
Effects of the support on bifunctional one-step synthesis of methylal via methanol oxidation catalysed by Fe–Mo-based bifunctional catalysts†
Meng
Yuan
 ad,
Ruiyuan
Tang
b,
Xiangyu
Sun
c,
Zhimei
Zhang
ad,
Yuanyu
Tian
*ade and
Yingyun
Qiao
*ad
ad,
Ruiyuan
Tang
b,
Xiangyu
Sun
c,
Zhimei
Zhang
ad,
Yuanyu
Tian
*ade and
Yingyun
Qiao
*ad
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao, Shandong 266580, China. Tel: +86-532-86057766
bResearch Center of Petroleum Processing & Petrochemicals, Xi'an Shiyou University, Xi'an, Shanxi 710065, China
cCollege of Petroleum Engineering, China University of Petroleum (East China), Qingdao 266580, PR China
dShandong Engineering and Technology Research Center of High Carbon Low Carbonization, China University of Petroleum (East China), Qingdao 266580, PR China
eKey Laboratory of Low Carbon Energy and Chemical Engineering, Shandong University of Science and Technology, Qingdao, Shandong 266590, China
First published on 4th November 2020
Abstract
In the one-step preparation of methylal from methanol, achieving high methanol conversion and high methylal yield is a huge challenge. To address this problem, in this study a new Fe–Mo-based catalyst was designed, and the effect of the type and amount of the support on catalytic activity was explored. The results showed that Mo:Fe(2)/HZSM-5(80 + 80), in which HZSM-5(80 + 80) was the catalytic support, had excellent catalytic performance, with the yield of methylal reaching 81.33%, much higher than other reported results of this process. Through XPS, NH3-TPD and PY-FTIR analyses it was found that the formation of Mo5+ promoted coordination of the two terminal oxygens with the Mo double bond in the Fe2(MoO4)3 octahedron, and the higher the B/L acid site ratio, the better the catalytic activity and the higher the selectivity of the target product. The apparent activation energy also further proved that Mo:Fe(2)/HZSM-5(80 + 80) was highly suitable for the one-step production of methylal from methanol.
1. Introduction
Methylal (DMM) is an important chemical solvent. It has many applications in the energy and chemical industries as it is environmentally friendly.1 DMM has been widely used as a very good organic solvent in the pharmaceutical and cosmetic industries, and as a diesel fuel additive.2 The most important aspect of DMM as a new diesel additive is that not only can it decrease emissions of NOx and particulate matter in the diesel exhaust, but also improve the combustion performance of the fuel, thereby increasing its thermal efficiency. If DMM can be produced at a low cost on a large scale, it can effectively improve the combustion quality of fuel and improve the current situation of fuel shortage. Therefore, there has been increasing research attention on the one-step conversion of methanol to DMM.3The conventional method of synthesising DMM includes two steps. First, methanol is selectively oxidised to obtain formaldehyde (FA) under the action of an oxidising catalyst (Ag or Fe–Mo catalysts; eqn (1)).4,5 Subsequently, methanol and formaldehyde are acetalised under the action of an acidic catalyst to obtain DMM (eqn (2)).6 The conventional DMM production method has the disadvantages of complicated procedures, high reaction temperature and the corrosion of equipment under high acidity.7,8 Therefore, the production of DMM in one step from methanol as a result of the coupling of two reaction steps has attracted considerable attention (eqn (3)).
 | (1) |
| HCHO + 2CH3OH → CH3OCH2OCH3 + H2O | (2) |
 | (3) |
The one-step process not only produces the target product, DMM, but also several by-products such as methyl formate (MF), dimethyl ether (DME), FA and carbon oxides.9 The product distribution of the one-step process is determined by the acidic and oxidation centers of the catalyst. Excessive redox sites may result in the enrichment of FA, MF and COx, while excessive acidic active centres lead to large amounts of DME formation.10,11 For a more efficient one-step process for the preparation of DMM, the bifunctional catalyst must have an appropriate number and strength of oxidising and acidic sites.12
There have been many reports on dual-function catalysts required for one-step processes, as shown in Table 1.13 These previous one-step bifunctional catalysts are mainly divided into four catalytic systems. Nikonova1 and Yuan14,15 reported on the one-step synthesis of DMM via methanol oxidation on a Re catalyst: at a reaction temperature of 513 K, the methanol conversion rate was 53.7% and the DMM selectivity was 83.1%. Royer16 reported on the selective oxidation of methanol to DMM on a heteropoly acid catalyst: at a reaction temperature of 553 K, the methanol conversion rate was 63% and the DMM selectivity was 89.2%. Kaichev,12 Lu,17 and Sun18 reported on the selective oxidation of methanol to DMM on V and Ti catalysts, respectively. Of all these studies, the results of Sun were the best: at a reaction temperature of 483 K, the methanol conversion rate was 66% and the DMM selectivity was 93%. Zhao11 reported on the selective oxidation of methanol to DMM on other V catalysts: at a reaction temperature of 488 K, the methanol conversion rate was 21.3% and the DMM selectivity was 82.4%. It can be seen that—regardless of the specific catalyst in this process—achieving a balance between the acidic and oxidation centers of the dual-function catalyst is the key to process optimisation.19 This is also the focus of this article.20
| Catalytic system | Researchers | Temperature/K | Methanol conversion (%) | Formaldehyde selectivity (%) | Yield (%) |
|---|---|---|---|---|---|
| Rhenium catalyst | Nikonova1 | 500 | 43.0% | 90.0% | 38.7% |
| Yuan15 | 513 | 53.7% | 83.1% | 44.6% | |
| Royer16 | 553 | 63.0% | 89.2% | 56.2% | |
| Heteropolyacid catalyst | Chen29 | 383 | 55.6% | 81.5% | 45.3% |
| Prado23 | 393 | 58.0% | 78.0% | 45.2% | |
| Xue6 | 373 | 48.4% | 98.2% | 47.5% | |
| Vanadium–titanium catalyst | Kaichev12 | 393 | 59.7% | 57.0% | 34.0% |
| Liu5 | 423 | 72.0% | 85.0% | 61.2% | |
| Lu17 | 423 | 49.0% | 93.0% | 45.6% | |
| Bennici43 | 423 | 58.0% | 58.0% | 33.6% | |
| Sun18 | 483 | 66.0% | 93.0% | 61.4% | |
| Other vanadium catalysts | Zhao11 | 488 | 21.3% | 82.4% | 17.6% |
Iron–molybdenum catalysts are generally used in the industry for the oxidation of methanol to a low concentration of FA (feed amount < 7.5 mol%).21 The conversion of methanol is 99% and selectivity for FA is 94%, while DMM forms in very small amounts. It was assumed that more DMM would be generated when the methanol feed was greatly increased.22,23 Thus, by fine-tuning the Fe–Mo catalyst and increasing the proportion of methanol in the reaction feed to change the selectivity of the catalyst, it was found that the yield of DMM could be increased to 81.3%.
