
Structural, mechanical, and biological characterization of hierarchical nanofibrous Fmoc-phenylalanine-valine hydrogels for 3D culture of differentiated and mesenchymal stem cells†
Haniyeh
Najafi
a,
Ali Mohammad
Tamaddon
*ab,
Samira
Abolmaali
 ab,
Sedigheh
Borandeh
abc and
Negar
Azarpira
d
ab,
Sedigheh
Borandeh
abc and
Negar
Azarpira
d
aPharmaceutical Nanotechnology Department, Shiraz University of Medical Sciences, Shiraz, Iran. E-mail: amtamadon@gmail.com; Fax: +98-713-2424126; Tel: +98-713-2424127
bCenter for Nanotechnology in Drug Delivery, Shiraz University of Medical Sciences, Shiraz, Iran
cPolymer Technology Research Group, Department of Chemical and Metallurgical Engineering, Aalto University, 02152 Espoo, Finland
dTransplant Research Center, Shiraz University of Medical Sciences, Shiraz, Iran
First published on 14th September 2020
Abstract
Fmoc-dipeptides are a class of short aromatic peptides featuring eminent supramolecular self-assembly, which is due to the aromaticity of the Fmoc group, which improves the association of peptide building blocks. This study aimed to introduce a new dipeptide hydrogel scaffold, Fmoc-phenylalanine-valine (Fmoc-FV), for 3D culture of various cells. Peptide hydrogel scaffolds were prepared by the pH-titration method in various concentrations and temperatures, and characterized by spectroscopic methods, including circular dichroism, attenuated total reflection FT-IR and fluorimetry. Mechanical behaviors such as thixotropy and temperature-sensitivity were investigated by oscillatory rheology. The Fmoc-FV hydrogels were then applied in 3D-culture of WJ-MSCs (mesenchymal stem cells), HUVECs (normal endothelial cells), and MDA-MB231 (tumor cell line) by live-dead fluorescence microscopy and Alamar blue viability assay experiments. The results confirmed that the β-sheet structure is principally interlocked by π–π stacking of the Fmoc groups and entangled nanofibrous morphologies as revealed by FE-SEM. Fmoc-FV self-assembly in physiologic conditions resulted in a thermo-sensitive and shear-thinning hydrogel. Notably, the Fmoc-FV hydrogel exhibited cell type-dependent biological activity, so higher cell proliferation was attained in HUVEC or MDA-MB231 cells than WJ-MSCs, indicating a possible need for incorporating cell-adhesion ligands in the Fmoc-FV hydrogel matrix. Therefore, the structural and biological properties of the Fmoc-dipeptide hydrogels are inter-related and can affect their applications in 3D cell culture and regenerative medicine.
1. Introduction
Molecular self-assembly is a phenomenon activated by non-covalent interactions such as π–π stacking, electrostatic interactions, hydrogen bonds, and hydrophobic interactions with basic building blocks, organized spontaneously into unique supramolecular nano-scale architectures.1–3 This bottom-up approach has been used in fabrication of numerous nanostructures with potential applications in 3D-cell culture,4,5 regenerative medicine,6,7 immune boosting,8,9 sensing,10,11 and drug delivery.12,13 Over the past decades, supramolecular peptide hydrogels have gained much attention in biomedical fields due to their numerous advantages, such as ease of synthesis, degradation over time into predictable metabolites without immunogenicity, easy integration into bioactive ligands, widely tuneable mechanical properties, cell signalling capacity, and most importantly high biocompatibility.2,14–16 Owing to their interesting properties, short peptide hydrogels containing 2–5 amino acid residues have attracted significant interest. Unlike many other nano-structured hydrogels, short peptide hydrogels form through the self-assembly process in response to changes in physiological stimuli including pH,17,18 ionic strength,19,20 organic solvent,21,22 or temperature.23 According to recent reports,24–30 efficient gelation of oligopeptides generally requires a large aromatic ligand, such as benzyloxy carbonyl (Cbz), 9-fluorenylmethoxycarbonyl (Fmoc), naphthalene (Nap), and pyrene or phenothiazine (PTZ), facilitating self-assembly of peptides into supramolecular hydrogels.Apart from aromatic ligands such as Fmoc, commonly used as an amine protecting group in peptide synthesis,31,32 the amino acid sequence can alter the physicochemical and biological properties of short peptide hydrogels. In general, the overall hydrophobicity determines the gelation property of the Fmoc-dipeptide. Ulijn et al. investigated the self-assembly behaviour of Fmoc-FG, Fmoc-GG, and Fmoc-GF in comparison to Fmoc-FF. Their results indicated a shift in the apparent pKa and thermo-sensitive gelation depending on the Fmoc-dipeptide hydrophobicity. It was also found that Fmoc-dipeptides such as Fmoc-GF could form precipitates rather than hydrogel networks in pH lower than their apparent pKa, which was confirmed by micrometre scale sheet-like structures in TEM.33 Tang et al. formed hydrogels from leucine-based Fmoc-dipeptide homologues, including Fmoc-LL, Fmoc-LG, and Fmoc-GL, to study the impact of the aromatic side chains on the self-assembly behaviour of Fmoc-dipeptides. Their results indicated that replacing the phenyl ring with an isobutyl chain did not affect the propensity of the molecule to self-assemble into extended fibrillar structures. However, no hydrogel structure was formed in the case of Fmoc-GL.24 Hence, aromatic amino acids such as L-phenylalanine have a synergistic effect with Fmoc on the self-assembly process through formation of π–π stacking interactions. All in all, the amino acid composition and sequence govern the self-assembly and gelation properties of Fmoc-dipeptides. On the other hand, an ideal scaffold for cell delivery should possess excellent mechanical properties and tailored structure with an interconnected pore network.34,35 The scaffold moduli and shear-thinning/self-healing kinetics are important determinants of the scaffold suitability for biomedical applications. Shear-thinning systems are developed to form network structures under physiological conditions, flow under moderate pressure (during injection), and self-heal (after injection).35 As addressed in the literature, hydrogels with low storage and loss moduli, low yield strain, and low viscosity are generally well suited for injection.36,37 Notably, the mechanical (i.e. stiffness, recovery rate, and injectability) and functional properties (i.e. permeability and aggregate modulus) of hydrogel scaffolds can play a crucial role in regulating the interactions between cells and the extracellular matrix and directing the cell phenotype.38,39 For example, mesenchymal stem cells (MSCs) proliferate on stiff hydrogels,40 whereas neuronal stem cells prefer soft hydrogels for their growth.41 Also, the scaffold microstructure may direct differentiation of stem cells. Cheng et al. showed that a porous scaffold derived from the articular cartilage can induce chondrogenic differentiation of adipose-derived adult stem cells without exogenous growth factors. In comparison with native cartilage, the designed scaffold exhibited suitable biomechanical properties such as the same aggregation modulus with 10-fold more permeability.42 Therefore, the present study aimed to introduce Fmoc-FV as a new dipeptide hydrogel scaffold for 3D-culture applications. To do so, we carried out synthesis of Fmoc-FV in the liquid phase and formed hydrogels by the pH-titration method. The gelation property and morphology of Fmoc-FV were compared to Fmoc-phenylalanine-glycine (Fmoc-FG). Consequently, the structural and mechanical properties of Fmoc-FV were investigated by spectroscopic methods and oscillatory rheometry. Importantly, 3D-cell growth in the supported hydrogel was realized for Wharton's jelly mesenchymal stem cells (WJ-MSCs), human umbilical vein endothelial cells (HUVECs), and the MDA-MB-231 breast cancer cell line. To the best of our knowledge, neither detailed mechanical nor biological studies were conducted on Fmoc-FV hydrogels, which will be discussed in this paper.
