vertically. The PU and PU composite samples were characterized using a cone calorimeter, FTT0030, UK device, according to ISO 5660; the sample dimension for these measurements was 10 mm length × 10 mm width × 10 mm thickness, samples were exposed to a heat flux of 35 kW m−2, and the data reported are the averages of triplicate experiments. Limiting oxygen index (LOI) values were measured according to the ASTM D2863-97 standard on a JF-3 oxygen index meter (Jingning Analysis Instrument Company, China), using samples of dimension 100 mm × 10 mm × 10 mm. Foam surface properties were determined by measuring the static water contact angle (WCA) with a DSA30 contact angle analyzer (KRUSS, Germany) using a 3 μL water droplet.
3. Results and discussion
3.1 Microstructure characterization and analysis
The microstructure and surface morphology of the pure foam and the foam composite samples were characterized by scanning electron microscopy (SEM). The pure PU foam was light yellow in appearance (photograph shown in Fig. 2a), and the surface of the pore network in the SEM images appeared relatively smooth and clean (Fig. 2b and c). Dip-coating with GO and exposure to reducing conditions generated the PU-RGO foam, which appeared dark (Fig. 2d). The RGO was coated on the foam network (Fig. 2e and f). The silicone resin (SiR) used in this study consisted of a mixture of dimethylsiloxane, methyphenylsiloxane, and monophenylsiloxane components. The processing conditions involved adding silica nano-particles to the resin, dip-coating the PU-RGO material, and then curing the resulting SiR layer with a polyamine cross-linking agent. The colorless SiR coating on the PU-RGO material led to a minor change in appearance (Fig. 2g). The pore surface of the resulting PU-RGO–SiR foam took on a mottled look in the SEM images (Fig. 2h and i) but the large pores of the original PU foam remained intact. The higher magnification SEM images revealed a roughened and cracked microstructure on the pore surface, attributed to the SiR coating layer (Fig. 2j). Elemental microanalysis performed in situ by energy dispersive X-ray spectroscopy (EDS) confirmed the presence of silicon in the PU-RGO–SiR samples, while no silicon was detected in the untreated PU-RGO samples (Fig. 2k and l). | ||
| Fig. 2 (a) photograph and (b and c) SEM images of the pristine PU foam; (d) photograph and (e and f) SEM images of PU-RGO; (g) photograph and (h–j) SEM images of PU-RGO–SiR; and EDS data of (k) PU-RGO and (l) PU-RGO–SiR. | ||
The form of the carbon element in the RGO coatings was assessed by Raman spectroscopy. The Raman D-band (breathing mode corresponding to the k-point phonons of A1g symmetry) is typically used to determine the density of the sp3-hybridized defects relative to pure graphite,59 while the G-band is ascribed to the E2g phonons associated with the sp2-hybridized carbon atoms. The intensity ratio of the D-band to the G-band, ID/IG, is often used to estimate the sp2 domain size in graphitic materials. As shown in Fig. 3a, the value of ID/IG decreased from 1.14 to 1.05 after the GO was subjected to reduction by N2H4, indicating the removal of the oxygenated functional groups and partial restoration of the sp2 conjugated carbon structure in the GO sheets.60 As shown in Fig. 3b, the X-ray diffraction (XRD) pattern of GO adsorbed on the PU foam surface exhibited a strong peak at 11.26°, corresponding to an interlayer spacing of 0.78 nm. It should be noted that the amorphous peak of the PU foam61 around 22° is quite difficult to observe in contrast to the strong peak of GO at 11.26°. After reduction, the diffraction peak of the material (PU-RGO) increased to 22.94°, and the amorphous peak of the PU foam overlapped. The shifted 2θ of the diffraction peak indicates that the interlayer spacing is 0.40 nm. These values are consistent with the prior reports on reduction of GO to RGO, indicating that the PU skeleton did not interfere with the GO reduction to form a graphene structure.62,63
 | ||
| Fig. 3 (a) Raman and (b) XRD spectra of PU-GO and PU-RGO. (c) FTIR spectra and (d) TGA curves of PU, PU-RGO and PU-RGO–SiR. | ||
The Fourier-transform infrared (FTIR) spectra of PU, PU-RGO and PU-RGO–SiR all displayed bands characteristic of PU. The band at 3281 cm−1 was assigned to the N–H stretching vibration; the bands at 2970 and 2875 cm−1 were assigned to the C–H stretching vibrations; the band at 1720 cm−1 was assigned to the amide I C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration; and the band at 1090 cm−1 was assigned to the asymmetric C–O–C stretching vibrations. In addition, the bands at 1540 and 1220 cm−1 can be assigned to the N–H deformation and the amide II C–N stretching vibrations, respectively. Similar spectra and assignments have been reported on PU and PU/graphene composites.64 For the PU-RGO samples, the most obvious absorption bands characteristic of RGO were the CC modes observed at 1650 cm−1. For the PU-RGO–SiR, additional bands associated with the silane coating were observed. The band at 1010 cm−1 was assigned to Si–O–Si, and the bands at 1240 cm−1 and 780 cm−1 were ascribed to the deformation and stretching vibrations of Si–CH3.65
O stretching vibration; and the band at 1090 cm−1 was assigned to the asymmetric C–O–C stretching vibrations. In addition, the bands at 1540 and 1220 cm−1 can be assigned to the N–H deformation and the amide II C–N stretching vibrations, respectively. Similar spectra and assignments have been reported on PU and PU/graphene composites.64 For the PU-RGO samples, the most obvious absorption bands characteristic of RGO were the CC modes observed at 1650 cm−1. For the PU-RGO–SiR, additional bands associated with the silane coating were observed. The band at 1010 cm−1 was assigned to Si–O–Si, and the bands at 1240 cm−1 and 780 cm−1 were ascribed to the deformation and stretching vibrations of Si–CH3.65
Thermogravimetric analysis (TGA) tests were used to characterize the composites (Fig. 3d). When heated under a nitrogen atmosphere, PU displayed a substantial (>70%) mass loss in the temperature range 260–400 °C, which is ascribed to the pyrolysis of the PU foam. The PU-RGO displayed a similar mass loss curve in this temperature range, but the mass loss rate was lower than that for pure PU. The residual mass after heating to 800 °C was 31.2% of the original. The PU-RGO–SiR composition displayed a distinctively different mass-temperature profile. The mass loss curve became smooth than that for PU, and the residual mass of the composite after heating to 800 °C was substantially greater (51.4% of the original) compared with those of either pure PU or PU-RGO formulation. As a reference, the TGA trace of pure SiR is given in Fig. S7 (ESI†).
