
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of sterically congested oxindole derivatives via visible-light-induced radical-coupling†
Yanling
Shen
,
Ning
Lei
,
Cong
Lu
,
Dailin
Xi
,
Xinxin
Geng
,
Pan
Tao
,
Zhishan
Su
 and
Ke
Zheng
*
and
Ke
Zheng
*
Key Laboratory of Green Chemistry & Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: kzheng@scu.edu.cn
First published on 17th November 2021
Abstract
The oxindole scaffold represents an important structural feature in many natural products and pharmaceutically relevant molecules. Herein, we report a visible-light-induced modular methodology for the synthesis of complex 3,3′-disubstituted oxindole derivatives. A library of valuable fluoroalkyl-containing highly sterically congested oxindole derivatives can be synthesized by a catalytic three-component radical coupling reaction under mild conditions (metal & photocatalyst free, >80 examples). This strategy shows high functional group tolerance and broad substrate compatibility (including a wide variety of terminal or non-terminal alkenes, conjugated dienes and enynes, and a broad array of polyfluoroalkyl iodide and oxindoles), which enables modular modification of complex drug-like compounds in one chemical step. The success of solar-driven transformation, large-scale synthesis, and the late-stage functionalization of bioactive molecules, as well as promising tumor-suppressing biological activities, highlights the potential for practical applications of this strategy. Mechanistic investigations, including a series of control experiments, UV-vis spectroscopy and DFT calculations, suggest that the reaction underwent a sequential two-step radical-coupling process and the photosensitive perfluoroalkyl benzyl iodides are key intermediates in the transformation.
Introduction
The oxindole scaffold is one of the most abundant structural units in a wide variety of natural products, agrochemicals, and pharmaceutical agents.1 In particular, 3,3′-disubstituted oxindoles with one or more all-carbon quaternary centers play a significant role in a number of biologically active2 and medicinal agents3 (scheme 1A), benefitting from numerous distinct properties arising from their fully substituted carbon centers. Therefore, extensive efforts are focused on developing an efficient strategy for the construction of these compounds.4 One classical method for accessing this class of compounds is nucleophilic substitution of 3-substituted oxindoles, taking advantage of their high nucleophilicity at the C3-position of oxindoles, and most of these methods can be rendered enantioselective through the action of asymmetric catalysts (Scheme 1B, left part).4c–g However, the practical utilization of these methods is largely limited to the unhindered substrates (primary or secondary), and the extension to tertiary substrates is still a challenge due to their increased steric hindrance, as well as the limitations of commercially available tertiary substrates.5 Despite extensive efforts made in substitution reactions, a small number of radical–radical coupling processes have been reported as an alternative protocol for the synthesis of 3,3′-disubstituted oxindoles. However, these strategies have major limitations in complex molecular settings (Scheme 1B, right part).6 Considering the increasing demand of drug discovery for flexible modulation of 3,3′-disubstituted oxindole derivatives with structural complexity, the discovery of new protocols to access these important frameworks from ubiquitous starting materials is highly desired.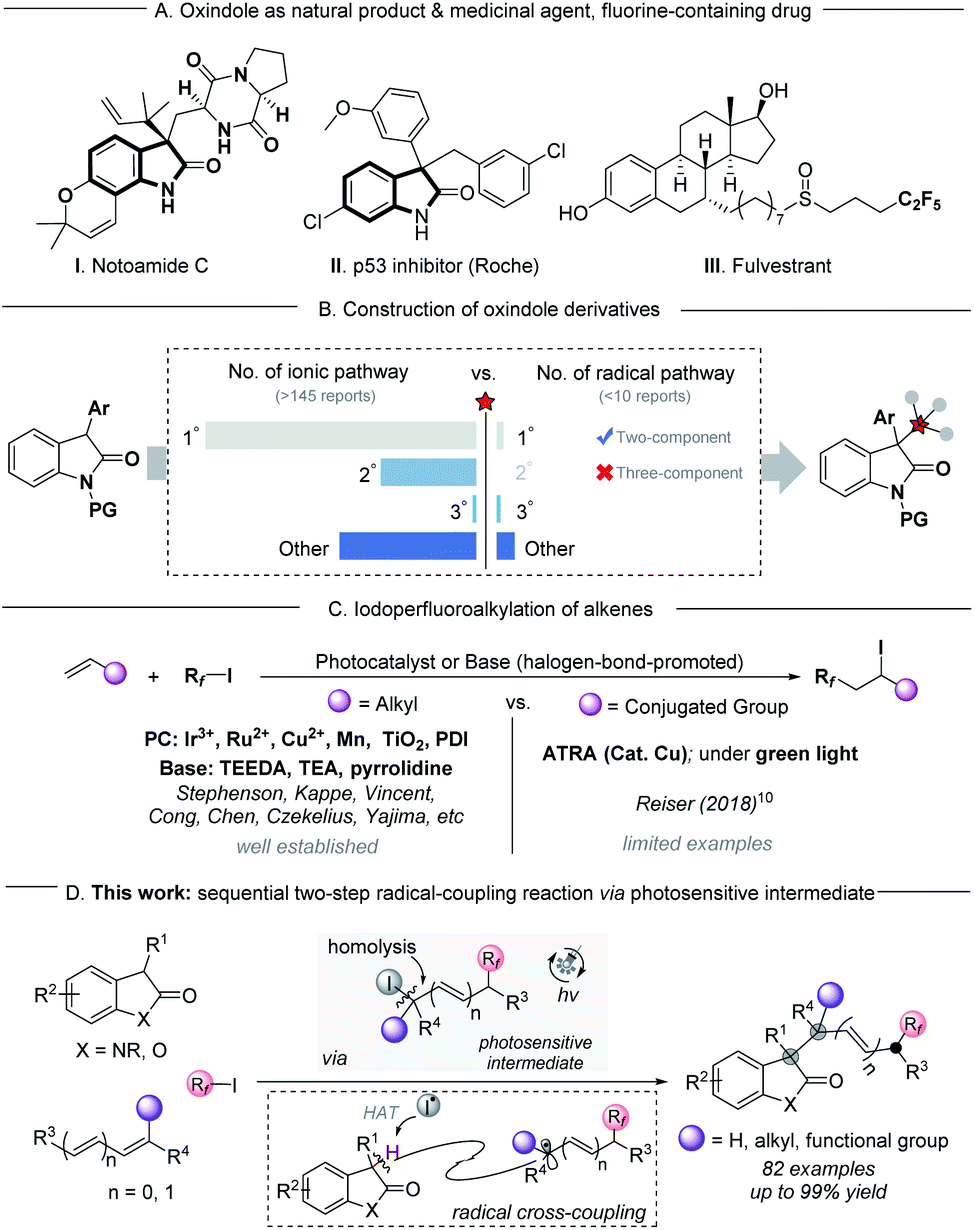 | ||
| Scheme 1 Sequential two-step radical-coupling reaction via photosensitive intermediates. | ||
Fluorine-containing groups in organic molecules serve as versatile and valuable motifs in the agrochemical industry, medicinal chemistry and materials science, and have the potential to exert profound changes in the lipophilicity, solubility, metabolic stability, and bioavailability properties of molecules in drug discovery.7 The iodoperfluoroalkylation of olefins is an efficient transformation for accessing this class of compounds relying on the use of photochemistry, in which the commercially available perfluoroalkyl halides (Rf–X) were used as powerful and versatile precursors to generate electrophilic perfluoroalkyl radicals.8 Numerous elegant reports of photocatalytic iodoperfluoroalkylation of alkenes using organic bases or photocatalysts have been well established.9 However, for most of them, the radical acceptors were limited to alkyl alkenes, and only a limited number of analogous transformations have been reported for conjugated alkenes (Scheme 1C).10 This was likely to originate from the instability of conjugated carbon radical intermediates,11,12 or iodoperfluoroalkylation products such as sensitive benzyl iodides, especially the tertiary benzyl iodides with steric hindrance.10a,13 We questioned whether or not such unstable, photosensitive benzyl iodides could be used as intermediates for the next transformations by secondary excitation under irradiation. If it works, it should be possible to design a sequential multistep radical-coupling process to construct functionally and structurally diverse products from ubiquitous starting materials under mild conditions.