In previous reports, we explored the effect of the Fe–Mo ratio on this type of catalyst and determined the optimal ratio. The purpose of this study was to explore the effects of the catalyst support on the one-step synthesis of methylal via methanol oxidation catalysed by Fe–Mo-based bifunctional catalysts based on the previously determined optimal ratio of Fe to Mo. The effect of oxidation of the bifunctional catalysts and regulation of acidic sites on the distribution of methanol selective oxidative polycondensation products was investigated. The microscopic and surface properties of the catalyst were determined by XRD, BET, XPS and SEM. The surface acidity of the catalyst was determined by NH3-TPD and PY-FTIR. Additionally, the reaction mechanism was studied to determine the effect of changes in oxidation and acidic sites on the distribution of products.
2. Experimental
2.1 Synthesis of Fe–Mo catalysts
Iron nitrate (Fe(NO3)3·9H2O), anhydrous methanol, absolute ethanol and ammonium molybdate tetrahydrate ((NH4)2MoO4·4H2O) were purchased from Sinopharm Pharmaceutical Co., Ltd. Commercial porous SiO2, Al2O3, Y zeolite and ZSM-5 molecular sieves with different silicon-to-aluminum ratios (China Industrial Chemical Water Purification Materials Factory) were calcined in air at 550 °C for 4 hours before use.The preparation process of the Fe–Mo-based bifunctional catalyst is described below. Typically, 8.08 g Fe(NO3)3·9H2O was dissolved in 120 mL of deionised water and stirred to dissolve, and then 7.692 g (NH4)2MoO4·4H2O was added. The mixture was stirred on a magnetic stirrer at 500 rpm for 30 min and then covered with a polyethylene plastic film and stirred at 20 °C for 4 hours. The surface of the film is punched with several small holes for ventilation, and heated and stirred at 60 °C for 1 hour, at 70 °C for 1 hour, and at 80 °C to a viscous state. Finally, it was put in an oven at 80 °C and slowly evaporated to dryness.
In order to regulate the distribution of methanol selective oxidative polycondensation products, we change the type and amount of the carrier to change the acidity of the catalyst to affect the catalytic effect. First, five carriers of SiO2, Al2O3, HY zeolite and (silica–aluminum ratio 80, 40) HZSM-5 molecular sieves were selected. A stoichiometric amount of five carriers was added to the above gel by impregnation to form a slurry which was vigorously stirred 12 hours at 20 °C. The mixture was then heated and stirred at 70 °C to a viscous state, dried at 105 °C for 8 hours, and finally calcined at 550 °C for 4 hours at a temperature increase rate of 10 degrees per minute in the air. The collected catalysts were expressed as Mo:Fe(2)/carrier (carrier: SiO2, Al2O3, HY zeolite, HZSM-5(40) and HZSM-5(80)), and the loadings of Mo and Fe were 10 wt% and 3 wt%, respectively.
After activity evaluation and a series of characterisation analyses, three carriers (HY zeolite, HZSM-5(40, 80)) with a superior catalytic effect were selected for compound optimization. The compounding process repeated the steps previously described for preparation of a single-supported catalyst, with the only difference being that the impregnated support was uniformly mixed with another fresh support in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio after drying at 105 °C. Subsequently, the mixture was heated to 300 °C for one hour at 10° min−1 in the air and then calcined at 550 °C for 4 h. The collected catalyst was expressed as Mo:Fe(2)/carrier (carrier = HY zeolite + HZSM-5(80), HY zeolite + HZSM-5(40), HY zeolite + HY zeolite; HZSM-5(80) + HY zeolite, HZSM-5(80 + 80) or HZSM-5(80 + 40); the term before ‘+’ represents the impregnated carrier and the term after represents the pure carrier without impregnation).
:1 ratio after drying at 105 °C. Subsequently, the mixture was heated to 300 °C for one hour at 10° min−1 in the air and then calcined at 550 °C for 4 h. The collected catalyst was expressed as Mo:Fe(2)/carrier (carrier = HY zeolite + HZSM-5(80), HY zeolite + HZSM-5(40), HY zeolite + HY zeolite; HZSM-5(80) + HY zeolite, HZSM-5(80 + 80) or HZSM-5(80 + 40); the term before ‘+’ represents the impregnated carrier and the term after represents the pure carrier without impregnation).
2.2 Catalyst characterization
The specific surface area and total pore volume were determined from an automatic specific surface and porosity analyzer ASAP 2460. After processing the sample according to the BET measurement requirements, the specific surface area is determined within the specified relative pressure range, and the pore size distribution and pore volume are calculated.24 X-ray diffraction (XRD) patterns are recorded on an X-ray diffractometer X′ Pert PRO MPD (PANalytical B.V. Netherlands) using an acceleration voltage of 40 kV and 40 mA.25 The diffraction angle (2θ) is scanned in the range 5–75 °C at 8° min−1. X-ray photoelectron spectroscopy ESCALab250 Xi-type XPS was used to determine and analyze the metal valence state and surface energy state distribution on the catalyst surface.26 A hitachi SU3500 tungsten filament scanning electron microscope was used to observe the microscopic morphology of the sample.27The sample was first dried in air at 300 °C, and then pyridine was adsorbed at 20 °C for 24 h. The sample was then evacuated in a vacuum at 150 °C for 2 h to remove the physically adsorbed pyridine. Subsequently, the wave number analysis was collected on a NEXUS FT-IR Fourier infrared spectrometer of Nicholis Corporation.20 Automated temperature-programmed chemisorption (TPD) Autochem 2950HP was used to detect the acidity of samples by temperature-programmed desorption of ammonia. First, the pre-screened catalyst (30–40 mesh) was pretreated at 550 °C for 2 hours, and then ammonia gas was absorbed for 40 minutes. Afterwards, the physically adsorbed ammonia was desorbed with a helium purge for 1 h. Finally, the temperature rising rate of 10 °C min−1 was increased to 700 °C for analysis and detection.28,2927Al MAS NMR was carried out with Bruker. An Avance II 400 spectrometer was operated at B0 = 9.4 T (Larmor frequency) equipped with a 2.5 mm Bruker dual-channel probe.30 The pulse width was set to 1.8 μs, and accumulate 50000 scans at a sample rotation rate of 10 kHz. It was determined that the relaxation delay of 0.1 s was long enough to allow quantitative analysis of the zeolite sample.
2.3 Catalytic activity measurements
The steady state activity of different catalysts under normal pressure was investigated in a micro-fluidised bed reactor. The specific reaction device is shown in Fig. 1. The main part of the micro-fluidised bed reactor was made of 304 stainless steel with a length of 80 cm and an inner diameter of 2.5 cm. Before the reaction, the catalyst was sieved through a standard test sieve to select a catalyst powder with particle size in the range of 45 to 75 μm to ensure the fluidisation state of the catalyst in the fluidised bed. The reaction was catalysed at 390 °C, 0.1 MPa, with methanol: air (v/v) = 11:9 and a reaction space velocity of 15000 h−1. To conduct the experiments, the fluidised bed was connected, and checked for airtightness, the reaction temperature was set and the reactor was preheated until the correct temperature for the reaction was reached. First, air was introduced into the reactor to promote the fluidisation of the catalyst particles, and then preheated methanol vapour was introduced. The product was then separated from the catalyst by the filtration unit of the enlarged section of the reactor, which also prevents the fine catalyst powder from entering the condensation separation system. Other gases were recorded by using a wet flow meter and the concentration of reactants and products was determined by gas chromatography (GC). Methanol, dimethoxymethane, MF and FA were detected with an FID detector using a SE-54 capillary column chromatograph (60 m × 0.25 mm × 0.25 μm). CO2, CO and O2 were determined using a column filled with carbon molecular sieves.
 | ||
| Fig. 1 Diagram of the experimental equipment for the evaluation of catalyst activity. | ||
The calculation equation are as follows:




Note: each test is reproduced at least twice, and the obtained mass balance is above 95.0%.
