
A self-humidifying proton exchange membrane embedded with phosphonic acid-functionalized mesoporous silica nanoparticles that has excellent dispersion and water retention†
Henghui
Huang
a,
Shaoyi
Xu
ab,
Li
Zhang
c,
Jiantao
Fan
 *ab,
Hui
Li
*a and
Haijiang
Wang
c
*ab,
Hui
Li
*a and
Haijiang
Wang
c
aDepartment of Materials Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China
bAcademy for Advanced Interdisciplinary Studies, Southern University of Science and Technology, Shenzhen, 518055, China
cDepartment of Mechanical and Energy Engineering, Southern University of Science and Technology, Shenzhen, 518055, China
First published on 11th November 2020
Abstract
Nanomaterials with a uniform size, large surface area, high adsorption capacity, and excellent dispersion are critical to proton conduction and cell performance when functionalized and incorporated into a proton exchange membrane. We designed and fabricated a novel composite PEM based on vinylphosphonic acid-functionalized mesoporous silica nanoparticles (VPA-MSNs), with the aim of improving the cell performance and proton conductivity of a perfluorosulfonic acid (PFSA) membrane under low relative humidity (RH) conditions. The experimental results show that the VPA-MSN(–NH2)/PFSA composite membrane provides a proton conductivity of up to 23.2 mS cm−1 at 30% RH and up to 9.6 mS cm−1 at 10% RH, which are respectively 3.2 and 8.6 times higher than those of a pristine PFSA membrane, and 5.6 and 10.4 times higher than those of a commercial Nafion membrane. In addition, the VPA-MSN(–NH2)/PFSA composite membrane exhibits superior fuel cell performance and low ohmic resistance at low relative humidity. Improvements in water absorption/retention, proton conductivity, and single-cell performance resulted from these effectively hydrophilic surfaces and mass transfer pathways, as the mesoporous structure and abundant functional groups in the membrane enhanced the connectivity of the hydrophilic zone, forming new proton transport channels. The incorporation of VPA-MSNs into the PFSA matrix also enhanced the water retention properties and mechanical strength, and adjusted the organic–inorganic interface and hydrophilic ionic domains inside the membranes simultaneously. The results indicate that this new type of VPA-MSN(–NH2)/PFSA composite membrane is promising for developing PEMs that tolerate variations in humidity.
1. Introduction
The water sorption behavior of proton exchange membranes is one of the most important factors affecting their structural,1 transport, and electrochemical properties. Materials with good water retention and excellent proton transport properties are urgently needed for proton exchange membranes (PEMs).2 Membranes based on PFSA yield excellent proton conduction, but adequate water is required to maintain PEMs in a hydrated state, making it a poor candidate for operating under low-humidity conditions,3 because proton conduction deteriorates severely.To enhance a membrane's water retention and adjust its water content, materials with high water sorption and low water release have been introduced into PEMs. Inorganic nanosized fillers such as SiO2, ZrO2, and TiO2 have abundant hydrophilic groups that retain a large number of water molecules through hydrogen bonding, ensuring that the PEM has a sufficiently high water content;4 therefore they have broad applications under low humidity operating conditions. However, the poor dispersion and low proton conductivity limited SiO2 nanoparticle application in high performance PEMs.5
The high surface free energy of SiO2 nanoparticles is the main reason for their poor dispersion. A strategy using electrostatic repulsion or steric hindrance has shown the potential to address these problems through functionalizing their surface, effectively preventing particle agglomeration and improving their dispersibility.6 In another approach, when more and stronger carrier sites (conducting groups), which are crucial determiners of proton transfer, were introduced into the surface of nanoparticles, proton conductivity was enhanced. To date, various SiO2-based materials with various functional groups have been incorporated into membranes to improve the proton conduction and cell performance of proton exchange membranes under low humidity and high temperature conditions. Amiinu et al. prepared a Nafion hybrid membrane including silica functionalized with imidazole groups using the sol–gel method.7 They claimed that the synthesized membrane had better thermo-mechanical properties and proton conductivity in a non-humidified environment. Kamol et al. prepared composite membranes by dispersing fluorinated polyoxadiazole oligomer (ODF)-functionalized silica nanoparticles in a Nafion matrix. A physical crosslinking effect and better water retention were observed due to the inclusion of these functionalized nanoparticles.8 Wang et al. prepared a type of bio-inspired Nafion membrane (Bio-Nafion) composed of biofunctional SiO2 (Bio-SiO2) nanofibers and a Nafion matrix; their results showed that the introduction of Bio-SiO2 nanofibers into composite membranes significantly improved water uptake, dimensional stability, and proton conductivity at low relative humidity.6 Sangaraju et al. synthesized new sulfonated silica (SSA) using tetraethyl orthosilicate and chlorosulfuric acid through calcination, which was used as a filler to create Nafion–SSA composite membranes for use in PEMFCs under low relative humidity (RH) conditions. High water uptake and ion exchange capacity values were observed, and the Nafion–SSA composite membrane showed better proton conductivity than recast Nafion.9 Zhao et al. investigated the effects of the –NH2 group and the –PO3H2 group in submicrospheres of silica as a filler for a Nafion membrane. Their work showed that –PO3H2 groups will perform well at low relative humidity. They mentioned that the lack of hydration can be resolved by adding a large number of –PO3H2 groups to the filler.10 The presence of silica in a Nafion matrix can help to overcome the limitations of Nafion by improving proton conductivity at elevated temperature and low RH due to the excellent water retention capacity of silica,11 which has significantly improved fuel cell performance. Table 1 shows a summary of silica application in Nafion membranes.
| Embedded nano-material | Proton conductivity (mS cm−1) | Water uptake (%) | Reference |
|---|---|---|---|
| Amino acid functionalized SiO2 nanofiber | 140.4 (20 °C, 100RH), 242.4 (80 °C, 100RH), in-plane | 47.6% (10 wt%, 20 °C) | 7 |
| Amino-acid functionalized hollow mesoporous silica microspheres | 119 (30 °C, 100RH), 210 (80 °C, 100RH), in-plane | 41.2% (4 wt%, 30 °C) | 12 |
| Sulfonated silica | 111 (100% RH, 80 °C), 84 (100% RH, 30 °C), through-plane | 24.9% (5 wt%, 25 °C) | 9 |
| Fluorinated polyoxadiazole oligomer (ODF)-functionalized silica nanoparticles | 65 (100% RH, 80 °C), 4.0 (15%, 80 °C), through-plane | 48.2% (5 wt%, 25 °C) | 8 |
| Silica functionalized with imidazole groups | 105 (100% RH, 80 °C), 0.65 (anhydrous condition, 80 °C), through-plane | — | 13 |
| The –NH2 group and the –PO3H2 group in submicrospheres of silica | 207(100% RH, 80 °C), 15 mS (40% RH, 80 °C), through-plane | 41.2% (5 wt%, 25 °C) | 10 |
| Phosphonic acid-functionalized mesoporous silica nanoparticles | 136 (90% RH, 80 °C), 19.6 (30% RH, 80 °C), 9.2 (10% RH, 80 °C), through-plane | 49.5 (10 wt%, 25 °C) | This study |
Among these potentially promising water-retentive materials for PEM applications at low RH, phosphonic acid-based materials meet the requirements. Phosphoric acid groups act not only as proton donors but also as proton acceptors, and they possess a relatively high dielectric constant and hydration energy,14 so phosphonic acid-based materials have higher water absorption and retention capacity,15 as well as the ability to transport protons at low humidity; hence, phosphonic acid-functionalized SiO2 nanoparticles may have better water-retention capacity and proton conductivity than unmodified ones. When phosphoric acid groups with the same type of electric charge were introduced into the surface of nanoparticles, the mutual repulsion among nanoparticles and between the nanoparticles and the PFSA molecular chain was enhanced,16 endowing them with excellent dispersion performance in a PFSA membrane.
The specific surface area and size of nanoparticles incorporated into a proton exchange membrane are critical to the number of proton conduction sites and proton conduction paths.17 Mesoporous materials have larger specific surface areas because their nanoparticles contain many micropores with diameters in the range of 2–50 nm. The size of synthesized nanoparticles is closely related to their precursors and surfactants. When precursors possess abundant ionic groups, they repel each other, making it difficult for nano-microspheres to agglomerate and thereby keeping the prepared nanoparticles small.18 The size and amount of micropores have a significant impact on the water-retention of composite membranes, the water in micropores has a lower saturated vapor pressure value (according to the Kelvin equation) and therefore materials with smaller pores have higher water retention.
We therefore designed and synthesized mesoporous silica nanoparticles (MSNs) with different functional groups and micropores, to study the types and number of functional groups as well as how the sizes and number of their mesopores influenced the nanoparticles' dispersion, water retention, mechanical properties, proton conduction, and single-cell performance of the composite membrane. Abundant water-sorption groups, numerous smaller size pores, and excellent dispersion ensured that the prepared VPA-MSN(–NH2)/PFSA composite membranes had better proton conductivity and single-cell performance than an unmodified PFSA membrane, and lower impedance under low-humidity conditions. The experimental results show that the prepared VPA-MSN(–NH2)/PFSA composite membrane in this paper has great potential for application at low relative humidity.
