
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton to hydride umpolung at a phosphonium center via electron relay: a new strategy for main-group based water reduction†
Takumi
Oishi
a,
Leonardo I.
Lugo-Fuentes
b,
Yichuan
Jing
a,
J. Oscar C.
Jimenez-Halla
b,
Joaquín
Barroso-Flores
 cd,
Masaaki
Nakamoto
a,
Yohsuke
Yamamoto
a,
Nao
Tsunoji
e and
Rong
Shang
*a
cd,
Masaaki
Nakamoto
a,
Yohsuke
Yamamoto
a,
Nao
Tsunoji
e and
Rong
Shang
*a
aDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-3-1 Kagamiyama, Higashi-Hiroshima, 739-8526, Japan. E-mail: rshang@hiroshima-u.ac.jp
bDepartment of Chemistry, Division of Natural and Exact Sciences, University of Guanajuato, Campus Gto, Noria Alta s/n, 36050 Guanajuato, Mexico
cCentro Conjunto de Investigación en Química Sustentable UAEM-UNAM, Unidad San Cayetano, 50200 Toluca de lerdo, México
dInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior S/N, Ciudad Universitaria, alcaldía de Coyoacán, CP 04510, Ciudad de México, México
eDepartment of Chemistry, Graduate School of Advanced Science and Engineering, Hiroshima University, 1-4-1 Kagamiyama, Higashi-Hiroshima, 739-8527, Japan
First published on 15th November 2021
Abstract
Generation of dihydrogen from water splitting, also known as water reduction, is a key process to access a sustainable hydrogen economy for energy production and usage. The key step is the selective reduction of a protic hydrogen to an accessible and reactive hydride, which has proven difficult at a p-block element. Although frustrated Lewis pair (FLP) chemistry is well known for water activation by heterolytic H–OH bond cleavage, to the best of our knowledge, there has been only one case showing water reduction by metal-free FLP systems to date, in which silylene (SiII) was used as the Lewis base. This work reports the molecular design and synthesis of an ortho-phenylene linked bisborane-functionalized phosphine, which reacts with water stoichiometrically to generate H2 and phosphine oxide quantitatively under ambient conditions. Computational investigations revealed an unprecedented multi-centered electron relay mechanism offered by the molecular framework, shuttling a pair of electrons from hydroxide (OH−) in water to the separated proton through a borane-phosphonium-borane path. This simple molecular design and its water reduction mechanism opens new avenues for this main-group chemistry in their growing roles in chemical transformations.
Introduction