2. Results and discussion
2.1. Synthesis and charcterization of Fmoc-FV
Fmoc-FV was synthesized with an acceptable recovery yield (70.6%). Fmoc-dipeptides were prepared in 2 consecutive steps: (1) coupling of the tert-butyl ester of valine or glycine to Fmoc-protected phenylalanine by carbodiimide chemistry, and (2) elimination of the tert-butyl protecting group under acidic conditions (Fig. 1).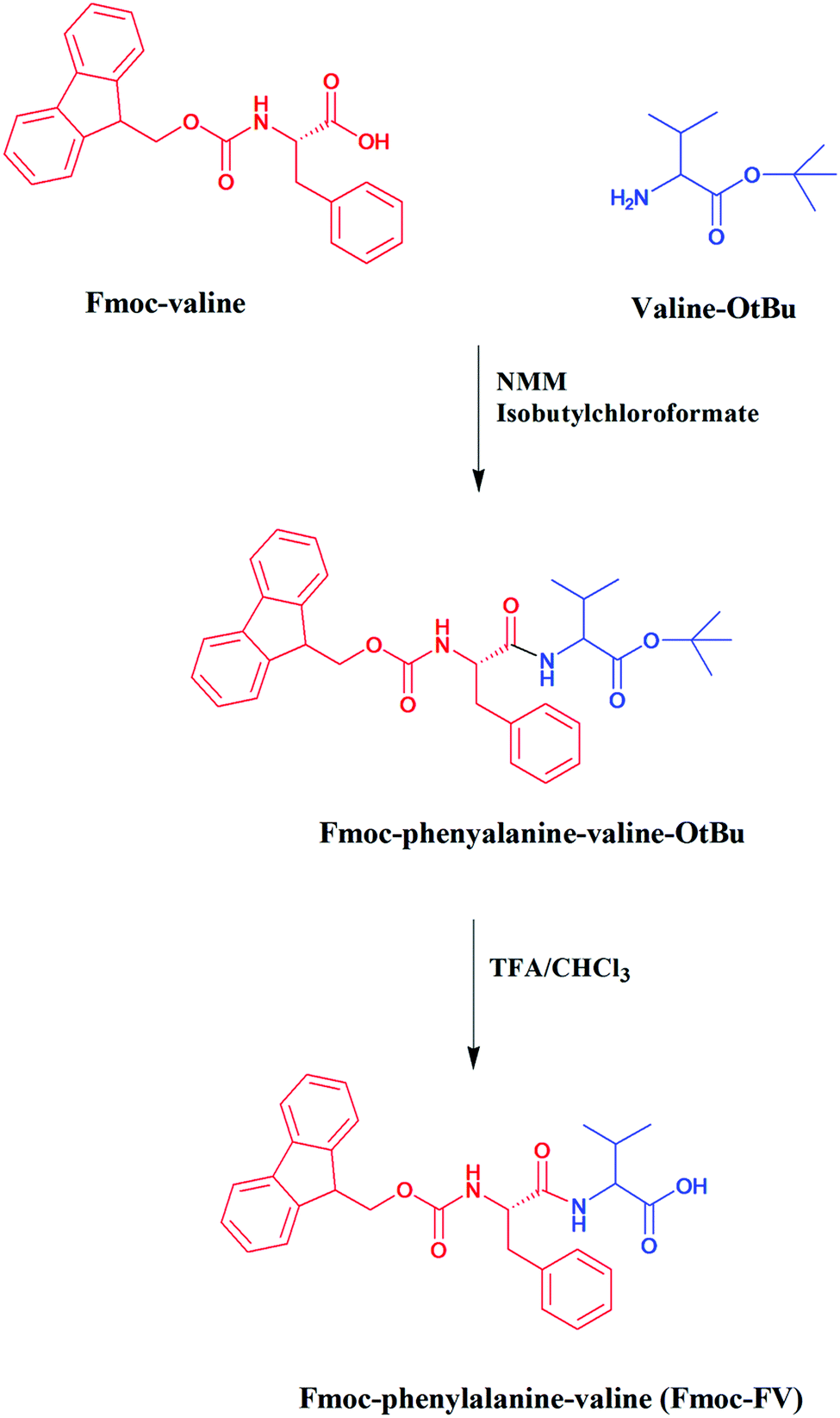 | ||
| Fig. 1 Schematic routes for synthesis of Fmoc-dipeptides (e.g. Fmoc-FV). | ||
The products were characterized by FT-IR and 1H-NMR spectroscopy. tert-Butyl esters of Fmoc dipeptides like Fmoc-FV-OtBu present peaks at 3302 cm−1 (N–H stretching), 3064, 3030, 2976, and 2932 cm−1 (C–H stretching), 1734 cm−1 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1656 cm−1 (amide CO stretching), and 1542 cm−1 (N–H bending). After the elimination of the tert-butyl protecting group by TFA, the ester CO stretching bond disappeared and the characteristic peak of the acidic CO stretching bond appeared at 1709 cm−1, similarly to Fmoc-FG. Moreover, the FT-IR spectra of Fmoc-dipeptides showed a peak around 1660 cm−1, confirming the presence of an amide bond between amino acids (ESI. 1,†).43
O stretching), 1656 cm−1 (amide CO stretching), and 1542 cm−1 (N–H bending). After the elimination of the tert-butyl protecting group by TFA, the ester CO stretching bond disappeared and the characteristic peak of the acidic CO stretching bond appeared at 1709 cm−1, similarly to Fmoc-FG. Moreover, the FT-IR spectra of Fmoc-dipeptides showed a peak around 1660 cm−1, confirming the presence of an amide bond between amino acids (ESI. 1,†).43
The 1H-NMR spectra of the short peptides in DMSO-d6 (ESI. 2,†) showed multiple peaks at 2–4 ppm, corresponding to the aliphatic carbon protons, and signals at 6.5–8 ppm, which refer to the phenyl protons. Also, the characteristic peak of the hydroxyl proton of the carboxylic group was observed at 12.5 ppm, confirming successful synthesis of dipeptides.
2.2. Fmoc-FV gelation and morphology
Dispersion of Fmoc-FV in deionized water can lead to hierarchical assembly and gelation, which depends on the concentration, pH, and temperature.2,23,44 To prepare hydrogels, Fmoc-FV was dissolved by alkalizing the aqueous dispersion through adding concentrated NaOH solution.45 At high pH, the carboxylic terminal group of Fmoc-FV was expected to be ionized in the solution. Since the Fmoc moiety can be removed at pH > 12, filtered NaOH solution was added dropwise until pH = 9.1 ± 0.1 was reached.46 Then, the Fmoc-dipeptide solutions were titrated with HCl until clear gels were formed. A vial inversion test confirmed the formation of a self-supporting gel without collapse. When the pH was reduced (above the critical gelation concentration), antiparallel β-sheets formed, the fibres entangled, and a self-supporting hydrogel was obtained as indicated in Sections 3.3 and 3.4.18,47 Gelation occurred at concentrations above 20 mM at various pH values ranging from 3.2–7.5. Similar gelation behaviours were reported elsewhere for Fmoc-dipeptides containing leucine and alanine.18,24,27 Unlike Fmoc-FG, which became viscous only in acidic pH (between 3 and 5) (data not shown), Fmoc-FV gelation readily occurred at physiologic pH (7.4). It has been shown that the gelation of Fmoc-dipeptides occurs at a pH below their apparent pKa. Moreover, depending on the hydrophobicity of Fmoc-dipeptides, the apparent pKa shifts from the theoretical value due to their self-assembly in aqueous medium.33 Similarly, Fmoc-FF revealed two distinct pKa values (5.2–6.2 and 9.5–10.2), resulting in structural transitions.18 Apart from the gelation pH, the appearance of the Fmoc-dipeptide hydrogels varies with the amino acid composition. Unlike Fmoc-FG, which became turbid by acid titration,45 the Fmoc-FV hydrogel exhibited a transparent hydrogel. Similarly, Fmoc-LF, unlike Fmoc-LA, was reported to form transparent hydrogels.45FE-SEM image of Fmoc-FV confirming a fibrous network with a fibril thickness of 30 ± 5 nm (Fig. 2A). In contrast, Fmoc-FG fibres were thin with a thickness of 10 ± 7 nm (ESI. 3,†), indicating that the fibre thickness depends on the amino acid composition. Interestingly, Fmoc-FV formed a “spaghetti-like” pile of thick fibres with favourable pores due to the high hydrophobicity and aggregation number of the Fmoc-FV precursor.48 As similarly shown elsewhere for Fmoc-FG or Fmoc-GF, glycine residues result in flexible fibres which do not favour β-sheet formation.33 On the other hand, the isopropyl side chain of valine can boost the thickness and consequently the overall rigidity of the peptide fibres. Also, Fmoc-GG and Fmoc-AA form nanofibers with an average thickness of 33 and 68 nm, respectively.27 These results suggest that the Fmoc moiety is playing the key role in self-assembly, and the peptide composition provides tunable physicochemical properties.24,33,49