2. Experimental
2.1 Materials and chemicals
Vinylphosphonic acid (VPA, 98%), tetraethyl orthosilicate (TEOS, 99.5%), (3-aminopropyl)triethoxysilane (APTES), cetyltrimethylammonium bromide (CTAB), sodium hydroxide (NaOH), Triton-100, and aqueous ammonia (NH3·H2O) were purchased from Shanghai Macklin Biochemical Co., Ltd. (China). A 25% Aquivion® dispersion was used as the PFSA matrix.2.2 Synthesis of MSNs with –OH and –NH2 groups
The mesoporous silica nanoparticles were prepared by the Stöber method.19 First, 3.0 g cetyltrimethylammonium bromide (CTAB), 0.5 g Triton X-100, and 2 mL aqueous ammonia were dissolved in 200 mL aqueous ethanol solution (160 mL H2O and 40 mL ethanol). Then 2 mL TEOS or APTES was added under vigorous stirring for 12 h at 80 °C. At the end of the reaction, a white suspension had formed. Next, a white powder was obtained by centrifuging the solution and washing the white precipitate with excess DI water and ethanol. The remaining surfactant template was removed by reflux for 12 h at 80 °C in a mixed solution containing 10 mL HCl (37%) and 100 mL ethanol. Finally, the resulting white solid was dried under vacuum at 70 °C overnight.2.3 Synthesis of phosphonic acid-functionalized MSNs
A large number of hydroxyl or amino groups were on the surface of the newly prepared MSNs, and phosphonic acid groups could be introduced into the MSNs through either esterification or Michael addition.20 Under basic conditions, amino groups can be converted to phosphonate groups via Michael addition by adding vinylphosphonic acid and excess NaOH. We therefore dispersed the prepared MSNs, which contained –NH2 groups, in a solvent composed of 50 wt% C2H5OH and 50 wt% water. Then, in an ice-water bath, we added vinylphosphonic acid (VPA-Na) and 1 mol L−1 NaOH solution dropwise into the solution. The reaction lasted 12 hours, after which the VPA-MSNs were obtained by acidifying VPA(Na)-MSNs using 0.1 mol L−1 H2SO4.2.4 Preparation of the VPA-MSN(–NH2)/PFSA composite membrane
PFSA and composite membranes with various nano-sized fillers were prepared using a precision coating method. In each case, the MSNs and a small amount of Triton X-100 were dispersed in a mixed solution of ethanol, isopropanol, and water, and then the PFSA dispersion was added into the mixture, forming a 10 wt% polymer dispersion. This was cast with a doctor blade, an ePTFE web was used as an enhancement layer, and then the wet membrane was dried slowly at 60 °C.21 Finally, the membrane was thermally treated at 195 °C and annealed for 40 min. The thickness of all the membranes was 15 ± 1 μm.2.5 Characterization of VPA-MSN(–NH2) and VPA-MSN(–NH2)/PFSA composite membranes
The molecular structure of VPA-MSN(–NH2) was characterized using a Raman spectrometer, and the surface hydrophilicity of all the membranes was characterized with a contact angle analyzer. The 13C-NMR (AM 400 MHz Nuclear Magnetic Resonance, Bruker, USA) spectrum of the APTES and VPA-APTES was recorded with D2O and CD3OD as a mixed solvent. The particles' zeta potential, size, and distribution were characterized using an SZ-100 Dynamic Light Scattering Analyzer (Horiba).The membranes' water uptake was calculated from the weight difference between the dry membrane and the wet membrane (eqn (2.1)), and their swelling ratio was tested by measuring changes in the length and thickness in dry and wet states (eqn (2.2)). The dry and wet membranes were prepared as previously reported.22
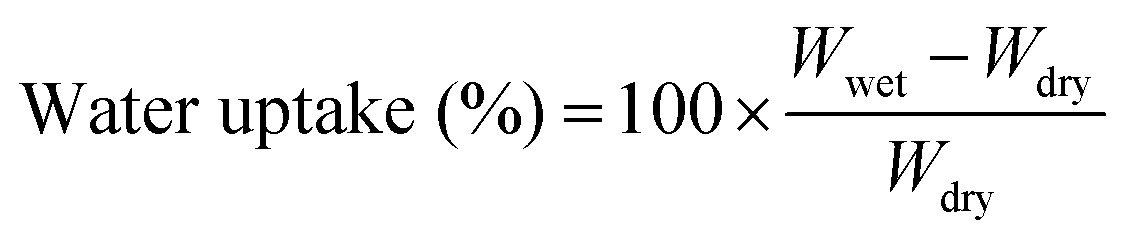 | (2.1) |
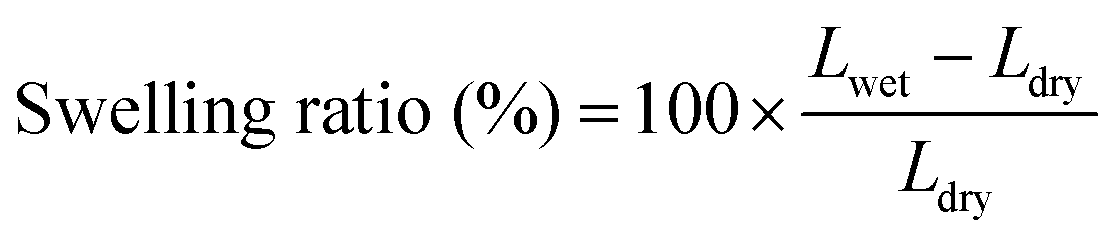 | (2.2) |
Water release testing was conducted on the PFSA membrane and VPA-MSN(–NH2)/PFSA composite membranes at 60 °C and 10% RH. The membranes were weighed (W) at various test times, and the water retention ratio was calculated with eqn (2.3):
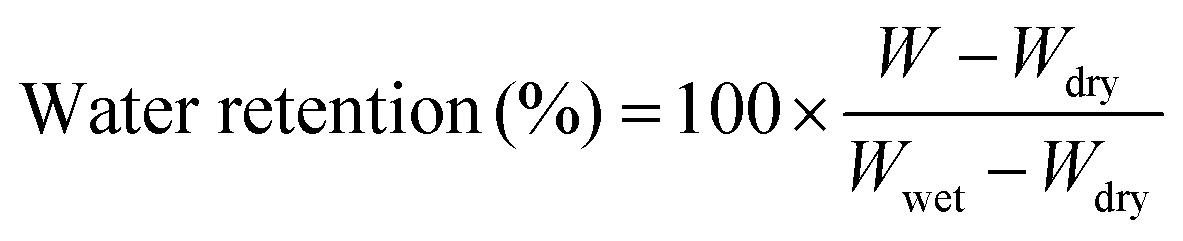 | (2.3) |
The mechanical properties of the PEMs were studied using the ASTM D882-02 standard test method. Membrane samples were die-cut into dumbbell-shaped specimens (40 mm × 5 mm gauge area), and then the specimens were tested with an Instron 5866 universal tester, using a tensile rate of 10 mm min−1 and at 25 ± 1 °C.23
2.6 SAXS test
The nanostructures of the membrane were probed by small-angle X-ray scattering (SAXS) using a Rigaku D/Max 2500 v/Pc (CuK 40 kV, 200 mV) at the rate of 1° min−1 in the range of 0.5–10°. The scattering spectra were plotted as a function of the scattering vector q, defined as q = (4π(sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ)/λ), where θ is the scattering angle.
θ)/λ), where θ is the scattering angle.
2.7 Proton conductivity measurement
Proton conductivity testing was carried out in a chamber with constant temperature and humidity, using a 740 Membrane Test System (Scribner Associates, MTS) with an impedance analysis interface and ZPlot® software. The membranes were preconditioned for 120 min at each test's RH, and then the pre-treated membranes were measured with an impedance analysis interface and analyzed using ZPlot. The MTS was used to measure the through-thickness conductivity (σ in S cm−1) of the PEMs, determined from the resistance (R, Ω), the membrane's thickness (L, cm), and the cross-sectional area through which the current passed (A, cm2), using eqn (2.4): | (2.4) |
2.8 Single-cell performance evaluation
The single-cell performance of the membranes was tested by assembling PEMs into membrane electrode assemblies (MEAs). MEAs based on PFSA, Gore 740, and PFSA/PEIMPA membranes were prepared as follows. First, the thermal transfer method was used to prepare the catalyst-coated membrane (CCM), the catalyst layers (CLs), and the PEM by hot-pressing for 3 min at 170 °C and 65 psi. The cathode and anode catalyst layers were prepared from 47.6 wt% commercial Pt/C and an Aquivion ionomer (EW = 790). The Pt loading was 0.30 mg cm−2 at the cathode CL and 0.1 mg cm−2 at the anode CL. The MEA was formed using two sheets of gas diffusion layers (GDLs) and the as-prepared CCM, and its active area was 50 cm2.An 885 Fuel Cell Potentiostat (Scribner Associates) was used to evaluate MEAs containing various PEMs: the PFSA, Gore 740, and PFSA/PEIMPA composite membranes. The operating temperature of the single-cell system was controlled by using a temperature control system, and the RH of the feed gas was controlled by using a humidifier. The feed gases (hydrogen and air) had stoichiometries of 1.3 and 2.0, respectively. The cell voltage at various current densities (scanned from 0 to 2 A cm−2) was tested, yielding polarization curves and power density curves. Before testing, the MEA needed to be activated at a high current density until the cell performance stabilized.24
The EIS of the MEAs was carried out using a frequency analyzer after the polarization curves were obtained. The scanning frequency of the EIS measurements was 106 to 0.1 Hz at a current density of 1.0 A cm−2.
3. Results and discussion
3.1 Preparation of MSN(–OH), VPA(–NH2) and VPA-MSN(–NH2)/PFSA
A schematic of the procedure for synthesizing the MSNs with –NH2 and –OH groups is shown in Fig. 1. Under alkaline conditions, the precursors (TOES and APTES) were hydrolyzed, dehydrated, and condensed, forming the nano-microsphere structure. Next, the template agent (CTAB) was adsorbed and dispersed in the nanospheres. Under heated reflux conditions, the template agent was dissolved in a strong acid solution (HCl), resulting in the formation of mesoporous silica nanoparticles. Because the –OH groups of MSN(–OH) would have been difficult to replace via esterification with HEDP, we did not choose HEDP-MSN(–OH) to fabricate the composite membranes.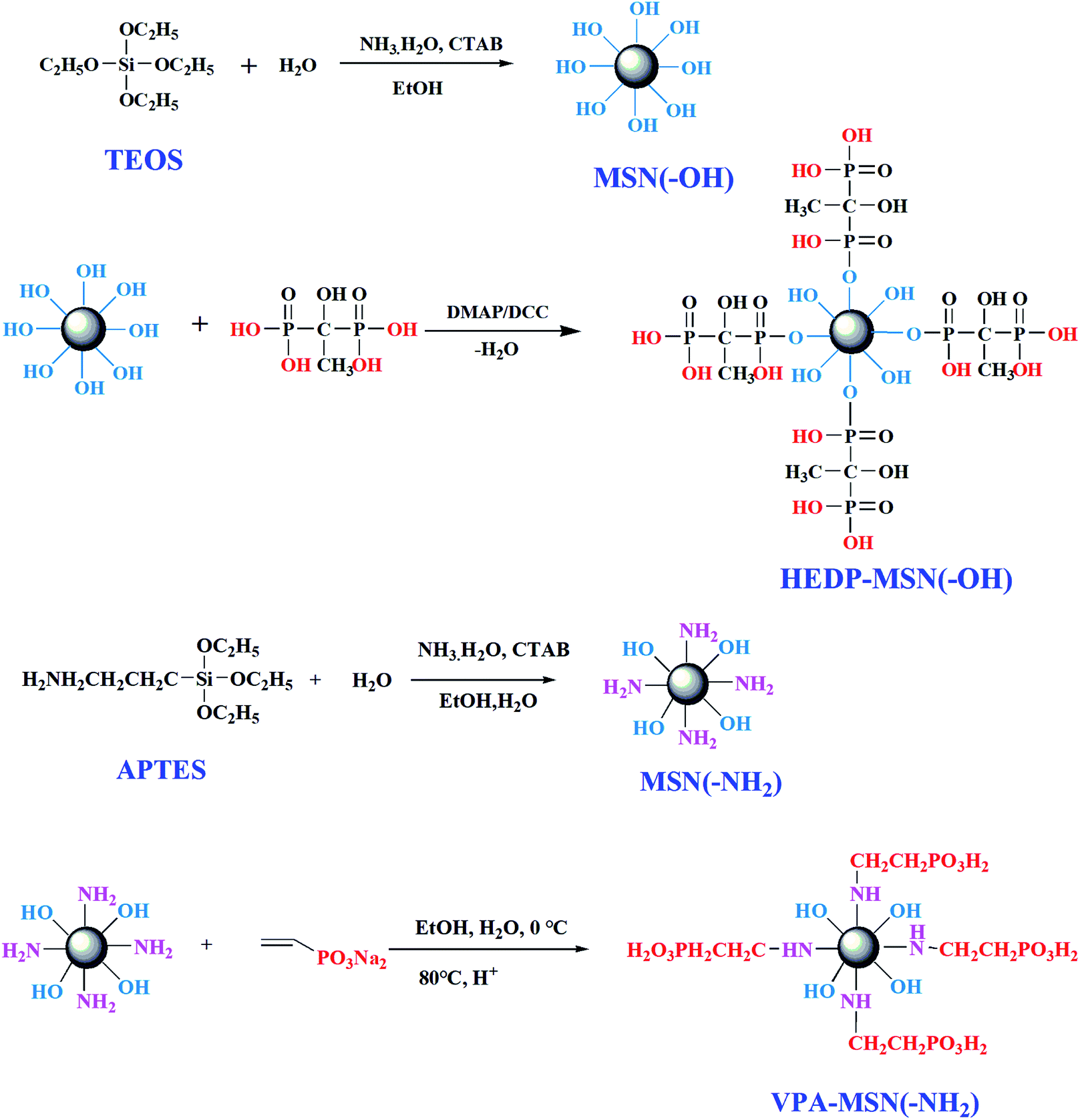 | ||
| Fig. 1 Procedure for synthesizing the MSNs with –OH or –NH2 groups and the phosphonic acid-functionalized MSNs. | ||
The structure of MNS(–OH), MSN(–NH2) and VPA-MSN(–NH2) was characterized using Raman spectra. As shown in Fig. 2(a), the peaks at 966 cm−1 and 2940 cm−1, which indicate the stretching vibrations of –NH– and –NH2, are weaker for PEIMPA than for MNS(–NH2), indicating that most of the amino groups were reacted and replaced. The peaks at 657 cm−1 and 820 cm−1 represent the bending vibrations of P–O, and the peak at 1060 cm−1 represents the bending vibration of P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, indicating that –PO3H2 groups were introduced into MNS(–NH2), some of the –NH2 groups were functionalized as phosphonic acid, and the phosphonic acid-functionalized MSNs were appropriately synthesized.