Continuing our interest in developing new photochemical methods,14 we present herein visible-light-driven highly selective three-component radical coupling reactions for the construction of fluoroalkyl-containing highly sterically congested oxindole derivatives in a single step, which would require multistep syntheses using other methods (>80 examples, Scheme 1D). This transformation could successfully be applied to a wide variety of conjugated alkenes, including terminal or non-terminal styrene derivatives, dienes and enynes. The potential efficacy of this new approach was evidenced by the success of gram-level synthesis, late-stage functionalization, and modular modification of known bioactive pharmaceuticals.
Results and discussion
On the basis of our hypothesis, we first selected 3-phenylindolin-2-one (1a), α-methyl-styrene (2a) and perfluorobutyl iodide (3a) as model substrates for optimization studies. After exploring different reaction conditions, the best results were accomplished with DIPEA as the base additive under white light irradiation in THF under N2, affording three-component coupling product 4 in 88% isolated yield with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr value (Table 1, entry 1). Among the light sources screened, white light proved to be the most effective at facilitating transformation and green light could not promote the reaction (Table 1, entries 2–3 and Table S1†). Several other common organic solvents led to slightly lower yields (Table 1, entries 4–5 and Table S2†). When using inorganic base Cs2CO3 or K2CO3 instead of DIPEA, lower yields were observed, and aniline completely shut down the reaction (Table 1, entries 6–8). Either higher or lower concentrations of the reaction mixture gave decreased yields (Table 1, entry 9). To our delight, the reaction time could be significantly shortened with a similar outcome at higher temperature (Table 1, entry 10). A series of control experiments indicated that the base additive DIPEA, light, and N2 atmosphere were all crucial for this reaction (Table 1, entries 11–12).
:1 dr value (Table 1, entry 1). Among the light sources screened, white light proved to be the most effective at facilitating transformation and green light could not promote the reaction (Table 1, entries 2–3 and Table S1†). Several other common organic solvents led to slightly lower yields (Table 1, entries 4–5 and Table S2†). When using inorganic base Cs2CO3 or K2CO3 instead of DIPEA, lower yields were observed, and aniline completely shut down the reaction (Table 1, entries 6–8). Either higher or lower concentrations of the reaction mixture gave decreased yields (Table 1, entry 9). To our delight, the reaction time could be significantly shortened with a similar outcome at higher temperature (Table 1, entry 10). A series of control experiments indicated that the base additive DIPEA, light, and N2 atmosphere were all crucial for this reaction (Table 1, entries 11–12).
|
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yieldb% |
|
a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), 3a (0.2 mmol) and DIPEA (0.2 mmol) in THF (1.0 mL) were irradiated with 10 W white LEDs under N2 at 25 °C.
b Yield of the isolated product; dr (diastereomeric ratio) of product was 1:1.
c Reaction time: 4 h. DIPEA = N,N-diisopropylethylamine.
|
||
| 1 | None | 88 |
| 2 | 395 nm LED | 75 |
| 3 | 525 nm LED | Trace |
| 4 | DMF as solvent | 69 |
| 5 | CH3OH as solvent | 66 |
| 6 | Cs2CO3 instead of DIPEA | 43 |
| 7 | K2CO3 instead of DIPEA | 73 |
| 8 | Aniline instead of DIPEA | Trace |
| 9 | 0.2 M or 0.05 M | 77 |
| 10 | Heat to 40 °C | 88c |
| 11 | Without DIPEA | Trace |
| 12 | In air or without light | Trace |
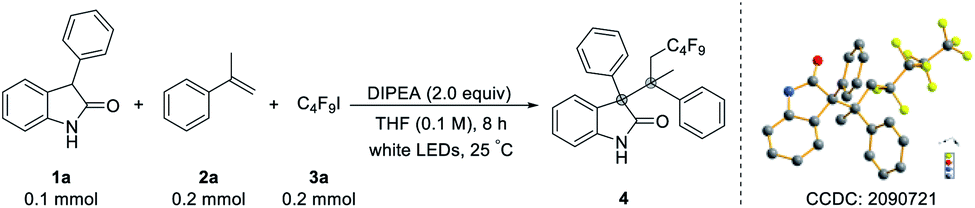
With the optimized reaction conditions in hand, we first explored the scope of 3-substituted oxindoles (Table 2). Remarkably, the reaction proved suitable for the synthesis of highly sterically congested quaternary centers even in good to excellent yields (5–29). The reactions performed well with a variety of 3-aryl substituted oxindoles with electron-neutral, electron-donating, and electron-withdrawing substituents, giving the corresponding products in excellent yields (5–16). The oxindole with 4-substituents on the aromatic ring produced the products with slightly lower yield (5) due to its steric crowding adjacent to the reactive site. Substituents on the aromatic ring of the 3-aryl oxindole with a variety of functional groups afforded the product in good to excellent yields (17–25). To our delight, 3-heterocyclic- and 3-naphthyl- substituted oxindoles were also well tolerated under optimized conditions (26 and 27). The substituted group on the nitrogen of oxindoles does not affect the outcome (86%, 28), and benzofuranone gave the desired product in high yield (95%, 29).