3.2 Electrical-conductivity analysis
The conductivity of the PU-RGO–SiR composite was 118 ± 5 S m−1 at room temperature. We performed several experiments with the aim to explore the conductivity stability of PU-RGO–SiR when subjected to mechanical and chemical stress. First, PU-RGO and PU-RGO–SiR were immersed in methylbenzene for 200 s, and the conductivity was monitored as a function of time (Fig. 4a). The conductivity of the PU-RGO–SiR formulation decreased to 86.6% of its initial value after immersion for 200 s. By contrast, the conductivity of the PU-RGO formulation decreased sharply within 15 s of immersion, and after 200 s the conductivity was only 0.3% of its initial value. Fig. 4b presents the conductivity histograms of the PU-RGO and PU-RGO–SiR samples soaked in hexane, phenixin, xylene and chloroform for 24 hours. The PU-RGO formulations displayed a marked decrease in the conductivity after treatment, falling to less than 0.3% for all the solvents tested. By contrast, the conductivity of the PU-RGO–SiR formulation was more than 75% after exposure to all the solvents tested. The strong degradation in the conductivity performance for the PU-RGO formulation is mainly ascribed to the following reasons. Firstly, these non-conductive solvents would easily fill the gaps between the RGO sheets, and this should be the dominant reason for the drastic decrease in conductivity. Furthermore, the filled non-conductive solvents would permeate into the PU skeleton and make the PU foam swell. The swollen foam thus made RGO's conductive network loose and not closed as before. Lastly, the interaction between the self-assembled RGO layer and the PU skeleton is van der Waals force, which is not strong enough to withstand the organic solvent, and the RGO layer would detach from the PU skeleton. Thus, under the combination of these effects, the conductivity of PU-RGO drastically decreased to less than 0.3% of the original. As to the PU-RGO–SiR, these non-conductive solvents could not permeate into the gap of the RGO layers under the protective effect of the SiR coating, due to its impermeable nature derived from the cross-linked, dense structure, which made the PU-RGO–SiR less prone to permeation and swelling. Under the protective effect of SiR coating, the conductivity of PU-RGO–SiR did not decrease substantially, and could still maintain more than 75% of conductivity even after soaking in organic solvents for 24 hours.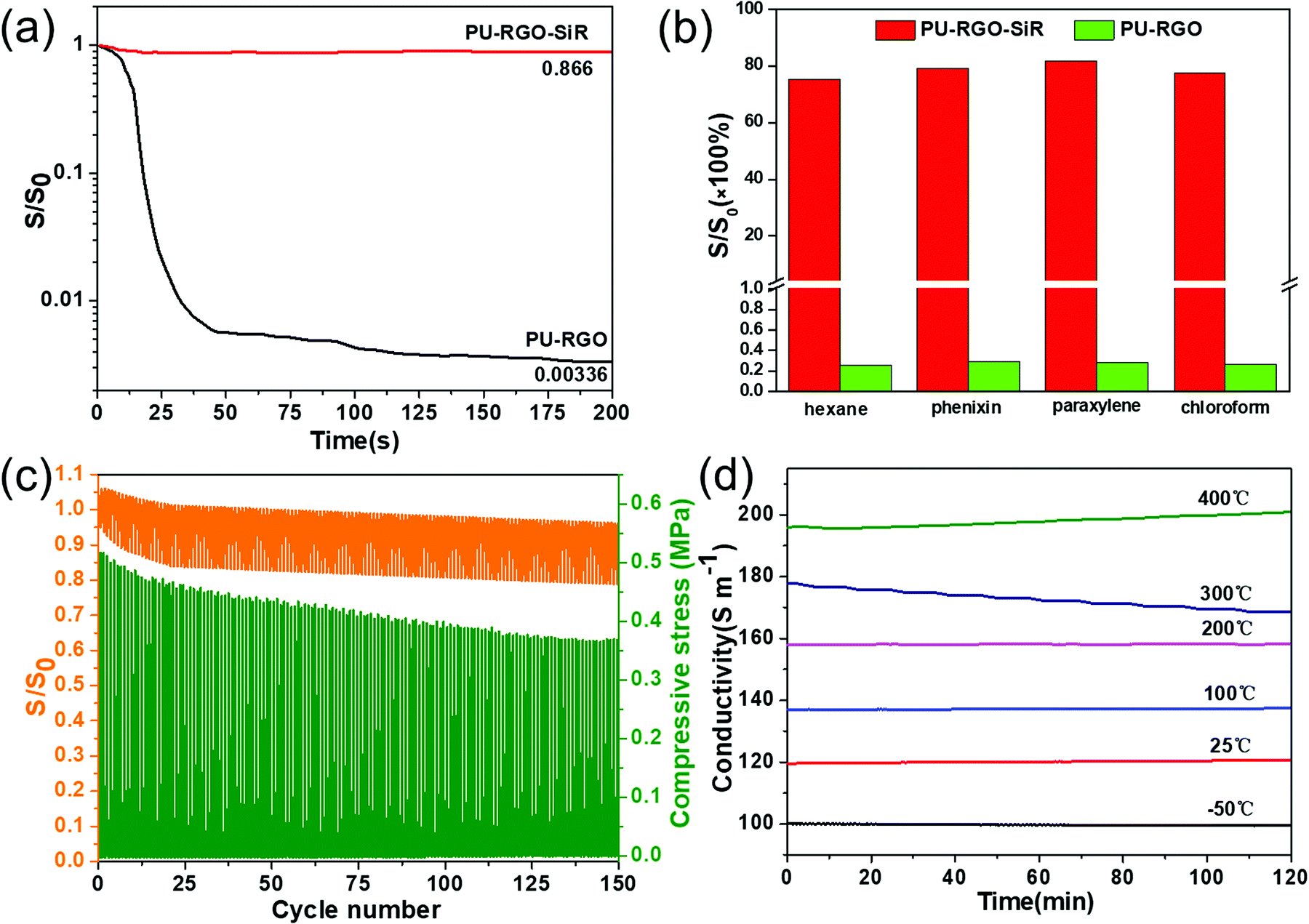 | ||
| Fig. 4 (a) Value of S/S0 for PU-RGO–SiR and PU-RGO immersed in methylbenzene as a function of time. (b) Values of S/S0 for PU-RGO and PU-RGO–SiR before and after soaking in the indicated liquids for 24 h. (c) Value of S/S0 for PU-RGO and PU-RGO–SiR subjected to repeated compression cycles. (d) Conductivity as a function of time for PU-RGO–SiR maintained at the indicated temperatures for 2 h. | ||
Furthermore, the SEM images in Fig. S8 (ESI†) further proved the protective effect of the SiR coating, where the RGO–SiR layer still adhered to the PU skeleton after immersion in hexane solvent for 24 h. Compared with the pristine PU-RGO–SiR (Fig. S8a, ESI†), the skeleton foam surface retained its intact structure (Fig. S8b, ESI†) without obvious detachment of the RGO–SiR layer. Moreover, in order to further verify the protective effect of SiR, a long-time immersion experiment of acidic and alkaline solution, as well as various organic solvents, was carried out. As shown in Fig. S9 (ESI†), severe phenomena of detachment were observed for the PU-RGO sample, while PU-RGO–SiR had no obvious shedding phenomena regardless of acidic or alkaline solution or organic solvents. This further verified that PU-RGO–SiR could remain intact under the protective effect of the RGO coating even after encountering harsh conditions. Hence, the stable behavior of PU-RGO–SiR was beneficial to retain the stable electrical conductivity, which was promising to find greater potential application.