O bonds, indicating that –PO3H2 groups were introduced into MNS(–NH2), some of the –NH2 groups were functionalized as phosphonic acid, and the phosphonic acid-functionalized MSNs were appropriately synthesized.
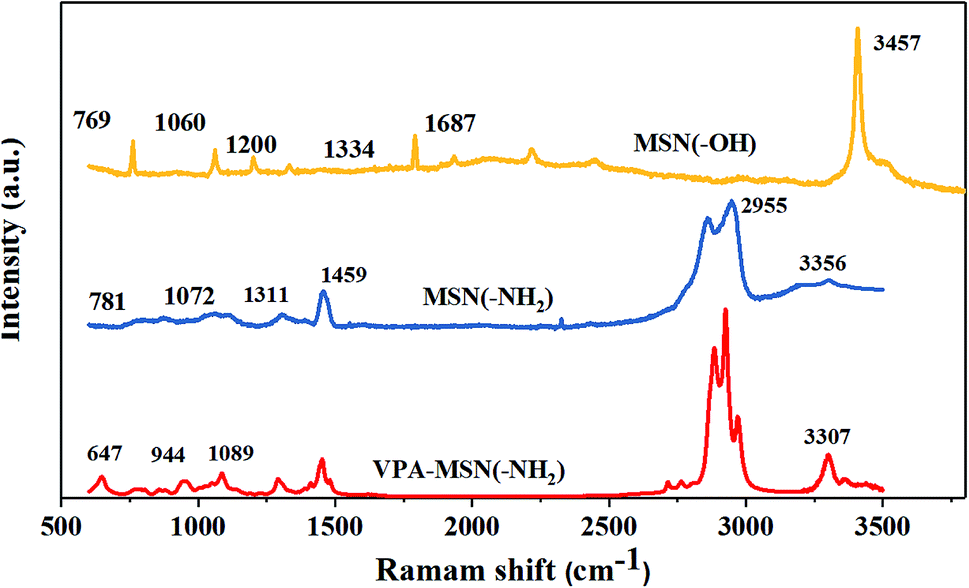 | ||
| Fig. 2 Raman spectra of the MSN(–OH), MSN(–NH2) and the VPA-MSN(–NH2). | ||
Fig. S1(a and b)† illustrates the 13C NMR spectrum of MSN(–NH2) and VPA-MSN(–NH2), and Fig. S2(a–c)† shows the 13C NMR spectrum of the MSN(–NH2), and a mixture of MSN(–NH2) and VPA at the beginning of the reaction and after 6 hours of reaction (the 12 hour reaction product mentioned in Section 2.3 is not used for the NMR test because of the poor solubility of final products for the NMR test). Compared with those for only the mixture of MSN(–NH2) and VPA, the intensities of the peaks at 131.2 and 137.4 ppm (assigned to CH2CH) have obviously decreased and new peaks have appeared at 58.7 and 41.2 ppm (assigned to –CH2–CH–NH–), implying that the –NH2 groups have reacted with the CC bonds, and CC bonds were converted to –C–C– bonds. Moreover, the peak at 41.2 ppm (assigned to –CH2–PO3H2) has appeared for obtained VPA-MSN(–NH2), indicating that the –PO3H2 groups were introduced. Besides, compared with the 1H NMR spectrum of MSN(–NH2) and the VPA-MSN(–NH2), the results show the –NH2 content decreased and new –PO3H2 appeared. Therefore, the 1H NMR, 13C NMR and Raman spectra demonstrated that the reaction between nucleophile NH2 and a double bond is successful and the prepared VPA-MSN(–NH2) was obtained.
3.2 Dispersion performance and size of phosphonic acid-functionalized MSNs
The dispersion performance of MSNs with –OH or –NH2 groups and of phosphonic-acid functionalized MSNs was characterized using the zeta potential of nanoparticles dispersed in an ethanol–water solution. A higher zeta potential meant more electric charge on the nanoparticles' surface, making it easier for them to disperse. As shown in Fig. 3, the MSNs with –OH and –NH2 groups had a lower zeta potential, and when the solution was allowed to stand, the particles tended to aggregate and slowly precipitate. But the phosphonic acid-functionalized MSNs had a higher zeta potential because their surface had abundant –PO3H2 groups with the same electric charge, so the repulsion between the nanoparticles enabled them to stably disperse in the solution.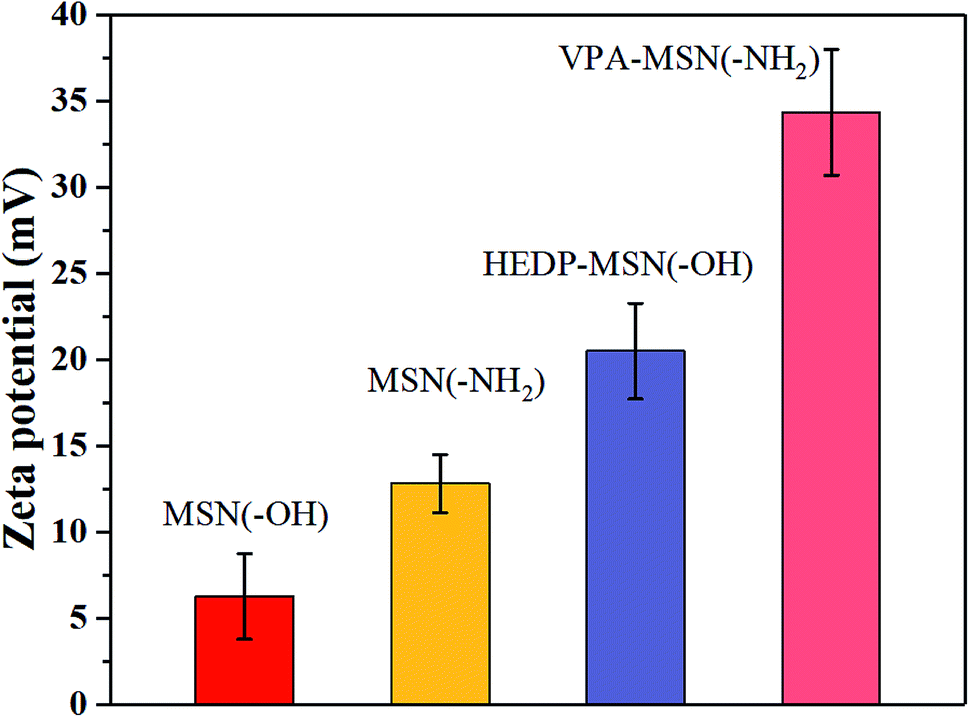 | ||
| Fig. 3 The zeta potential of the MSNs with various functional groups, dispersed in an ethanol–water solution. | ||
The particle size and distribution of the MSNs with –OH, –NH2, and –PO3H2 groups were determined by dynamic light scattering (DLS). The effective diameter and polydispersity of the MSNs are presented in Fig. 4(a and b). The MSNs with –PO3H2 groups were smaller and had a more uniform distribution than those with –OH. The size of the MSNs with –PO3H2 groups was also observed using scanning electron microscopy (SEM) and transmission electron microscopy (TEM), and the surface morphology of the nanospheres was clearly observed. As can be seen in the SEM and TEM images (Fig. 3(c and d)), the diameter of most microspheres was in the range of 30–80 nm, and the nanoparticles' distribution was uniform.