Main group compounds have shown growing potential for transition metal-like reactivities, and thus offer an economical alternative to metal-based chemical transformations.1–8 One fundamentally important process related to sustainable energy production is the direct generation of high-energy dihydrogen molecules by water splitting (water reduction).9,10 Currently, besides direct electrolysis,9–15 artificial water reduction strategies usually require a reduced form of (s- or d-block) metal as a reactant or catalyst, where heterolytic O–H bond cleavage during water splitting occurs through oxidative addition over one or two metal centres (Chart 1A).16–18 The electron-rich metal centre accepts electrons from the electronegative oxygen and transfers it to a proton (in H2O) to form a hydride, which then recombines with the second proton on oxygen to generate dihydrogen.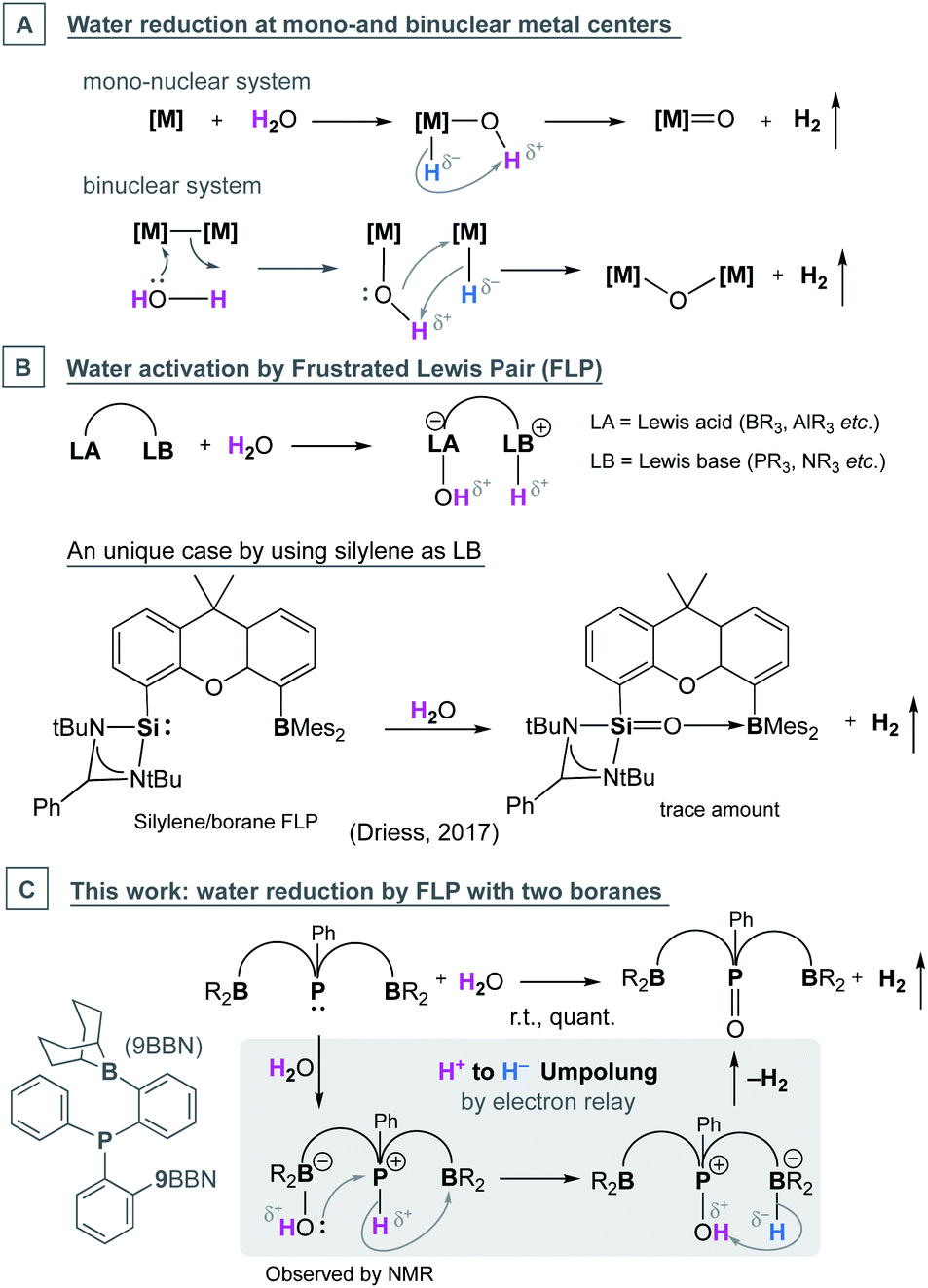 | ||
| Chart 1 Metal- (A) and p-block element (B and C)-based mechanisms for water activation and reduction. | ||
This general heterolytic mechanism of water cleavage (Chart 1A) has proven difficult to replicate by main-group elements. Although water HO–H bond cleavage, to generate element–hydride (E–H) bonds, has been achieved by oxidative addition at low valent elements (e.g. Al/Ga(I)19–23 and Si/Ge(II)24 species) and 1,2-addition across main-group multiple bonds (e.g. disilenes),25–34 a p-block E–H bond is usually less reactive than an s-/d-block M–H bond. Their reduced reactivity and large steric bulk (usually employed for their isolation) tend to favor the activation product instead of liberation of dihydrogen. A remarkable p-block example of water reduction was reported by Nielsen and Skrydstrup, which took advantage of the reducing power of the B–B bond in an sp3–sp3 diboron(4) system,35 resembling a binuclear metal system.
Another main-group strategy for facile H2O activation involves a bulky Lewis acid and base pair (frustrated Lewis pair, FLP, Chart 1B). In this case, group 15 functionalities, such as phosphines with electron-donating substituents, are usually employed as a Lewis base to deprotonate the Lewis acid-coordinated water molecule. However, the conversion of a proton, in a water molecule, into hydride cannot be achieved and thus the reaction stops at the HO–H bond cleavage step.36–44 By using both low-valent p-block elements and FLP chemistry, Driess and coworkers reported a metal-free water reduction example, from an intramolecular silylene/borane system (Chart 1B).45 However, since silylene is in a highly reduced form, this water reduction is not entirely unexpected. The necessity of borane in the water reduction was not discussed explicitly in the report, though its involvement in H2 generation was proposed based on the calculated reaction mechanism.