In order to obtain the apparent activation energy of the methylal industry in a one-part process of methanol and ensure the accuracy of the experimental calculation of the apparent activation energy, we controlled the methanol conversion rate below 10%, the reaction conditions: atmospheric pressure, reaction temperature 180–240 °C, and total gas flow 50, 100 and 200 mL min−1. The rate calculation formula is as follows:31
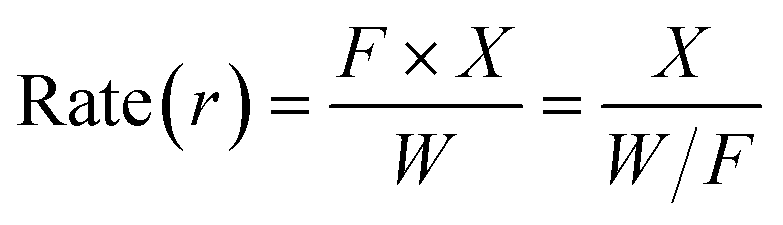
The activation energy was calculated using the Arrhenius equation.
3. Results and discussion
3.1 Preliminary exploration of a bifunctional catalyst
| Catalyst | Methanol conversion (%) | Product selectivity (%) | |||||
|---|---|---|---|---|---|---|---|
| DMM | FA | MF | DME | COx | Yield | ||
| Mo:Fe(2)/HY zeolite | 17.27 | 70.8 | 7.6 | 19.8 | 1.02 | 0.78 | 12.23 |
| Mo:Fe(2)/Al2O3 | 20.50 | 55.0 | 28.1 | 15.2 | 1.61 | 0.19 | 11.28 |
| Mo:Fe(2)/HZSM-5(40) | 28.50 | 58.0 | 7.81 | 31.7 | 1.91 | 0.58 | 16.53 |
| Mo:Fe(2)/HZSM-5(80) | 22.56 | 68.2 | 9.81 | 20.7 | 1.09 | 0.20 | 15.39 |
| Mo:Fe(2)/SiO2 | 21.94 | 8.30 | 79.7 | 8.70 | 0.00 | 3.29 | 1.82 |
In summary, the difference in the type of catalyst carrier had a considerable influence on the catalytic activity in the one-step methanol method. It is suspected that this difference is mainly due to the direct influence of the different oxidative and acidic characteristics of the Fe–Mo-based catalysts on the distribution of the catalytic products. To verify this hypothesis, a series of characterisation analyses on five catalysts were performed.
 | ||
| Fig. 2 N2 sorption isotherms (A) and PSD curves (B) of the catalysts. | ||
The pore size distribution is often related to the adsorption capacity of the adsorbent and the activity of the catalyst. Micropores are more conducive to the small molecule adsorption process of catalytic reactions, and mesopores facilitate the adsorption and separation of macromolecules. Combined with the results of Table 1, it could be inferred that the coexistence of micropores and mesopores in a catalyst is more conducive to the formation of the target product DMM in the one-step methanol process. Table S1† shows the specific surface area, pore volume and average pore size of different catalysts. The results in Tables 1 and S1† show that methanol conversion was the lowest with the Mo:Fe(2)/HY zeolite, which had the smallest specific surface area, and was the highest with Mo:Fe(2)/HZSM-5(40), which had the largest specific surface area. The larger the specific surface area, the more active sites are present and the higher the activity of the catalyst. The catalyst optimisation process in the next step focused on a catalytic carrier having a relatively large specific surface area and the presence of both micropores and mesopores.
Fig. 3 shows the XRD patterns of the catalysts. All samples exhibited a typical carrier structure. Mo in the form of MoO3 was observed as peaks at 2θ = 12.5°, 23.8°, 27.5°, 30.1° and 62.2°, and the characteristic peaks of Fe2(MoO4)3 were observed at 15.6°, 20.9°, 24.8°, 37.5°, 55° and 68°. Only two characteristic peaks of Fe2O3 were found, at 45° and 54.9°. These diffraction peaks matched the characteristic diffraction peaks of the compounds in their respective powder diffraction cards.32 The above results indicated that there were three compounds—Fe2O3, MoO3 and Fe2(MoO4)3—on the prepared bifunctional catalyst. Fe2(MoO4)3 could enable the redox cycle by electron transfer between the lattice oxygen and the metal ion, enhancing the oxidising ability of the catalyst and providing an oxidation centre for the one-step production of DMM from methanol. Bifunctional catalysts prepared using different feedstocks did not affect the metal loading of the final catalyst, except that the different peak intensities differed slightly.
 | ||
| Fig. 3 XRD patterns of catalysts with different carriers. | ||
The micro-morphology of the bifunctional catalysts prepared with different supports is shown in Fig. 4. From Fig. 4(a), it can be seen that the Mo:Fe(2)/HY zeolite bifunctional catalyst had regular spherical particles with different particle sizes between 40 and 60 μm, and large pore structures. Some small particles on the surface had combined to form small area accumulations, which may have been formed by the loading of metal oxides on the surface. From Fig. 4(b) it can be seen that the Mo:Fe(2)/Al2O3 bifunctional catalyst had an irregular lattice structure with a high pore density; under greater magnification it could also be observed that particles were attached to the surface. It can be seen from Fig. 4(c) that the Mo:Fe(2)/HZSM-5 bifunctional catalyst was composed of a collection of cubic lattices of different sizes, and that the channels were very dense. These aggregates help to increase the specific surface area of the catalyst and provide great benefit for its activity. It can be seen from Fig. 4(d) that the Mo:Fe(2)/SiO2 bifunctional catalyst had dense particles with different shapes and sizes, and that the shapes were very irregular.33 When the magnification was increased, it could be seen that the surface of the catalyst was densely covered with various aggregated particles; such polymerisation is beneficial in increasing the specific surface area of the catalyst.
 | ||
| Fig. 4 SEM images of (a) a Mo:Fe(2)/HY zeolite (b) Mo:Fe(2)/Al2O3 (c) Mo:Fe(2)/HZSM-5 and (d) Mo:Fe(2)/SiO2. Elemental mapping images of (e) O, (f) Al, (g) Fe and (h) Mo. | ||
Fig. 4(e–h) provides elemental mapping images of the Mo:Fe(2)/Al2O3 catalyst, showing the distribution of O, Al, Fe and Mo. The images show that the catalyst had the most O content, similar Al and Fe content, and sparse Mo content, and that O, Al, Fe, and Mo were uniformly dispersed on the entire catalyst. From SEM images of the five catalysts, it was found that the most suitable carriers were HZSM-5 and HY zeolite.