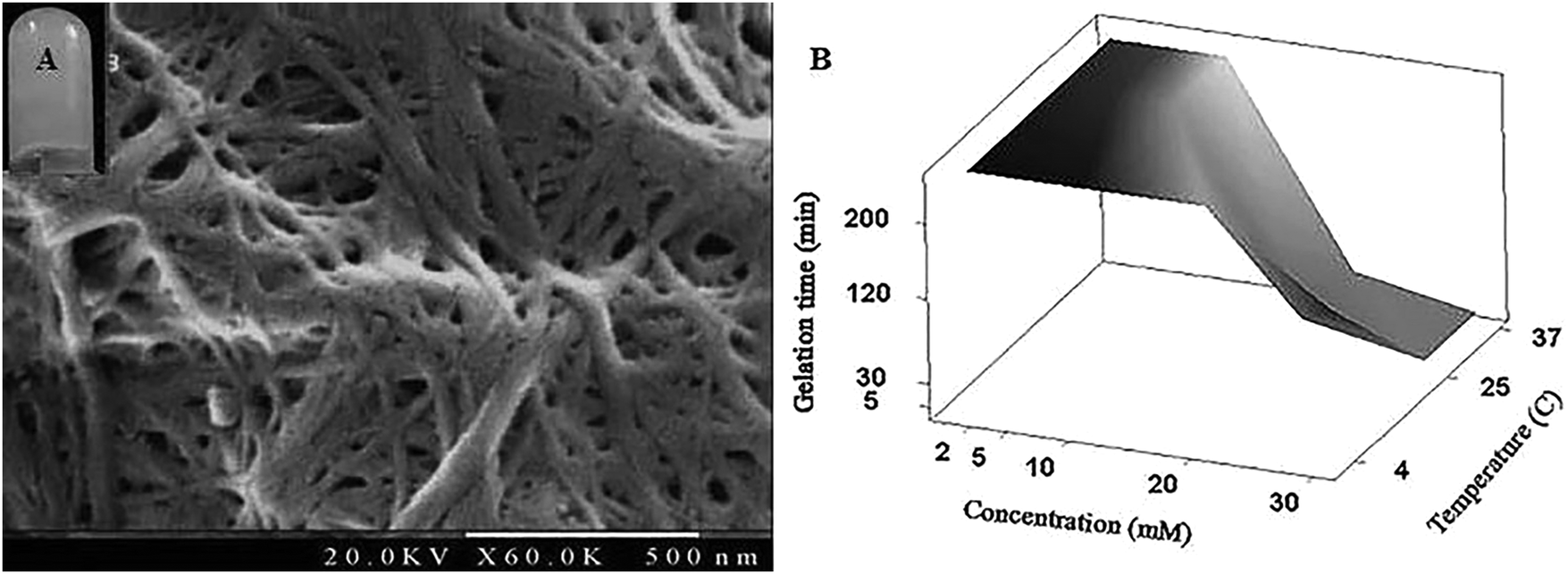 | ||
| Fig. 2 (A) FE-SEM image of the self-assembled Fmoc-FV hydrogel (thickness = 30 ± 5 nm). (B) Combined effects of temperature and concentration on the gelation time for the Fmoc-FV hydrogel at physiologic pH = 7.4. | ||
A possible interplay of the effects of concentration and temperature on the gelation time of Fmoc-FV was investigated by the tube inversion method. As shown in Fig. 2B, by raising the temperature from 4 °C to 37 °C, the gelation time decreased. In addition, gelation occurred at concentrations lower than 20 mM. Also, more transparent hydrogels of Fmoc-FV were attained upon heating. Therefore, the temperature can alter the gelation property of Fmoc-FV, which will be explained in the Oscillatory rheology section.
2.3. Supramolcular assembly of Fmoc-FV
To understand the mechanism by which gelation occurs, the secondary structure of Fmoc-FV was characterized by fluorescence pyrene assay, ATR-FTIR, fluorescence spectroscopy, and CD. Due to the availability of hydrophilic and hydrophobic moieties, the Fmoc-dipeptide can undergo assembly at concentrations above the CAC. To investigate the critical concentration of fibre formation, the fluorescence intensities of samples were determined for increasing concentrations of Fmoc-dipeptide in the presence of a fixed amount of pyrene. The critical concentration at the intersection of the flat regions was observed at 0.5 mM (Fig. 3). Similarly, Fmoc-FF showed a CAC value of 0.337 mM with a lower propensity to self-assemble compared to the Fmoc-dipeptides comprising α-methyl-L-phenylalanine.50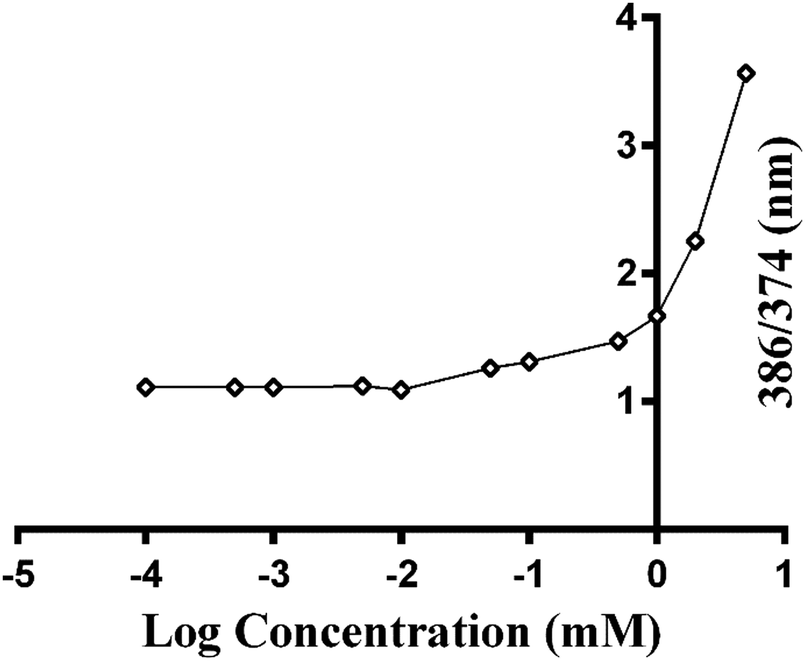 | ||
| Fig. 3 Pyrene fluorescence intensity ratios (I386/I374) as a function of the logarithm of various concentrations of Fmoc-FV in PBS buffer at pH = 7.4 (data represent mean ± SD for triplicate measurements). | ||
ATR-FTIR spectra of Fmoc-FV at different pH values were collected in the wavenumber range from 1500–1800 cm−1. After acid titration of Fmoc-FV alkaline solution, two dominant peaks appeared in the amide I region (around 1600 cm−1 and 1690 cm−1) (Fig. 4A), suggesting that the main secondary structure was antiparallel β-sheets. However, the peaks around 1600 cm−1 were weak; hence, a lower proportion of β-sheet structures was present. In contrast, a broad peak around 1655 cm−1 was observed, indicating abundance of random coil structure. π–π stacking interaction was proposed as a driving force triggering self-assembly of aromatic dipeptides.49,51 For further characterization of Fmoc-dipeptide association, the arrangements of fluorenyl groups in the supramolecular structures were observed by fluorescence spectroscopy (Fig. 4B). At a pH around 9, the maximum emission wavelength was observed at 316 nm. Upon adding hydrochloric acid, the emission maximum wavelength immediately shifted to 326 nm, and fluorescence quenching occurred, indicating anti-parallel π–π stacking of fluorenyl rings.24 Complementary to the ATR-FTIR and fluorescence spectroscopy experiments, the CD spectrum of Fmoc-FV was collected in the wavelength range from 190–260 nm (Fig. 4C). A ‘v’-shaped spectrum with a negative peak around 218 nm and positive peaks around 190 nm and 240 nm was obtained, indicating an anti-parallel structure with a β-sheet arrangement. In addition, the negative peak between 210 and 230 nm was weak; hence, a lower amount of β-sheet structures was present, as it was similarly shown in the ATR-FTIR investigation. It should be noted that this measurement was carried out at low concentration since the scattered light arising from the gel structure interferes with CD data collection.33,51 Altogether, the spectroscopic data suggested that the Fmoc-FV peptide moiety should be in an anti-parallel β-sheet arrangement with interlocked chains through π–π stacking of the Fmoc moieties.