Herein we report a new FLP design for metal-free water reduction based on phosphine and borane functionalities. While triphenyl phosphine does not react with water under ambient conditions, we found that a bisborane-functionalized triaryl phosphine can instantaneously and quantitatively reduce water to generate dihydrogen and phosphine oxide.46–49 This reaction features an electron relay mechanism among the borane-phosphine-borane centers, which allowed an umpolung of a proton in water to a hydride on borane driven at a phosphonium center. This then led to formation of phosphine oxide and H2, leaving both Lewis acidic boranes intact after the reaction.
Discussion
During the systematic development of group 13 ligands for transition metal complexes, we set out to synthesize an ortho-phenylene linked bis(borane)-phosphine (BPB) ligand. We envisioned that the rigid and π-rich aryl linker would increase the stability and reactivity in comparison to previously reported systems.50 We were able to obtain the air-stable, colorless crystalline pinacolboranyl derivative (BPB-pin), 1a, from bis(2-bromophenyl)(phenyl)-phosphine51via Miyaura–Ishiyama borylation with bis(pinacolato)diboron (B2pin2) in 45% yield (Scheme 1, see the ESI† for full structural and spectroscopic data).52 Following the strategy developed by Wagner,531a was converted to the dialkyl-substituted 9BBN derivative 1b in 34% yield. The formation of the lithium boron hydride intermediate (1-Int) was detected by a proton-coupling 11B NMR experiment of the reaction mixture, which showed a broad quartet at −26.1 ppm, confirming the three hydrides bound to the boron centre (1JBH = 59 Hz, ESI 4.2†). Single-crystals of 1-Int were also obtained, which revealed a (1-Int)4LiCl cluster structure (see the ESI†).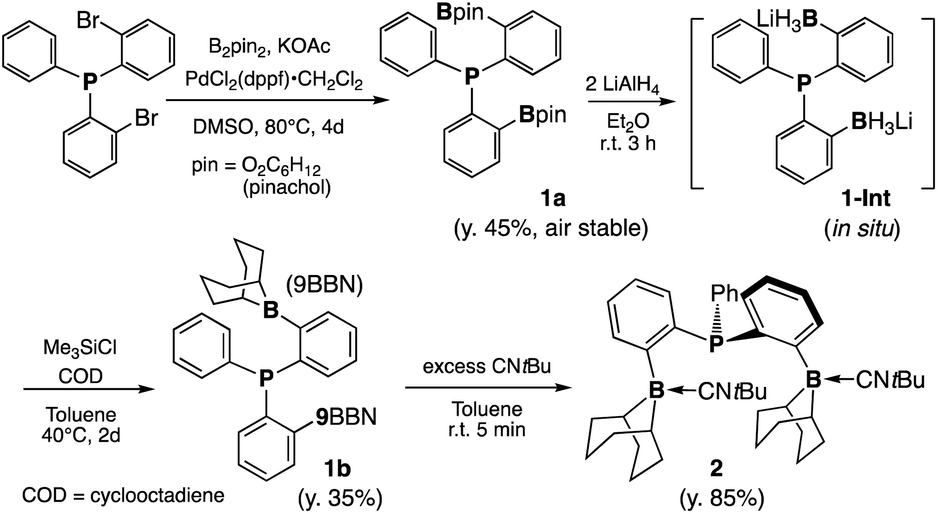 | ||
| Scheme 1 Synthetic pathway to bis(boranylphenyl)phenyl phosphine derivatives 1a, 1b and 2. | ||
Compound 1b is a highly reactive crystalline solid. Its 31P{1H} NMR spectrum showed a sharp singlet at −7.0 ppm. The 11B{1H} NMR spectrum showed a broad singlet at 79.2 ppm, similar to that of Bourissou's phosphorus/borane ambiphilic ligand bearing the cyclohexylborane moiety.54 The down-field 11B NMR signal suggests a highly Lewis acidic borane center with no strong intra- or intermolecular coordination from the phosphine moiety. Attempts to obtain single crystals of 1b were not successful in our hands. However, its isocyanide-Lewis adduct 2 could be obtained as colorless crystals in 85% yield (Scheme 1). X-ray analysis of a single crystal of 2 showed the molecular structure of 1b with each borane coordinated by an isocyanide moiety (Fig. 1b). The solution NMR spectrum of 2 was fully consistent with its solid-state structure, showing no dissociation of the Lewis base in solution at room temperature.