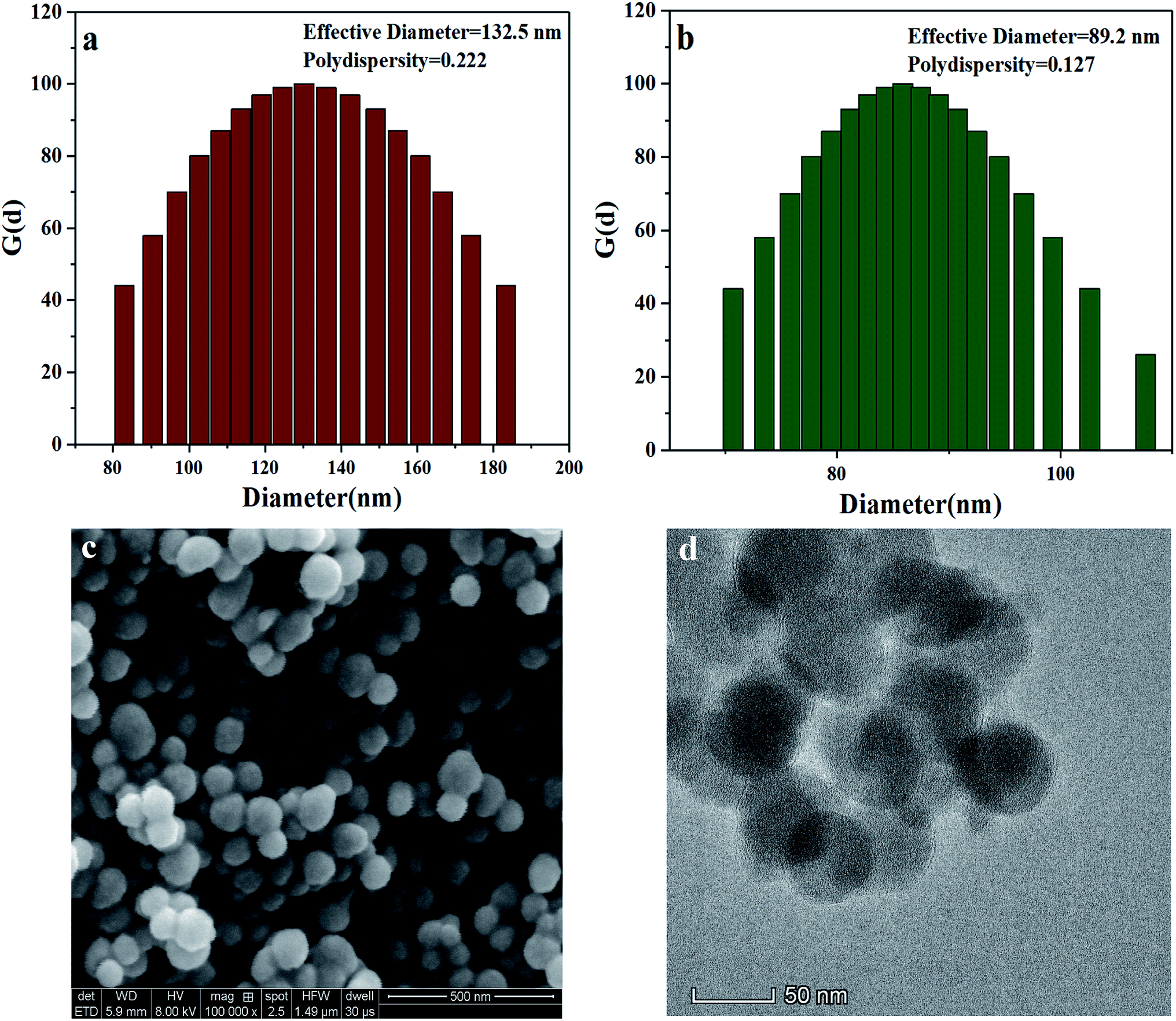 | ||
| Fig. 4 Effective diameter and polydispersity of (a) MSN(–OH) and (b) VPA-MSN(–NH2); (c) SEM and (d) TEM images of VPA-MSN(–NH2). | ||
Because of their excellent dispersion and small size, the VPA-MSN(–NH2) nanoparticles had a larger adsorption capacity and specific surface area.25 The nitrogen adsorption–desorption isotherms for the MSNs are shown in Fig. 5. Based on the BJH theory, we tested and calculated the Brunauer–Emmet–Teller (BET) surface area, adsorption capacity, total pore volume, and mean pore diameter of the MSNs (see Table 1). The MSN(–OH) had a BET surface area of 329.66 m2 g−1, compared with up to 1092.4 m2 g−1 for the VPA-MSN(–NH2). It is known that functional groups exist mainly on the surface of nanospheres, so MSNs possess more active sites for functionalization or proton transfer when the particles have a larger specific surface area. The volume adsorbed in the nitrogen adsorption–desorption isotherm of the MSN nanometer microspheres increased sharply at a relative pressure (p/p0) of approximately 0.50, indicating capillary condensation in the microspheres' uniform structure. Although the pore size distributions (Fig. 4(c and d)) and mean pore diameters (Table 2) were similar for MSN(–OH) and VPA-MSN(–NH2), the proportion of micropores (less than 2 nm) in MSN(–NH2) was greater than that in MSN(–OH), indicating that the presence of the amino groups enhanced the dispersion of the template agent (CTAB).
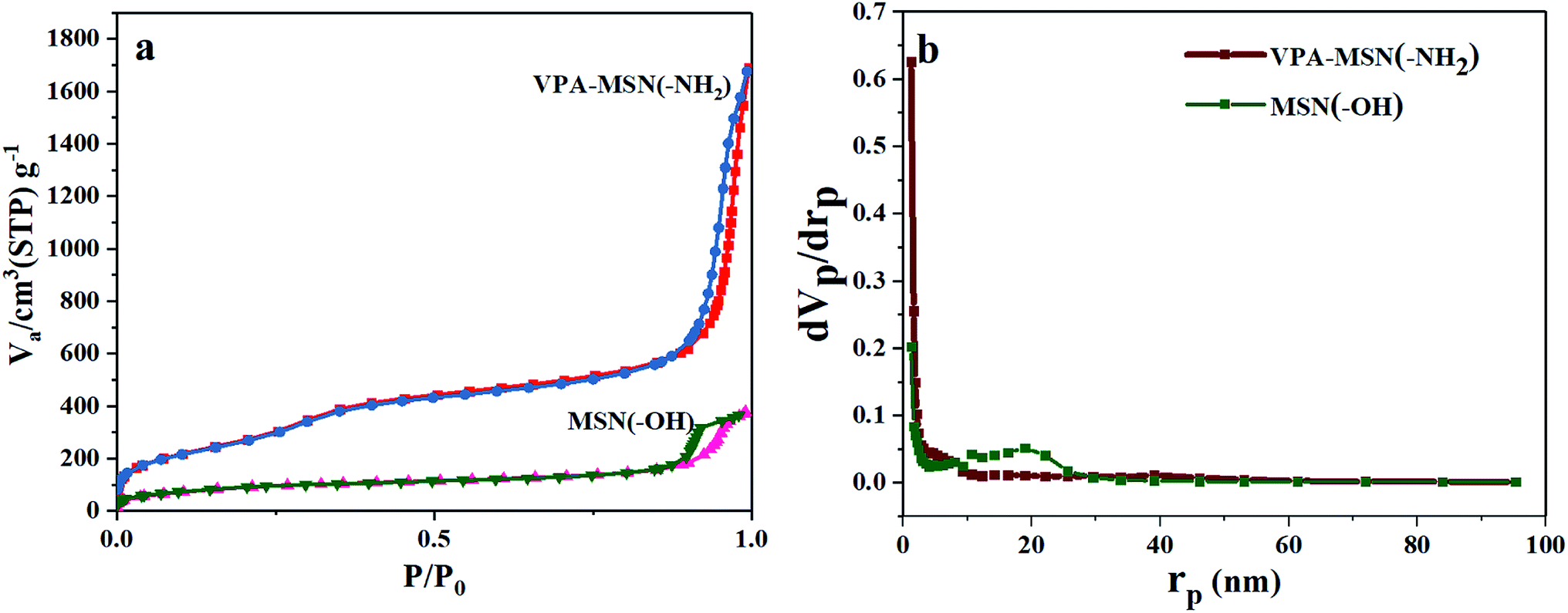 | ||
| Fig. 5 (a) BET adsorption isotherms (N2) and (b) pore size distributions of MSN(–OH) and VPA-MSN(–NH2). | ||
| Sample | BET s.a. (m2 g−1) | V m (cm3(STP) g−1) | Total pore volume (p/p0 = 0.990) | Mean pore diameter (nm) |
|---|---|---|---|---|
| MSN | 329.66 | 75.741 | 0.5816 | 7.0573 |
| VPA-MSN(–NH2) | 1092.4 | 250.99 | 2.4787 | 6.0762 |
The cross-sectional and surface SEM images of MSN(–OH)/PFSA and VPA-MSN(–NH2)/PFSA are shown in Fig. 6. The VPA-MSN(–NH2)/PFSA composite membranes were smaller and had excellent dispersion. The nanoparticles were uniformly distributed inside the membrane, no defects or particle aggregates were detected on the surface or in the cross-section, and they were slightly larger than pristine nanoparticles. But when the MSN(–OH) was incorporated into the composite membrane, it showed particle aggregates in the cross-sectional and surface SEM images. The EDS mapping for the surface and cross-section of MSN(–OH)/PFSA and VPA-MSN(–NH2)/PFSA composite membranes also showed that the additive (VPA-MSN(–NH2)) was uniformly distributed throughout the membrane (see Fig. 7). The incorporated particles' large size and uneven dispersion weakened the membrane's water retention performance and lengthened the proton conduction paths, hampering proton conductivity and failing to improve the PEM's single-cell performance.26
 | ||
| Fig. 6 SEM images of the VPA-MSN(–NH2)/PFSA composite membrane: (a) surface, and (b) cross-section. SEM images of the MSN(–OH)/PFSA composite membrane: (c) surface and (d) cross-section. | ||
 | ||
| Fig. 7 EDS mapping for the surface of the (a) VPA-MSN(–NH2)/PFSA composite membrane and (b) MSN(–OH)/PFSA, and (c and d) cross-section of the VPA-MSN(–NH2)/PFSA composite membrane. | ||
3.3 Mechanical properties and dimensional stability of the membranes
It is well known that a higher water content is helpful for transporting protons but is detrimental to a membrane's mechanical properties and structural stability. The VPA-MSN(–NH2) nanoparticles had a rigid structure and abundant active groups, and the membrane's mechanical properties were enhanced by these two key factors. The mechanical properties of the membranes were tested as described in Section 2.5, and the corresponding stress–strain curves are presented in Fig. 8. The results indicated that the composite membrane with more VPA-MSN(–NH2) nanoparticles enhanced the tensile strength and Young's modulus but decreased the elongation at break, indicating that the –NH2 groups formed a tighter structure with PFSA than the –OH groups. In addition, more VPA-MSN(–NH2) was incorporated into the composite membrane, increasing its tensile strength and Young's modulus, which revealed that the MSNs played an enhancing role in the composite membrane. When it was subjected to external forces, the stress concentration effect occurred due to the presence of the MSNs; at the same time, the propagation of cracks was inhibited by the rigid structure of the inorganic nanoparticles, effectively enhancing the composite membranes' mechanical properties.27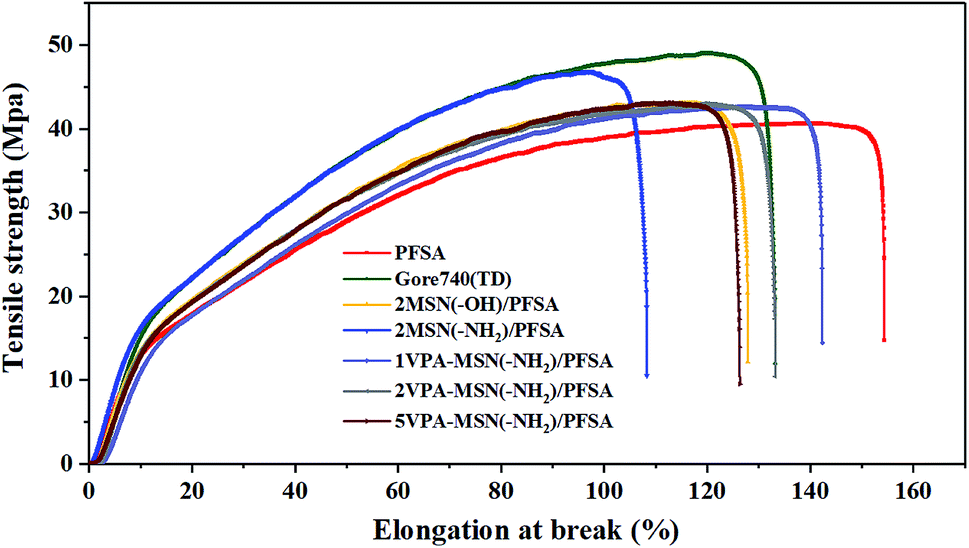 | ||
| Fig. 8 Mechanical properties of the PFSA membrane, Gore 740 membrane, and VPA-MSN(–NH2)/PFSA composite membranes with various contents. | ||
The dimensional stability of the membranes was characterized using their swelling ratios. As seen in Table 3, the swelling ratios of the PFSA membrane and Gore 740 membrane were 3.54% and 2.86%. In comparison, the values for the MSN(–OH)/PFSA and VPA-MSN(–NH2)/PFSA composite membranes with various contents were higher than that for the Gore 740 membrane but lower than that for the PFSA. This was because on the one hand, more water molecules were absorbed by the hydrophilic groups, increasing the PEM's swelling ratio, and on the other hand, the PFSA matrix was fixed by nanoparticles with rigid structures.28
| Sample | Water uptake (%) | Water contact angle (°) | Swelling ratio (%) |
|---|---|---|---|
| PFSA | 38.9 ± 1.2 | 76.2 ± 1.16 | 3.54 ± 0.16 |
| Gore 740 | 37.6 ± 0.8 | 84.23 ± 1.29 | 2.68 ± 0.12 |
| 2MSN(–OH)/PFSA | 41.2 ± 1.3 | 69.26 ± 0.86 | 3.12 ± 0.23 |
| 2MSN(–NH2)/PFSA | 43.2 ± 0.6 | 67.25 ± 1.37 | 2.57 ± 0.09 |
| 1VPA-MSN(–NH2)/PFSA | 42.3 ± 1.7 | 71.53 ± 0.54 | 3.34 ± 0.24 |
| 2VPA-MSN(–NH2)/PFSA | 45.5 ± 1.3 | 68.45 ± 0.97 | 3.06 ± 0.34 |
| 5VPA-MSN(–NH2)/PFSA | 48.7 ± 2.1 | 64.23 ± 1.15 | 2.95 ± 0.27 |
Our findings demonstrated that the MSN(–NH2) was effective for simultaneously improving the mechanical strength and dimensional stability of the composite membranes, guaranteeing them under practical fuel cell operating conditions.