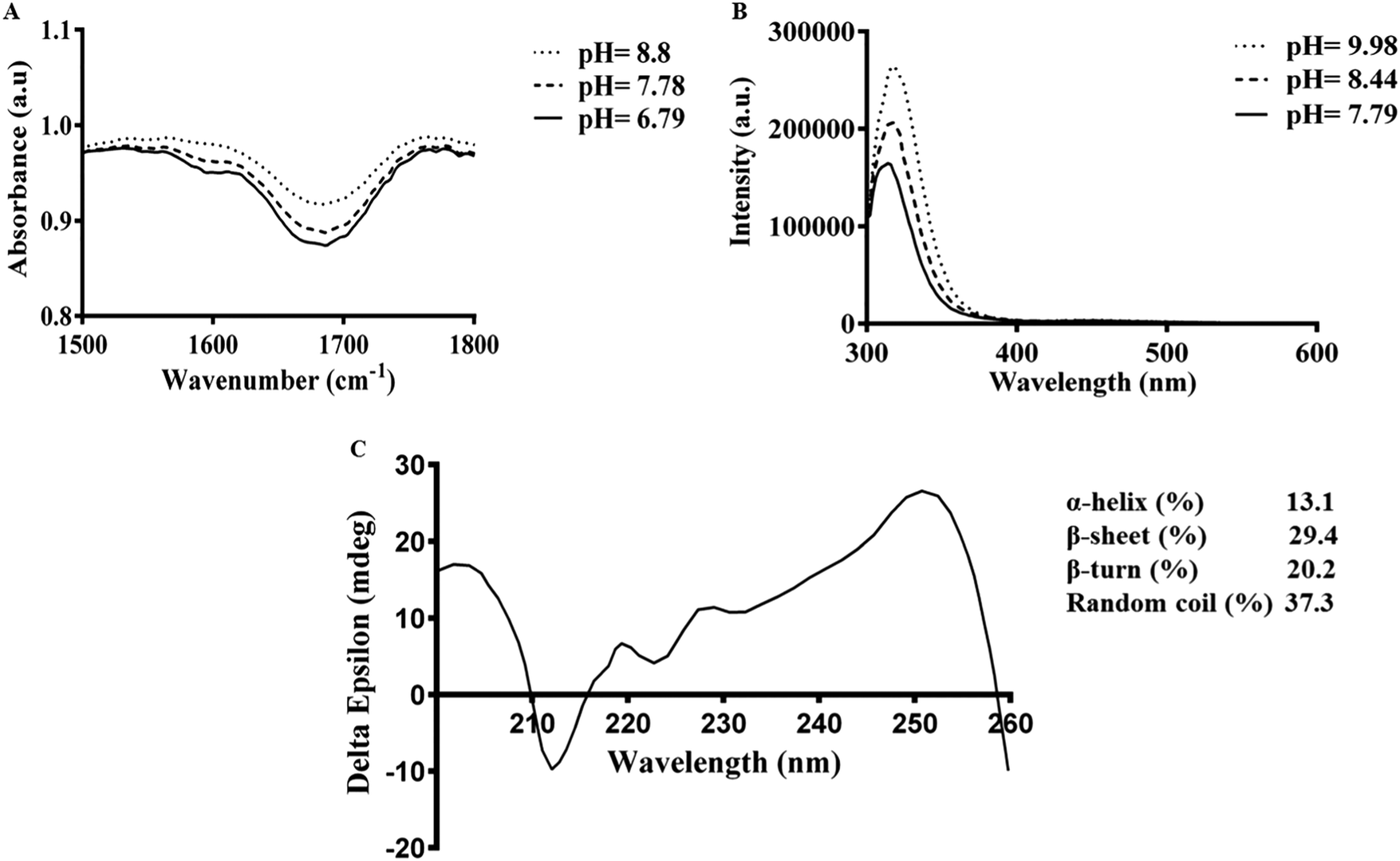 | ||
| Fig. 4 Spectroscopic characterization of Fmoc-FV: (A) FTIR-ATR spectra of Fmoc-FV showing peaks in the 1700–1600 cm−1 wavenumber regions, indicating formation of antiparallel β-sheets, (B) fluorescence emission spectra of Fmoc-FV at excitation λmax = 265 nm, and (C) CD spectrum of Fmoc-FV showing a peak at 218 nm indicating a β-sheet structure and higher proportion of random coil structures. | ||
2.4. Oscillatory rheology
Apart from the microscopic alteration in the supramolecular structure of Fmoc-dipeptides, the peptide composition can macroscopically influence the visco-elasticity of the hydrogels. As shown in Fig. 6A, the elastic modulus (G′) appeared 2–3 times higher than the viscous modulus (G′′) at pH 8.5 until a crossover of the moduli was observed at around 1 rad s−1. Both G′ and G′′ showed strong frequency dependence between 0.1 and 100 rad s−1, indicating the shear-thinning property and transition of gel to liquid-like materials by shear stress.33 By decreasing the pH to 7.4 (Fig. 5A), G′ was mainly higher (4–6 times) than G′′ between 0.1 and 100 rad s−1, so both moduli displayed weak frequency dependence, confirming the stable gel-like structure with an entangled fibrillar network as shown in the FE-SEM image.27 Moreover, by comparing the modulus G′ at various pH values, Fmoc-FV exhibited a stiffer hydrogel at pH 7.4 (G′ = 12–26 kPa) than pH 8.5 (G′ = 0.0003–3.5 Pa).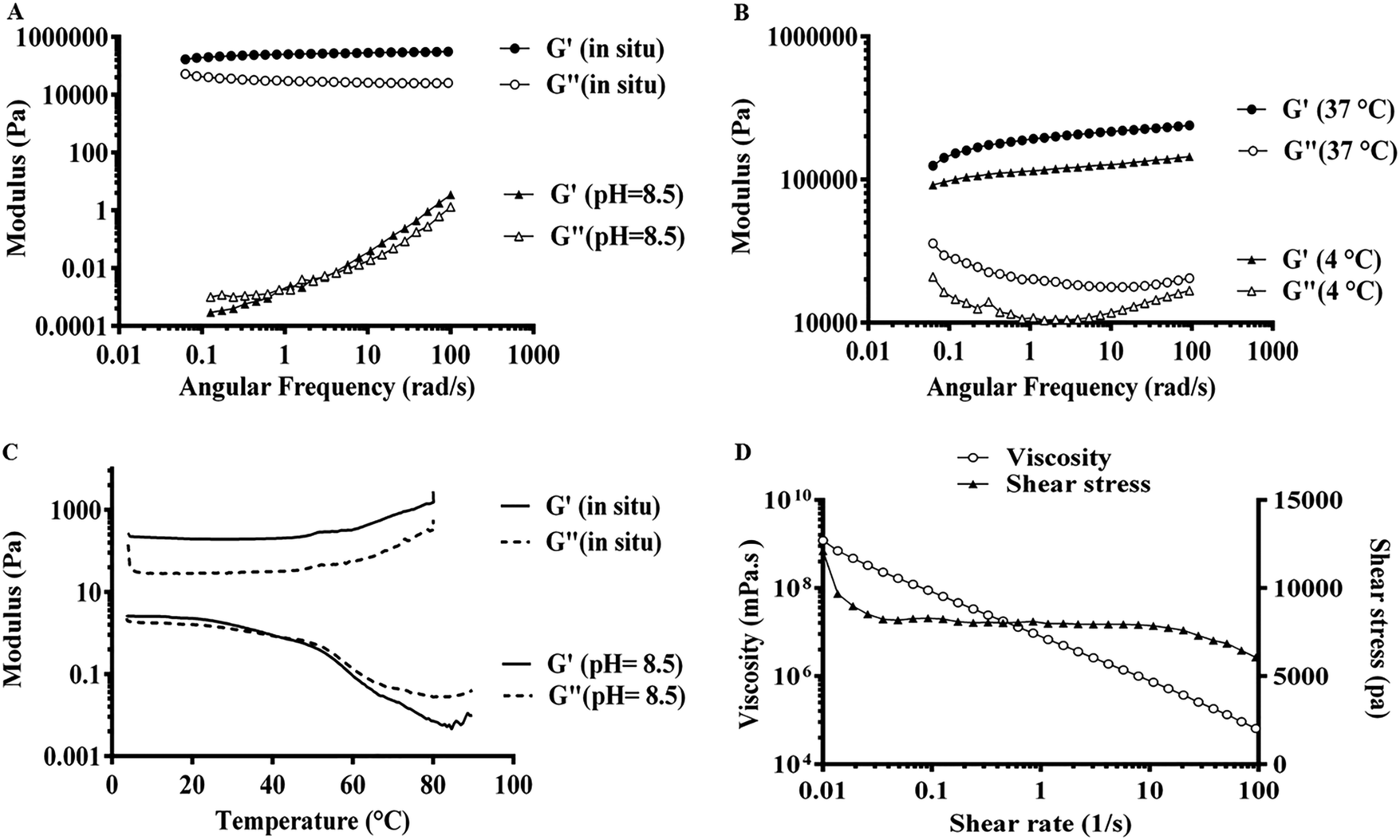 | ||
| Fig. 5 Mechanical characterization of 20 mM Fmoc-FV: (A) dynamic frequency sweep of Fmoc-FV before (pH = 8.5) and after gelation (in situ pH = 7.4), (B) dynamic frequency sweep at 4 °C and 37 °C, (C) dynamic temperature sweep from 4 to 90 °C, and (D) shear stress and viscosity data. | ||
There are only a few reports on the mechanical properties of Fmoc-dipeptide hydrogels. Hence, a dynamic frequency sweep was performed first at 4 °C and 37 °C. As shown in Fig. 5B, the G′ values at 37 °C were found to be 1.5–2 times higher than the G′ values at 4 °C, which showed that the hydrogel became stiffer upon heating. Then, a dynamic temperature sweep was performed at pH 8.5 and 7.4. Fig. 5C shows the temperature-dependency of the mechanical moduli, so the G′ values appeared about 2 times higher than the G′′ values until a crossover of the moduli around 50 °C (Tgel) occurred at pH 8.5. At pH 7.4, G′ was several times higher than G′′, indicating that gelation occurred at a temperature lower than the studied range. Moreover, the high stiffness of the Fmoc-FV hydrogel was confirmed by the relatively high G′ values. The gel stiffness could be causally related to the intermolecular interactions associated with the amino acids (phenylalanine and valine). On the other hand, during the heating step, aggregated fibres were re-dissolved and locked in the self-assembled structures, resulting in an increase in the effective fibre concentration and gel stiffness. These results clearly revealed that the Fmoc-FV hydrogels became stiffer with raising the temperature, which is in line with our previous results (Section 3.2) and visual observations.24,33 As shown in Fig. 5D, by increasing the shear rate, a modest degeneration of the three-dimensional hydrogel network occurred, which in turn led to a decrement in the viscosity of the Fmoc-FV hydrogel.23 All in all, these results confirmed that Fmoc-FV formed a shear-thinning hydrogel from the self-assembling amphiphiles at physiologic pH.
2.5. WJ-MSC isolation and characterization
The plastic-adherent cells from Wharton's jelly formed a monolayer with fibroblast-like morphology (Fig. 6A). After differentiation toward adipogenic and osteogenic lineages, the presence of lipid vacuoles was confirmed by Oil Red O, and calcium deposition was revealed with Alizarin Red (Fig. 6B). The flow cytometry results showed that the WJ-MSCs expressed stromal markers (CD90 and CD44), while they were negative for the hematopoietic markers (CD34 and CD45) (Fig. 6C). It has been reported that WJ-MSCs proliferated like plastic adherent cells under in vitro culture conditions. As similarly shown, they are positive for the expression of CD105, CD44, CD73, and CD90, and negative for the expression of hematopoietic cell surface markers such as CD34, CD19, CD45, CD11a, and HLA DR (human leukocyte antigen).52–54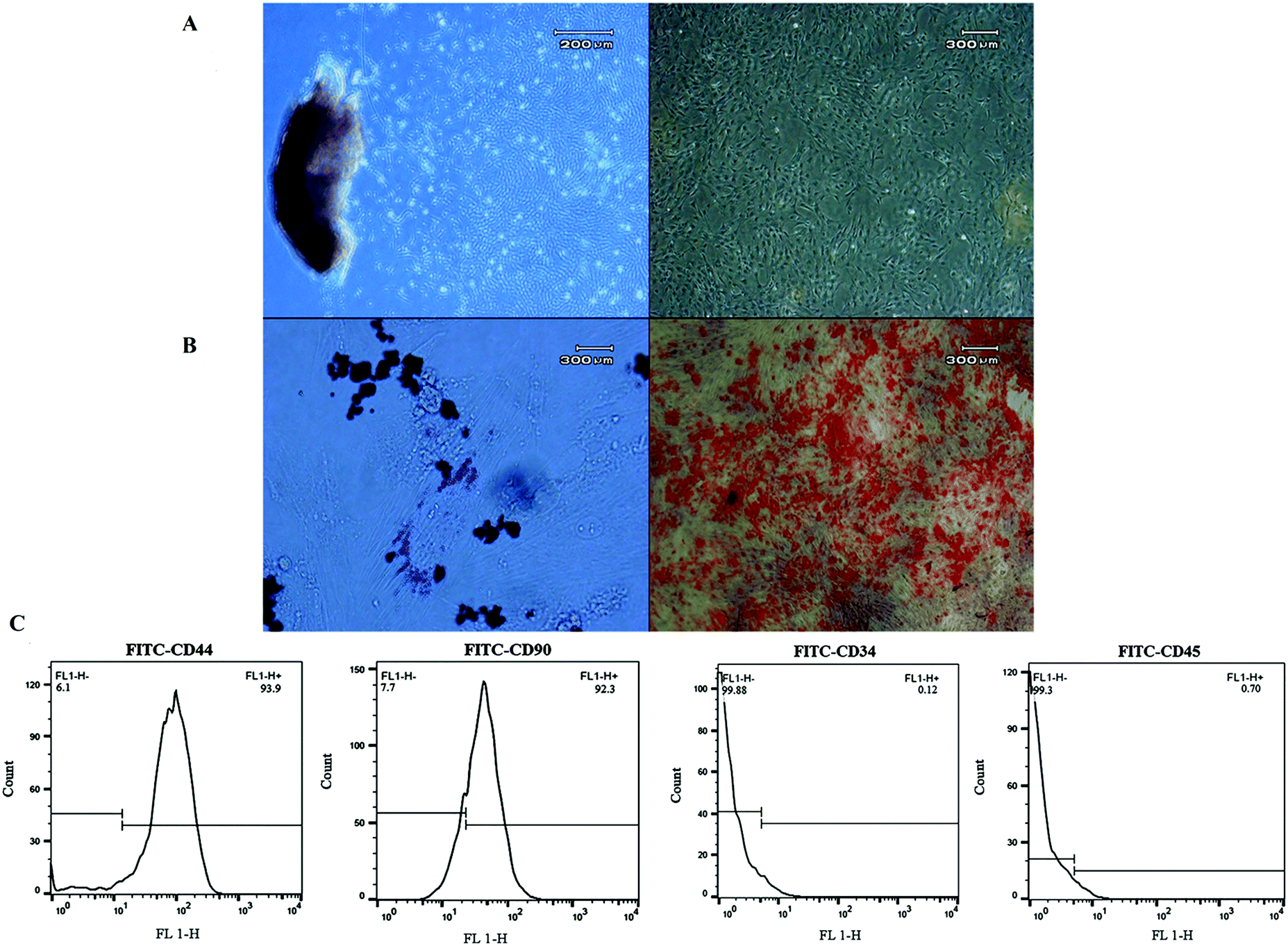 | ||