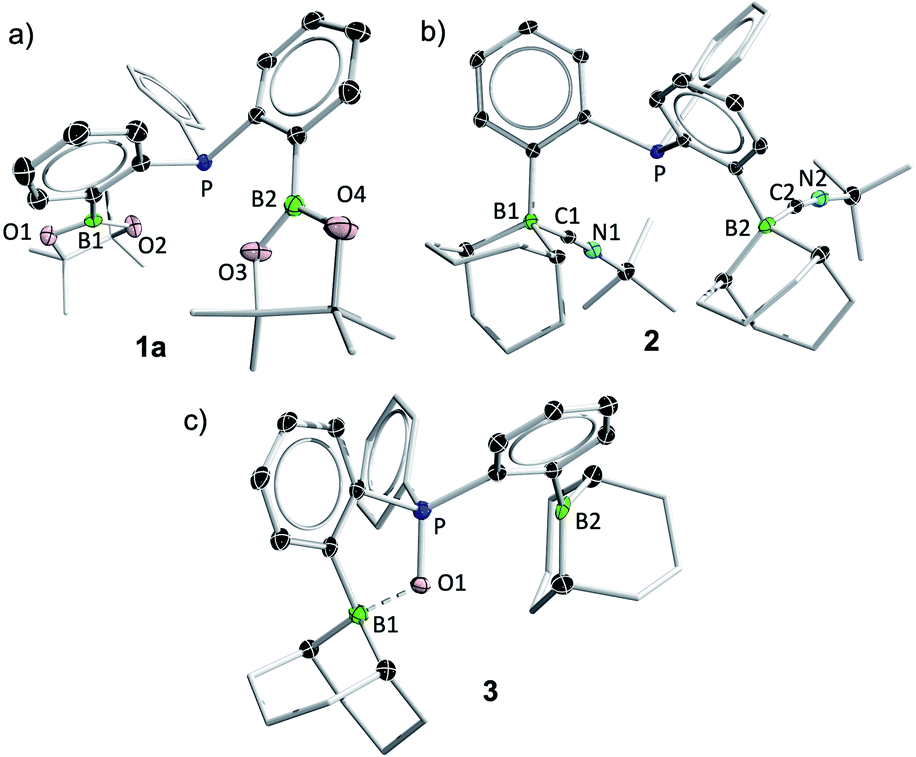 | ||
| Fig. 1 Solid-state structures of 1a (a), 2 (b) and 3 (c). Thermal ellipsoids are drawn at 30% probability. Peripheral ellipsoids are omitted for clarity. CCDC: 2096462 (1a), 2096464 (2), 2096465 (3). See the ESI for 1-int, CCDC 2096463 and 3·H2O, CCDC 2109052.† | ||
Compound 1b did not react with H2 under ambient conditions. However, it reacted with H2O at room temperature with vigorous evolution of gas (Fig. 2). The 1H NMR spectrum of the reaction mixture showed a singlet at 4.47 ppm, assignable to H2. An analogous reaction with D2O in toluene led to clean generation of D2, detected by 2H NMR. These results confirmed that both hydrogen atoms in the generated dihydrogen are from the water molecule (Fig. 2). In addition, phosphine oxide 3 was isolated in 97% yield. The solid-state structure of 3 showed a POBCC 5-membered ring,55 with a short P–O distance of 1.5322(13) Å and a remarkably long B1–O distance of 1.633(2) Å. The tetrahedral geometry at B1 (ΣB1CCC = 339.9°) suggests a definite B–O interaction. In solution however, only one boron signal at 48.2 ppm could be observed at room temperature, which broadened into the baseline below 0 °C (see spectra in the ESI†). The 1H NMR spectrum also suggested a symmetrical structure in solution at room temperature. These revealed a fluxional coordination of the oxide to both borane centres. The 31P{1H} NMR signal of compound 3 showed a sharp signal at 60.4 ppm. Interestingly, compound 3 does not react with additional H2O under an inert atmosphere but decomposes readily in air at room temperature.