The oxidation states of Fe and Mo atoms in the Fe:Mo(2)/CAR (CAR included Al2O3, HZSM-5(80) and HY zeolite, with relatively good catalytic effect, as shown in Table 2) composite bifunctional catalyst were studied by XPS.34 The results of peak fitting are shown in Fig. 5. As shown in Fig. 5(A), the binding energy split peaks appearing at 712.3 and 726.2 eV corresponded to Fe3 +, and the Fe 2p orbit splitting peaks at 713.5, 716.9 and 732.8 eV corresponded to Fe2+. The XPS results of Mo 3d electrons are shown in Fig. 5(B). The binding energy peaks of Mo 3d5/2 and Mo 3d3/2 at 233.2 and 235.5 eV, respectively, corresponded to Mo6+ in MoO3 and Fe2(MoO4)3 respectively. The smaller peak of Mo 3d5/2 at 232.1 eV corresponded to Mo4+. It can be seen from the analysis that Mo6+ and Fe3+ were the main valence states of Mo and Fe in the elemental distribution on the catalyst. This was consistent with the results shown in the XRD spectra. In previous studies we determined that the best ferromolybdenum ratio was two and also discussed in detail the synergy between Mo and Fe active sites.13 The XPS spectra of Fe and Mo in Fig. 5 show that Mo existed in substantially the same form, and that only Mo:Fe(2)/HZSM-5(80) and the Mo:Fe(2)/HY zeolite showed an Fe2+peak. In combination with the results in Table 2, it was found that the selectivity of these two catalysts for DMM was higher. Therefore, we speculate that the presence of Fe2+ promotes the synergy between Mo and Fe, thereby increasing selectivity for the target product. Accordingly, optimisation of the next dual-function catalyst focused on the HZSM-5 and HY zeolite carriers.
 | ||
| Fig. 5 XPS spectra of (A) Fe 2p and (B) Mo 3d of the Fe–Mo bifunctional catalysts on different carriers. | ||
 | ||
| Fig. 6 NH3-TPD profiles of the Fe–Mo bifunctional catalysts on the different carriers. | ||
Qualitative analysis of the acidic sites of the catalyst was performed using PY-FTIR. As shown in Fig. 7, the characteristic peak of pyridine adsorbed on the B acid sites appeared at 1540 cm−1, and the characteristic peak of pyridine adsorbed on the L acid appeared at 1450 cm−1. The 1490 cm−1 peak represents the sum of the two acid centres.20 It can be seen in Fig. 7 that the presence of B acids in the Mo:Fe(2)/SiO2 catalyst could hardly be detected, whereas in the Mo:Fe(2)/Al2O3, Mo:Fe(2)/HZSM-5(40), Mo:Fe(2)/HZSM-5(80) and Mo:Fe(2)/HY zeolite catalysts both B and L acids were detected. The FTIR peak areas and corresponding relative acidity were roughly calculated according to the Lambert–Beer law.35 From the results shown in Table S3,† it can be seen that the Mo:Fe(2)/HZSM-5(40) catalyst had the most L acid content, and the B acid content was also moderate to high. Olesya1 reported that too much L acid promoted the dehydration condensation reaction of methanol and formic acid, promoting the formation of by-product MF. This observation also explains why the Mo:Fe(2)/ZSM-5(40) catalyst had the highest selectivity for the by-product MF (Table 2). The Mo:Fe(2)/HZSM-5(80) catalyst had the lowest L acid content, and the B acid content was moderate. The Mo:Fe(2)/HY zeolite catalyst had the most B acid content, followed by L acid. The Mo:Fe(2)/SiO2 catalyst had only L acid and the Mo:Fe(2)/Al2O3 catalyst had moderate content of both B and L acids. Combined with the results in Table 2, it was determined that Mo:Fe(2)/SiO2 had the lowest DMM selectivity and a high selectivity for by-products. Therefore, the acidic active centres of the bifunctional catalyst are derived from the synergistic effect of L acid and B acid sites, and the absence of B acid will affect the production of methanol polycondensation product DMM.
 | ||
| Fig. 7 FTIR spectra of pyridine adsorbed on the different catalysts. | ||
Studies have found that the distribution of Al sites on the molecular sieve framework will affect the concentration of catalyst B acid and L acid sites, thereby affecting the synergistic effect of these sites and thus affecting the production of target products. Combining the results shown in Tables 2 and S3,† the Mo:Fe(2)/ZSM-5(80) and Mo:Fe(2)/HY zeolite catalysts with higher B/L band ratios had higher DMM selectivity, whereas Mo:Fe(2)/ZSM-5(40) and Mo:Fe(2)/Al2O3 had high selectivity for MF and FA by-products. This proves that the distribution of Al sites on the molecular sieve framework influenced the concentration of catalyst B acid and L acid sites by increasing the B/L band ratio, thereby promoting the synergistic effect of active catalyst sites to improve catalytic activity. In the Mo:Fe(2)/HZSM-5(80) and Mo:Fe(2)/HY zeolite catalysts, both L acids and B acids were present and the B/L band ratio was moderate, which was the key consideration for optimisation of the bifunction catalyst.
It is well known that when Fe or Mo is loaded onto a zeolite catalyst and then calcined, the metals diffuse into the pores and undergo ion exchange.36 The metals then influence the distribution of B acid and L acid sites on the surface and inside the zeolite. To characterise these effects, we used PY-FTIR to characterise the molecular sieves before and after loading the metals; the results are shown in Fig. S2 and Table S3.† As can be seen, B acid sites were dominant in HZSM-5; however, after loading with Fe or Mo, the B/L ratio was significantly altered: the content of L acid increased significantly, which may be the result of ion exchange between Fe or Mo metal ions and Al outside the molecular sieve framework, resulting in an overall increase in acidity.9 In comparison, the content of B acid sites decreased slightly, indicating that during the heat treatment MOx migrated to the hydroxyl sites of the HZSM-5 tetrahedral framework channel through the surface thermal diffusion mechanism, leading to a decrease in the strength of B acid, consistent with previous reports. Nevertheless, after metal loading B acid was still the dominant acid in HZSM-5.37 The ion exchange of metal ions may affect the acid distribution of the zeolite, so the oxidation and acidic centres of the iron–molybdenum-based bifunctional catalyst influence each other and coordinate the catalytic activity together.