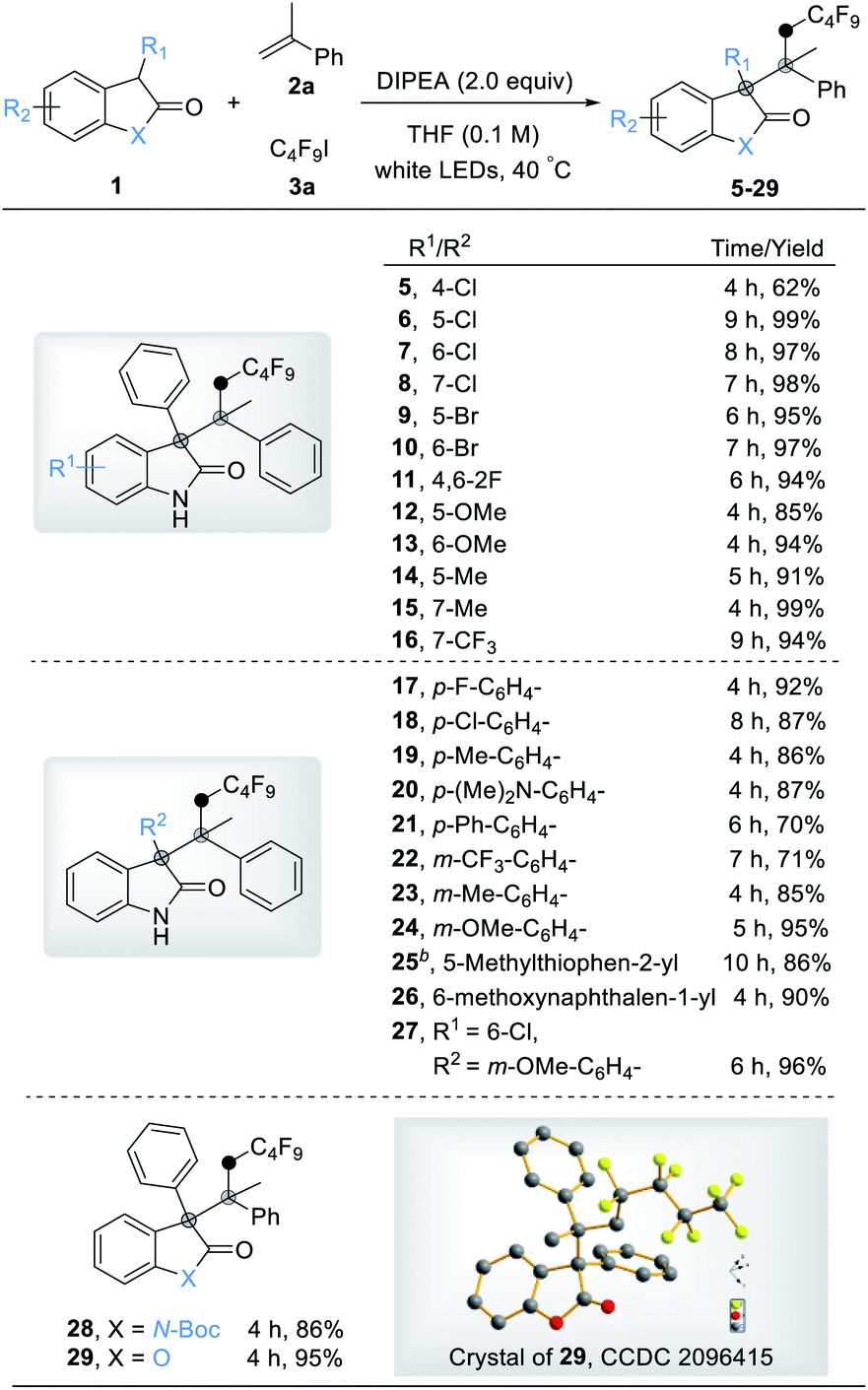
Having established a broad scope for oxindoles, our attention was turned to the scope with respect to alkenes. As shown in Table 3, both simple and functionalized alkenes were good coupling partners under these conditions (30–66). Substituted styrenes with electron-donating and electron-withdrawing groups gave the corresponding products in good yields (30–36). Even more electron-deficient pentafluoro-substituted styrene (37) could be used effectively, producing the desired product in moderate yield. 2-Vinylnaphthalene and 9-vinylanthracene underwent the reaction smoothly to give the corresponding products in high yields (38 and 39). The boronic acid (40) and carboxyl group (41) were well tolerated, thus making this protocol suitable for synthesis of oxindole derivatives without protective groups. Disubstituted alkenes were also well-tolerable with excellent yields (42–43). Additionally, the product 43 gave a higher dr value (10:1) due to its steric hindrance. Heterocyclic olefins gave the products in moderate yields due to the lability of the heterocyclic system (44–45). Notably, the reaction could accommodate thiophene ethylene, and a mixture of 1,2-addition and 1,6-addition products was observed in acceptable yield (46). To our delight, a wide range of highly functionalized 1,1-disubstituted alkenes such as α-bromo (47), α-boronic ester (48), α-trifluoromethyl (49), and α-carboxyl (50) could be successfully used in this radical-based process to give three-component coupling products in synthetically useful yields, and an excellent dr was obtained for product 50 (dr > 20:1), opening avenues for further synthetic operations. Meanwhile, a highly selective reaction occurred at the aryl alkene position of the substrate, which contained both an aryl alkene and an alkyl alkene moiety, to obtain an exclusive product in excellent yield (51). Non-terminal styrenes such as 1,2-dihydronaphthalene (52), 1-cyclobuten-1-ylbenzene (53), and 1H-indene (54) also performed well to give congested products in moderate yields.
|
a Reaction conditions: 1 (0.1 mmol), 2a (0.2 mmol), 3a (0.2 mmol), and DIPEA (0.2 mmol) in THF (1.0 mL) were irradiated with 10 W white LEDs under N2 at 40 °C. Yield of the isolated product. The dr (diastereomeric ratio) of the product was 1:1 unless otherwise noted.
b
dr = 2:3.
c
dr = 10:1.
d Detected by 1H NMR, Z:E = 2:1 (1,6 addition).
e
dr = 4:1.
f
dr > 20:1.
g
dr = 5:2:3.