 | ||
| Fig. 2 Reaction schemes and NMR spectra of 1b with H2O and D2O at room temperature (a) and with frozen water at −80 °C (b). | ||
Although FLPs are well known for heterolytic bond cleavages in water36–44 and dihydrogen,56–61 to the best of our knowledge, there is only one exmple of water reduction by metal-free FLP systems (Chart 1C). A control reaction of free PPh3 and BCy3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio at 50 °C in benzene did not yield any H2 overnight (see spectra in the ESI†), which means the architecture of the rigid phenylene backbone and the proximity of two boranes are essential for this reaction. In addition, numerous examples of phenylene linked phosphine/borane FLPs for small molecule activation studies have been reported previously. However, water reduction has not been described.62–76
:2 ratio at 50 °C in benzene did not yield any H2 overnight (see spectra in the ESI†), which means the architecture of the rigid phenylene backbone and the proximity of two boranes are essential for this reaction. In addition, numerous examples of phenylene linked phosphine/borane FLPs for small molecule activation studies have been reported previously. However, water reduction has not been described.62–76
In the search for possible HO–H bond splitting intermediates like those observed in FLP chemistry,36–44 a toluene-d8 solution of 1b was frozen while water was added by syringe. The 31P{1H} NMR measurement of the reaction mixture at −80 °C revealed a small singlet at 18.3 ppm which splits into a doublet (1JPH = 474 Hz) in a subsequent 31P NMR measurement, confirming the generation of a phosphonium ([Ar3PH]+) intermediate (Fig. 2b). This signal could not be observed at temperatures higher than −30 °C, where the reaction proceeds too quickly. Attempts to isolate a similar O–H bond cleavage product from the reaction of 1b and methanol were not successful. A large excess of methanol (ca. 50 equiv.) was needed for an observable change in the NMR reactions with 1b, where the 31P{1H} NMR spectrum of the reaction mixture revealed a singlet which splits into a doublet in a proton-coupled 31P NMR experiment (see spectra in the ESI†). Removal of solvent led to a complete recovery of 1b. This suggested that the cleavage of the MeO–H bond by 1b is slightly uphill energetically, and 1b and the activation product (4, see the ESI†) are in equilibrium in solution.
To obtain more insights into the mechanistic details on the water reduction by 1b, we performed theoretical calculations at the SMD(benzene):ω-B97XD/6-311G(d) level (see the ESI† for computational details). The calculated reaction mechanism (Fig. 3) suggests that the water activation starts with its coordination to one borane to give adduct H1 (ΔGR1 = +4.7 kcal mol−1). Then the phosphine serves as a base to deprotonate the coordinated HO–H bond (via transition state TSH1, ΔG‡1 = +7.7 kcal mol−1) which generates intermediate H2 (observed by NMR at −80 °C as 3-I, ΔGR2 = −1.2 kcal mol−1). The resulting phosphonium cation now behaves as an electrophile77 to receive the pair of electrons of the hydroxyl group while transferring the first hydrogen as a hydride to the second boron (TSH2, ΔG‡2 = +14.5 kcal mol−1) in a concerted fashion to form H3 (ΔGR3 = +8.6 kcal mol−1). This is followed by the decoordination of hydroxyl from the first boron (TSH3, ΔG‡3 = +1.0 kcal mol−1) generating rotamer H4 (ΔGR4 = −2.5 kcal mol−1) which keeps closer the hydrogens to finally lead to formation of H2 (viaTSH4, ΔG‡4 = +1.2 kcal mol−1) and the phosphine oxide 3 (ΔGR5 = −31.7 kcal mol−1). Overall, this reaction is exergonic (ΔG0R = −22.1 kcal mol−1). The calculated total energy barrier of +18.0 kcal mol−1 is in line with our reported reaction conditions. H2 was also observed experimentally at −80 °C by NMR experiments.
 | ||
| Fig. 3 Proposed reaction mechanism for the formation of 3 calculated at the SMD(benzene): ω-B97XD/6-311G(d) level (298 K) (a) and NBO charges of the phosphorus and transfer of hydrogen atoms from H2 (observed in the NMR spectrum as 3-I) to H3 (b). | ||
The natural bond orbital (NBO) charges of the phosphorus atom and the transferred hydrogen atom between H2 and H3 reflected the charge transfer corresponding to oxidation and reduction respectively, showing a simultaneous increase in the charge of P and decrease in that of H (Fig. 3b). Also, our theoretical calculations suggest that there is a fluxional coordination of the oxygen to each boron atom in 3 (see Fig. S5†) with a low barrier energy of 8.4 kcal mol−1, indicating a weak O–B coordination which is consistent with the experimental observations.