3.2 Optimisation of bifunctional catalysts
| Catalyst | Methanol conversion (%) | Product selectivity (%) | |||||
|---|---|---|---|---|---|---|---|
| DMM | FA | MF | DME | COx | Yield | ||
| Mo:Fe(2)/HY zeolite + HZSM-5(80) | 34.11 | 73.69 | 14.81 | 8.7 | 2.02 | 0.78 | 25.14 |
| Mo:Fe(2)/HY zeolite + HZSM-5(40) | 28.5 | 66.1 | 11.7 | 18.9 | 2.51 | 0.79 | 18.84 |
| Mo:Fe(2)/HY zeolite + Y zeolite | 24.10 | 68.80 | 10.61 | 17.11 | 2.77 | 0.71 | 16.58 |
| Mo:Fe(2)/HZSM-5(80 + 80) | 87.44 | 93.00 | 2.05 | 4.17 | 0.21 | 0.57 | 81.32 |
| Mo:Fe(2)/HZSM-5(8 + 40) | 85.43 | 81.3 | 3.91 | 11.11 | 1.91 | 0.77 | 69.45 |
| Mo:Fe(2)/HZSM-5(80) + HY zeolite | 40.29 | 70.61 | 12.11 | 12.41 | 3.16 | 0.71 | 28.45 |
| Mo:Fe(2)/HZSM-5(40 + 80) | 85.18 | 80.11 | 7.11 | 10.17 | 2.01 | 0.60 | 68.24 |
| Mo:Fe(2)/HZSM-5(40 + 40) | 78.45 | 77.11 | 4.81 | 11.77 | 5.81 | 0.58 | 60.49 |
| Mo:Fe(2)/HZSM-5(40) + HY zeolite | 54.29 | 77.83 | 10.61 | 6.84 | 3.85 | 0.87 | 42.25 |
As can be seen from Table 3, the methanol conversion of the nine groups of optimisation catalysts were all improved compared with the previous five groups of catalysts. Among these, the catalyst with HZSM-5 as the support had the most obvious increase in methanol conversion: Mo:Fe(2)/HZSM-5(40) had a highest methanol conversion rate of 85.18% and Mo:Fe(2)/HZSM-5(80) had a maximum methanol conversion rate of 87.44%. However, the maximum conversion rate after compounding the Mo:Fe(2)/HY zeolite was only 34.11%, and the effect was not very good. The Mo:Fe(2)/HZSM-5(80 + 80) catalyst had a DMM conversion rate of 93% and a yield of 81.32%. This result was greatly superior to that obtained using the other catalysts in Table 1 for this process. The methanol conversion with the Mo:Fe(2)/HZSM-5(80 + 40) catalyst was also 85.43% higher, the selectivity for DMM was 81.3% and the yield was 69.45%. The Mo:Fe(2)/HZSM-5(80) + HY zeolite catalyst afforded a methanol conversion rate of only 40.29%, and the Mo:Fe(2)/HZSM-5(40) + HY zeolite catalyst had a methanol conversion rate of 54.29%, which were lower than other catalysts in the same group. The catalytic differences were mainly due to the fact that the combined HY zeolite support did not perform as well as the two types of HZSM-5 catalysts in terms of acidity and acid type. To further investigate the specific role of the Si/Al ratio in the reaction, we evaluated Mo:Fe(2)/HZSM-5(X) (X = 20, 60, 100) catalysts. From the results in Table S7† it can be seen that the methanol conversion rate of the single-supported Fe–Mo-based catalyst was low, but the DMM selectivity gradually increased as the Si/Al ratio increased. From the HZSM-5(80) data, the optimised Fe–Mo bifunctional catalyst increased the methanol conversion rate from 21.89% to 84.41%, and the DMM selectivity also increased. According to the previous characterisation and analysis, it is known that with increasing the Si/Al ratio, the concentration of Al sites on the molecular sieve framework decreases, the concentrations of B and L acid sites gradually decreases, and the B/L band ratio gradually increases. From the results in Tables 3 and S6† it can be seen that the larger the B/L band ratio, the higher the DMM selectivity; therefore, the increase in the B/L band ratio promotes the production of the target product. We speculate that the key factors affecting the catalytic performance is the synergistic effect of the carrier's L acid and B acid sites, and the synergy between the carrier and Fe–Mo elements. In order to verify this speculation, a further series of characterisation analyses were performed on the catalyst.
 | ||
| Fig. 8 (A) N2 sorption isotherms and (B) PSD curves of the catalysts. | ||
The XRD spectrum of the catalyst before and after compounding optimization is shown in Fig. 9. We have selected representative Mo:Fe(2)/Y zeolite + Y zeolite, Mo:Fe(2)/HZSM-5(40 + 80), Mo:Fe(2)/HZSM-5(80 + 80) catalysts for comparison. As can be seen from Fig. 9, the characteristic peaks of the carrier did not change before and after compounding. The characteristic absorption peaks of Fe2O3, MoO3 and Fe2(MoO4)3 crystals have not changed. This result proved that optimisation of the catalyst did not change the electron transfer between the oxygen and metal ions of the Fe–Mo oxide lattice. This also showed that the optimised combination of the dual-function catalyst did not affect the metal loading of the catalyst, nor was it the root cause of the catalytic effect.
 | ||
| Fig. 9 XRD patterns of catalysts with different carriers. | ||
XPS spectra were used to compare the effects of catalyst optimisation on the oxidation states of Fe and Mo before and after the catalyst combination optimisation. The fitted XPS spectra are shown in Fig. 10; Fig. 10(A) shows that the main valence states of Fe before and after the catalyst optimisation were Fe2+ and Fe3+, respectively. It can be seen in Fig. 10(B) that the valence states of Mo before the catalyst optimisation were Mo6+ and Mo5+, which also coexisted in the optimised catalyst.38 Therefore, it was speculated that optimisation of the catalyst prompted the emergence of Mo5+. Combining the previous discussion of the synergistic effect of the active sites of Fe and Mo oxidation, it is proposed that the appearance of Mo5+ promoted the two terminal oxygens to coordinate with the Mo double bond in the octahedron.39 This activated the hydrogen of the methanolic hydroxyl group to produce a methoxyl species, which was the intermediate in the formation of FA and allowed rapid further reaction.40 This is one of the reasons why the catalytic activity was significantly improved after optimisation.
 | ||
| Fig. 10 XPS spectra of (A) Fe 2p and (B) Mo 3d of the Fe–Mo bifunctional catalysts, on the different carriers. | ||
 | ||
| Fig. 11 NH3-TPD profiles of the Fe–Mo bifunctional catalysts on the different carriers. | ||
Qualitative analysis of the acid sites of the optimised catalyst was performed using pyridine infrared, and the results are shown in Fig. 12. It can be seen in Fig. 12 that the three catalysts have the characteristic peaks of pyridine of the B acid and L acid. The FTIR area peak and its relative acidity were roughly calculated according to Lambert–Beer's law. The result is shown in Table S6.† It can be seen from Table S6† that the three types of catalysts have the highest L acid content of Mo:Fe(2)/HZSM-5(40 + 80). Mo:Fe(2)/HZSM-5(80 + 80) has the highest B acid content. According to the results shown in Tables 3 and S6,† the two groups of catalysts with the highest and lowest amounts of L acid sites and B acid sites did not have the best catalytic activity. When B:L = 3.82, the Mo:Fe(2)/HZSM-5(80 + 80) catalyst had the best catalytic effect. This also indicated that the synergistic effect of L acid and B acid was most conducive to the dehydration condensation of methanol and FA to obtain DMM. Benito,42 Gayubo,43 and Abdullah44 all reported that as the Si/Al ratio increases, the ratio of B/L sites also increases. As the concentration of the Al sites on the molecular sieve framework decreases, the concentrations of B acid and L acid gradually decreases, but the rate of decrease of B acid sites is lower than that of L acid sites, so the B/L ratio gradually increases. This trend can be attributed to the limited dehydroxylation of B sites compared to L sites that occurs at high SiO2/Al2O3 fractions. Table S3† shows that when the Si/Al ratio was from 40 to 80, the concentrations of L acid and B acid sites both decreases, but the B/L ratio increased. These findings were in general agreement with the results of others. From the results of Table 3 it can also be seen that as the B/L ratio increased, the target product selectivity also increased, which was due to the shape-selective catalytic behaviour of the catalyst. An important step in the one-step synthesis of DMM via the methanol oxidation process is to control the reaction at the methanol acetalisation stage, where the B acid sites of the catalyst play a crucial role. It is difficult for methanol to undergo acetalisation to form DMM when utilising bifunctional catalysts without B acid sites.45 This hypothesis could be tested by investigating this reaction utilising Mo:Fe(2)/SiO2 catalysis. The Mo:Fe(2)/HZSM-5(80 + 80) catalyst had the highest B acid site content and the best catalytic effect. Many studies have proved that the B acid sites are the key active sites of the catalyst in the methanol conversion process. For example, the selectivity of the MTG and MTO (methanol to olefin) reaction to form gasoline increases with an increase of the B/L ratio.37 In contrast, the higher the ratio of L acid sites, the higher the selectivity of the by-product MF. Therefore, the appropriate ratio of the B acid and L acid was also one of the main factors in improving the yield of the target product. Meng et al.46 found that the Al outside the framework of the zeolite catalyst was lost after the acid treatment, which resulted in loss of the Si–O–Al structure and thus a partial reduction in the number of B acid sites. It was later discovered, however, that in the process of removing Al, although the Al outside the framework was removed, it was transferred to the framework in one step. The loss of B acid sites led to a decrease in the B/L ratio, increasing selectivity for by-products. Almutairi et al.47 found that the steam treatment process causes Al species to be re-inserted into the superstructure, therefore making the B acid sites closer to the outer surface more accessible. At the same time, according to the catalytic results, the higher the B/L ratio, the better the catalytic activity and the higher the selectivity for the target product. This also verified the previous theoretical analysis that the higher the B acid site content, the greater the DMM selectivity and—by contrast—that the higher the L acid site content, the higher the selectivity of the by-product MF.