| Fig. 6 Characterization of human Wharton's jelly-derived mesenchymal stem cells. (A) The morphology of mesenchymal stem cells (MSCs), (B) differentiation of MSCs into adipocytes and osteocytes, confirmed by Oil Red O and Alizarin Red, respectively, and (C) the flow cytometry results showed that cells were positive for CD44 (93.9%) and CD90 (92.3%) and negative for CD34 (0.12%) and CD45 (0.7%). | ||
2.6. 3D culture and live/dead assay
The potential application of the Fmoc-FV hydrogel as a 3D scaffold was investigated for culturing WJ-MSCs (mesenchymal stem cells), HUVECs (primary cells), and MDA-MB231 (tumour cell line). To encapsulate the cells, an equal volume of Fmoc-FV solution was mixed with cell suspensions prepared in serum-free DMEM. Interestingly, the self-supporting hydrogels were formed rapidly at 37 °C; hence, homogeneity of the cell distribution within the hydrogel matrix was ensured. It seems that the DMEM medium components such as the buffering agents and metal ions play a vital role in accelerating self-assembly and gelation of Fmoc-FV. As shown in Fig. 7, the gelation process did not change the cell viability according to the live–dead assay performed 4 h post incubation. No dead cells (red) were detected within the gels and the green (live) cells were visualized with well-defined round contours. Within the first 24 h of culture, cell proliferation was noticed in the Fmoc-FV scaffold for all cell cultures. In addition, the cells adopted a polyhedral shape with fine filopodia. After 72 h incubation, the cells formed a 3D-network, which was more pronounced in MDA-MB231. As expected, the Fmoc-FV hydrogel showed cell type-dependent growth, so only a few dead cells appeared in the hydrogel containing HUVEC and MDA-MB231 cells. In contrast, the number of dead cells increased for WJ-MSCs after 72 h post-incubation. Interestingly, WJ-MSCs almost maintained a spherical or ellipsoidal shape in the Fmoc-FV scaffold. This event can be attributed to low cell–matrix attachment of WJ-MSCs to the Fmoc-FV scaffold as similarly shown elsewhere.33 Zhou et al. reported that the Fmoc-FF hydrogel incorporated with the Fmoc-RGD ligand promoted cell adhesion with subsequent spreading of human adult dermal fibroblasts.55 | ||
| Fig. 7 Fluorescence microscopy images of 3D culture of WJ-MSC, HUVEC, and MDA-MB231 cells incorporated in the Fmoc-FV scaffold. Green live cells showed normal morphology while red showed dead cells. Unlike the hydrogel promoting cell spreading and proliferation of HUVEC and MDA-MB231 cells, less cell proliferation was attained in WJ-MSCs due to a possible lack of cell–matrix attachments. | ||
2.7. MTT and Alamar blue assays
To investigate the cytocompatibility of the hydrogel construct and a disentangled hydrogel nanofiber, the cell viability was determined by (A) MTT and (B) Alamar blue cytotoxicity assay, respectively.As shown in Fig. 8A, Fmoc-FV nanofibers showed no significant cytotoxicity at concentrations as low as 0.05 mM in the HUVEC and MDA-MB231 cell lines for 24 h (P > 0.05). However, their cytotoxicity increased significantly with the nanofiber concentration, so the cell viability reduced significantly at 0.001, 0.005, and 0.01 mM in both cell lines (P < 0.05). As discussed earlier, the Fmoc-FV scaffold showed cell-dependent behaviour. It was also revealed that the cytotoxicity of Fmoc-FV nanofibers at high concentrations in HUVEC and MDA-MB231 cells was significantly lower than WJ-MSCs, which can be explained according to the higher sensitivity of WJ-MSCs to nanofiber-induced toxicity.
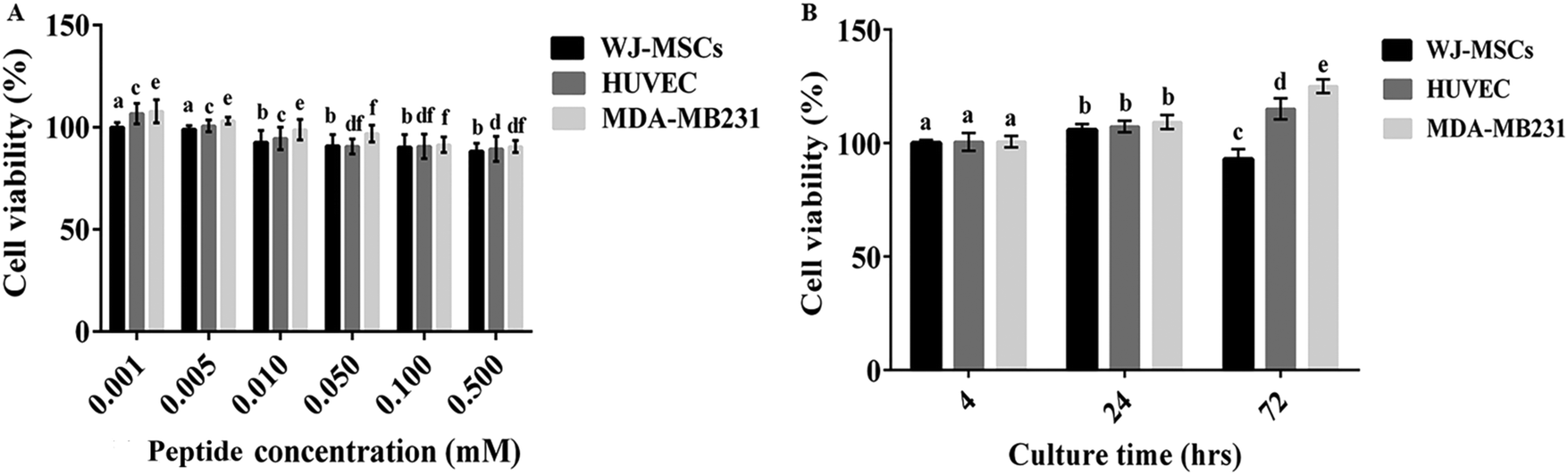 | ||
| Fig. 8 Viability of WJ-MSC, HUVEC, and MDA-MB231 cells at various Fmoc-FV concentrations (A) and after encapsulation in the 3D scaffold for different incubation times (B). a, b, c, d, e, and f: inter-group comparisons according to the Tukey follow-up test. Groups with the same superscripts were not significantly different at α = 0.05 (P > 0.05); however, dissimilar letters indicate a significant difference (P < 0.05). | ||