Following the same mechanistic route as described above, the reaction of 1b with MeOH (see Fig. S7†) was also investigated computationally. The coordination of methanol to the first boron gives adduct Me1 (ΔGR1 = −0.4 kcal mol−1) followed by the proton transfer leading to intermediate 4 (Me2 in Fig. S7,† ΔGR2 = 4.1 kcal mol−1) through TSMe1 (ΔG‡1 = +6.5 kcal mol−1) which are accessible at room temperature. However, the following reaction steps are very high in energy and the reaction stops at this point. In general, the reduction of MeOH by 1b to produce CH4 and 3 must overcome a total energy barrier of 58.4 kcal mol−1 and, therefore, is practically unreachable. The reaction to afford the MeO–H bond cleavage intermediate (4) is exergonic, and because of the excess of methanol, this intermediate could be detected by NMR spectroscopy but could not be isolated.
Conclusions
This work reports a 9-BBN derivative of a bisborane-functionalized phosphine which reduces water to dihydrogen at room temperature in a stoichiometric reaction to produce phosphine oxide as the sole by-product. Computational investigations revealed that phosphine initially serves as a Lewis base to split water by deprotonation cooperatively with one borane. The resulting phosphonium then serves as a Lewis acid to accept the borane-bound hydroxide to facilitate a hydride transfer to the 2nd borane.Although triphenyl phosphine and trialkyl/aryl boranes are not particularly reactive in comparison to other derivatives often employed in FLP chemistry, the preorganization of the borane-phosphine-borane framework in 1b is very important to coordinate the phosphine/phosphonium bifunctionality, taking advantage of phosphorus' ability of hypervalent bonding and its non-polar P–H bond for the umpolung of proton to hydride. This simple design of a bisborane-functionalized phosphine FLP has demonstrated a new working paradigm for metal-free water reduction, showing a new strategy for generation of hydride at a main-group element from water that was previously only possible at metal centres.
Data availability
Crystallographic data for compounds 1a, 1-Int, 1b, 2 and 3·H2O has been deposited at the CCDC under 2096462–2096465, and 2109052. They can be obtained from [https://www.ccdc.cam.ac.uk/structures/?]. Computational details, spectroscopic data supporting this article have been uploaded as part of the supplementary information.Author contributions
RS came up with the molecular design and supervised the development of the synthesis and reactivity studies with help from YY and MN. YJ developed the synthesis of 1a. TO improved the synthesis of 1a, developed the synthesis of 1b and discovered the water reduction reaction. TO carried out the synthesis and characterization of all the compounds reported in this paper. LIL-F carried out all the computational studies under close supervision of JOCJ-H with additional help from JBF. NT carried out GC analysis of reaction mixtures to confirm the presence of H2. RS and TO wrote the initial MS draft and LIL-F and JOCJ-H added the computational part of the study. All authors contributed to the discussion of the results and finalization of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Ms. Kawata for management of the X-ray diffractometer (N-BARD, Hiroshima University) and her advice on the X-ray analysis. We also thank the Center for Collaborative Research & Community Cooperation, Hiroshima University, and Japan Society for Promotion of Science, Grant-in-Aid for Early-Career Scientists (19K15544 to R. S.) for financial support of this work. L. I. L.-F. acknowledges CONACyT for financial support through his MSc fellowship #1080463. J. B-F. thanks DGTIC-UNAM for granting access to their supercomputer known as ‘Miztli’ and Mrs Citlali Martínez-Soto for keeping our local computing facilities running.Notes and references
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- S. Yao, Y. Xiong and M. Driess, Organometallics, 2011, 30, 1748–1767 CrossRef CAS.
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed.
- T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- S. Kundu, Chem.–Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363, 2290–2309 CrossRef CAS.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- P. Nikolaidis and A. Poullikkas, Renewable Sustainable Energy Rev., 2017, 67, 597–611 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, PNAS, 2006, 103, 15729–15735 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- V. Artero, M. Chavarot-Kerlidou and M. Fontecave, Angew. Chem., Int. Ed., 2011, 50, 7238–7266 CrossRef CAS PubMed.
- V. S. Thoi, Y. Sun, J. R. Long and C. J. Chang, Chem. Soc. Rev., 2013, 42, 2388–2400 RSC.
- O. V. Ozerov, Chem. Soc. Rev., 2008, 38, 83–88 RSC.
- W. E. Piers, Organometallics, 2011, 30, 13–16 CrossRef CAS.