As previously stated, the coated RGO–SiR layer was beneficial for PU-RGO–SiR to retain the structural integrity in the case of solvents. Meanwhile, it also contributed greatly to enhance the mechanical properties of PU-RGO–SiR. The compressive stress–strain curves of the PU-RGO–SiR composite and pure PU presented that the compressive stress of PU-RGO–SiR increased to 0.48 MPa at 60% strain (Fig. S10, ESI†), which was much higher than that of the pure PU foam (∼2.29 kPa at 60% strain). This can be ascribed to the robust structure of the RGO–SiR coating layer. Besides the inherent mechanical properties of the RGO nano-sheets that are helpful to enhance the compressive stress of PU-RGO–SiR, the π–π interaction between SiR and RGO is another key factor. The phenyl groups in SiR are in good affinity with the graphene sheet via π–π interaction. When PU-RGO was immersed into the SiR solution, the SiR chains would intercalate into the layer spacing of the RGO sheets. The formation of π–π stacking could thus significantly enhance the mechanical properties.
Additionally, in order to investigate the effect of compression on the conductivity of PU-RGO–SiR, the real-time electrical-conductivity (S) was monitored and recorded during the 150 cyclic compression tests. The S/S0 could be reckoned as the index reflecting the stability of electrical conductivity, and the resultant S/S0 changes and compressive stress are plotted in Fig. 4c. During the 150 cyclic compression tests, the compressive stress gradually decreased from 0.48 Mpa to 0.38 Mpa with about 20.8% loss, while the conductivity change (S/S0) remained comparatively stable, and no evident decrease but with very small loss was observed. When the pressure was completely released, the PU-RGO–SiR conductivity could be recovered to 90.5% of the initial value, indicating stable electrical conductivity under compression. In addition, SEM analysis of the PU-RGO–SiR sample after the compression cycle was performed, and compared with the pristine PU-RGO–SiR (Fig. S8a, ESI†), the skeleton foam surface remained intact without evident detachment of the RGO–SiR layer (Fig. S8c, ESI†). It should be noted that the skeleton surface was comparatively smooth than that before the compression test; this might be caused by strong mechanical stress causing some particles to fall off. However, the RGO–SiR coating remains intact without cracks being observed, indicating that the PU-RGO–SiR still retains good mechanical cycle stability and stable electrical conductivity under hundreds of compression tests, which could be an outstanding candidate facing the harsh practical conditions.
As shown in Fig. 4d, the conductivity of the PU-RGO–SiR composites remained constant at −50 °C, 25 °C, 100 °C, and 200 °C for 2 h. This indicated that the PU-RGO–SiR composites exhibited quite stable electrical conductivity at temperatures below 200 °C. At 300 °C, the conductivity of PU-RGO–SiR decreased from 177.7 S m−1 to 167.69 S m−1, approximately about 5.6% loss. At 400 °C, the conductivity of PU-RGO–SiR gradually increased from 195.8 S m−1 to 200.9 S m−1, about 2.6% increase. To summarize, compared with the constant conductivity at −50 °C, 25 °C, 100 °C, and 200 °C, even the electrical conductivity of PU-RGO–SiR vibrated with time elapsing at 300 °C and 400 °C, but their vibration range was not beyond 5.6%. These results implied that the PU-RGO–SiR composites exhibited stable electrical conductivity in a temperature range from −50 °C to 400 °C, and were efficient as electric materials at different temperatures.
A particular apparatus was designed to test the electrical conductivity in the combustion process, and is shown in Fig. 5a (a schematic diagram of the apparatus used to measure the sample conductivity changes during exposure to flame is shown in Fig. S11, ESI†). The sample connected with the electrical source, the resistance tester or the LED by a wire, and an alcohol blast burner was used to generate the flame. The brightness of the LED indicated the changes in the electrical current, which reflected the changes in the electrical conductivity indirectly.
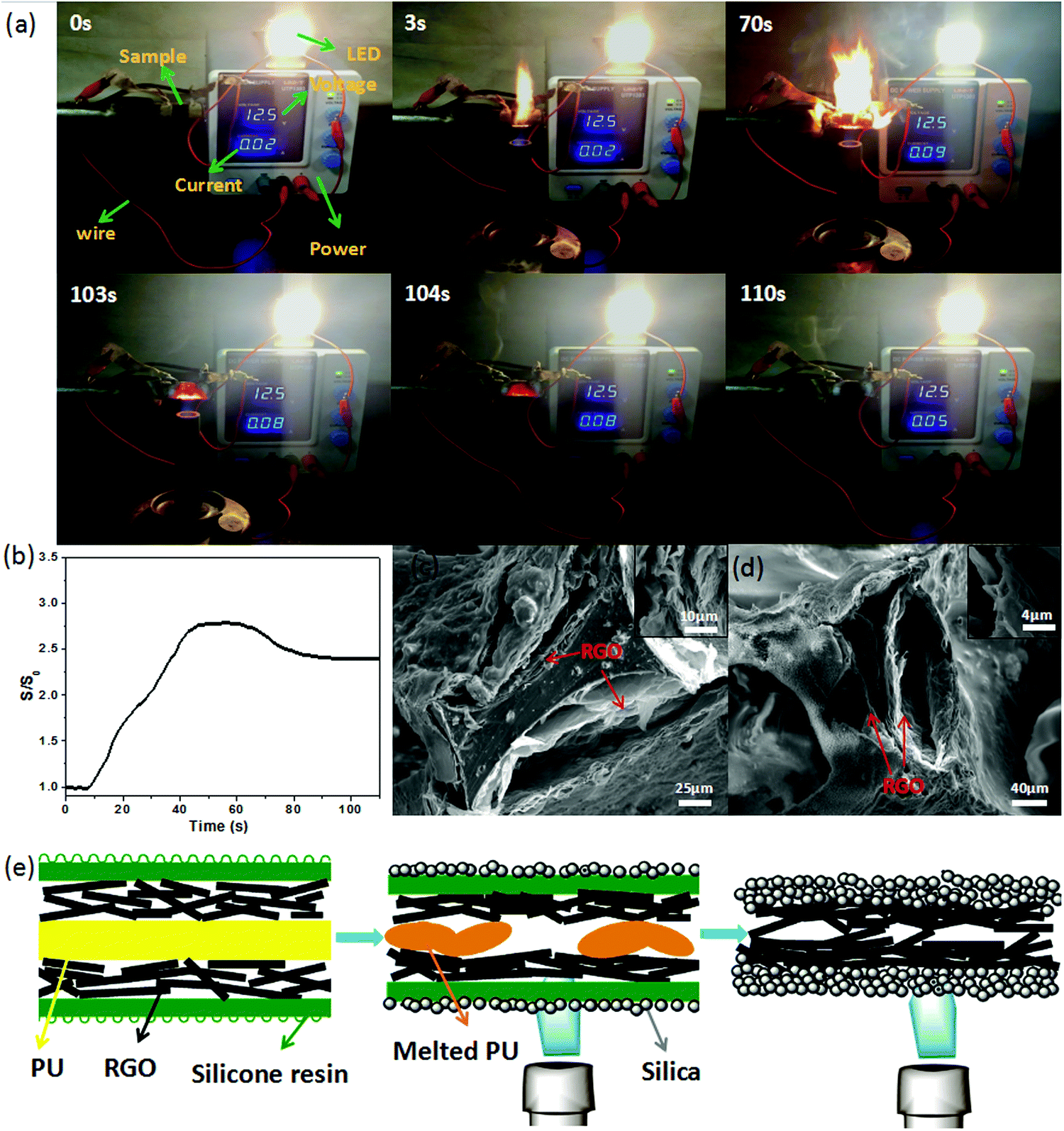 | ||
| Fig. 5 (a) Photographs of PU-RGO–SiR exposed to the direct flame source for 110 s; the digital display screen presents the current changes of the burning process. (b) S/S0 change of PU-RGO–SiR during combustion. Typical SEM images of (c) the RGO layer and SiR coating of PU-RGO–SiR and (d) the RGO layer and the silica crust of PU-RGO–SiR after the flame-retardant test. (e) Mechanism for the conductivity increase during combustion. | ||