3.4 Water uptake, water retention, and surface hydrophilicity of the membranes
For most PEMs, water acts as the proton carrier, so a high water content can increase the diameter of the ion clusters in the membrane, inducing the formation of continuous proton transport channels.29 The water content is closely related to the membrane's water uptake and retention. Compared with the pristine PFSA membrane (39.2%) and the Gore 740 membrane (37.6%), the 5MSN(–OH)/PFSA, 5MSN(–NH2)/PFSA, and 5VPA-MSN(–NH2)/PFSA membranes had higher water uptake, reaching 41.2%, 45.7%, and 48.7%, respectively; this indicated that all of the hydrophilic groups (–OH, –NH2, and –PO3H2) possessed water sorption ability, but the –PO3H2 groups' was significantly better. Because these MSNs' larger specific surface area meant they had more hydrophilic groups, more water was absorbed in this way.The water uptake of the PFSA and VPA-MSN(–NH2)/PFSA composite membranes under 10–100% RH conditions and immersion in water is exhibited in Fig. 9(a). The water absorption ratio of various membranes demonstrated that the VPA-MSN(–NH2) and MSN(–OH) added into composite membranes can effectively improve the water sorption of the membrane across the entire humidity range, and the water uptake of the VPA-MSN(–NH2)/PFSA composite membrane is significantly higher than that of PFSA at lower RH. Further, the composite membranes of the VPA-MSN(–NH2)/PFSA with a higher content (i.e., with more –PO3H2 groups) also have higher water uptake, indicating that the water molecules were adsorbed by a larger number of –PO3H2 groups and numerous smaller pores.
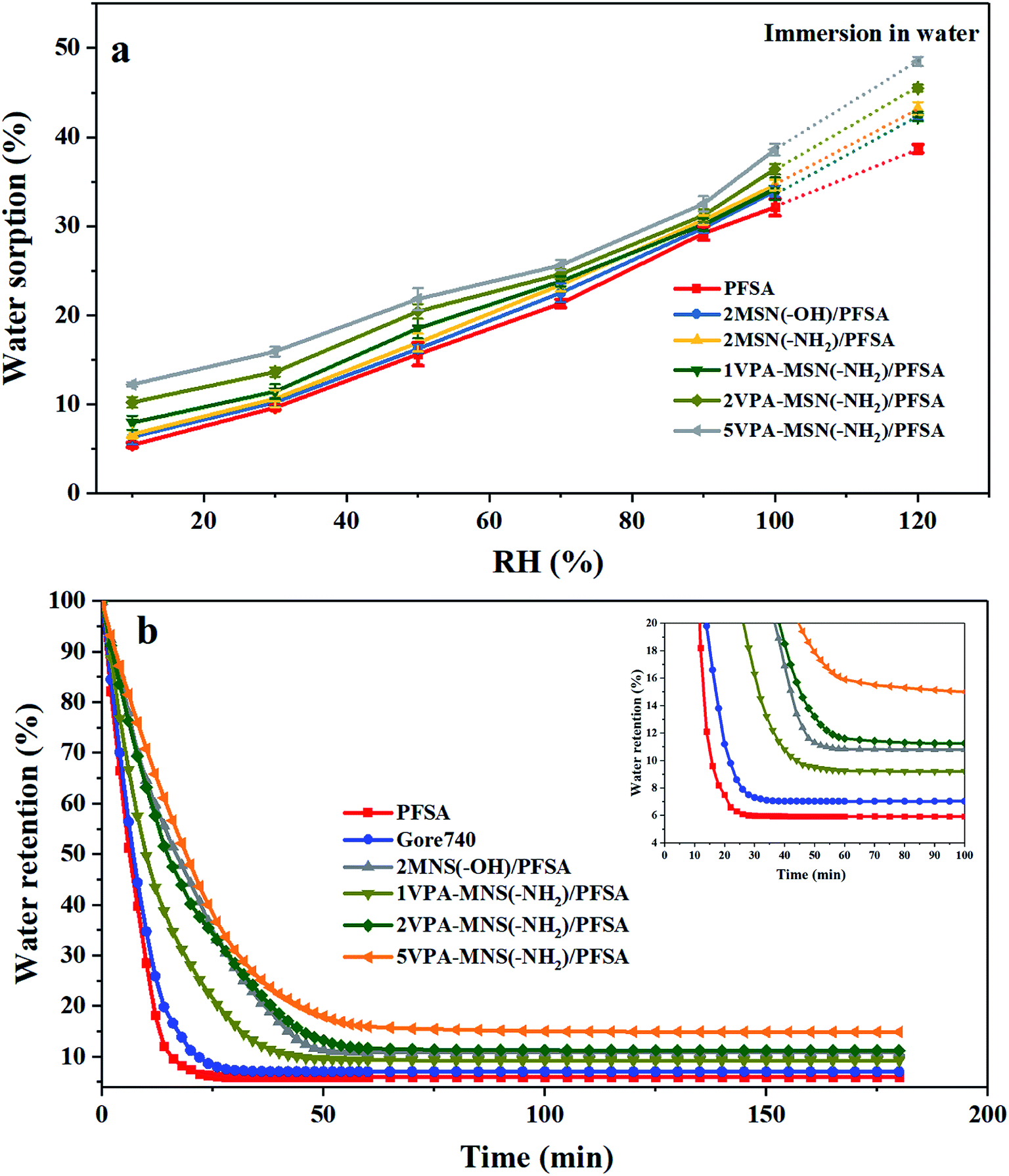 | ||
| Fig. 9 (a) The water uptake of the VPA-MSN(–NH2)/PFSA composite membrane and PFSA at various RHs. (b) Water retention from the VPA-MSN(–NH2)/PFSA composite membranes and the PFSA and Gore 740 membrane at various test times. | ||
Water retention was characterized using the water release ratios of the membranes at low RH. Fig. 9(b) presents the water retention at 10% RH and various test times. The first stage was characterized by high water release via the evaporation of free water, while the second stage saw more moderate water release through the slower evaporation of bound water. Hence, the incorporation of MSN(–NH2)/PFSA or VPA-MSN(–NH2)/PFSA significantly reduced the composite membranes' water release ratios in these initial stages. In the first stage, the rate of water loss was mainly related to the hydration energy of the existing groups within the membrane; the decreasing slope of the water release curve and the decrease in the rate of water loss indicated that the –NH2 and –PO3H2 groups had a stronger affinity for water than the –OH groups. In the second stage, the abundant mesopores and micropores in the added materials were key factors affecting the water content in the equilibrium state, because they could store and retain more water. According to the Kelvin equation, the saturated vapor pressure of water is significantly reduced in smaller channels, which is why the composite membranes remained wet at a low RH.30
The surface hydrophilicity of the VPA-MSN(–NH2)/PFSA composite membrane was also enhanced (as measured from the membranes' water contact angles; see Table 2) because more hydrophilic groups had been introduced into the composite membrane. The membranes with higher surface hydrophilicity could effectively decrease the interfacial resistance to water and proton transfer in the membrane.
3.5 Proton conductivity
Thanks to the nanoparticles' excellent dispersibility and the high water absorption capacity of the phosphonic acid groups, the VPA-MSN(–NH2)/PFSA composite membranes exhibited greatly increased proton conduction at RHs less than 70%, and their advantages became particularly obvious under lower RH conditions. Fig. 10 shows that the PFSA membrane yielded a conductivity of 150.6 mS cm−1 and the Gore 740 membrane yielded 114.2 mS cm−1 at 90% RH. By comparison, the VPA-MSN(–NH2)/PFSA with 1, 2, and 5 wt% contents yielded slightly lower proton conduction, reaching 136.2, 124.7, and 106.6 mS cm−1 under identical conditions. This was because the protons were more easily disassociated from –SO3H groups than from –PO3H2. It is worth noting that at 30% and 10% RH, the Gore 740 membrane yielded conductivities of only 11.2 and 1.8 mS cm−1, and the PFSA membrane yielded 8.6 and 1.8 mS cm−1, whereas the VPA-MSN(–NH2)/PFSA with a 5 wt% content reached 19.6 and 9.2 mS cm−1. This significant increase occurred because the added VPA-MSN(–NH2) existed in an ionic nanophase, and the resulting additional hydrophilic zones enhanced the proton transport channels' continuity.31 Further, the abundant water-retaining groups in the composite membrane ensured that it had sufficient proton transfer carriers at low humidity,32 and additional hydrogen bonds were provided by the functional groups of VPA-MSN(–NH2).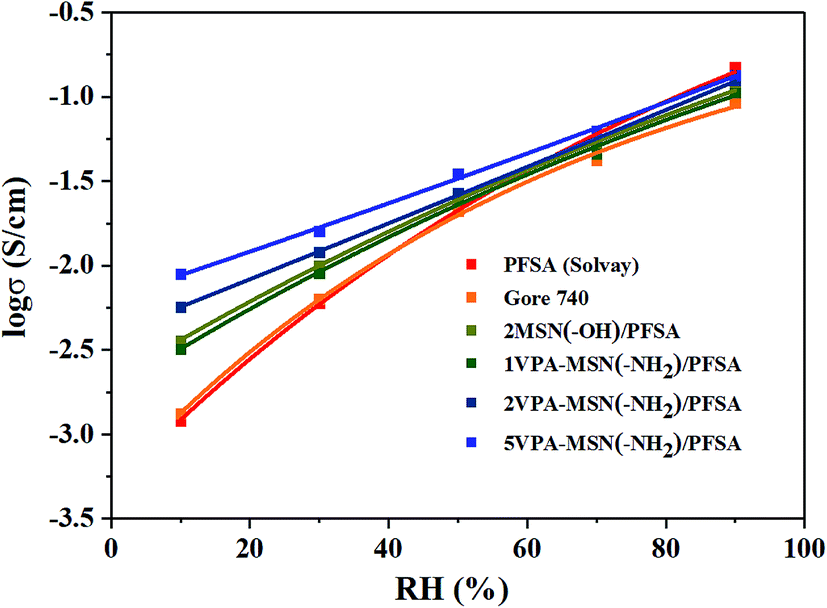 | ||
| Fig. 10 Proton conductivity of the PFSA membrane and VPA-MSN(–NH2)/PFSA composite membranes with 1, 2, and 5 wt% contents at various RHs and 80 °C. | ||
From the temperature-dependence of proton conductivity, the activation energy (Ea) for proton conductivity was calculated using the Arrhenius equation, and the values are presented in Fig. 11(a and b). At 80% RH, the Ea for the PFSA membrane was 11.74 kJ mol−1 and Gore 740 membrane was 12.89 kJ mol−1. The incorporation of VPA-MSN and MNN with –NH2 or –OH groups slightly increased the Ea of the composite membrane to 13.76 kJ mol−1 and 16.56 kJ mol−1, implying that the Grotthuss mechanism predominated in proton transport. This is because sulfonic acid groups are more susceptible to proton ionization than phosphate groups. Under low humidity conditions (RH = 20%), compared with PFSA (25.18 kJ mol−1) and Gore 740 membranes (26.56 kJ mol−1) with a high energy barrier, the VPA-MSN(–NH2)/PFSA membranes had a low conductivity barrier, and the Ea values for the 1VPA-MSN(–NH2)/PFSA, 2VPA-MSN(–NH2)/PFSA, and 5VPA-MSN(–NH2)/PFSA composite membranes were 21.38, 20.35, and 18.78 kJ mol−1, respectively. This implies that the VPA-MSN(–NH2) efficiently helped the membrane transmit protons, and that its high VPA-MSN(–NH2) loading made the 5VPA-MSN(–NH2)/PFSA composite membrane more favorable for proton transfer due to its low energy barrier. These experimental results indicated that the PFSA membrane had stronger humidity dependence, and the proton-conduction Ea increased from 11.74 kJ mol−1 to 25.18 kJ mol−1 when the RH dropped from 80% to 20%. The addition of VPA-MSN(–NH2) effectively reduced the humidity dependence of the PEMs: for the 5VPA-MSN(–NH2)/PFSA composite membrane, the proton conduction Ea only increased from 14.47 to 18.78 kJ mol−1. The lower proton-conduction energy barrier and enhanced proton conductivity of VPA-MSN(–NH2)/PFSA occurred mainly because VPA-MSN(–NH2) adsorbed and retained water molecules, increased the number of carriers for proton conduction and favored the Grotthuss mechanism transport pathway.