- M. Klahn and T. Beweries, Rev. Inorg. Chem., 2014, 34, 177–198 CAS.
- R. Noyori, I. Tomino, M. Yamada and M. Nishizawa, J. Am. Chem. Soc., 1984, 106, 6717–6725 CrossRef CAS.
- C. Jones, D. P. Mills and R. P. Rose, J. Organomet. Chem., 2006, 691, 3060–3064 CrossRef CAS.
- X. Li, X. Cheng, H. Song and C. Cui, Organometallics, 2007, 26, 1039–1043 CrossRef CAS.
- G. Linti and A. Seifert, Z. Anorg. Allg. Chem., 2008, 634, 1312–1320 CrossRef CAS.
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem.–Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- S. Masamune, Y. Eriyama and T. Kawase, Angew. Chem., Int. Ed. Engl., 1987, 26, 584–585 CrossRef.
- J. E. Mangette, D. R. Powell and R. West, Organometallics, 1994, 13, 4097–4104 CrossRef CAS.
- N. Wiberg, W. Niedermayer, K. Polborn and P. Mayer, Chem.–Eur. J., 2002, 8, 2730–2739 CrossRef CAS.
- H. Kobayashi, T. Iwamoto and M. Kira, J. Am. Chem. Soc., 2005, 127, 15376–15377 CrossRef CAS PubMed.
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023–1031 CrossRef CAS PubMed.
- B. Pampuch, W. Saak and M. Weidenbruch, J. Organomet. Chem., 2006, 691, 3540–3544 CrossRef CAS.
- P. D. N. Tokitoh, D. A. Yuasa and P. T. Sasamori, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 1217–1219 CrossRef.
- J. A. Hardwick and K. M. Baines, Chem.–Eur. J., 2015, 21, 2480–2488 CrossRef CAS PubMed.
- J. A. Hardwick and K. M. Baines, Angew. Chem., Int. Ed., 2015, 54, 6600–6603 CrossRef CAS PubMed.
- D. Nieder, L. Klemmer, Y. Kaiser, V. Huch and D. Scheschkewitz, Organometallics, 2018, 37, 632–635 CrossRef CAS.
- M. Flinker, H. Yin, R. W. Juhl, E. Z. Eikeland, J. Overgaard, D. U. Nielsen and T. Skrydstrup, Angew. Chem., Int. Ed., 2017, 56, 15910–15915 CrossRef CAS PubMed.
- R. Duchateau, R. A. van Santen and G. P. A. Yap, Organometallics, 2000, 19, 809–816 CrossRef CAS.
- R. Roesler, W. E. Piers and M. Parvez, J. Organomet. Chem., 2003, 680, 218–222 CrossRef CAS.
- A. L. Travis, S. C. Binding, H. Zaher, T. A. Q. Arnold, J.-C. Buffet and D. O'Hare, Dalton Trans., 2013, 42, 2431–2437 RSC.
- T. Xu and E. Y.-X. Chen, J. Am. Chem. Soc., 2014, 136, 1774–1777 CrossRef CAS PubMed.
- É. Rochette, M.-A. Courtemanche, A. P. Pulis, W. Bi and F.-G. Fontaine, Molecules, 2015, 20, 11902–11914 CrossRef PubMed.
- B. R. Barnett, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2014, 51, 541–544 RSC.
- T. Wang, G. Kehr, L. Liu, S. Grimme, C. G. Daniliuc and G. Erker, J. Am. Chem. Soc., 2016, 138, 4302–4305 CrossRef CAS PubMed.
- R. Tirfoin, J. Gilbert, M. J. Kelly and S. Aldridge, Dalton Trans., 2018, 47, 1588–1598 RSC.
- J. Lam, S. Sampaolesi, J. H. W. LaFortune, J. W. Coe and D. W. Stephan, Dalton Trans., 2018, 48, 133–141 RSC.
- Z. Mo, T. Szilvási, Y.-P. Zhou, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 3699–3702 CrossRef CAS PubMed.
- L. A. Körte, S. Blomeyer, S. Heidemeyer, A. Mix, B. Neumann and N. W. Mitzel, Chem. Commun., 2016, 52, 9949–9952 RSC.
- L. A. Körte, S. Blomeyer, S. Heidemeyer, J. H. Nissen, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2016, 45, 17319–17328 RSC.
- L. A. Körte, S. Blomeyer, J.-H. Peters, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Organometallics, 2017, 36, 742–749 CrossRef.