Before the PU-RGO–SiR sample was exposed to the flame, the current was 0.02 A. Once the sample was treated with flame, the LED grew brighter and the electrical current reached the maximum 0.09 A at 70 s. After we removed the flame, the current gradually decreased from 0.08 A to 0.05 A (Video S1, ESI†). Fig. 5b shows the S/S0 change of PU-RGO–SiR monitored during the combustion process. The PU-RGO–SiR conductivity started to increase in the initial ∼50 s, then remained stable from 50 s to 75 s, and finally decreased from 75 s to 103 s. At 104 s, the conductivity stopped decreasing once the flame was removed. After completely cooled at 110 s, the conductivity was still higher than the original. The increasing conductivity rate after burning may be ascribed to the particular structures. A SEM image of the initial PU-RGO–SiR shown in Fig. 5c revealed that the foam skeleton was covered with loosely stacked RGO lamellas. After directly exposing the PU-RGO–SiR to the flame for a long time, the structure of the PU foam was destroyed and the contracted RGO was stacked closely due to the high temperature, as shown in Fig. 5d and Fig. S12 (ESI†). These dense RGO layers were beneficial to reduce the electrical resistance. Schematic diagrams of the micro-structural changes of PU-RGO–SiR in the combustion process are shown in Fig. 5e. More SEM images of PU-RGO–SiR after complete combustion are shown in Fig. S13 (ESI†); the silica crust was generated during the burning process, which could protect the RGO layer. In addition, after complete combustion, the residue could retain the integrity of the structure and exhibited good mechanical properties as shown in Fig. S14 (ESI†), indicating that PU-RGO–SiR could act as electrically conductive materials even after complete combustion. The stable conductivity of PU-RGO–SiR under harsh conditions enables potential applications in a wider range.
3.3 Flame retardancy analysis
While detecting the PU-RGO–SiR conductivity during combustion, we found that PU-RGO–SiR exhibited flame retardancy, which should be ascribed to the incorporated RGO and SiR layers. Previous work had reported that both RGO and SiR could enhance the flame retardancy.66 Here, to evaluate the RGO and SiR effects on improving the flame retardancy, we compared the PU, PU-RGO, PU-SiR and PU-RGO–SiR flame retardancy through a vertical burning test. As shown in Fig. 6a, the pure PU foam was directly exposed to the fire source for only 1 s and then removed from the fire immediately (Video S2, ESI†); the flame rapidly spread throughout the full sample with molten dripping and a lot of smoke, and the pure PU foam combusted completely after 8 s. The PU-RGO vertical test is shown in Fig. 6b; when the sample was directly exposed to the fire source for 4 s (Video S3, ESI†), flames spread fast throughout the full sample but without molten dripping, it could retain the original shape even after complete combustion, which should be attributed to the covered RGO. Compared with 8 s burning time of the pure PU foam, RGO could effectively improve the flame retardancy of the PU foam. Fig. 6c shows the burning process of PU-SiR, where the specimen was directly exposed to the fire source for 10 s and then removed (Video S4, ESI†). It was ignited quickly with rapid flame propagation, while melt-dripping with fire could be observed and the burning time prolonged to 52 s. The flame retardancy of PU-SiR was improved due to the formation of a porous and dense nano-silica layer, which could prevent heat transfer and protect the PU foam.55 PU-RGO–SiR caught fire for 10 s after ignition (Video S5, ESI†), as shown in Fig. 6d; PU-RGO–SiR self-extinguished after the flame source was removed at 13 s, and a silica layer was generated on the sample surface. The PU-RGO–SiR exhibited enhanced flame retardancy compared to both PU-RGO and PU-SiR, and this might be due to the synergistic effect of the silica layer (produced by the SiR coating) and the RGO layer on improving the flame retardancy.55 | ||
| Fig. 6 Images of (a) PU, (b) PU-RGO, (c) PU-SiR and (d) PU-RGO–SiR during vertical flame tests. | ||
Cone calorimetry tests were carried out to investigate how RGO and SiR could increase the flame retardancy of the PU foam through a series of parameters such as time to ignition (ti), heat release rate (HRR), peak heat release rate (pHRR), time to pHRR, total heat release (THR), mass loss rate (MLR) and total smoke release (TSR). Cone calorimetry testing results are shown in Fig. 7. Fig. 7a shows the HRR curves of the pure PU foam and its composites. The pure PU foam burnt very fast and exhibited a sharp HRR peak where the peak HRR was 208 kW m−2. Compared with pure PU, the PU-RGO also exhibited a similar curve of HRR, but the peak width increased and the pHRR decreased. Similarly, the peak width in the HRR curves of PU-SiR and PU-RGO–SiR became bigger and pHRR decreased further, and the time to pHRR was also much longer than that of the PU foam. This indicated that the flame burning rate of the PU foam was reduced by coating RGO or SiR individually or together. The pHRR values of the PU-RGO, PU-SiR and PU-RGO–SiR composites decreased from 208 kW m−2 for the pure PU foam to 130, 101 and 73 kW m−2 (Table 1), respectively. Compared with the pure PU foam, the HRR of PU-RGO, PU-SiR and PU-RGO–SiR was greatly decreased by 37.5%, 51.4% and 65.0%, respectively.