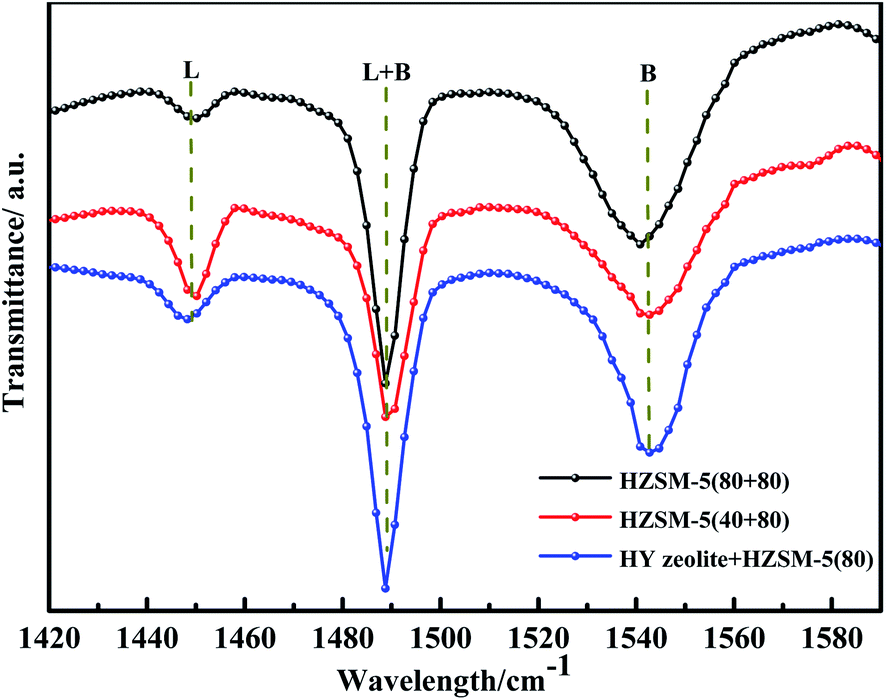 | ||
| Fig. 12 FTIR spectra of pyridine adsorbed on the different catalysts. | ||
The polycondensation reaction of methanol and FA is an acid-catalysed shape-selective reaction, for which the acidity and diffusion properties of zeolites are especially important.48 The number of B acid sites affects the selectivity of DMM, and the intrinsic diffusion properties also need to be considered. The diffusion capacity is another factor affecting catalytic activity.49 The high specific surface area and pore volume reduce diffusion resistance and prevent the formation of coke that would block the pores. On the other hand, a larger specific surface area, outer surface area and mesopore volume enable more acidic centres to be exposed to the reactants and promotes the reaction.30 Additionally, the increased number of acidic sites also reduces the rate of carbon deposition.
The 27Al MAS NMR spectra of the three composite catalysts are shown in Fig. 13. Previous literature data has indicated that the signal at 0 ppm in the 27Al MAS NMR spectrum corresponds to the octahedral-coordinated Al atoms not incorporated into the zeolite framework, with the signal at 45–65 ppm corresponding to the tetrahedral-coordinated Al atoms within the zeolite framework.41 As shown in Fig. 13, the three catalysts all had tetrahedral-coordinated Al atoms at 54.66 ppm. However, Mo:Fe(2)/HZSM-5(40 + 80) and Mo:Fe(2)/HZSM-5(80 + 80) had weak peaks at 0 ppm, whilst Mo:Fe(2)/HY zeolite + HZSM-5(80) had a stronger ion peak at 0 ppm. Therefore, for the Mo:Fe(2)/HY zeolite + HZSM-5(80) ion-exchange catalyst, only a fraction of the Al was incorporated into the zeolite framework, with the incomplete combination of Al resulting in a decrease in the amount of solid acid.50 This explains why the NH3-TPD spectrum showed a low content of strong acid and medium-strong acid for Mo:Fe(2)/HY zeolite + HZSM-5(80), as well as its low methanol conversion rate. It is well known that aluminosilicate is mainly dependent on the acidity of active zeolite. For ZSM-5, the B acid site directly corresponds to the tetrahedral-coordinated Al atoms.51Fig. 13 shows that the 54.66 ppm peak intensity of Mo:Fe(2)/HZSM-5(80 + 80) was high, so its B acid content was relatively large; this was consistent with the results in Table S6.† The increase in B acid content also promoted the formation of target products, so Mo:Fe(2)/HZSM-5(80 + 80) had the greatest selectivity for DMM.
 | ||
| Fig. 13 27Al MAS NMR spectra of the three composite catalysts. | ||
3.3 Apparent activation energies of different carriers catalysts
From the methanol oxidation pathway (Fig. S1†)52 it can be seen that methanol is dehydrogenated in one step to obtain FA, which is then oxidised to obtain formic acid, which further oxidises to form DMM. When methanol interacts with the acidic position of the catalyst, it dehydrates and condenses to form DME. FA and methanol will also undergo aldol condensation when encountering the acidic position of the catalyst to form DMM.52 Additionally, when formic acid and methanol interact with the acidic site of the catalyst, dehydration condensation occurs to form MF. Therefore, differences in the active site of the catalyst directly affect the resulting catalysis products. The effect of different catalytic properties on catalytic activity was explored through a series of characterisation analyses. To investigate whether any factors other than the oxidation and acidity characteristics of the catalysts impacted their catalytic activity, the activation energy with different catalysts was measured. These Arrhenius plots are shown in Fig. 14 and the detailed results of the methanol conversion are provided in Fig. S3.† | ||
| Fig. 14 Arrhenius plots for different carrier catalysts. | ||
As shown in Fig. 14, the lowest activation energy of Mo:Fe(2)/HZSM-5(80 + 80) is 45.06 kJ mol−1, Mo:Fe(2)/HZSM-5(40 + 80) also has a lower activation energy of 47.14 kJ mol−1. Mo:Fe(2)/HY zeolite + HZSM-5(80) has the highest activation energy of 62.77 kJ mol−1. The results in Table 3 show that the methanol conversion rates of the two groups of catalysts with similar activation energy were also very close. The catalyst with the highest activation energy had low catalytic activity and the methanol conversion rate was only 34.11%. This showed that the lower activation energy was also one of the reasons for the higher catalytic activity of the Fe–Mo-based bifunctional catalysts. It also revealed that the Mo:Fe(2)/ZSM-5(80 + 80) bifunctional catalyst was the most suitable for the one-step production of DMM from methanol using the same preparation method and reaction process.