|
|---|
|
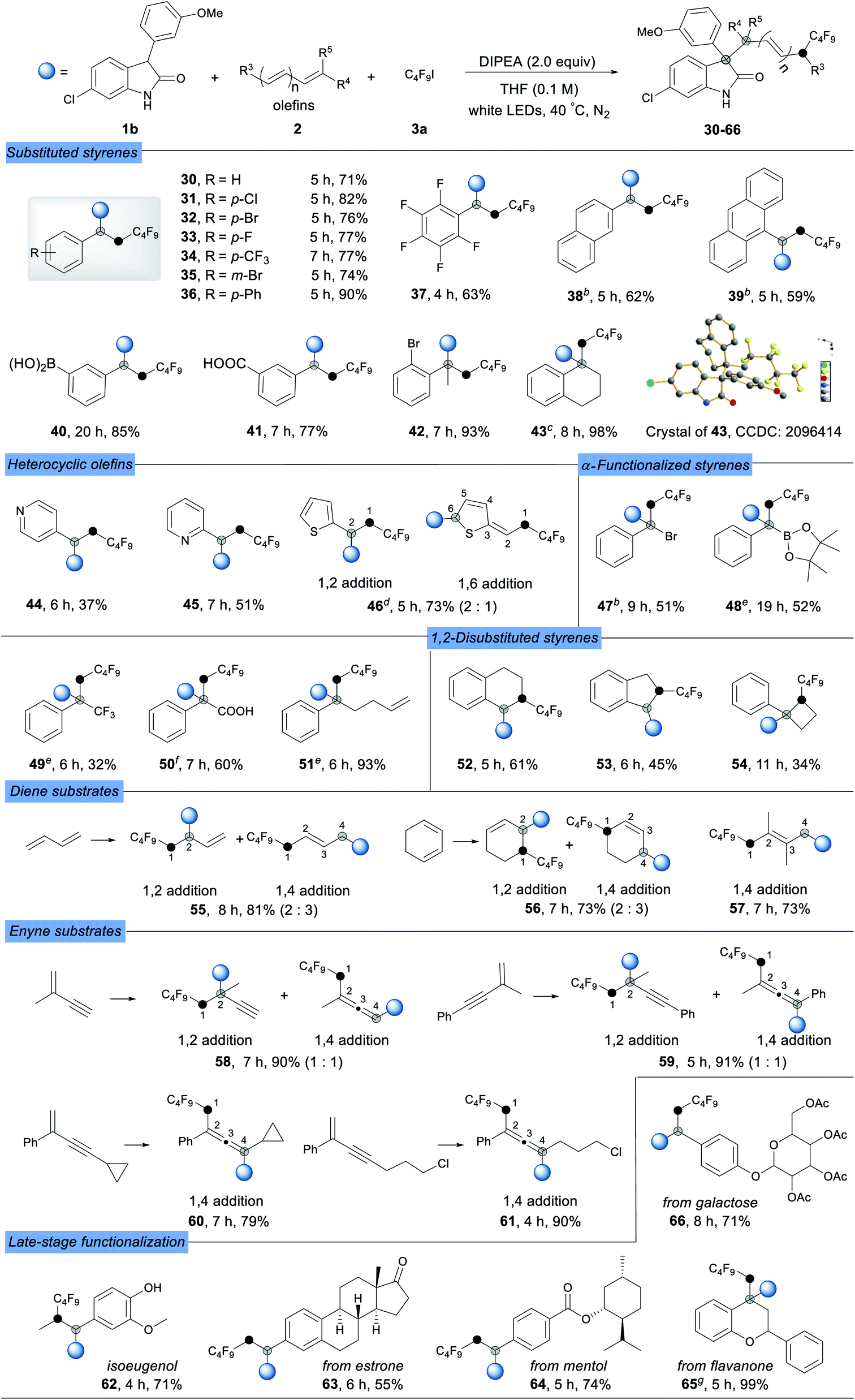
It was worth noting that conjugate olefins such as 1,3-dienes (55–57) proved to be competent partners in the reaction, highlighting the extraordinary substrate scope of this strategy. When 1,3-butadiene and cyclohexa-1,3-diene were subjected to this protocol, a mixture of 1,2-addition and 1,4-addition products was obtained in high yields (55 and 56), while only 1,4-addition product 57 was obtained for 2,3-dimethylbuta-1,3-diene. Notably, 1,3-enynes could also be used as radical acceptors to form substituted allenyl (1,4-adducts) or alkynyl (1,2-adducts) derivatives under the optimized conditions, which are important structural motifs found in pharmaceuticals and considered as key intermediates in the synthesis of complex molecules. Compared to the product mixture obtained with 2-methyl substituted enynes (58 and 59), 2-phenyl substituted enynes gave the 1,4-addition product allenes (60 and 61) in excellent yields exclusively. The excellent functional group compatibility and broad substrate scope of this strategy encouraged us to evaluate its application in the late-stage functionalization of bioactive molecules. Several drugs and natural product derivatives such as isoeugenol, estrone, menthol, flavanone and galactose were subjected to the optimized reaction conditions, resulting in the desired complex products in good to excellent yields (62–66).
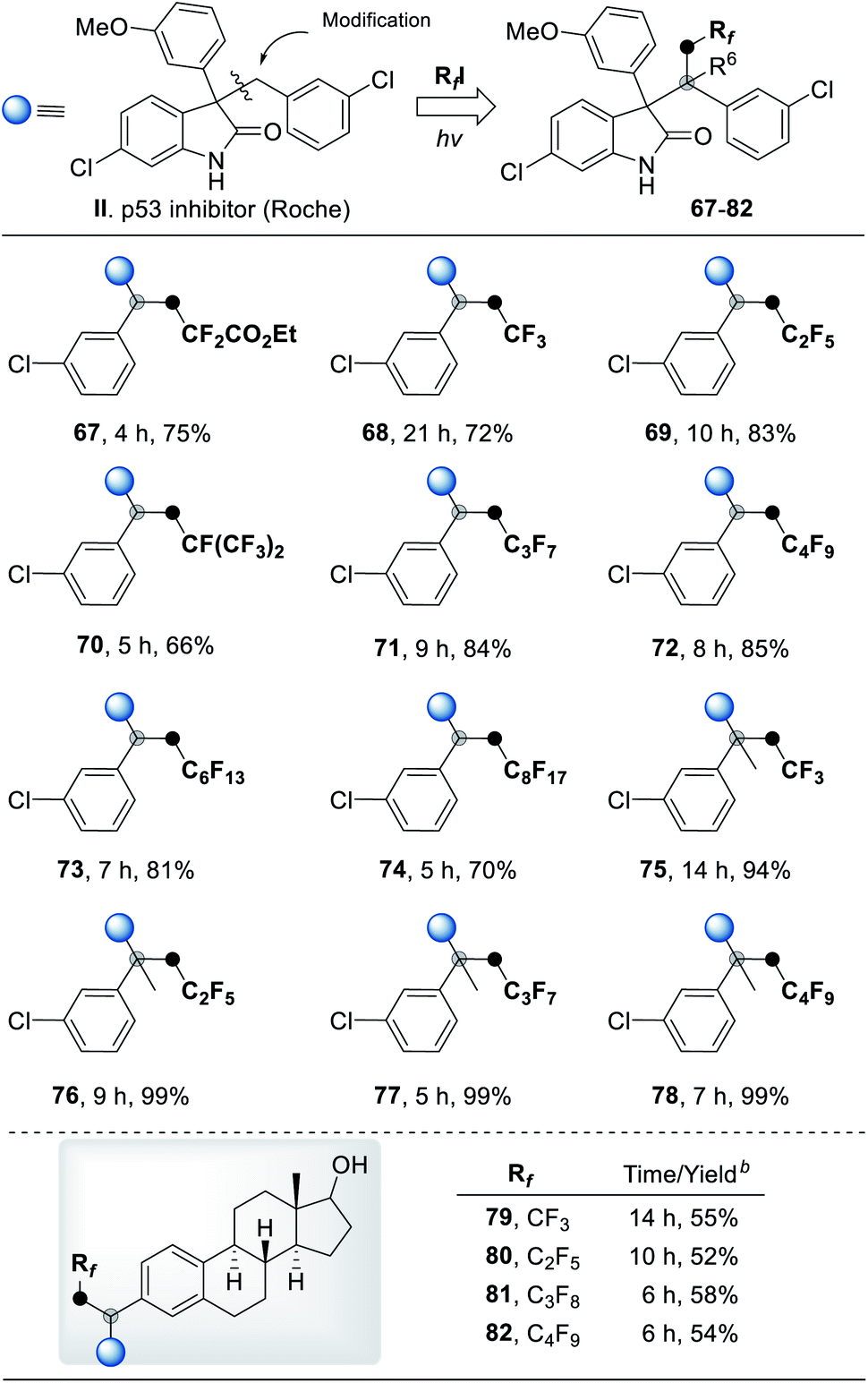
To illustrate the utility of this new transformation with respect to drug discovery, we applied this radical three-component coupling reaction to modify p53 inhibitor II (Roche). Using a variety of polyfluoroalkyl halides as the fluorinating reagent, a series of polyfluorinated analogues (67–78) of inhibitor II were furnished in high yields under mild conditions (Table 4). The estrogen derivatives were also tolerable in our system to deliver corresponding products in good yields after a two-step reaction (79–82). The success of sunlight-driven transformation (78, 84% yield) and gram-scale synthesis (78, 1.05 g, 65% yield) revealed the potential of this strategy in industrial applications (for more details see ESI, Fig. S6†). To demonstrate the biological application of these analogues, some of the polyfluorinated oxindole derivatives were assayed for growth inhibitory activities on the human cancer cell line MCF-7 (breast cancer); compared to p53 inhibitor II, the anti-tumor activity of fluorine-modified compounds was significantly improved (Table 5 and S5†). Further SAR of these new scaffolds is currently in progress in our groups.
|
a Reaction conditions: 1b (0.1 mmol), 2 (0.2 mmol), 3a (0.2 mmol), and DIPEA (0.2 mmol) in THF (1.0 mL) were irradiated with 10 W white LEDs under N2 at 40 °C. Yield of the isolated product. The dr (diastereomeric ratio) of the product was 1:1.
b Two steps, see the ESI† for exact experimental procedures.
|
|---|
|
| Compounds | p53 inhibitor II | 32 | 56 | 68 | |
| IC50 (MCF-7) | 38.95 | 21.16 | 21.99 | 16.73 | |
| Compounds | 69 | 70 | 71 | 72 | 76 |