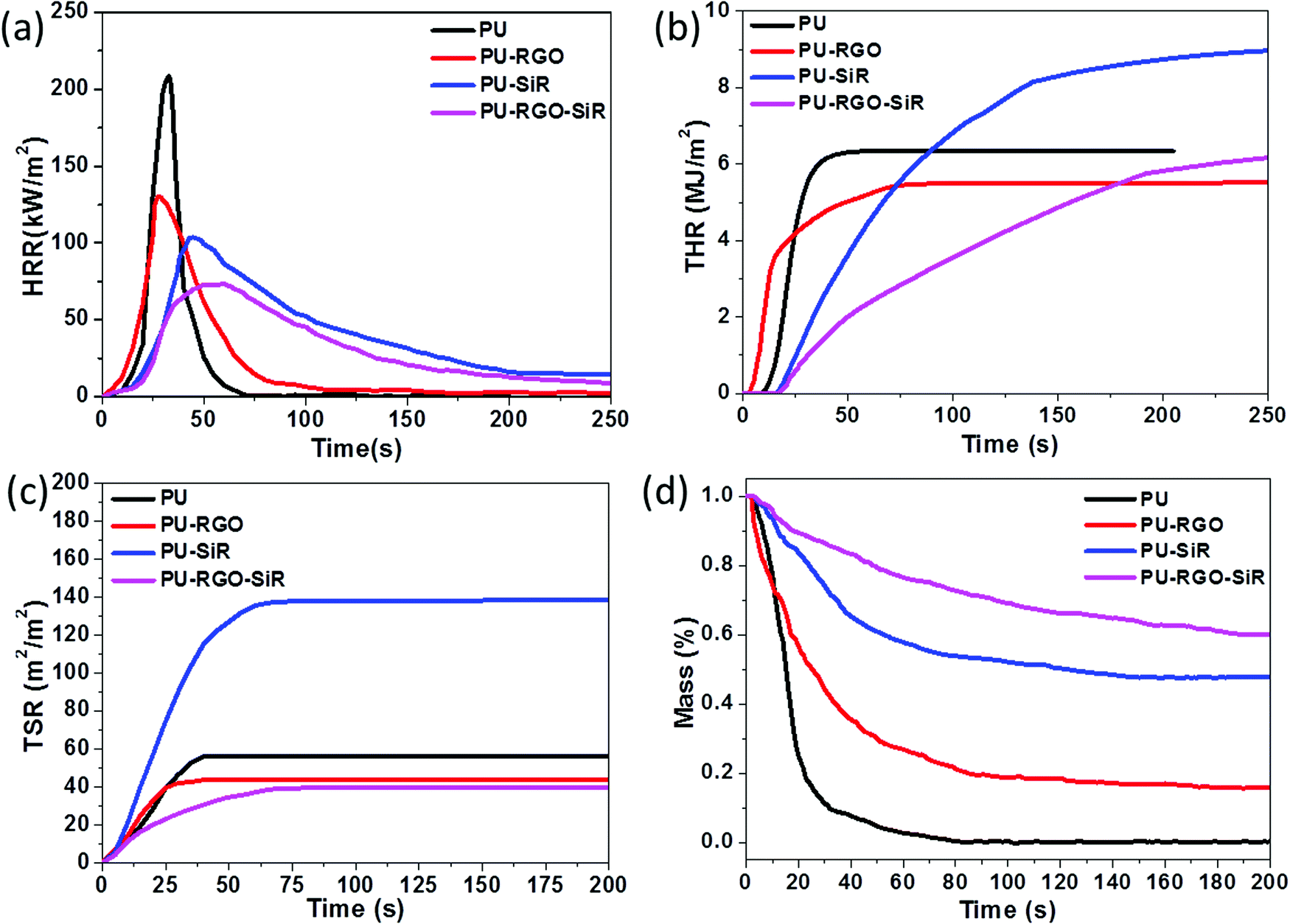 | ||
| Fig. 7 (a) HRR, (b) THR, (c) TSR and (d) MLR curves of the PU foam and composites. | ||
| Sample | pHRR (kW m−2) | Time to pHRR (s) | Char (%) | TSR (m2 m−2) | LOI (%) |
|---|---|---|---|---|---|
| PU | 208 | 33 | 0 | 56.5 | 14.7 ± 0.3 |
| PU-RGO | 130 | 28 | 16 | 43.7 | 19.3 ± 0.2 |
| PU-SiR | 101 | 55 | 48 | 138.4 | 26.7 ± 0.3 |
| PU-RGO–SiR | 73 | 66 | 60 | 39.4 | 31.5 ± 0.3 |
Additionally, the THR curves of the PU foam and the foam composite are shown in Fig. 7b. It only took 25 s for the THR curve of PU to reach a plateau due to its easy flammability. For PU-RGO, about 75 s was required to reach a plateau where the combustion was completed. It should be noted that the time to ignition represented the starting point for the sample to combust and release heat, and the ignition time of PU-RGO was shortened compared to that of PU. It seemed that the incorporation of RGO facilitated the foam combustion. But after careful analysis, it could be found that the ending ignition time of PU-RGO was prolonged compared to that of pure PU. Moreover, the THR of PU-RGO was decreased by about 20% compared to that of PU. The prolonged combustion time and decreased THR implied that the incorporation of RGO had enhanced the flame retardancy. These phenomena could be explained as follows. Firstly, as we known the RGO exhibited superior thermal conductivity;67 once the PU-RGO was ignited, the heat would transfer faster and make the sample combust earlier, and thus the starting ignition time was shortened compared to that of PU. Furthermore, the presence of RGO coating would facilitate and even accelerate the char formation during combustion, and thus the heat and smoke release were suppressed during combustion. The prolonged ending ignition time also indicated that the combustion rate was suppressed due to the barrier effect of RGO. This was consistent with the reported work in which the RGO layer played a great role in facilitating the char layer formation.68 As to PU-SiR and PU-RGO–SiR, at least 250 s were required to complete the combustion. It should be noted that the THR of PU-SiR was higher than that of pure PU. Because there were many combustible groups like methyl, phenyl in SiR, it was reasonable that the PU-SiR exhibited a higher THR than PU during the cone calorimetry test, which was consistent with the reported work.55 A further comparison was made between PU-SiR and PU-RGO–SiR; the THR of PU-RGO–SiR was decreased compared to that of PU-SiR, and this also further verified that the incorporation of RGO enhanced the flame retardancy.
PU foams usually burnt very fast and produced toxic smoke and gases, which were the major reasons for fire-related deaths.69 The TSR curves (shown in Fig. 7c) of PU and its composites revealed smoke issues. Compared with the pure PU foam, after coating SiR, the TSR of PU-SiR drastically increased from 56.5 m2 m−2 to 138.4 m2 m−2, and this should be ascribed to the flammability of SiR. Once SiR was ignited, the smoke and heat would be released during the combustion process, and a similar phenomenon had been reported previously.55 Considering that the weight of the coated SiR was 3 times of the PU weight, it was reasonable that the TSR curve of PU-SiR exhibited a much higher behavior than that of PU due to the inherent flammability of SiR. But as to the sample of PU-RGO, it presented a lower TSR curve than those of pure PU and PU-SiR, indicating that the toxic smoke of PU-RGO decreased during the combustion process. This should be ascribed to the barrier effect of GO, which showed highly unusual permeation properties, and provided a so-called “tortuous path” effect70,71 that could significantly alter the diffusion path of the pyrolysis products. Therefore, the resulting RGO nano-sheets could provide high-quality barriers and block all gases and liquids.72 Furthermore, RGO could absorb polycyclic aromatic hydrocarbon (PAH) species. Serving as a template of micro-char, these active PAH species would propagate on graphene and they are eventually converted to carbon on graphene.73 Under the combination of these two actions, the production of toxic smoke decreased in the presence of the RGO layer.