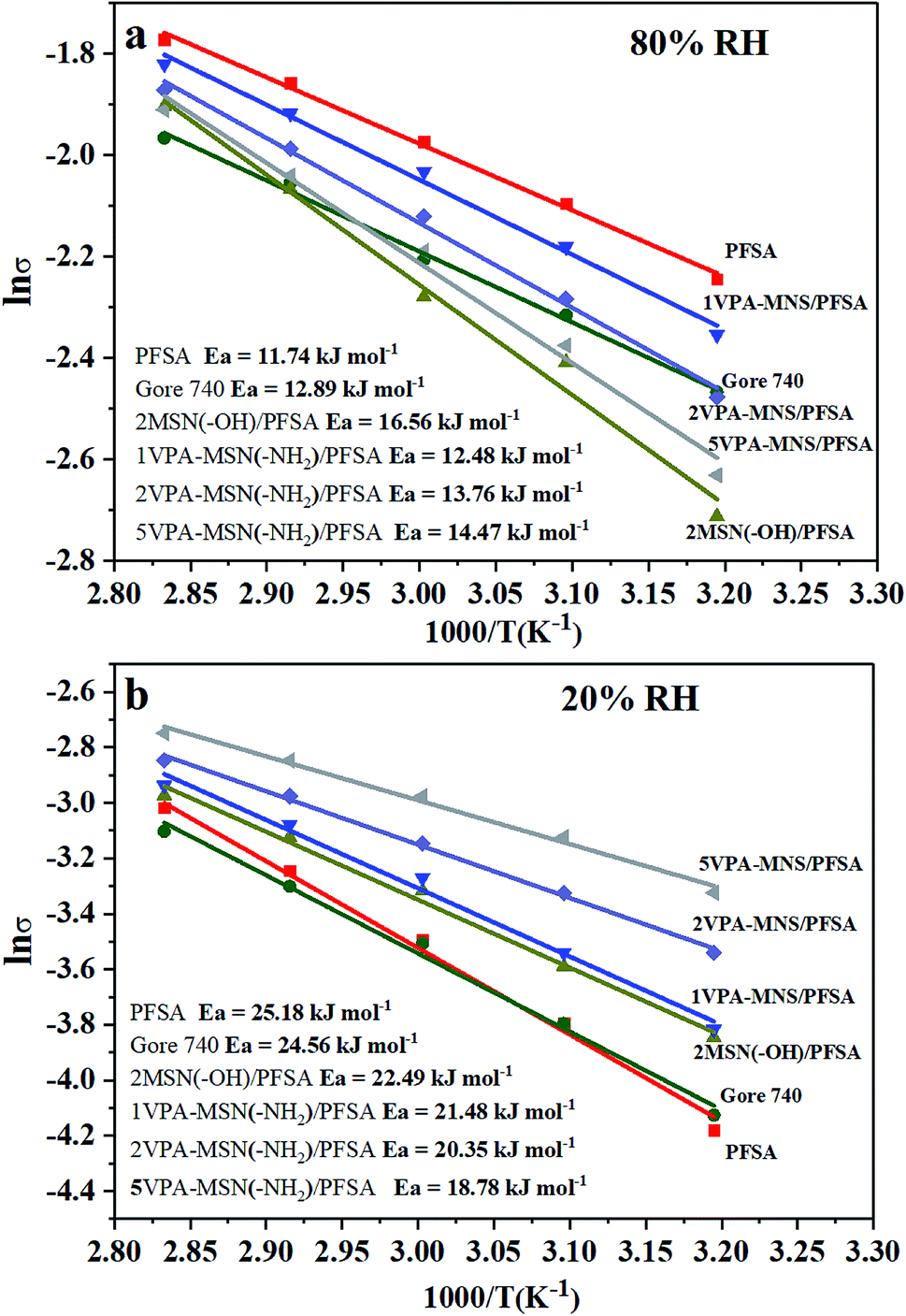 | ||
| Fig. 11 Arrhenius plot of the activation energy for proton conductivity at (a) 80% RH and (b) 20% RH for the PFSA membrane, Gore 740 membrane and VPA-MSN(–NH2)/PFSA composite membranes. | ||
Improving the connectivity of the hydrophilic zone in the composite membrane is the key approach for enhancing proton conductivity. This hydrophilic region is formed by microphase separation of the ionomer, so we investigated the microphase separation in the VPA-MSN(–NH2)/PFSA composite membranes using SAXS.33 The results for the PFSA membrane and VPA-MSN(–NH2)/PFSA composite membranes with various contents are presented in Fig. 12, which shows a change in the spacing of the hydrophilic zone after the introduction of VPA-MSN(–NH2) and MSN(–OH). The q values (scattering vector) of PFSA, MSN(–OH)/PFSA, 1VPA-MSN(–NH2)/PFSA, 2VPA-MSN(–NH2)/PFSA and 5VPA-MSN(–NH2)/PFSA were 1.29, 1.72, 2.24, 2.78 and 3.56 nm−1, respectively. The Bragg spacing d, which refers to the distance between two water clusters, can be calculated using d = 2π/q. The d value shifted from 5.04 to 1.77 nm after 5 wt% VPA-MSN(–NH2) was incorporated into the PFSA. Less space between water clusters meant that the VPA-MSN(–NH2)/PFSA composite membrane network connected adjacent hydrophilic domains within the PFSA matrix, thereby enhancing the continuity of the proton channels. Fig. 12 also shows that the scattering peaks of the VPA-MSN(–NH2)/PFSA composite membrane are wider than those for the PFSA membrane. The reason for these results is that a part of separated hydrophilic areas merged into a large hydrophilic area, and therefore the size of the hydrophilic zone was not uniform and the boundary between hydrophilic and hydrophobic domains did not appear clearly, as well as the addition of the VPA-MSN into the PFSA matrix reduced their phase separation. Therefore the small q value of the VPA-MSN(–NH2)/PFSA membrane was attributed to the formation of large ionic clusters and enhancement of the continuity of hydrophilic domains, and the broader SAXS profile was attributed to the low phase separation and the hydrophilic zones with various sizes. Although the phase separation of the membrane was decreased, the ion conductivity increased with the VPA-MSN(–NH2) content because of the large ionic domains and a large number of ion-conducting functional groups.