| IC50 (MCF-7) | 22.63 | 7.23 | 20.60 | 21.04 | 20.99 |
Having established a broad scope for the synthesis of polyfluorinated oxindole derivatives, our attention was focused on the mechanism of this three-component radical-coupling reaction. UV-vis spectroscopic measurements on various combinations of 1b, 2b, and 3a with different bases were first performed. Compared to the marked yellow color and red-shift of the UV-vis absorption observed by using strong bases such as Cs2CO3 or DUB as additives (the Rf˙ was initiated by the photoconductive EDA complex of enolate with C4F9I, see Fig. S7 and S8†), no color change and no detectable red-shift with a mixture of DIPEA, 1b and C4F9I were observed. These results indicated that the possibility of the EDA complex of substrates with DIPEA as the base additive was excluded (for more control experiments see ESI, Tables S6 and S7†). Moreover, the iodoperfluoroalkylation product 85 was obtained during the reaction course, even without oxindole under the standard conditions (Scheme 2a). We monitored this process by 1H NMR and found that benzyl iodide 85 was unstable and would rapidly decompose under white light irradiation (see ESI, Fig. S9†). This outcome was different from Reiser's work, in which the iodoperfluoroalkylation product 85 can be obtained in high yield due to its stability under green light.10 Meanwhile, the results of light on/off experiments suggested that the formation of 85 did not occur by the chain process (see ESI, Fig. S9†), which was consistent with the previous reports.10a,15 We considered that in our process, this unstable, photosensitive benzyl iodide 85 might be the key intermediate for the next coupling step.
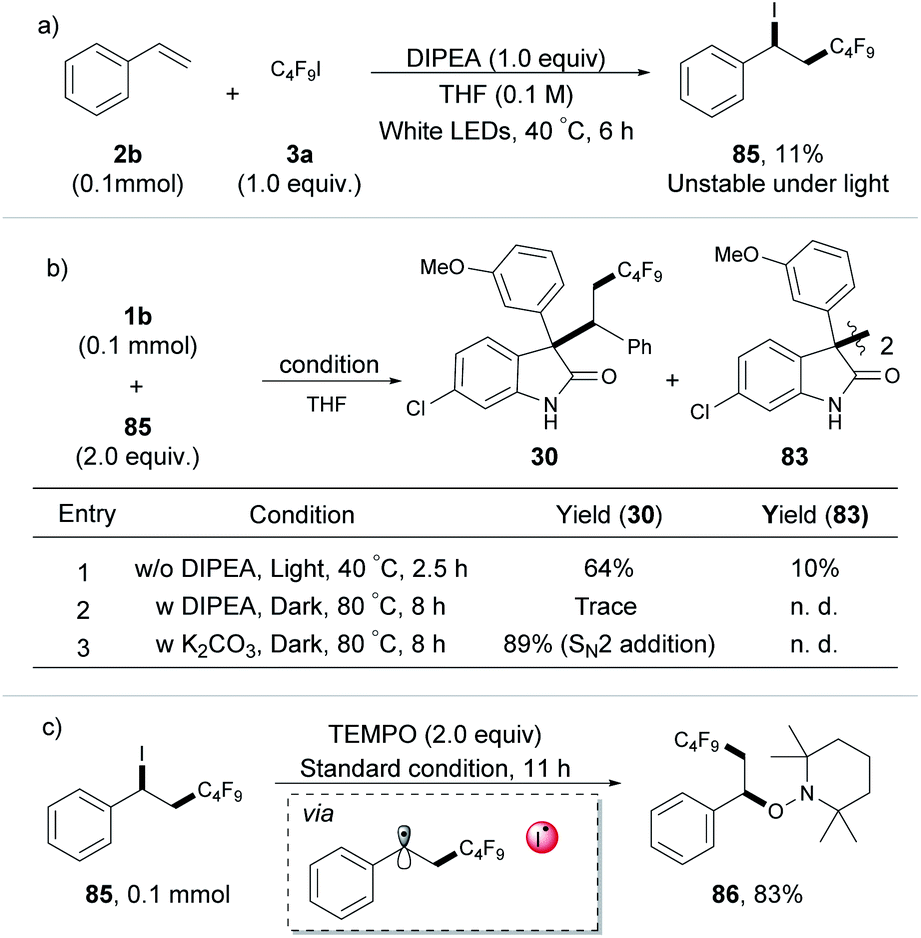 | ||
| Scheme 2 Control experiments. | ||
To gather direct experimental evidence, the intermediate 85 was synthesized and subjected to the optimized reaction conditions. To our delight, compound 85 showed a noticeable reactivity toward 1b and product 30 was obtained in good yield with 10% dimerization product 83 of oxindole. In the absence of light, compared to the high yield obtained with K2CO3 (SN2 addition), only a trace amount of the desired product 30 was detected in the presence of DIPEA at 80 °C in the dark for 8 h (Scheme 2b),16 indicating that 1b and 85 were more likely to undergo a radical coupling process than a nucleophilic process under standard conditions. Additionally, the benzyl radical captured product 86 was obtained in 83% yield (Scheme 2c) when the radical scavenger TEMPO and 85 were irradiated directly in THF. These results provided a piece of direct evidence for the formation of the benzyl radical and I˙ radical as key intermediates by homolysis of the C–I bond of photosensitive benzyl iodide 85 under irradiation during the transformation, as well as the reaction undergoing a radical-coupling pathway. These can also explain why a low yield was observed for the iodoperfluoroalkylation product 85 in Scheme 2a (due to the photolysis of 85 under light irradiation).