The barrier effect of RGO further verified that the TSR curve of PU-RGO–SiR was lower than that of PU-SiR, indicating that the incorporation of RGO coating could effectively suppress the smoke production and release. Under the barrier effect of RGO, the TSR of PU-RGO–SiR is reduced by 30.2% compared with that of the pristine PU foam. In addition, the MLR curves shown in Fig. 7d demonstrated that the foam composites had better fire resistance properties than the pure PU foam after coating RGO and SiR individually or together. The pure PU foam almost had no residue after the combustion test, while for the samples of PU-RGO, PU-SiR and PU-RGO–SiR, their residue increased from 0 to 16%, 48% and 60%, respectively. Besides the HRR, THR TSR and MLR, the LOI of the foam composite materials greatly increased compared to that of the pure PU foam from 14.7% to PU-RGO 19.3%, PU-SiR 26.7% and PU-RGO–SiR 31.5%, respectively. The characteristic parameters reflecting the flame retardancy are summarized in Table 1. It could be concluded that after coating RGO–SiR, the LOI of PU-RGO–SiR greatly increased from 14.7% to 31.5%, and the PU-RGO–SiR composites exhibited a great reduction of 65% in pHRR and a reduction of 30.2% of TSR, while the char residue drastically increased to 60%. The testing curves of HRR, THR, TSR, and MLR and the time to pHRR showed that the foam had better flame retardancy through coating RGO–SiR than the individual RGO or SiR, indicating that RGO and SiR had a synergistic effect on improving the flame retardancy.
Based on the positive effect of the RGO–SiR layer on enhancing the flame retardancy of the PU-RGO–SiR composites, we compared the flame retardancy of RGO–SiR coating in this work with the reported literature in recent years, and the results are summarized in Table S1 (ESI†). It could be found that PU-RGO–SiR exhibited the biggest pHRR reduction and char increment; the flame retardancy of the RGO–SiR coating was superior to most reports in recent years, and this should be contributed to the synergistic effect of RGO and SiR. On the other hand, it could not be ignored that the involvement of repeatedly dip-coating the RGO–SiR layer greatly increased the PU foam weight, and even under the negative effect of the high content of RGO and SiR, the PU-RGO–SiR composites were good candidates as flame-retardant materials.
Fig. 8 presents the residual chars of the pure PU foam and PU foam composites after the flame test. No residue was left for the pure PU foam after combustion, while the foam composites showed different features of the residue. PU-RGO showed a black/white residue, PU-SiR cracked into grey multi-pieces, and PU-RGO–SiR exhibited a grey/black intact residue. These results proved that the RGO–SiR coated foam had better flame retardancy than RGO and SiR individually.
 | ||
| Fig. 8 Photographs of the residual chars of the PU foam and composites after cone calorimetry tests from (a) pure PU, (b) PU-RGO, (c) PU-SiR and (d) PU-RGO–SiR. | ||
In order to investigate the synergistic effect of RGO and SiR to clarify the possible flame-retardant mechanism, micro-morphology analysis and element content analysis of different samples before and after combustion were performed. The residue was carefully characterized by FTIR and SEM in order to discover the possible mechanisms of PU-RGO–SiR, and the PU-RGO–SiR image after combustion (shown in the inset of Fig. 9a) revealed that a gray crust layer was generated and FTIR spectral analysis was conducted as shown by the orange curve in Fig. 9a. Compared with underneath matter (green curve), the peak at ∼1000 cm−1 was assigned to the Si–O–Si bond stretching vibration, and the peak at ∼760 cm−1 was assigned to the Si–C bond stretching vibration, C–H bond absorption peak at ∼2900 cm−1 and Si–CH3 symmetric deformation vibration or rocking vibration absorption peaks at ∼1260 cm−1 and ∼700 cm−1 were not found in the orange curve. That indicated that the white crust was mainly composed of silicon dioxide and a small amount of silicon carbide without any organic matter. The SEM images of the crust layer (shown in Fig. 9e–h) represented that nanometer porous particle structures were produced on the foam skeleton due to the thermal degradation of SiR coating after combustion. The SEM images of the silica protective layer underneath (Fig. 9b–d) confirmed that the inner foam retained the initial structure due to the protection of the crust.
 | ||
| Fig. 9 (a) FT-IR spectra of the PU-RGO–SiR foam after extinguishing the silica crust (orange curve) and the bulk (green curve) underneath, and the inset shows the photograph of the PU-RGO–SiR foam cross-section after extinguishing. (b–d) SEM images of the PU-RGO–SiR foam of the silica crust underneath. (e–h) SEM images of the silica crust. | ||
The SEM-EDX spectra of the white crust and the inner matter are shown in Fig. S15 (ESI†) and the detailed element contents are listed in Table 2. The increased Si and decreased C elements in the silica layer implied that SiR was decomposed into a compact silica crust during combustion. The compact silica layer could provide a barrier effect to inhibit the transfer of heat and oxygen from the outside into the inside, as well as to suppress the combustible gases from the inside to the outside to avoid further combustion, and thus can prevent the foam from combustion and preserve the foam structure integrity.
| Sample element | PU-RGO–SiR | Silica layer | Silica layer underneath | |||
|---|---|---|---|---|---|---|
| wt% | atom% | wt% | atom% | wt% | atom% | |
| C | 54.17 | 66.36 | 3.29 | 6.31 | 38.04 | 50.99 |
| O | 24.32 | 22.37 | 54.01 | 61.18 | 31.17 | 31.37 |
| Si | 21.51 | 11.27 | 42.70 | 32.51 | 30.78 | 17.64 |
As stated before, the as-prepared PU-RGO–SiR presented stable electrical conductivity under harsh conditions like solvent, compression and even flame attack. Besides these harsh conditions, the foam composites were also used under specifically wet conditions like water vapor or droplets, and thus the surface properties should be taken into account to evaluate the stability of electrical conductivity for foam composites. Here, the surface behavior of the as-prepared PU-RGO–SiR was characterized by measuring the static water contact angle (WCA) with a DSA30 contact angle analyzer using a 3 μL water droplet. As shown in Fig. 10a, the PU-RGO–SiR foam displayed a WCA of ∼156 ± 3°, indicating that the foam surface exhibited a superhydrophobic behavior. Fig. 10b shows the standing status of the water droplet on the foam surface, where the water droplets could remain standing for 5 minutes and did not permeate into the foam. The superhydrophobic behavior of the PU-RGO–SiR surface should be ascribed to the rough structure as shown in Fig. 10c, where was mainly constructed by the dual-sized hydrophobic silica nano-particles in the SiR coating. Thereby, the PU-RGO–SiR surface with a special superhydrophobic behavior could protect the water droplets from permeation, which was beneficial for PU-RGO–SiR to retain the stable electrical conductivity under harsh conditions.