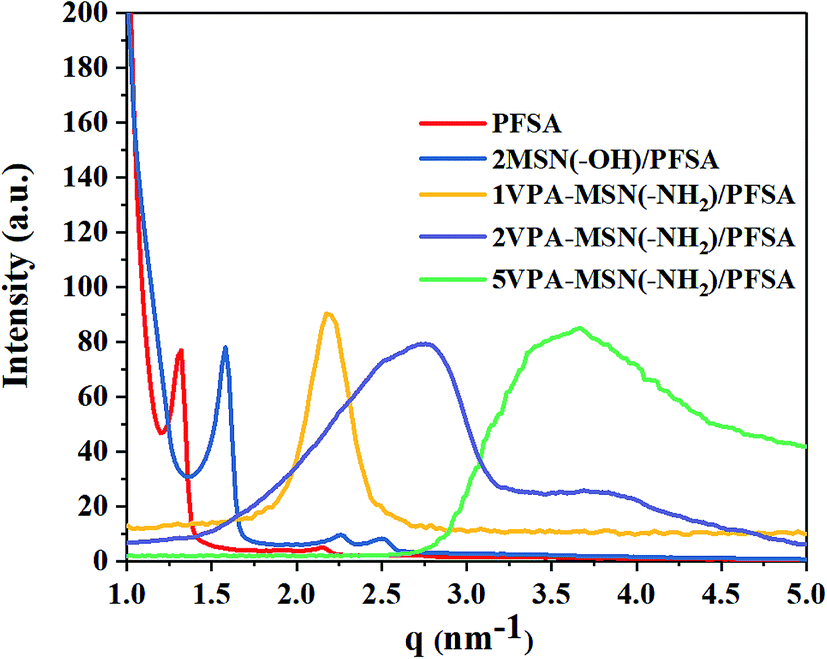 | ||
| Fig. 12 SAXS patterns of PFSA and VPA-MSN(–NH2)/PFSA composite membranes. | ||
3.6 H2 crossover current density
The dispersion, size and content of the inorganic nanoparticles incorporated obviously affect the gas permeability of the composite membrane. Hydrogen crossover is the diffusion of hydrogen from the anode to the cathode; the hydrogen that crosses over can directly react with oxygen, resulting in reduced cell voltage also may generate hydrogen peroxide which could lead to chemical degradation. The H2 crossover current density of the PFSA, Gore 740, MNS(–OH)/PFSA and VPA-MSN(–NH2)/PFSA composite membranes was tested using linear sweep voltammetry (LSV), and the results are shown in Fig. 13. The results illustrated that as more VPA-MSN(–NH2) was added into the composite membrane, the membranes had higher H2 crossover current density. Besides, the VPA-MSN(–NH2)/PFSA composite membranes (3.2 mA cm−1) had significantly lower H2 crossover current density than MNS(–OH)/PFSA (4.5 mA cm−1) at the same content. The reason for this result is that the VPA-MSN(–NH2) possesses a smaller size, which inhibited the permeation of hydrogen into the PEM. Besides, the VPA-MSN(–NH2) is uniformly and stably distributed throughout the composite inhibiting the agglomeration of nanoparticles, which had a larger size and therefore inhibited the penetration of hydrogen. In this way, the H2 crossover channels were blocked and the VPA-MSN(–NH2)/PFSA composite membranes exhibited lower H2 crossover current density.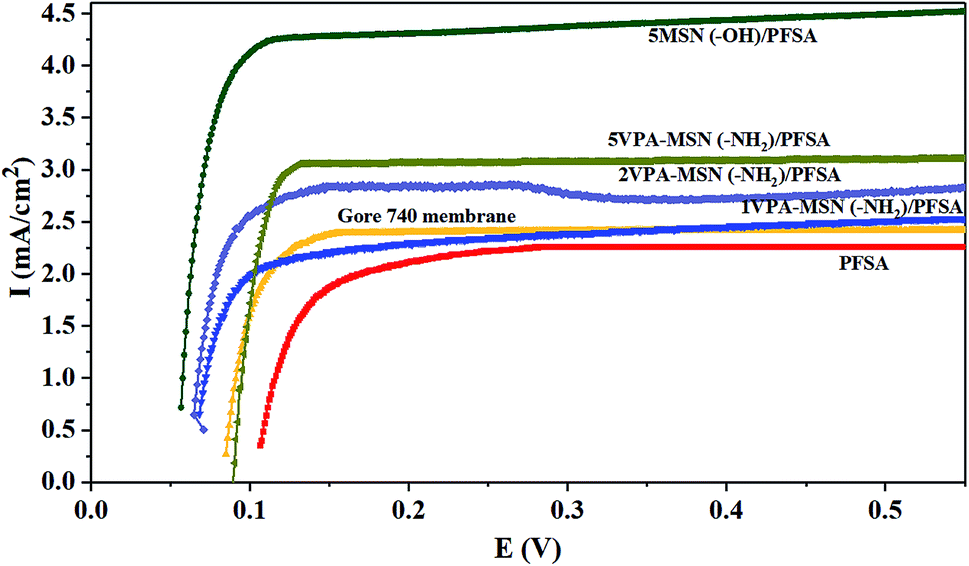 | ||
| Fig. 13 H2 crossover current density of Gore 740, PFSA MNS(–OH)/PFSA and VPA-MSN(–NH2)/PFSA composite membranes in LSV tests. | ||
3.7 Single-cell performance
As shown above, the VPA-MSN(–NH2)/PFSA composite membranes exhibited higher proton conduction and better water retention capacity, both of which were beneficial for single-cell performance. We investigated this performance at 70 °C and various RHs using a fuel cell test system. Fig. 14(a–e) present the cell voltage and power densities at various current densities of MEAs based on the VPA-MSN(–NH2)/PFSA composite membranes with different contents; the PFSA and Gore 740 membrane results are also shown in Fig. 14(a and b). All of the membranes exhibited similar fuel cell performance and open circuit voltage at high RH because they had a sufficient water content and enough proton transport carriers to meet the requirements of the cell's electrochemical reactions. However, Fig. 13(a) shows that the cell voltages and power densities of the MEA based on the PFSA membrane decreased significantly when the RH dropped from 90% to 10%. Fig. 15 presents the cell voltages of the MEAs based on all the membranes at 1.0 A cm−2. The cell voltages of the PFSA membrane when the RH was 50%, 20%, and 0% were 0.652, 0.628, and 0.602 V – obviously lower than those of the benchmark membrane. Compared with the PFSA membrane, the PFSA/PEIMPA-70kDa-100 composite membrane at these RHs yielded 0.661, 0.650, and 0.632 V, and the 5VPA-MSN(–NH2)/PFSA composite membrane yielded 0.658, 0.648, and 0.632 V, clearly indicating that the composite membranes achieved better cell performance under low-humidity conditions. Similarly, the 5VPA-MSN(–NH2)/PFSA composite membranes had a higher power density at low RHs. Their superior cell performance and excellent energy output were mainly attributable to their tailored water-retention capacity and proton transport capabilities via the formation of hydrogen bonds with water; some of the water moved from the water-rich side to the water-poor side by counter-diffusion, ensuring that the PEM had sufficient carrier (water) to transport protons and thus retained high cell performance at low RHs.34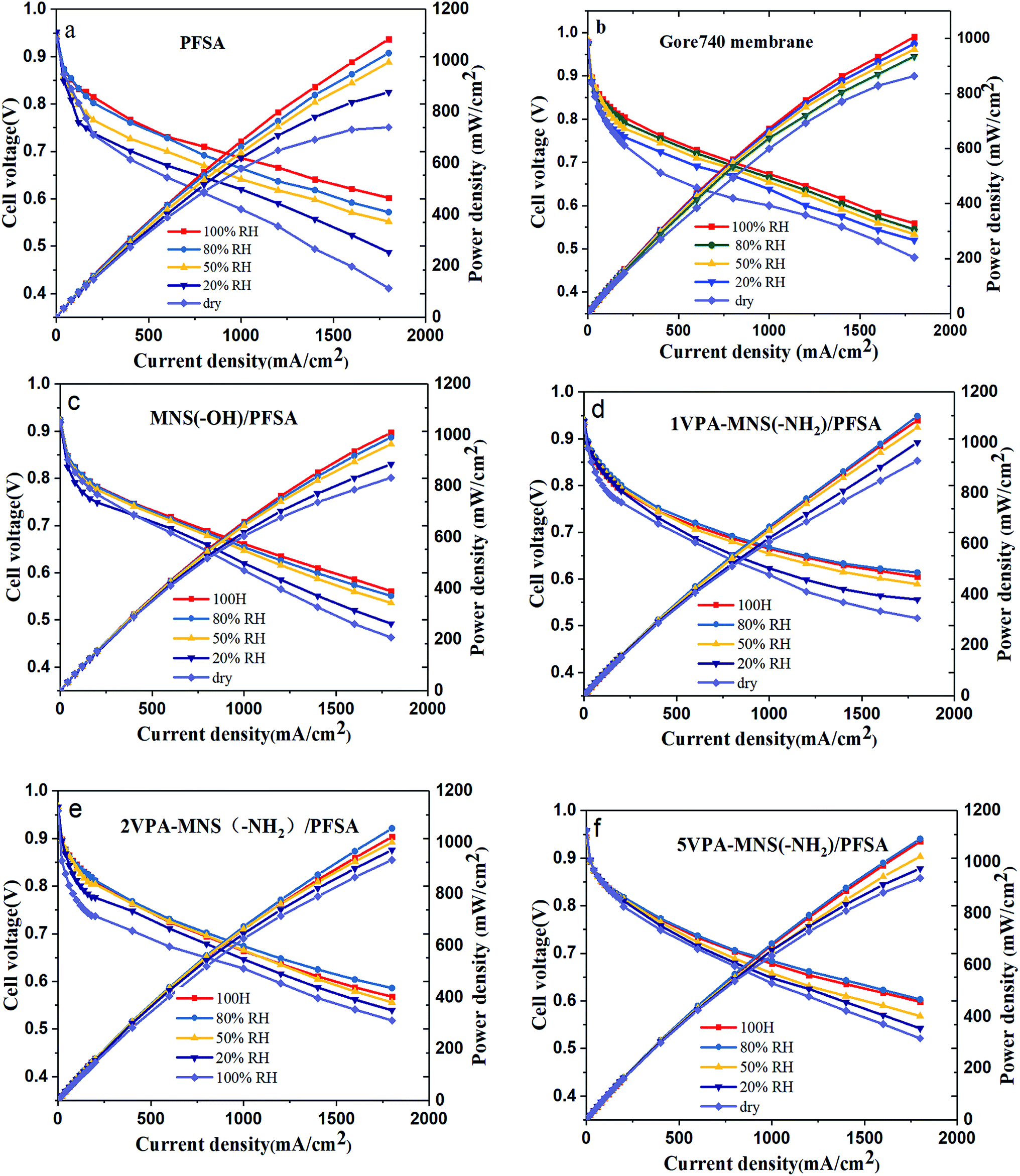 | ||
| Fig. 14 The polarization curves and power densities of MEAs based on (a) PFSA, (b) Gore 740, (c) MSN(–OH)/PFSA, (d) 1VPA-MSN(–NH2)/PFSA, (e) 2VPA-MSN(–NH2)/PFSA, (f) 5VPA-MSN(–NH2)/PFSA operated at 80 °C. | ||
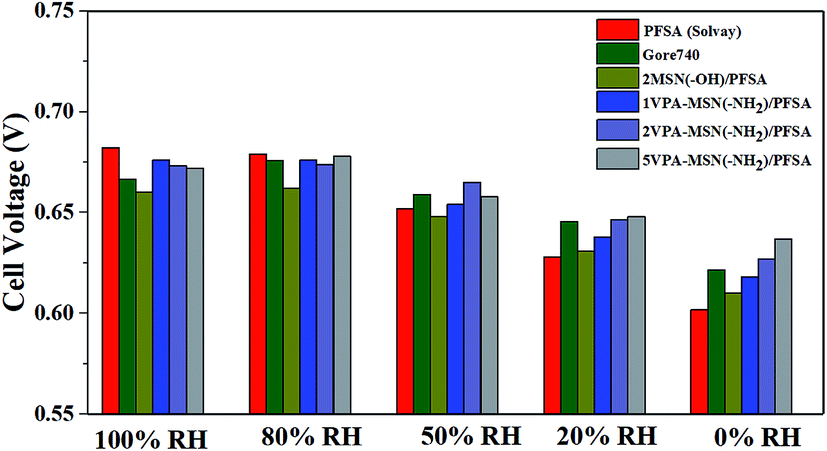 | ||
| Fig. 15 The cell voltages of MEAs based on Gore 740, PFSA, and VPA-MSN(–NH2)/PFSA composite membranes at 100, 80, 50, 20, and 0% RH (current density = 1 A cm−2). | ||
To further understand the effect that incorporating VPA-MSN(–NH2) and MSN(–OH) had on resistance to proton transport, Fig. 16 shows the EIS analyses of the MEAs based on VPA-MSN(–NH2)/PFSA and MSN(–OH)/PFSA composite membranes with various contents at 80%, 50%, 20%, and 0% RH. The results indicated that the cell voltage at 1 A cm2 is consistent with EIS of the MEA; as more VPA-MSN(–NH2) was incorporated, the composite membrane was better able to retain its lower resistance when the RH dropped from 80% to 0%, and the MEA with more VPA-MSN(–NH2) maintained a higher cell voltage at low RH. This was because these composite membranes had better water-retention ability, so a wetter state was maintained even at low or no RH, resulting in lower resistance to proton transport. In this way, more connected proton transport channels were preserved in the composite membrane than in the PFSA membrane, improving proton conduction and single-cell performance at lower RHs.