In addition, the proliferation of the WJ-MSC, HUVEC, and MDA-MB231 cells in the Fmoc-FV scaffolds was determined by Alamar blue assay (Fig. 8B). Alamar blue assay determines the intracellular metabolic activity; however, the enzymatically produced resorufin in the Alamar blue assay is water soluble and non-toxic, which enables kinetic measurements on the same culture construct without solubilisation, and the process is accompanied by a change in colour from indigo blue to pink56 (Fig. 8B). Within 24 h, it was shown that the hydrogel did not show significant cytotoxicity and supported cell proliferation for all cell cultures. After 72 h incubation, the cell density enhanced significantly for HUVEC and MDA-MB231 cells; however, the cell proliferation decreased significantly ∼15% for WJ-MSCs. Hence, Fmoc-FV is suggested as cell-supporting scaffolds for HUVEC and MDA-MB231 cells. However, cell–matrix adhesive factors such as RGD might be required for WJ-MSC growth.
3. Experimental
3.1. Chemicals
Fmoc-phenylalanine and amino acid tert-butyl esters (NH2-valine-OtBu and NH2-glycine-OtBu) were purchased from Chempep (Wellington, Florida, USA) and Aapptec (Louisville, Kentucky, USA). tert-Butyl methyl ether, chloroform (CHCl3), and potassium bromide (KBr) were supplied by Merck (Darmstadt, Germany). The Alamar blue viability assay kit was purchased from G-biosciences (St Louis, MO, USA). Trifluoroacetic acid (TFA) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Alfa Aesar (Haverhill, Massachusetts, USA) and Daejung (Busan, South Korea). CD90-FITC, CD45-FITC, CD44-FITC, and CD34-FITC were purchased from BioLegend (San Diego, California, USA). Fluorescein diacetate (FDA), propidium iodide (PI), N-methyl morpholine (NMM), isobutyl chloroformate, pyrene, and other solvents and chemicals were supplied by Sigma-Aldrich (St Louis, MO, USA). All aqueous solutions were freshly prepared from deionized water (Direct Q UV3, Millipore, USA).3.2. Fmoc-FV synthesis and characterization
Fmoc-FV synthesis was carried out via tert-butyl ester reaction chemistry according to the published method with a slight modification.47 Briefly, 1.22 mmol isobutyl chloroformate was added to a mixture of Fmoc-phenylalanine-OH (472.6 mg) and NMM (1.22 mmol) dissolved in CHCl3 for 5 min while stirring at −20 °C. Then, 2.5 ml valine-OtBu (255.834 mg) or glycine-OtBu (204 mg) was added in CHCl3, and the mixture was stirred overnight. The organic layer was partitioned between CHCl3 and water and washed sequentially with HCl, NaHCO3 and brine. After drying with Na2SO4, CHCl3 was removed in a rotary evaporator. The tert-butyl protecting group was removed by dissolving the Fmoc-dipeptide in a CHCl3/TFA mixture and incubated overnight while stirring. The product was precipitated after adding 3 volumes of cold tert-butyl methyl ether, followed by centrifugation at 3000 × g for 15 min. The product was processed by lyophilization (Christ alpha 1–2 LD, Germany). The identity and purity of the products were confirmed by FT-IR (Vertex, Bruker, Germany) and 1H-NMR spectroscopy (Bruker-ARX 350 MHz, Germany).3.3. Gelation by pH-titration
A pre-defined weight of Fmoc-FV was suspended into 2 ml fresh deionized water to obtain different concentrations ranging from 0.0001 to 30 mM. Filtered NaOH was added dropwise until pH = 9.1 ± 0.1 was reached.45 The samples were sonicated until a clear solution was obtained. Dilute HCl (0.2 M) was then gradually added while the peptide solution was sonicated until gelation occurred.To monitor the effect of temperature and peptide concentration on the gelation time, preparations were made to concentrations ranging from 2–30 mM in deionized water at pH = 7.4, which were incubated at 4 °C, 25 °C and 37 °C at various times (5 min, 30 min, 1 h, 3 h, 6 h, and 24 h). Then, the sol–gel state was determined using the tube inversion test.44,57 All the experiments were run in triplicate.