After demonstrating that iodoperfluoroalkylation product 85 was a key intermediate during the transformation, subsequent efforts were aimed at confirming the essential HAT substance that formed the oxindole radical in the HAT process (Scheme 3a and b). The two possible HAT reagents for abstracting the hydrogen atom from the oxindole as described here were DIPEA˙+ (from photoexcitation of the XB adduct of DIPEA and C4F9I) and I˙ (from homolysis of photosensitive intermediate 85). As shown in Scheme 3a, no dimerization product 83 was observed when the mixture of oxindole 1b and C4F9I was subjected to the optimized reaction conditions, indicating that DIPEA˙+ was not a HAT reagent in our strategy. This was further supported by the DFT calculation of radical I˙ with a lower energy barrier in the HAT process (6.6 kcal mol−1 for I˙ vs. 33.9 kcal mol−1 for DIPEA˙+). The radical clock experiment of α-cyclopropylstyrene 2d gave the ring-opening product 87 in 87% yield with no dimerization product 83, suggesting that no radical I˙ was produced in the transformation because the product 87 was stable under light irradiation (Scheme 3b). This was further supported by DFT calculations.17 Together with DFT calculations and control experiments, we considered that the I˙ radical was the HAT reagent in this process, which was generated from the photolysis of photosensitive intermediate 85.
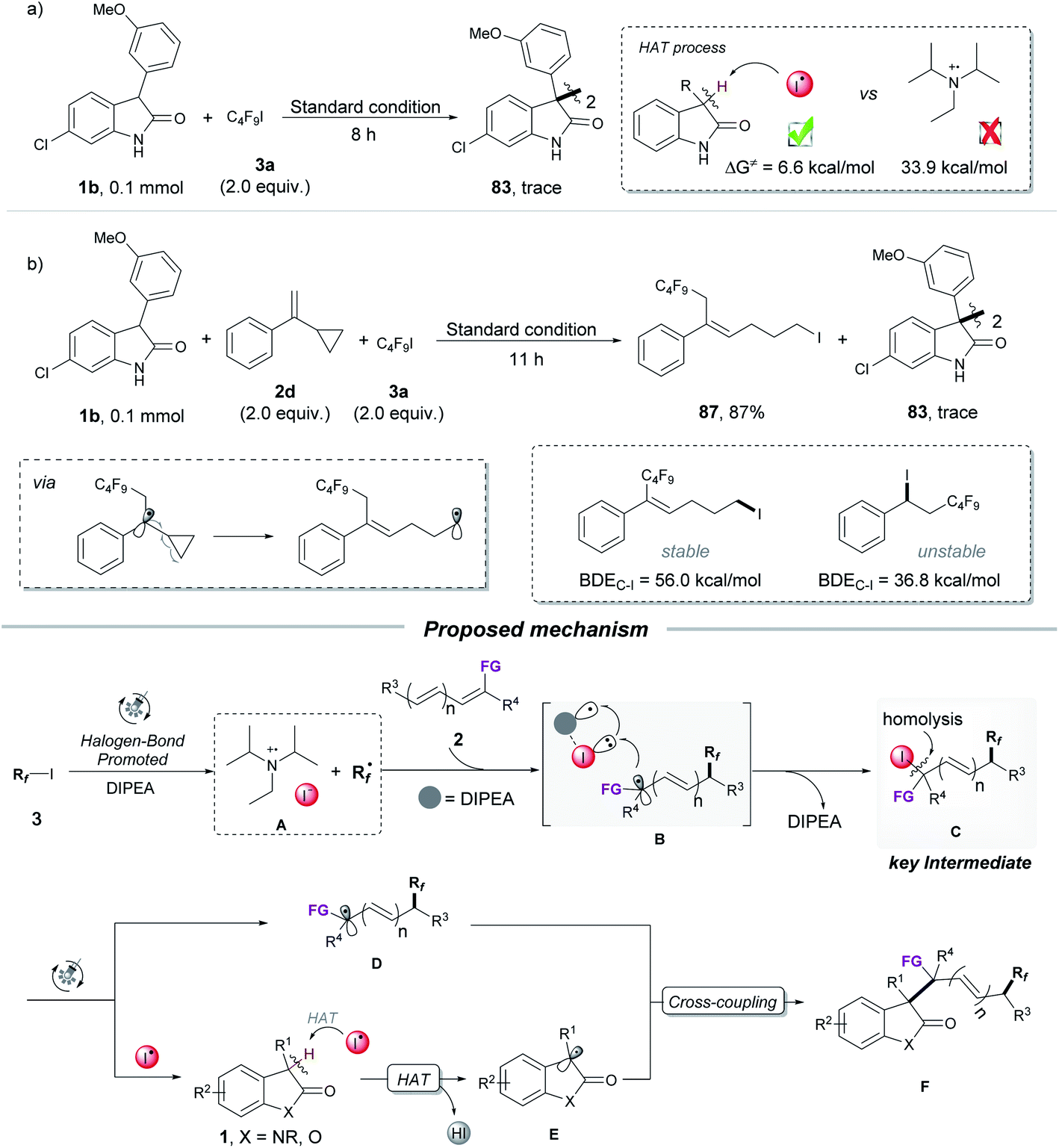 | ||
| Scheme 3 Control experiments and proposed mechanism. | ||
Based on the above mechanistic results and previous reports, the proposed mechanism for the radical three-component coupling reaction is depicted in Scheme 3. Upon visible-light irradiation, the XB (halogen bond) complex of DIPEA and perfluoroalkyl iodide 3 is excited and generates the fluoroalkyl radical Rf˙ and complex A (DIPEA˙+![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) I−).9f,i,18 The fluoroalkyl radical Rf˙ adds to the olefin 2 to form a new radical, which reacts with complex A, leading to the key photosensitive intermediate C (iodoperfluoroalkylation product). The iodine radical I˙ and radical D are generated by photolysis of intermediate C under light irradiation. The resulting radical I˙ acts as a HAT reagent, abstracting the hydrogen atom from oxindole 1 to generate the persistent oxindole radical E. Finally, the combination of the radical D with the oxindole radical E affords the three-component coupling product F.