3.4 Discussion
After screening and characterising five kinds of carriers, three more suitable carriers were selected and optimised. It was found that the methanol conversion rate of the optimised bifunctional catalyst increased from 22.56% to 87.44%. The highest yield of Mo:Fe(2)/HZSM-5(80 + 80) bifunctional catalyst was 81.3%. Through a series of characterisation analyses, it was found that there were three main reasons for the higher activity of this optimised bifunctional catalyst, under the same reaction conditions. First, the appearance of Mo5+ after optimisation promoted the coordination of the two terminal oxygens with the Mo double bond in the octahedron to activate the hydrogen of the methanolic hydroxyl group, thereby producing a methoxyl species that was the intermediate in the formation of FA and allowed rapid further reaction. Second, the increase in the acidity of the catalyst enhanced its activity, with its medium-strong acidity conducive to increasing the methanol conversion rate. However, if the strong acid content was too high it promoted formation of the by-product formic acid, which then reacted with methanol to form MF and decreased selectivity for the target product. As the second step in DMM synthesis is the acetalisation reaction of FA and CH3OH, the increased number of acidic sites on Fe–Mo-based catalysts might be favourable for DMM synthesis. Therefore, enhancement of the catalyst's weak acidity and mid-strength acid was one of the fundamental reasons for the increased catalyst activity after optimisation. Third, through the study of metal-ion exchange, changes of the Si/Al ratio and the diffusion capacity of zeolite, it was found that the B acid sites were the key active sites of the catalyst in the methanol conversion process. The higher the content of B acid sites, the greater the DMM selectivity; conversely, the higher the content of L acid sites, the higher the selectivity for the by-product MF. By studying the apparent kinetics, it was found that the bifunctional catalyst optimised with HZSM-5 as the carrier had the lowest apparent activation energy and the best catalytic effect. In previous reports, the highest yield of DMM from methanol using actinide and V–Ti bifunctional catalysts studied by Royer16 and Sun18 was 61.4%. The optimised Fe–Mo-based dual-functional catalyst had a maximum yield of 81.3%, and its catalytic performance far exceeded that of the traditional dual-functional catalyst.4. Conclusion
This study explored the effects of the catalyst support on the one-step synthesis of DMM via methanol oxidation catalysed by Fe–Mo-based bifunctional catalysts based on the previously determined optimal ratio of Fe to Mo. The acidity of the bifunctional catalyst was adjusted by changing the type of support, and the resulting Fe–Mo-based catalysts were tested in the one-step conversion of methanol to DMM in a fluidised bed reactor. The results showed that Mo:Fe(2)/HZSM-5(80 + 80), in which HZSM-5(80 + 80) was the catalytic support, had excellent catalytic performance with the yield of DMM reaching 81.33%, much higher than other reported results of this process. Additionally, in the process of preliminary screening and optimisation, we not only determined the best carrier but also explored the fundamental reasons for improved catalytic outcomes as follows: the appearance of Mo5+, which promoted the coordination of the two terminal oxygens with the Mo double bond in the Fe2(MoO4)3 octahedron; and, the critical role of the B acid sites in the shape-selective catalysis of the target product, with a higher B/L ratio improving the catalytic activity and selectivity for the target product. The effects of catalyst oxidation and acidic site regulation on the distribution of methanol selective oxidation polycondensation products were also explored. The apparent activation energy also further proved that bifunctional catalyst Mo:Fe(2)/HZSM-5(80 + 80) was highly suitable for the one-step methanol process. These results open up new possibilities for future DMM production via a simpler and more economical process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was conducted with THE National Natural Science Foundation of China (No. 21878335, 21576293 and 21576294), Major scientific and technological innovation projects in Shandong Province of China (No. 2018CXGC0301), and the Fundamental Research Funds for the Central Universities (18CX02121A), supported by the State Key Laboratory of Heavy Oil Processing (SKLOP201901001), the Postgraduate Innovation Funding Project of China University of Petroleum (East China) (YCX2020040) and the Shandong Natural Science Foundation (ZR2017QEE006).References
- O. A. Nikonova, M. Capron, G. Fang, J. Faye, A.-S. Mamede, L. Jalowiecki-Duhamel, F. Dumeignil and G. A. Seisenbaeva, J. Catal., 2011, 279, 310–318 CrossRef CAS.
- S. Damiri, H. R. Pouretedal and O. Bakhshi, Chem. Eng. Res. Des., 2016, 112, 155–162 CrossRef CAS.
- H. Huang, Y. Chen, J. Zhu, Y. Chen, D. Lv, Z. Zhu, L. Wei and Y. Wei, Energy Fuels, 2019, 33, 3504–3517 CrossRef CAS.
- H. Liu and E. Iglesia, J. Phys. Chem. B, 2003, 107, 10840–10847 CrossRef CAS.
- J. Liu, Q. Sun, Y. Fu and J. Shen, J. Colloid Interface Sci., 2009, 335, 216–221 CrossRef CAS.
- Z. Xue, H. Shang, Z. Zhang, C. Xiong, C. Lu and G. An, Energy Fuels, 2016, 31, 279–286 CrossRef.
- S. Chen, Y. Meng, Y. Zhao, X. Ma and J. Gong, AIChE J., 2013, 59, 2587–2593 CrossRef CAS.
- L. R. Merte, M. Ahmadi, F. Behafarid, L. K. Ono, E. Lira, J. Matos, L. Li, J. C. Yang and B. Roldan Cuenya, ACS Catal., 2013, 3, 1460–1468 CrossRef CAS.
- A. Kostyniuk, D. Key and M. Mdleleni, J. Energy Inst., 2020, 93, 552–564 CrossRef CAS.
- Y. Wu, Z. Li and C. Xia, Ind. Eng. Chem. Res., 2016, 55, 1859–1865 CrossRef CAS.
- Y. Zhao, Z. Qin, G. Wang, M. Dong, L. Huang, Z. Wu, W. Fan and J. Wang, Fuel, 2013, 104, 22–27 CrossRef CAS.
- V. V. Kaichev, G. Y. Popova, Y. A. Chesalov, A. A. Saraev, D. Y. Zemlyanov, S. A. Beloshapkin, A. Knop-Gericke, R. Schlögl, T. V. Andrushkevich and V. I. Bukhtiyarov, J. Catal., 2014, 311, 59–70 CrossRef CAS.