3.4. Structural analysis
The secondary structure of Fmoc-FV was investigated in the pH range of 7–10 by attenuated total reflectance-Fourier transform infrared (ATR-FTIR), fluorescence spectroscopy, and circular dichroism according to the succeeding sections. In addition, pyrene fluorescence assay and field emission scanning electron microscopy (FESEM) were carried out to monitor the Fmoc-dipeptide assembly.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The fluorescence intensities were determined at excitation λmax = 340 and emission λmax = 374 and 386 nm (Infinite M200, Tecan, Austria). The intensity ratio (I386/I374) of each sample was plotted against the logarithm of the nanofiber concentration (mM).
000 rpm. The fluorescence intensities were determined at excitation λmax = 340 and emission λmax = 374 and 386 nm (Infinite M200, Tecan, Austria). The intensity ratio (I386/I374) of each sample was plotted against the logarithm of the nanofiber concentration (mM).
3.5. Oscillatory rheology
The mechanical properties of the Fmoc-FV hydrogel were determined, using dynamic frequency sweep experiments on an Anton Paar (MCR-302) rheometer equipped with cone-plate (24.983 mm/1.002° diameter) or cup-bob (16.661–18.088 mm diameter) accessories. To ensure that the measurements were done in the linear viscoelastic region, an amplitude sweep was carried out and the results presented no variation in G′ and G′′ up to a strain of 1%. The elastic (G′) and viscous (G′′) moduli were recorded as a frequency function between 0.1 and 100 rad s−1. A solvent trap and an integrated temperature controller were utilized to keep the sample hydrated. These measurements were repeated at an alkaline pH = 8.5 (before gelation occurred) and physiologic pH (in situ) at 4 °C and 37 °C. Moreover, the hydrogel was tested at pH 8.5 and 7.4 as a function of temperature. To do so, the temperature was raised first from 4 to 90 °C at a rate of 5 °C min−1, and maintained for 1.5 min at 90 °C. Then, it was reduced from 90 to 4 °C over 1 min, and finally kept for 8 min at 4 °C. A viscosity measurement was done by monitoring the viscosity and shear stress over controlled shear rates from 0.01–100 s−1 at 25 °C. All the experiments were performed in triplicate.3.6. Cell culture
Cellular studies were carried out on WJ-MSCs isolated from fresh human umbilical cords (UCs), HUVECs, and the MDA-MB-231 breast cancer cell line purchased from Pasteur Institute (Tehran, Iran). The cells were cultured in DMEM-F12 and DMEM media supplemented with 10% FBS (PAA, Austria) and 1% antibiotic–antimycotic solution (penicillin–streptomycin, PAA, Austria) at 37 °C with 5% CO2.3.6.1.1. Mesodermal lineage differentiation assay. For the evaluation of the cell differentiation potential, commercial osteocyte and adipocyte differentiation media (Bonyakhteh, Iran) were used. After 14 and 21 days, the samples were fixed and stained with Alizarin Red and Oil Red O staining to confirm the cell differentiation potential.
3.6.1.2. Immunophenotyping by flow cytometry. To determine the phenotype of cell-surface antigens, 3rd-passage cells were stained with FITC conjugated antibodies specific for the following human antigens: CD34 and CD45 as hematopoietic lineage markers, and CD90 and CD44 as stromal surface biomarkers. Stained cells were re-suspended in PBS and analysed using a FACSCalibur flow cytometer (Becton Dickinson, USA). For each sample, at least 10
000 events were recorded as similarly reported elsewhere.54
3.7. Statistics
Data were expressed as mean ± standard deviation (SD) and were analysed using Prism Software ver. 6.0 (GraphPad, USA). Design Expert software ver. 6.0 (Statease, USA) was utilized for analysis of the gelation data. The nanofiber thickness and inter-fibre spacing were calculated using the ImageJ website (https://imagej.nih.gov/ij/). P < 0.05 was considered as statistically significant.4. Conclusion
Fmoc-FV is suggested as a new low-molecular weight hydrogelator for preparation of shear-thinning hydrogel scaffolds. In the present study, the structural, mechanical, and biological properties of Fmoc-FV were investigated in culture of three different cells, WJ-MSCs, HUVECs and MDA-MB-231. This study clearly revealed that substitution of the isopropyl side chain (in valine) with a hydrogen atom (in glycine) as well as alteration of the concentration and temperature significantly impacts Fmoc-dipeptides’ self-assembly. Such modification affects the Fmoc-dipeptide overall rigidity. Indeed, the hydrophobicity of the dipeptide tail is an important parameter for self-assembly.33 Also, the Fmoc group plays a dominant role in the self-assembly process through anti-parallel π–π stacks interlocking the β-sheets.18 Fmoc-FV self-assembled at physiologic conditions with a “spaghetti-like” pile of an entangled fibrillar network to form a stable, rigid, and viscous gel-like structure. Fmoc-FV exhibited also thermo-sensitivity, so the hydrogel became stiffer and transparent upon heating. Accordingly, the Fmoc-FV hydrogel was applied in 3D culture of different cell types such as WJ-MSCs, HUVECs, and MDA-MB-231. Based on the cellular experiments, the Fmoc-FV hydrogel showed cell type-dependent behaviour. Higher cytocompatibility and cell proliferation were attained in HUVEC and MDA-MB231 cells than WJ-MSCs, indicating the need for incorporating cell-adhesion ligands such as RGD or IKVAV in the Fmoc-FV hydrogel matrix. Altogether, Fmoc-FV can be proposed as a potential cell-supporting scaffold for 3D cell culture and as a basis for developing injectable carriers for dual cell therapy and drug delivery.Compliance with ethical standards
Informed consent was obtained from mothers, and the study was approved by the institutional ethics committee at Shiraz University of Medical Sciences, Shiraz, Iran.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This study was supported financially by Shiraz University of Medical Sciences (Grant No. SUMS-1396-01-05-14550). The authors gratefully acknowledge use of the facilities of the Center for Nanotechnology in Drug Delivery and Transplant Research Center, at Shiraz University of Medical Sciences. The authors wish to thank Mr H. Argasi and Dr L. Khojasteh at the Research Consultation Center (RCC) of Shiraz University of Medical Sciences for their invaluable assistance in editing this manuscript.References
- X. Yuan, B. He, Z. Lv and S. Luo, RSC Adv., 2014, 4, 53801–53811 RSC.
- S. Eskandari, T. Guerin, I. Toth and R. J. Stephenson, Adv. Drug Delivery Rev., 2017, 110, 169–187 CrossRef.
- W. Xu, Y. Hong, A. Song and J. Hao, Colloids Surf., B, 2020, 185, 110567 CrossRef CAS.
- X. Q. Dou and C. L. Feng, Adv. Mater., 2017, 29, 1604062 CrossRef.
- P. Worthington, D. J. Pochan and S. A. Langhans, Front. Radiat. Oncol., 2015, 5, 92 Search PubMed.