I−).9f,i,18 The fluoroalkyl radical Rf˙ adds to the olefin 2 to form a new radical, which reacts with complex A, leading to the key photosensitive intermediate C (iodoperfluoroalkylation product). The iodine radical I˙ and radical D are generated by photolysis of intermediate C under light irradiation. The resulting radical I˙ acts as a HAT reagent, abstracting the hydrogen atom from oxindole 1 to generate the persistent oxindole radical E. Finally, the combination of the radical D with the oxindole radical E affords the three-component coupling product F.
Conclusions
In summary, we have reported a photocatalytic three-component radical coupling reaction for the modular assembly of fluoroalkyl-containing oxindole derivatives. This procedure employs readily accessible or commercially available materials to deliver a series of functional and structural oxindoles with adjacent all-carbon quaternary centers in one chemical step, which are difficult to access by other methods. A wide variety of alkenes including terminal or non-terminal alkenes and conjugated dienes and enynes are successfully applied to this strategy. Moreover, we demonstrate the utility of this strategy in rapid and scalable synthesis of the known pharmaceutical analogues. Detailed mechanistic studies disclose that the photosensitive fluorobenzyl iodides are the key intermediates for this sequential multistep radical-coupling reaction. We anticipate that this simple and mild approach will likely be of interest to chemists both in academic and industrial fields. Further explorations of the biological activity of these new fluorine-modified oxindole compounds are underway in our laboratory.Data availability
All experimental and computational data in this manuscript are available in the ESI.† Crystallographic data for compound 4, 29 and 43 have been deposited at the CCDC 2090721, 2096415 and 2096414, respectively.Author contributions
K. Z. and Y. L. S. conceived the idea for this work and designed the experiments. Y. L. S., C. L., and N. L. performed and analyzed the experiments. Z. S. S. and Y. L. S. performed the theoretical study. P. T., D. L. X., and X. X. G. synthesized some of the substrates. K. Z. supervised the entire project. All authors discussed the results and helped in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Xiaoming Feng laboratory (SCU) for access to equipment and Yuqiao Zhou for X-ray structural analysis. We also thank the comprehensive training platform of the Specialized Laboratory in the College of Chemistry at Sichuan University for compound testing. Funding: we acknowledge support from the National Natural Science Foundation of China (Nos. 21602142), Sichuan Science and Technology Program (Nos. 2020YJ0301), and the Fundamental Research Funds for the Central Universities.Notes and references
- (a) N. H. Greig, X. F. Pei, T. T. Soncrant, D. K. Ingram and A. Brossi, Med. Res. Rev., 1995, 15, 3–31 CrossRef CAS PubMed; (b) J. Song, D.-F. Chen and L.-Z. Gong, Natl. Sci. Rev., 2017, 4, 381–396 CrossRef CAS; (c) A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084 RSC.
- H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Angew. Chem., Int. Ed., 2007, 46, 2254–2256 CrossRef CAS PubMed.
- (a) C. Hulme, G. B. Poli, F.-C. Huang, J. E. Souness and S. W. Djuric, Bioorg. Med. Chem. Lett., 1998, 8, 175–178 CrossRef CAS PubMed; (b) M. K. Christensen, K. D. Erichsen, C. Trojel-Hansen, J. Tjornelund, S. J. Nielsen, K. Frydenvang, T. N. Johansen, B. Nielsen, M. Sehested, P. B. Jensen, M. Ikaunieks, A. Zaichenko, E. Loza, I. Kalvinsh and F. Bjorkling, J. Med. Chem., 2010, 53, 7140–7145 CrossRef CAS PubMed.
- (a) Y. Ping, Y. Li, J. Zhu and W. Kong, Angew. Chem., Int. Ed., 2019, 58, 1562–1573 CrossRef CAS PubMed; (b) A. D. Marchese, E. M. Larin, B. Mirabi and M. Lautens, Acc. Chem. Res., 2020, 53, 1605–1619 CrossRef CAS PubMed; (c) F. Zhou, Y.-L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381–1407 CrossRef CAS; (d) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC; (e) K. Shen, X. Liu, L. Lin and X. Feng, Chem. Sci., 2012, 3, 327–334 RSC; (f) R. Dalpozzo, Adv. Synth. Catal., 2017, 359, 1772–1810 CrossRef CAS; (g) Z. Y. Cao, F. Zhou and J. Zhou, Acc. Chem. Res., 2018, 51, 1443–1454 CrossRef CAS PubMed.
- (a) C. D. Grant and M. J. Krische, Org. Lett., 2009, 11, 4485–4487 CrossRef CAS PubMed; (b) B. M. Trost, W. H. Chan and S. Malhotra, Chem.–Eur. J., 2017, 23, 4405–4414 CrossRef CAS PubMed.
- (a) H. Y. Huang, R. Wu, F. Wei, D. Wang and L. Liu, Org. Lett., 2015, 17, 3702–3705 CrossRef CAS PubMed; (b) T. Bleith, Q. H. Deng, H. Wadepohl and L. H. Gade, Angew. Chem., Int. Ed., 2016, 55, 7852–7856 CrossRef CAS PubMed; (c) H. Y. Huang, L. Cheng, J. J. Liu, D. Wang, L. Liu and C. J. Li, J. Org. Chem., 2017, 82, 2656–2663 CrossRef CAS PubMed; (d) J. J. Liu, H. Y. Huang, L. Cheng, Q. Liu, D. Wang and L. Liu, Org. Biomol. Chem., 2018, 16, 899–903 RSC; (e) K. Liang, N. Li, Y. Zhang, T. Li and C. Xia, Chem. Sci., 2019, 10, 3049–3053 RSC; (f) R. Ohnishi, M. Sugawara, M. Akakabe, T. Ezawa, H. Koshino, Y. Sohtome and M. Sodeoka, Asian J. Org. Chem., 2019, 8, 1017–1023 CrossRef CAS; (g) H. Wu, C. Qiu, Z. Zhang, B. Zhang, S. Zhang, Y. Xu, H. Zhou, C. Su and K. P. Loh, Adv. Synth. Catal., 2019, 362, 789–794 CrossRef; (h) G. Hong, P. D. Nahide and M. C. Kozlowski, Org. Lett., 2020, 22, 1563–1568 CrossRef CAS PubMed; (i) M. Sugawara, R. Ohnishi, T. Ezawa, M. Akakabe, M. Sawamura, D. Hojo, D. Hashizume, Y. Sohtome and M. Sodeoka, ACS Catal., 2020, 10, 12770–12782 CrossRef CAS; (j) Y. Sohtome, K. Kanomata and M. Sodeoka, Bull. Chem. Soc. Jpn., 2021, 94, 1066–1079 CrossRef CAS.