 | ||
| Fig. 10 (a) Water contact angle testing image of PU-RGO–SiR with a water contact angle of about 156°, indicating that the surface is superhydrophobic. (b) Standing status of water droplets on PU-RGO–SiR and (c) the SEM image of the PU-RGO–SiR surface from which the textured structure is clearly observed. | ||
4. Conclusions
Stable electrically conductive PU-RGO–SiR was prepared using a dip-coating method, which exhibited excellent flame retardancy, good mechanical performance and superhydrophobicity. The PU-RGO–SiR exhibited stable electrical conductivity under a compression test, upon exposure to flame or solvents, and even in a wider temperature range from −50 °C to 400 °C. The LOI greatly increased from 14.7% for the pure foam to 31.5% for PU-RGO–SiR. The cone calorimetry test for PU-RGO–SiR exhibited a great reduction of 65% in pHRR and a reduction of 30% of TSR. The testing results of HRR, THR, TSR, and MLR and the time to pHRR showed that the foam had better flame retardancy through coating RGO–SiR than RGO or SiR alone, indicating that RGO and SiR had a synergistic effect on improving the flame retardancy. The SEM images of the chars after extinguishing revealed that the SiR was transformed into a silica layer due to the inorganic barrier effect, preserving the RGO layer and the foam shape. Thereby, this polymer foam composite would find application in various areas such as electronics due to its stable electrical conductivity and high flame retardancy, even in a variety of harsh conditions like high temperature, flame, organic solvents and external compression.Conflicts of interest
We declare no conflicts of interest.Acknowledgements
This work was supported by the National Key Research and Development Program (2017YFB0307700), the Department of Scientific and Technology of Zhejiang Province (LGG19E030007 and LGG18E030007), and the Project for the Innovation of High Level Returned Overseas Scholars (or team) in Hangzhou. We also acknowledge the support from the Collaborative Innovation Center of Zhejiang Province for the Manufacture of Fluorine and Silicone Fine Chemicals and Materials (FSi2018B004, FSi2018A028, and FSi2019A012).References
- H. Shi, D. Shi, L. Yin, Z. Yang, S. Luan, J. Gao, J. Zha, J. Yin and R. K. Li, Nanoscale, 2014, 6, 13748–13753 RSC.
- H. B. Zhang, Q. Yan, W. G. Zheng, Z. He and Z. Z. Yu, ACS Appl. Mater. Interfaces, 2011, 3, 918–924 CrossRef CAS.
- R. M. Hodlur and M. K. Rabinal, Compos. Sci. Technol., 2014, 90, 160–165 CrossRef CAS.
- N. V. Gama, B. Soares, C. S. R. Freire, S. Rui, C. P. Neto, A. Barros-Timmons and A. Ferreira, Mater. Des., 2015, 76, 77–85 CrossRef CAS.
- N. Nazeran and J. Moghaddas, J. Non-Cryst. Solids, 2017, 461, 1–11 CrossRef CAS.
- N. Shahabudin, R. Yahya and S. N. Gan, Polymers, 2016, 8, 125 CrossRef.
- Q. Wu, L. X. Gong, Y. Li, C. F. Cao, L. C. Tang, L. Wu, L. Zhao, G. D. Zhang, S. N. Li and J. Gao, ACS Nano, 2017, 12, 416–424 CrossRef.
- T. K. Gupta, B. P. Singh, S. Teotia, V. Katyal, S. R. Dhakate and R. B. Mathur, J. Polym. Res., 2013, 20, 1–7 CAS.
- K. Y. Park, S. E. Lee, C. G. Kim and J. H. Han, Compos. Sci. Technol., 2006, 66, 576–584 CrossRef CAS.
- F. Qiang, L. L. Hu, L. X. Gong, L. Zhao, S. N. Li and L. C. Tang, Chem. Eng. J., 2018, 334, 2154–2166 CrossRef CAS.
- Q. Fang, Y. Shen and B. Chen, Chem. Eng. J., 2015, 264, 753–771 CrossRef CAS.
- W. T. He, P. A. Song, B. Yu, Z. P. Fang and H. Wang, Prog. Mater. Sci., 2020, 114, 100687 CrossRef CAS.
- K. Y. Guo, Q. Wu, M. Mao, H. Chen, G. D. Zhang, L. Zhao, J. F. Gao, P. G. Song and L. C. Tang, Composites, Part B, 2020, 193, 108017 CrossRef CAS.
- H. T. Yang, B. Yu, P. G. Song, C. Maluk and H. Wang, Composites, Part B, 2019, 176, 107185 CrossRef CAS.
- G. B. Huang, S. Q. Huo, X. D. Xu, W. Chen, Y. X. Jin, R. R. Li, P. A. Song and H. Wang, Composites, Part B, 2019, 177, 107377 CrossRef CAS.
- B. Lin, A. C. Y. Yuen, A. Li, Y. Zhang, T. B. Y. Chen, B. Yu, E. W. M. Lee, S. H. Peng, W. Yang, H. D. Lu, Q. N. Chan, G. H. Yeoh and C. H. Wang, J. Hazard. Mater., 2020, 381, 120952 CrossRef CAS.
- Y. X. Jin, G. B. Huang, D. M. Han, P. G. Song, W. Y. Tang, J. S. Bao, R. R. Li and Y. L. Liu, Composites, Part A, 2016, 86, 9–18 CrossRef CAS.
- Y. Shi, C. Liu, Z. Duan, B. Yu, M. Liu and P. Song, Chem. Eng. J., 2020, 399, 125829 CrossRef CAS.
- P. Bertrand, A. Jonas, A. Laschewsky and R. Legras, Macromol. Rapid Commun., 2015, 21, 319–348 CrossRef.
- Q. Q. Fan, J. Z. Ma, Q. N. Xu, W. An and R. J. Qiu, Prog. Org. Coat., 2018, 125, 215–221 CrossRef CAS.
- H. Vahabi, M. R. Saeb, K. Formela and J. M. L. Cuesta, Prog. Org. Coat., 2018, 119, 8–14 CrossRef CAS.
- P. Haifeng, W. Wei, P. Ying, S. Lei, H. Yuan and L. Kim Meow, ACS Appl. Mater. Interfaces, 2015, 7, 101–111 CrossRef.
- X. Ji, Y. Xu, W. Zhang, L. Cui and J. Liu, Composites, Part A, 2016, 87, 29–45 CrossRef CAS.
- M. J. Nine, M. A. Cole, D. N. H. Tran and D. Losic, J. Mater. Chem. A, 2015, 3, 12580–12602 RSC.
- L. Li, X. Zhao, C. Ma, X. Chen, S. Li and C. Jiao, J. Therm. Anal. Calorim., 2016, 126, 1821–1830 CrossRef.
- Y. Pan, H. Pan, B. Yuan, N. Hong, J. Zhan, B. Wang, L. Song and Y. Hu, Mater. Chem. Phys., 2015, 163, 107–115 CrossRef CAS.
- H. Pan, S. Qi, Z. Zhang, B. Yu and Y. Lu, J. Mater. Sci., 2018, 53, 9340–9349 CrossRef CAS.
- M. Haile, S. Fomete, I. D. Lopez and J. C. Grunlan, J. Mater. Sci., 2016, 51, 375–381 CrossRef CAS.
- A. A. Cain, M. G. B. Plummer, S. E. Murray, L. Bolling, O. Regev and J. C. Grunlan, J. Mater. Chem. A, 2014, 2, 17609–17617 RSC.