- L. Wang, S. Zhang, Y. Hasegawa, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Commun., 2017, 53, 5499–5502 RSC.
- R. Shang, S. Saito, J. O. C. Jimenez-Halla and Y. Yamamoto, Dalton Trans., 2018, 47, 5181–5188 RSC.
- T. Agou, J. Kobayashi and T. Kawashima, Org. Lett., 2005, 7, 4373–4376 CrossRef CAS.
- J. Wen, D. Wang, J. Qian, D. Wang, C. Zhu, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 2078–2082 CrossRef CAS PubMed.
- D. Franz, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2011, 40, 2433–2440 RSC.
- S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056–12057 CrossRef CAS.
- J. M. Breunig, F. Lehmann, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2014, 33, 3163–3172 CrossRef CAS.
- J. Paradies, Angew. Chem., Int. Ed., 2014, 53, 3552–3557 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS.
- D. W. Stephan, Chem, 2020, 6, 1520–1526 CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, Chem. Soc. Rev., 2017, 46, 5689–5700 RSC.
- J. Lam, K. M. Szkop, E. Mosaferi and D. W. Stephan, Chem. Soc. Rev., 2019, 48, 3592–3612 RSC.
- M. W. P. Bebbington, S. Bontemps, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2007, 46, 3333–3336 CrossRef CAS PubMed.
- T. W. Hudnall, Y.-M. Kim, M. W. P. Bebbington, D. Bourissou and F. P. Gabbaï, J. Am. Chem. Soc., 2008, 130, 10890–10891 CrossRef CAS PubMed.
- S. Moebs-Sanchez, G. Bouhadir, N. Saffon, L. Maron and D. Bourissou, Chem. Commun., 2008, 3435–3437 RSC.
- S. Porcel, G. Bouhadir, N. Saffon, L. Maron and D. Bourissou, Angew. Chem., 2010, 122, 6322–6325 CrossRef.
- S. Moebs-Sanchez, N. Saffon, G. Bouhadir, L. Maron and D. Bourissou, Dalton Trans., 2010, 39, 4417–4420 RSC.
- O. Baslé, S. Porcel, S. Ladeira, G. Bouhadir and D. Bourissou, Chem. Commun., 2012, 48, 4495–4497 RSC.
- Z. Li, K. Chansaenpak, S. Liu, C. R. Wade, P. S. Conti and F. P. Gabbaï, MedChemComm, 2012, 3, 1305 RSC.
- M.-A. Courtemanche, M.-A. Légaré, L. Maron and F.-G. Fontaine, J. Am. Chem. Soc., 2014, 136, 10708–10717 CrossRef CAS PubMed.
- R. Declercq, G. Bouhadir, D. Bourissou, M.-A. Légaré, M.-A. Courtemanche, K. S. Nahi, N. Bouchard, F.-G. Fontaine and L. Maron, ACS Catal., 2015, 5, 2513–2520 CrossRef CAS.
- R. Tirfoin, J. A. B. Abdalla and S. Aldridge, Chem.–Eur. J., 2015, 21, 11813–11824 CrossRef CAS PubMed.
- J. Möbus, T. vom Stein and D. W. Stephan, Chem. Commun., 2016, 52, 6387–6390 RSC.
- T. Özgün, G.-Q. Chen, C. G. Daniliuc, A. C. McQuilken, T. H. Warren, R. Knitsch, H. Eckert, G. Kehr and G. Erker, Organometallics, 2016, 35, 3667–3680 CrossRef.
- M. Boudjelel, E. D. Sosa Carrizo, S. Mallet−Ladeira, S. Massou, K. Miqueu, G. Bouhadir and D. Bourissou, ACS Catal., 2018, 8, 4459–4464 CrossRef CAS.
- T. Krachko, E. Nicolas, A. W. Ehlers, M. Nieger and J. C. Slootweg, Chem.–Eur. J., 2018, 24, 12669–12677 CrossRef CAS PubMed.
- C. A. Theulier, Y. García-Rodeja, N. Saffon-Merceron, K. Miqueu, G. Bouhadir and D. Bourissou, Chem. Commun., 2021, 57, 347–350 RSC.
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details, structural and spectroscopic data of 1a–b, 1-int, 2 and 3. Computational details. CCDC 2096462–2096465 and 2109052. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05135k |
| This journal is © The Royal Society of Chemistry 2021 |