- M. Yuan, Y. Che, R. Tang, S. Li, Y. Zhang, Y. Tian, Y. Qiao, Q. Liu and D. Li, Fuel, 2020, 261, 116416 CrossRef CAS.
- Y. Yuan, H. Liu, H. Imoto, T. Shido and Y. Iwasawa, J. Catal., 2000, 195, 51–61 CrossRef CAS.
- Y. Yuan, T. Shido and Y. Iwasawa, Chem. Commun., 2000, 1421–1422, 10.1039/b003870i.
- S. Royer, X. Secordel, M. Brandhorst, F. Dumeignil, S. Cristol, C. Dujardin, M. Capron, E. Payen and J. L. Dubois, Chem. Commun., 2008, 865–867, 10.1039/b714260a.
- X. Lu, Z. Qin, M. Dong, H. Zhu, G. Wang, Y. Zhao, W. Fan and J. Wang, Fuel, 2011, 90, 1335–1339 CrossRef CAS.
- Q. Sun, J. Liu, J. Cai, Y. Fu and J. Shen, Catal. Commun., 2009, 11, 47–50 CrossRef CAS.
- Y.-H. Yeh and R. J. Gorte, Ind. Eng. Chem. Res., 2016, 55, 12795–12805 CrossRef CAS.
- D. Yang, D. Li, H. Yao, G. Zhang, T. Jiao, Z. Li, C. Li and S. Zhang, Ind. Eng. Chem. Res., 2015, 54, 6865–6873 CrossRef CAS.
- R. Thattarathody and M. Sheintuch, Ind. Eng. Chem. Res., 2019, 58, 11902–11909 CrossRef CAS.
- H. Song, F. Jin, M. Kang and J. Chen, RSC Adv., 2019, 9, 40662–40669 RSC.
- P. Tan, Appl. Catal., A, 2019, 580, 111–120 CrossRef CAS.
- D. Zhou, X. Huang, H. Wen, R. Shen, Y. Liu, X. Guo and B. Li, Sustainable Energy Fuels, 2020, 4, 3677–3686 RSC.
- Z. W. Tian, Q. Liu and B. Bian, Sustainable Energy Fuels, 2020, 4, 2396–2403 RSC.
- Y. Lou, J. Ma, W. Hu, Q. Dai, L. Wang, W. Zhan, Y. Guo, X.-M. Cao, Y. Guo, P. Hu and G. Lu, ACS Catal., 2016, 6, 8127–8139 CrossRef CAS.
- L. Zhang, L. Chen, Y. Li, Y. Peng, F. Chen, L. Wang, C. Zhang, X. Meng, H. He and F.-S. Xiao, Appl. Catal., B, 2017, 219, 200–208 CrossRef CAS.
- C. Brookes, P. P. Wells, G. Cibin, N. Dimitratos, W. Jones, D. J. Morgan and M. Bowker, ACS Catal., 2013, 4, 243–250 CrossRef.
- S. Chen, S. Wang, X. Ma and J. Gong, Chem. Commun., 2011, 47, 9345–9347 RSC.
- Z. Han, F. Zhou, J. Zhao, Y. Liu, H. Ma and G. Wu, Microporous Mesoporous Mater., 2020, 302, 110194 CrossRef CAS.
- Q. Liu, J. Gao, F. Gu, X. Lu, Y. Liu, H. Li, Z. Zhong, B. Liu, G. Xu and F. Su, J. Catal., 2015, 326, 127–138 CrossRef CAS.
- Z. Zhu, G. Lu, Z. Zhang, Y. Guo, Y. Guo and Y. Wang, ACS Catal., 2013, 3, 1154–1164 CrossRef CAS.
- D. P. DePuccio, L. Ruíz-Rodríguez, E. Rodríguez-Castellón, P. Botella, J. M. López Nieto and C. C. Landry, J. Phys. Chem. C, 2016, 120, 27954–27963 CrossRef CAS.
- L. Wang, L. Chao, W. Qu, S. Xu, L. Zhang, J. Peng and X. Ye, Ultrason. Sonochem., 2018, 49, 24–32 CrossRef CAS.
- C. A. Emeis, J. Catal., 1993, 141, 347–354 CrossRef CAS.
- X. Cheng, P. Yan, X. Zhang, F. Yang, C. Dai, D. Li and X.-X. Ma, Mol. Catal., 2017, 437, 114–120 CrossRef CAS.
- J. Li, D. Han, T. He, G. Liu, Z. Zi, Z. Wang, J. Wu and J. Wu, Fuel Process. Technol., 2019, 191, 104–110 CrossRef CAS.
- G. Jin, W. Weng, Z. Lin, N. F. Dummer, S. H. Taylor, C. J. Kiely, J. K. Bartley and G. J. Hutchings, J. Catal., 2012, 296, 55–64 CrossRef CAS.
- V. M. Shinde and G. Madras, AIChE J., 2014, 60, 1027–1035 CrossRef CAS.
- H. Xia, Z. Liu, Y. Xu, J. Zuo and Z. Qin, Catal. Commun., 2016, 86, 72–76 CrossRef CAS.
- H. Fujitsuka, S. Oshima, Y. Matsumura and T. Tago, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.04.007.
- A. G. Gayubo, P. L. Benito, A. T. Aguayo, O. Martin and J. Bilbao, J. Chem. Technol. Biotechnol., 1996, 66, 183–191 CrossRef.
- P. L. Bennici, A. G. Gayubo, A. T. Aguayo, M. Olazar and J. Bilbao, J. Chem. Technol. Biotechnol., 1996, 65, 186–192 CrossRef.
- A. S. Al-Dughaither and H. de Lasa, Ind. Eng. Chem. Res., 2014, 53, 15303–15316 CrossRef CAS.
- A. Galadima and O. Muraza, J. Nat. Gas Sci. Eng., 2015, 25, 303–316 CrossRef CAS.
- X. Meng, Z. Lian, X. Wang, L. Shi and N. Liu, Fuel, 2020, 270, 117426 CrossRef CAS.
- S. M. T. Almutairi, B. Mezari, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, J. Catal., 2013, 307, 194–203 CrossRef CAS.
- Y. Xue, J. Li, P. Wang, X. Cui, H. Zheng, Y. Niu, M. Dong, Z. Qin, J. Wang and W. Fan, Appl. Catal., B, 2021, 280, 119391 CrossRef CAS.
- M. Rostamizadeh and F. Yaripour, Fuel, 2016, 181, 537–546 CrossRef CAS.
- J. Valecillos, E. Epelde, J. Albo, A. T. Aguayo, J. Bilbao and P. Castaño, Catal. Today, 2020, 348, 243–256 CrossRef CAS.
- C. Li, A. Vidal-Moya, P. J. Miguel, J. Dedecek, M. Boronat and A. Corma, ACS Catal., 2018, 8, 7688–7697 CrossRef CAS.
- R. Wojcieszak, E. M. Gaigneaux and P. Ruiz, ChemCatChem, 2013, 5, 339–348 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se01194k |
| This journal is © The Royal Society of Chemistry 2021 |