- X. Yao, Y. Liu, J. Gao, L. Yang, D. Mao, C. Stefanitsch, Y. Li, J. Zhang, L. Ou and D. Kong, Biomaterials, 2015, 60, 130–140 CrossRef CAS.
- A. S. Mao and D. J. Mooney, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14452–14459 CrossRef CAS.
- A. H. Badawi and T. J. Siahaan, Clin. Immunol., 2012, 144, 127–138 CrossRef CAS.
- J. Wiesner and A. Vilcinskas, Virulence, 2010, 1, 440–464 CrossRef.
- A. J. Barbosa, A. R. Oliveira and A. C. Roque, Trends Biotechnol., 2018, 36, 1244–1258 CrossRef CAS.
- H. Do, N. Makthal, A. R. VanderWal, M. O. Saavedra, R. J. Olsen, J. M. Musser and M. Kumaraswami, Nat. Commun., 2019, 10, 1–14 CrossRef CAS.
- Y. Tsume, A. Drelich, D. Smith and G. Amidon, Molecules, 2017, 22, 1322 CrossRef.
- F. Raza, Y. Zhu, L. Chen, X. You, J. Zhang, A. Khan, M. W. Khan, M. Hasnat, H. Zafar and J. Wu, Biomater. Sci., 2019, 7, 2023–2036 RSC.
- G. B. Qi, Y. J. Gao, L. Wang and H. Wang, Adv. Mater., 2018, 30, 1703444 CrossRef.
- N. Habibi, N. Kamaly, A. Memic and H. Shafiee, Nano Today, 2016, 11, 41–60 CrossRef CAS.
- Z. Jiang, J. Guan, J. Qian and C. Zhan, Biomater. Sci., 2019, 7, 461–471 RSC.
- Q. Wang, X. Zhang, J. Zheng and D. Liu, RSC Adv., 2014, 4, 25461–25469 RSC.
- C. Tang, A. M. Smith, R. F. Collins, R. V. Ulijn and A. Saiani, Langmuir, 2009, 25, 9447–9453 CrossRef CAS.
- A.-P. Xu, P.-P. Yang, C. Yang, Y.-J. Gao, X.-X. Zhao, Q. Luo, X.-D. Li, L.-Z. Li, L. Wang and H. Wang, Nanoscale, 2016, 8, 14078–14083 RSC.
- J. B. Matson, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2012, 8, 3586–3595 RSC.
- C. Veerman, K. Rajagopal, C. S. Palla, D. J. Pochan, J. P. Schneider and E. M. Furst, Macromolecules, 2006, 39, 6608–6614 CrossRef CAS.
- N. A. Dudukovic and C. F. Zukoski, Langmuir, 2014, 30, 4493–4500 CrossRef CAS.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015–2022 CrossRef CAS.
- C. Tang, R. V. Ulijn and A. Saiani, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 111 CrossRef.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS.
- I. Alfonso, M. Bolte, M. I. Burguete and S. V. Luis, Crystals, 2011, 1, 163–170 CrossRef.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- I. Sasselli, C. Pappas, E. Matthews, T. Wang, N. Hunt, R. Ulijn and T. Tuttle, Soft Matter, 2016, 12, 8307–8315 RSC.
- Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15029 CrossRef CAS.
- C. Han, J. Zhou, C. Liang, B. Liu, X. Pan, Y. Zhang, Y. Wang, B. Yan, W. Xie and F. Liu, Biomater. Sci., 2019, 7, 2920–2933 RSC.
- L. A. Carpino, D. Sadat-Aalaee, H. G. Chao and R. H. DeSelms, J. Am. Chem. Soc., 1990, 112, 9651–9652 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- C. Tang, R. V. Ulijn and A. Saiani, Langmuir, 2011, 27, 14438–14449 CrossRef CAS.
- T. Russo, U. D’Amora, A. Gloria, M. Tunesi, M. Sandri, S. Rodilossi, D. Albani, G. Forloni, C. Giordano and A. Cigada, Procedia Eng., 2013, 59, 233–239 CrossRef CAS.
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- M. H. Chen, J. J. Chung, J. E. Mealy, S. Zaman, E. C. Li, M. F. Arisi, P. Atluri and J. A. Burdick, Macromol. Biosci., 2019, 19, 1800248 CrossRef.
- M. H. Chen, L. L. Wang, J. J. Chung, Y.-H. Kim, P. Atluri and J. A. Burdick, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- A. Vedadghavami, F. Minooei, M. H. Mohammadi, S. Khetani, A. R. Kolahchi, S. Mashayekhan and A. Sanati-Nezhad, Acta Biomater., 2017, 62, 42–63 CrossRef CAS.
- R. A. Marklein and J. A. Burdick, Soft Matter, 2010, 6, 136–143 RSC.
- A. Banerjee, M. Arha, S. Choudhary, R. S. Ashton, S. R. Bhatia, D. V. Schaffer and R. S. Kane, Biomaterials, 2009, 30, 4695–4699 CrossRef CAS.
- N.-C. Cheng, B. T. Estes, H. A. Awad and F. Guilak, Tissue Eng., Part A, 2009, 15, 231–241 CrossRef CAS.
- S. S. Abolmaali, A. Tamaddon, H. Najafi and R. Dinarvand, J. Inorg. Organomet. Polym. Mater., 2014, 24, 977–987 CrossRef CAS.
- R. Vegners, I. Shestakova, I. Kalvinsh, R. M. Ezzell and P. A. Janmey, J. Pept. Sci., 1995, 1, 371–378 CrossRef CAS.
- D. J. Adams, L. M. Mullen, M. Berta, L. Chen and W. J. Frith, Soft Matter, 2010, 6, 1971–1980 RSC.
- T. W. Greene and P. G. Wuts, Protective groups in organic synthesis, Wiley, 1999 Search PubMed.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- D. Myers, Surfactant science and technology, John Wiley & Sons, 2020 Search PubMed.
- W. Y. Seow and C. A. Hauser, Mater. Today, 2014, 17, 381–388 CrossRef CAS.
- H. Arakawa, K. Takeda, S. L. Higashi, A. Shibata, Y. Kitamura and M. Ikeda, Polym. J., 2020, 1–8 Search PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- F. S. Sarvestani, M. A. Zare, F. Saki, F. Koohpeyma, I. H. Al-Abdullah and N. Azarpira, Iran. J. Basic Med. Sci., 2020, 23, 214 Search PubMed.
- T. Pirjali, N. Azarpira, M. Ayatollahi, M. Aghdaie, B. Geramizadeh and T. Talai, Int. J. Organ. Transplant. Med., 2013, 4, 111 CAS.
- M. Dominici, K. Le Blanc, I. Mueller, I. Slaper-Cortenbach, F. Marini, D. Krause, R. Deans, A. Keating, D. Prockop and E. Horwitz, Cytotherapy, 2006, 8, 315–317 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS.
- F. Barneh, M. Moshayedi, H. Mirmohammadsadeghi, S. Haghjooy-Javanmard, A. M. Sabzghabaee and S. Badri, Dis. Markers, 2013, 35, 857895 Search PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19 CrossRef CAS.
- M. Shigematsu, Carbohydr. Res., 1996, 292, 165–172 CrossRef CAS.
- F. Sabet Sarvestani, M. A. Zare, F. Koohpeyma, I. H. Al-Abdullah and N. Azarpira, Iran. J. Basic Med. Sci., 2020, 23, 214–223 Search PubMed.
- U. Goyal, C. Jaiswal and M. Ta, Stem Cells, 2018, 8, 2735–2740 Search PubMed.
- H. Najafi, S. S. Abolmaali, B. Owrangi, Y. Ghasemi and A. M. Tamaddon, Macromol. Res., 2015, 23, 618–627 CrossRef CAS.
- D. Gerlier and N. Thomasset, J. Immunol. Methods, 1986, 94, 57–63 CrossRef CAS.
- A. Raza and C.-C. Lin, J. Visualized Exp., 2012, e50081 Search PubMed.
- A. Scelsi, B. Bochicchio, A. Smith, V. L. Workman, L. A. Castillo Diaz, A. Saiani and A. Pepe, J. Biomed. Mater. Res., Part A, 2019, 107, 535–544 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sm01299h |
| This journal is © The Royal Society of Chemistry 2021 |