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) B. M. Johnson, Y. Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed; (c) R. Britton, V. Gouverneur, J.-H. Lin, M. Meanwell, C. Ni, G. Pupo, J.-C. Xiao and J. Hu, Nat. Rev. Methods Primers, 2021, 1, 47 CrossRef.
- (a) S. Barata-Vallejo, S. M. Bonesi and A. Postigo, Org. Biomol. Chem., 2015, 13, 11153–11183 RSC; (b) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284–2294 CrossRef CAS PubMed; (c) S. Barata-Vallejo, M. V. Cooke and A. Postigo, ACS Catal., 2018, 8, 7287–7307 CrossRef CAS; (d) S. Barata-Vallejo and A. Postigo, Chem.–Eur. J., 2020, 26, 11065–11084 CrossRef CAS PubMed; (e) C. Czekelius, L. Helmecke, M. Spittler and B. M. Schmidt, Synthesis, 2020, 53, 123–134 CrossRef.
- (a) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (b) C. J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed; (c) R. Beniazza, R. Atkinson, C. Absalon, F. Castet, S. A. Denisov, N. D. McClenaghan, D. Lastécouères and J.-M. Vincent, Adv. Synth. Catal., 2016, 358, 2949–2961 CrossRef CAS; (d) E. Yoshioka, S. Kohtani, T. Jichu, T. Fukazawa, T. Nagai, A. Kawashima, Y. Takemoto and H. Miyabe, J. Org. Chem., 2016, 81, 7217–7229 CrossRef CAS PubMed; (e) L.-L. Mao and H. Cong, ChemSusChem, 2017, 10, 4461–4464 CrossRef CAS PubMed; (f) Y. Wang, J. Wang, G. X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442–1445 CrossRef CAS PubMed; (g) T. Yajima and M. Ikegami, Eur. J. Org. Chem., 2017, 15, 2126–2129 CrossRef; (h) R. Beniazza, L. Remisse, D. Jardel, D. Lastecoueres and J. M. Vincent, Chem. Commun., 2018, 54, 7451–7454 RSC; (i) L. Helmecke, M. Spittler, K. Baumgarten and C. Czekelius, Org. Lett., 2019, 21, 7823–7827 CrossRef CAS PubMed; (j) C. Rosso, G. Filippini, P. G. Cozzi, A. Gualandi and M. Prato, ChemPhotoChem, 2019, 3, 193–197 CrossRef CAS; (k) C. Rosso, G. Filippini and M. Prato, Chem.–Eur. J., 2019, 25, 16032–16036 CrossRef CAS PubMed; (l) C. Rosso, J. D. Williams, G. Filippini, M. Prato and C. O. Kappe, Org. Lett., 2019, 21, 5341–5345 CrossRef CAS PubMed; (m) E. Zhu, X. X. Liu, A. J. Wang, T. Mao, L. Zhao, X. Zhang and C. Y. He, Chem. Commun., 2019, 55, 12259–12262 RSC; (n) Y. X. Ji, L. J. Wang, W. S. Guo, Q. Bi and B. Zhang, Adv. Synth. Catal., 2020, 362, 1131–1137 CrossRef CAS; (o) T. Mao, M. J. Ma, L. Zhao, D. P. Xue, Y. Yu, J. Gu and C. Y. He, Chem. Commun., 2020, 56, 1815–1818 RSC; (p) T. Yajima, M. Murase and Y. Ofuji, Eur. J. Org. Chem., 2020, 2020, 3808–3811 CrossRef CAS.
- (a) T. Rawner, E. Lutsker, C. A. Kaiser and O. Reiser, ACS Catal., 2018, 8, 3950–3956 CrossRef CAS; (b) S. Engl and O. Reiser, Eur. J. Org. Chem., 2020, 2020, 1523–1533 CrossRef CAS.
- (a) J. Cao, G. Wang, L. Gao, H. Chen, X. Liu, X. Cheng and S. Li, Chem. Sci., 2019, 10, 2767–2772 RSC; (b) H. B. Yang, Z. H. Wang, J. M. Li and C. Wu, Chem. Commun., 2020, 56, 3801–3804 RSC.
- (a) Q. Guo, M. Wang, Y. Wang, Z. Xu and R. Wang, Chem. Commun., 2017, 53, 12317–12320 RSC; (b) Q. Guo, M. Wang, Q. Peng, Y. Huo, Q. Liu, R. Wang and Z. Xu, ACS Catal., 2019, 9, 4470–4476 CrossRef CAS; (c) M. Israr, H. Xiong, Y. Li and H. Bao, Adv. Synth. Catal., 2020, 362, 2211–2215 CrossRef CAS.
- N. J. Straathof, S. E. Cramer, V. Hessel and T. Noel, Angew. Chem., Int. Ed., 2016, 55, 15549–15553 CrossRef CAS PubMed.
- (a) D. Jing, C. Lu, Z. Chen, S. Jin, L. Xie, Z. Meng, Z. Su and K. Zheng, Angew. Chem., Int. Ed., 2019, 58, 14666–14672 CrossRef CAS PubMed; (b) C. Lu, Z. Su, D. Jing, S. Jin, L. Xie, L. Li and K. Zheng, Org. Lett., 2019, 21, 1438–1443 CrossRef CAS PubMed.
- X.-C. Guo and Q.-Y. Chen, J. Fluorine Chem., 1999, 93, 81–86 CrossRef CAS.
- No conversion of 85 was observed at 80 °C in the dark for 8 h, indicating that 85 was stable at high temperature, and a radical pathway mediated by the thermal decomposition of 85 can be ruled out..
- DFT calculation showed that the C–I bond dissociation energy (BDE) of 87 (56.0 kcal mol−1) is higher than that of 85 (36.8 kcal mol−1)..
- X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638–4641 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, computational details, and spectroscopic data for all new compounds. CCDC 2090721, 2096415 and 2096414. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05273j |
| This journal is © The Royal Society of Chemistry 2021 |