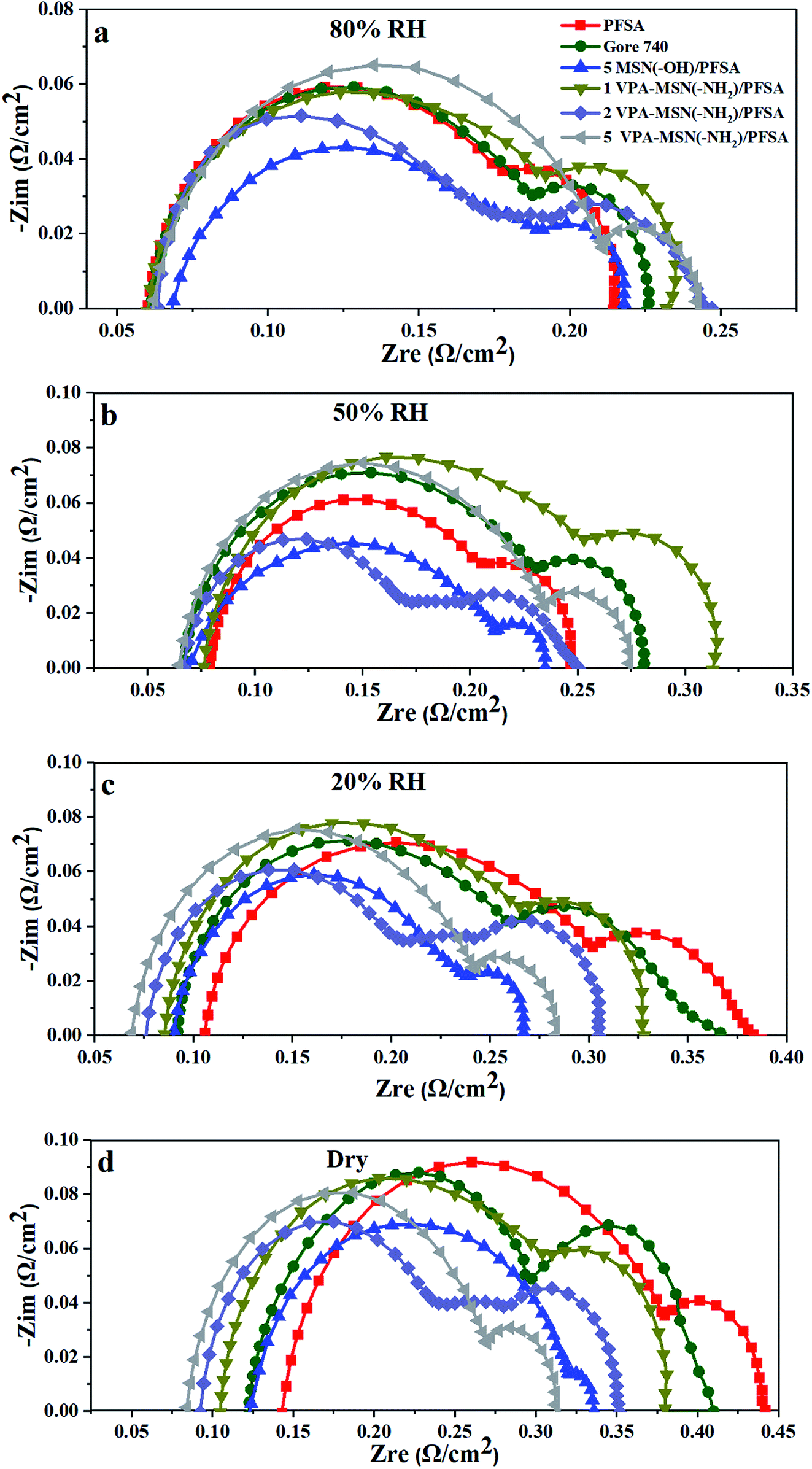 | ||
| Fig. 16 EIS of PFSA and VPA-MSN(–NH2)/PFSA composite membranes at (a) 80% RH, (b) 50% RH, (c) 20% RH, and (d) 0% RH (current density = 1 A cm−2). | ||
4. Conclusions
Novel composite membranes based on phosphonic acid-functionalized mesoporous silica nanoparticles were designed and fabricated. The prepared MSNs had a uniform size, large surface area, high adsorption capacity, and excellent dispersion. The incorporation of VPA-MSN(–NH2) into a PFSA matrix enhanced the membrane's water-retention properties, dimensional stability, and mechanical strength. The VPA-MSN(–NH2) had abundant hydrophilic groups, high adsorption capacity, and numerous micropores, endowing the composite membrane with high water uptake and water retention. The rigid structure and abundant active groups of VPA-MSN(–NH2) ensured that the membrane possessed excellent mechanical strength and dimensional stability. With its excellent dispersion performance and water retention properties, the VPA-MSN(–NH2) significantly enhanced the proton conductivity and cell performance of membranes under low-RH conditions. This study provides a promising approach to improve dispersion and adsorption capacity, and to enhance cell performance at low RHs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program of China (2018YFB1502503), the Guangdong Innovative and Entrepreneurial Research Team Program (2016ZT06N500), the Shenzhen Peacock Plan (KQTD2016022620054656), the Shenzhen Key Laboratory of Hydrogen Energy (ZDSYS2016033110134898), and the Development and Reform Commission of Shenzhen Municipality (2017, No. 1106).References
- Z. Wang, S. Guo, B. Zhang and L. Zhu, J. Membr. Sci., 2019, 592, 117375 CrossRef.
- C. H. Park, S. Y. Lee, D. S. Hwang, D. W. Shin, D. H. Cho, K. H. Lee, T. W. Kim, T. W. Kim, M. Lee, D. S. Kim, C. M. Doherty, A. W. Thornton, A. J. Hill, M. D. Guiver and Y. M. Lee, Nature, 2016, 532(7600), 480–483 CrossRef.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS.
- Y. Liu, J. Yin, Y. Fu, P. Zhao, Y. Zhang, B. He and P. He, Chem. Eng. J., 2020, 382, 122925–122933 CrossRef CAS.
- C. Bi, H. Zhang, Y. Zhang, X. Zhu, Y. Ma, H. Dai and S. Xiao, J. Power Sources, 2008, 184(1), 197–203 CrossRef CAS.
- H. Wang, X. Li, X. Zhuang, B. Cheng, W. Wang, W. Kang, L. Shi and H. Li, J. Power Sources, 2017, 340, 201–209 CrossRef CAS.
- I. S. Amiinu, W. Li, G. Wang, Z. Tu, H. Tang, M. Pan and H. Zhang, Electrochim. Acta, 2015, 160, 185–194 CrossRef CAS.
- Y. Treekamol, M. Schieda, L. Robitaille, S. M. MacKinnon, A. Mokrini, Z. Shi, S. Holdcroft, K. Schulte and S. P. Nunes, J. Power Sources, 2014, 246, 950–959 CrossRef CAS.
- K. Oh, O. Kwon, B. Son, D. H. Lee and S. Shanmugam, J. Membr. Sci., 2019, 583, 103–109 CrossRef CAS.
- Y. Zhao, H. Yang, H. Wu and Z. Jiang, J. Power Sources, 2014, 270, 292–303 CrossRef CAS.
- Y. P. Ying, S. K. Kamarudin and M. S. Masdar, Int. J. Hydrogen Energy, 2018, 43, 16068–16084 CrossRef CAS.
- Y. Yin, W. Deng, H. Wang, A. Li, C. Wang, Z. Jiang and H. Wu, J. Mater. Chem. A, 2015, 3, 16079–16088 RSC.
- C.-W. Yang, K.-H. Chen and S. Cheng, RSC Adv., 2016, 6, 111666–111680 RSC.
- X. Li, H. Ma, P. Wang, Z. Liu, J. Peng, W. Hu, Z. Jiang and B. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 30735–30746 CrossRef CAS.
- J. Joseph, C.-Y. Tseng and B.-J. Hwang, J. Power Sources, 2011, 196, 7363–7371 CrossRef CAS.
- W. Zhiwei, Z. Hao, C. Qiang, Z. Sumei, Y. Feng, K. Jian, C. Jinyao, C. Ya and X. Ming, J. Polym. Res., 2019, 26, 1–8 CrossRef.
- A. K. Sahu, S. Meenakshi, S. D. Bhat, A. Shahid, P. Sridhar, S. Pitchumani and A. K. Shukla, J. Electrochem. Soc., 2012, 159, F702–F710 CrossRef CAS.
- L. Fan, J. Shi and T. Gao, Energies, 2020, 13, 1383–1394 CrossRef CAS.
- C. Wang, Y. Liang, J. Miao, B. Wu, M. M. Hossain, M. Cao, Q. Ge, L. Su, Z. Zheng, B. Yang, P. Chen, R. Xia and J. Qian, J. Membr. Sci., 2019, 592, 117388–117396 CrossRef.
- S. Wang, J. Li, J. Suo and T. Luo, Appl. Surf. Sci., 2010, 256, 2293–2298 CrossRef CAS.
- H. Huang, L. Ni, J. Xu, X. Xie, L. Zhang, C. Yang, J. Fan, H. Li and H. Wang, Sustainable Energy Fuels, 2020, 4, 2859–2868 RSC.
- H. Huang, J. Xu, Q. Feng, L. Ni, L. Zhang, C. Yang, J. Fan, H. Li and H. Wang, J. Membr. Sci., 2020, 606, 118144 CrossRef CAS.
- S. Maity and T. Jana, ACS Appl. Mater. Interfaces, 2014, 6, 6851–6864 CrossRef CAS.
- S. Shi, A. Z. Weber and A. Kusoglu, J. Membr. Sci., 2016, 516, 123–134 CrossRef CAS.
- E. Labalme, G. David, P. Buvat, J. Bigarre and T. Boucheteau, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1308–1316 CrossRef CAS.
- J. Zeng, B. He, K. Lamb, R. De Marco, P. K. Shen and S. P. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 11240–11248 CrossRef CAS.
- S. Shin, J.-H. Jin and J. Jung, J. Ind. Eng. Chem., 2020, 88, 278–284 CrossRef CAS.
- H. Wang, X. Wang, T. Fan, R. Zhou, J. Li, Y. Long, X. Zhuang and B. Cheng, Solid State Ionics, 2020, 349, 115300 CrossRef CAS.
- Y. Liu, J. Yin, Y. Fu, P. Zhao, Y. Zhang, B. He and P. He, Chem. Eng. J., 2020, 382, 122925–122933 CrossRef CAS.
- J. Li, J. Lou, Z. Wang, L. Wang, F. Liu, X. Pu, J. Hu, S. Wang and C. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 5880–5890 CrossRef CAS.
- J. Li, J. Lou, Z. Wang, L. Wang, F. Liu, X. Pu, J. Hu, S. Wang and C. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 5880–5890 CrossRef CAS.
- X. Zhang, S. Yu, Q. Zhu and L. Zhao, Int. J. Hydrogen Energy, 2019, 44, 6148–6159 CrossRef CAS.
- I. Martens, A. Vamvakeros, R. Chattot, M. V. Blanco, M. Rasola, J. Pusa, S. D. M. Jacques, D. Bizzotto, D. P. Wilkinson, B. Ruffmann, S. Heidemann, V. Honkimäki and J. Drnec, J. Power Sources, 2019, 437, 226906–226912 CrossRef CAS.
- K. H. Lee, J. Y. Chu, A. R. Kim and D. J. Yoo, Int. J. Energy Res., 2019, 43, 5333–5345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se01339k |
| This journal is © The Royal Society of Chemistry 2021 |