- H. B. Chen, P. Shen, M. J. Chen, H. B. Zhao and D. A. Schiraldi, ACS Appl. Mater. Interfaces, 2016, 8, 32557–32564 CrossRef CAS.
- K. M. Holder, M. E. Huff, M. N. Cosio and J. C. Grunlan, J. Mater. Sci., 2015, 50, 2451–2458 CrossRef CAS.
- A. Lorenzetti, S. Besco, D. Hrelja, M. Roso, E. Gallo, B. Schartel and M. Modesti, Polym. Degrad. Stab., 2013, 98, 2366–2374 CrossRef CAS.
- C. Liu, Y. Fang, X. Miao, Y. Pei, Y. Yan, W. Xiao and L. Wu, Sep. Purif. Technol., 2019, 229, 115801 CrossRef CAS.
- G. Laufer, C. Kirkland, A. A. Cain and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2012, 4, 1643–1649 CrossRef CAS.
- L. Li, B. Li, L. Wu, X. Zhao and J. Zhang, Chem. Commun., 2014, 50, 7831–7833 RSC.
- D. Patra, P. Vangal, A. A. Cain, C. Cho, O. Regev and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2014, 6, 16903–16908 CrossRef CAS.
- X. Zhang, Q. Shen, X. Zhang, H. Pan and Y. Lu, J. Mater. Sci., 2016, 51, 10361–10374 CrossRef CAS.
- J. N. Gavgani, H. Adelnia and M. M. Gudarzi, J. Mater. Sci., 2014, 49, 243–254 CrossRef CAS.
- S. Stankovich, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342–3347 CrossRef CAS.
- S. H. Liao, P. L. Liu, M. C. Hsiao, C. C. Teng, C. A. Wang, M. D. Ger and C. L. Chiang, Ind. Eng. Chem. Res., 2012, 51, 4573–4581 CrossRef CAS.
- G. Huang, S. Chen, S. Tang and J. Gao, Mater. Chem. Phys., 2012, 135, 938–947 CrossRef CAS.
- K. Y. Li, C. F. Kuan, H. C. Kuan, C. H. Chen, M. Y. Shen, J. M. Yang and C. L. Chiang, Mater. Chem. Phys., 2014, 146, 354–362 CrossRef CAS.
- X. Wang, W. Xing, X. Feng, B. Yu, L. Song and Y. Hu, Polym. Chem., 2014, 5, 1145–1154 RSC.
- C. Hu, J. Xue, L. Dong, Y. Jiang, X. Wang, L. Qu and L. Dai, ACS Nano, 2016, 10, 1325 CrossRef CAS.
- Y. Ji, G. Chen and T. Xing, Appl. Surf. Sci., 2019, 474, 203–210 CrossRef CAS.
- Y. Ji, Y. Li, G. Chen and T. Xing, Mater. Des., 2017, 133, 528–535 CrossRef CAS.
- H. Pan, B. Yu, W. Wang, Y. Pan, L. Song and Y. Hu, RSC Adv., 2016, 6, 114304–114312 RSC.
- Y. Shi, C. Liu, L. Fu, F. Yang, Y. Lv and B. Yu, Composites, Part B, 2019, 179, 107541 CrossRef CAS.
- W. Yang, Y. Xia, X. Liu, J. Yang and Y. Liu, Polym. Compos., 2018, 39, 3841–3848 CrossRef CAS.
- X. Qiu, C. K. Kundu, Z. Li, X. Li and Z. Zhang, J. Mater. Sci., 2019, 54, 13848–13862 CrossRef CAS.
- X. Chen, J. Zhuo, W. Song, C. Jiao, Y. Qian and S. Li, Polym. Adv. Technol., 2015, 25, 1530–1537 CrossRef.
- X. Chen, M. Li, J. Zhuo, C. Ma and C. Jiao, J. Therm. Anal. Calorim., 2016, 123, 439–448 CAS.
- S. Hamdani, C. Longuet, D. Perrin, J. M. Lopez-Cuesta and F. Ganachaud, Polym. Degrad. Stab., 2009, 94, 465–495 CrossRef CAS.
- P. Jia, H. Liu, Q. Liu and X. Cai, Polym. Degrad. Stab., 2016, 134, 144–150 CrossRef CAS.
- Q. Wu, Q. Zhang, L. Zhao, S. N. Li, L. B. Wu, J. X. Jiang and L. C. Tang, J. Hazard. Mater., 2017, 336, 222–231 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- V. M. Gun'Ko, M. S. Vedamuthu, G. L. Henderson and J. P. Blitz, J. Colloid Interface Sci., 2000, 228, 157–170 CrossRef.
- L. Liu, Z. Xiong, D. Hu, G. Wu and P. Chen, Chem. Commun., 2013, 49, 7890–7892 RSC.
- L. Yan, M. Lin, C. Zeng, Z. Chen, S. Zhang, X. Zhao, A. Wu, Y. Wang, L. Dai and J. Qu, J. Mater. Chem., 2012, 22, 8367–8371 RSC.
- X. Zhang, D. Liu, Y. Ma, J. Nie and G. Sui, Appl. Surf. Sci., 2017, 422, 116–124 CrossRef CAS.
- I. K. Moon, J. Lee, R. S. Ruoff and H. Lee, Nat. Commun., 2010, 1, 1–6 Search PubMed.
- S. Some, Y. Xu, Y. Kim, Y. Yoon, H. Qin, A. Kulkarni, T. Kim and H. Lee, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- Y. Liu, J. Ma, T. Wu, X. Wang, G. Huang, Y. Liu, H. Qiu, Y. Li, W. Wang and J. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 10018–10026 CAS.
- C. Liu, T. Chen, C. H. Yuan, C. F. Song, Y. Chang, G. R. Chen, Y. T. Xu and L. Z. Dai, J. Mater. Chem. A, 2016, 4, 3462–3470 CAS.
- J. Hu and F. Zhang, J. Therm. Anal. Calorim., 2014, 118, 1561–1568 CrossRef CAS.
- W. Bernd, K. Andra, S. A. German, C. Federico, C. Giovanni, A. Markus and B. Lennart, Nat. Nanotechnol., 2015, 10, 277–283 CrossRef.
- H. Kim, D. W. Kim, V. Vasagar, H. Ha, S. Nazarenko and C. J. Ellison, Adv. Funct. Mater., 2018, 28, 1803172 CrossRef.
- M. Blais and K. Carpenter, Fire Technol., 2015, 51, 3–18 CrossRef.
- R. R. Nair, H. A. Wu, P. N. Jayaram, I. V. Grigorieva and A. K. Geim, Science, 2012, 335, 442–444 CrossRef CAS.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS.
- Y. Su, V. G. Kravets, S. L. Wong, J. Waters, A. K. Geim and R. R. Nair, Nat. Commun., 2014, 5, 4843 CrossRef CAS.
- S. Jia, X. Lu, S. Luo, Y. Qing, N. Yan and Y. Wu, Chem. Eng. J., 2018, 348, 212–223 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01540g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |