
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe mechanism of Raf activation through dimerization†
Mingzhen
Zhang
 a,
Ryan
Maloney
a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
a,
Ryan
Maloney
a,
Hyunbum
Jang
a and
Ruth
Nussinov
*ab
aComputational Structural Biology Section, Frederick National Laboratory for Cancer Research in the Laboratory of Cancer Immunometabolism, National Cancer Institute, Frederick, MD 21702, USA. E-mail: NussinoR@mail.nih.gov; Tel: +1-301-846-5579
bDepartment of Human Molecular Genetics and Biochemistry, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel
First published on 18th November 2021
Abstract
Raf, a threonine/serine kinase in the Raf/MEK/ERK pathway, regulates cell proliferation. Raf's full activation requires dimerization. Aberrant activation through dimerization is an important therapeutic target. Despite its clinical importance, fundamental questions, such as how the side-to-side dimerization promotes the OFF-to-ON transition of Raf's kinase domain and how the fully activated ON-state kinase domain is stabilized in the dimer for Raf signaling, remain unanswered. Herein, we decipher an atomic-level mechanism of Raf activation through dimerization, clarifying this enigma. The mechanism reveals that the replacement of intramolecular π–π stacking by intermolecular π–π stacking at the dimer interface releases the structural constraint of the αC-helix, promoting the OFF-to-ON transition. During the transition, the inhibitory hydrophobic interactions were disrupted, making the phosphorylation sites in A-loop approach the HRD motif for cis-autophosphorylation. Once fully activated, the ON-state kinase domain can be stabilized by a newly identified functional N-terminal basic (NtB) motif in the dimer for Raf signaling. This work provides atomic level insight into critical steps in Raf activation and outlines a new venue for drug discovery against Raf dimerization.
Introduction
Raf belongs to the family of serine/threonine kinases in the mitogen-activated protein kinase (MAPK) pathway, regulating cell proliferation.1–3RAF is an oncogene.4,5 Mammals express three Raf isoforms, A-Raf, B-Raf, and C-Raf (Raf-1).6 Among them, B-Raf is mutated in ∼50–60% of malignant melanomas, ∼80% of which are V600E.7–9 The Raf isoforms share the conserved N-terminal tandem Ras binding domain (RBD) and cysteine-rich domain (CRD) as well as the C-terminal kinase domain.10–15 The autoinhibition of Raf is maintained by the interactions between RBD-CRD/KD domains and 14-3-3 proteins.16–18 Raf is a downstream effector of Ras. Active GTP-bound Ras binds to Raf's RBD, releasing its autoinhibition and recruiting it to the plasma membrane.2,12,19,20 The gathering of Raf proteins on the membrane by Ras nanoclusters/dimers promotes the dimerization of Raf's kinase domain, leading to full activation.21–23 The activated Raf transmits the signals through the MAPK cascade by phosphorylating the downstream effector, mitogen-activated protein kinase (MEK) which, in turn, phosphorylates the extracellular signal-regulated kinase (ERK).24The full activation of Raf requires the dimerization of the kinase domain.25–27 Raf can homo- and hetero-dimerize.28,29 Dimerization can also occur with the kinase domain of the closely related kinase suppressor of Ras (KSR) proteins.30,31 The B-Raf/B-Raf homodimer and B-Raf/C-Raf heterodimer appear to be dominant.29 Disruption of Raf dimerization impairs MAPK signalling.29 ERK-induced feedback phosphorylation interferes with Ras/Raf interactions, reducing Raf dimerization.32,33 The aberrant activation of Raf through dimerization modulates the clinical outcomes.34–37 Drug resistance in the p61 spliced B-Raf mutant is due to constitutive Raf dimerization.38 Raf inhibitors are more sensitive to Raf monomers than to dimers.39 B-Raf/C-Raf heterodimerization causes paradoxical activation of the MAPK pathway in melanoma treatment, resulting in secondary skin carcinoma.22,32,40,41 These observations suggest that the dimer interface of Raf proteins is a potential therapeutic target against cancer,42,43 supporting the development of the next generation paradox breakers targeting the Raf dimerization.44,45
Raf kinase domain has the conserved protein kinase fold, with the N- and C-lobes.15,46–48 However, the dimerization of Raf is unique. Different from other protein kinases, Raf is the only known kinase that dimerizes in a transverse side-to-side manner for cis-autophosphorylation.15 The dimerization in other protein kinases via face-to-face (symmetric) or face-to-side (asymmetric) can direct the phosphorylation sites in one kinase domain to another, promoting the trans-autophosphorylation.24,49 The side-to-side dimerization in Raf puts the phosphorylation sites in the A-loop in the protomers further apart. It has been elusive as to why Raf proteins evolved the transverse side-to-side dimerization strategy and, importantly, how this strategy promotes Raf's cis-autophosphorylation for full activation. Efforts focused on Raf dimer interface. It has been shown that B-Raf mutations R509H and W450A at the interface reduce dimerization, while E586K promotes it.22,29 Another (potential) key component for Raf dimerization involves the phosphorylated N-terminal acidic (NtA) motif (pSSDD in B-Raf) shown to generate electrostatic interactions with the basic RKTR segment in the αC-helix at the interface.22,50
Despite the biological and clinical significance of knowledge of the interface, fundamental questions relating to Raf dimerization, i.e., how the side-to-side dimerization promotes the OFF-to-ON transition of Raf's kinase domain and how the fully activated ON-state kinase domain is stabilized in the dimer for Raf signaling, remain unresolved. In this work, we modeled and simulated B-Raf monomers, C-Raf monomers, B-Raf/B-Raf homodimers, and B-Raf/C-Raf heterodimers, striving to understand Raf dimerization. We decipher a detailed mechanism, showing that the disruption of the intramolecular π–π stacking by intermolecular π–π stacking at the dimer interface can induce significant conformational changes in the kinase domain for the OFF-to-ON transition. The disruption of the intramolecular π–π stacking between Y566 and H510 releases the structural constraint of the αC-helix in the kinase domain. This promotes (i) the disruption of the inhibitory hydrophobic interactions of the OFF-state conformation and (ii) the movement of the A-loop phosphorylation sites towards the catalytic HRD motif (574–576 in B-Raf) for cis-autophosphorylation. These collective conformational changes drive the Raf kinase domain towards the fully activated ON-state conformation. We also show that once the kinase domain is fully activated, the ON-state conformation can be stabilized by the N-terminal basic (NtB) motif (435–444 in B-Raf). This NtB motif is adjacent to the NtA motif, is highly basic, and is conserved in the Raf family. The overall conformational changes upon dimerization seen in these simulations are consistent with experiments,38 and explain drug resistance by constitutive Raf dimerization. This work outlines a new venue for Raf drug discovery against dimerization.
Results
The monomer is OPEN and becomes CLOSED upon dimerization
Like other protein kinases, the inactive OFF conformation of the Raf kinase domain features an OUT αC-helix and collapsed A-loop. In the active ON conformation, the αC-helix is IN and the A-loop is extended (Fig. S1†). In the fully activated ON state, the A-loop is phosphorylated. Two phosphorylation sites in the A-loop are conserved in Raf isoforms (pT599 and pS602 in B-Raf, pT491 and pS494 in C-Raf). The NtA motif in B-Raf is constitutively phosphorylated at pS446, while that in C-Raf is unphosphorylated prior to full activation.51 Following these conventions, we modeled and simulated the B-Raf monomers, C-Raf monomers, B-Raf/B-Raf homodimers, and B-Raf/C-Raf heterodimers. Configurations that began with an active conformation are denoted as BA (B-Raf with active conformation) and CA (C-Raf with active conformation), while inactive configurations are denoted as BI (B-Raf with inactive conformation) and CI (C-Raf with inactive conformation), where B and C stand for B-Raf and C-Raf respectively, as summarized in Table S1.†Raf kinase domain consists of two lobes, the N-lobe composed of a β-sheet with five stacking β-strands and αC-helix, and the C-lobe containing a bundle of α-helices (Fig. S1†). The center of mass distances between the N-lobe and C-lobe (NC distances) were measured to characterize the overall conformational changes upon the dimerization (Fig. 1A and B). In the calculations, the center of masses of the N-lobe β-sheet and C-lobe helices involving αE, αF, αH, and αI were considered, since these regions were conserved during the simulations. In contrast, other regions, such as the αG region in the C-lobe, showed high flexibility. The NC distances in the monomers are larger than those in the dimers, which appears to be more significant for B-Raf systems than C-Raf systems (Fig. 1B). The residue-based distances between the N-lobe (K483 in B-Raf, K375 in C-Raf) and C-lobe (αE, αF, αH, and αI helices) were also calculated. These suggest the rotational motions of the αE, αF, αH, and αI helices in the C-lobe relative to the N-lobe (Fig. S2†). Conformational switches between the OPEN conformation with the longer N-lobe and C-lobe distance and the CLOSED conformation with the shorter N-lobe and C-lobe distance during the activation cycle have been observed in other protein kinases.52,53 We sampled the similar conformational changes, suggesting that Raf proteins also follow the structural conversion of the kinase domain between the OPEN conformation in the monomers and the CLOSED conformation upon dimerization.
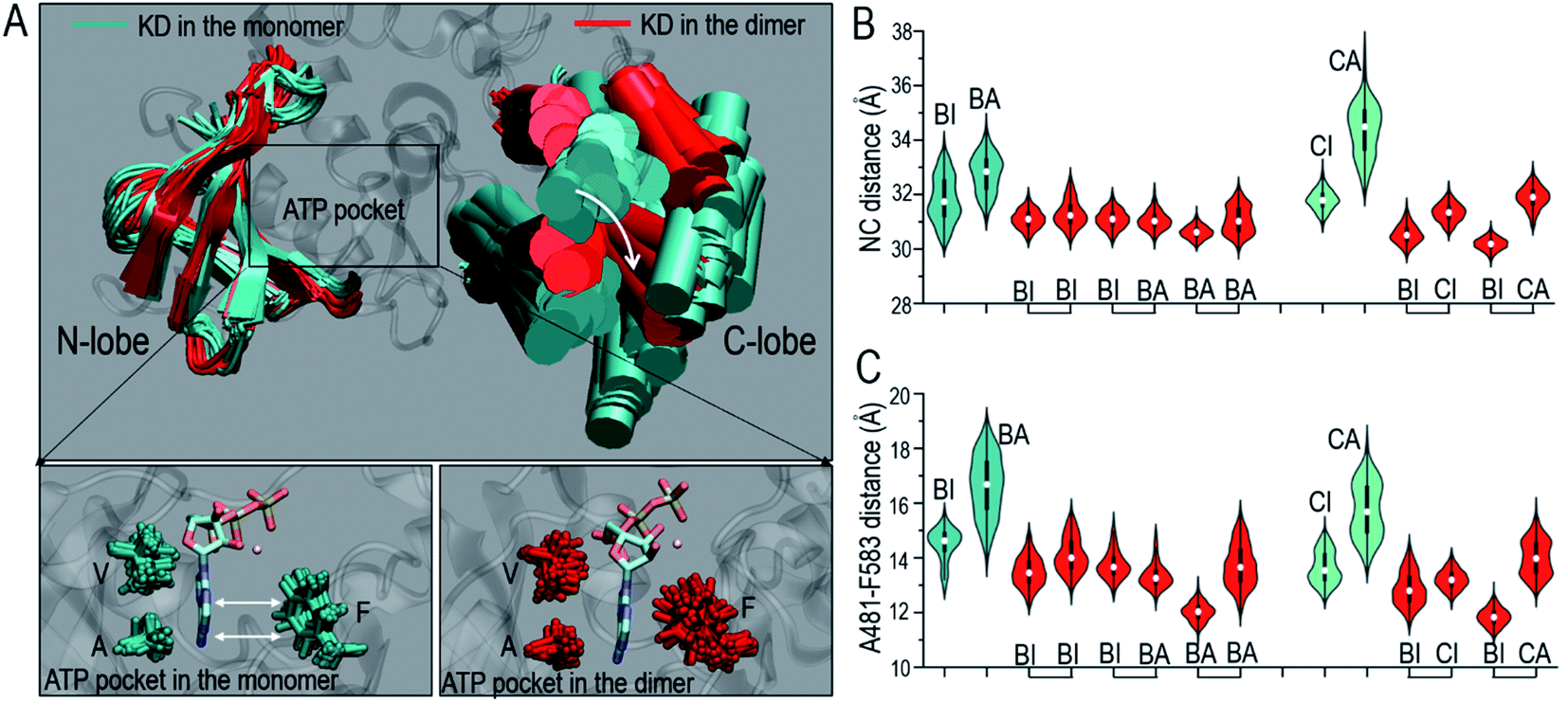 | ||
| Fig. 1 Overall conformational change of Raf kinase domain upon dimerization. (A) The superimposed structures of the inactive B-Raf kinase domains in the monomeric (BI) and dimeric state (BI in BI/BI). Violin plots representing (B) the center of mass distance between the N-lobe and C-lobe (NC distance) of the Raf kinase domain and (C) the Cα distance between C-spine residues, A481 in the N-lobe and F583 in the C-lobe. In the violin plots, the white dots denote the median, the thick black lines denote the data ranging from 25% to 75%, and the curves denote the probability density of the data. Staples in x-axis denote the dimeric configurations. BI refers to B-Raf with inactive conformation, CI refers to C-Raf with inactive conformation, where B and C stand for B-Raf and C-Raf respectively. BA, CA refer to the respective active conformations. | ||
The overall conformational changes upon dimerization help assemble the catalytic spine (C-spine) in the kinase domain. The C-spine regulates the ATP binding in the kinase domain.54 In the C-spine, the VA residues, V471 and A481 (V363, A373 in C-Raf) in the N-lobe, and F583 (F475 in C-Raf) in the C-lobe stabilize the ATP's adenosine ring in the ATP pocket (Fig. 1A). The A481–F583 and V471–F583 distances were measured to characterize the C-spine upon dimerization. The results show that the VF and AF distances in the C-spine were considerably reduced upon dimerization (Fig. 1C and S3A†). The larger residue distances indicate that the VA residues are further apart from the F residues in the Raf monomers than in the dimers. The opening of the active site cleft indicates that the ATP pockets in the monomers are less compact than those in the dimers. In the B-Raf monomer (BI), the ATP molecule was still loaded in the pocket, but the adenosine ring was not fully buried (Fig. 1A). In the dimers, the distances between the VA and F residues in the C-spine were smaller. ATP can be confined tightly by well assembled C-spine residues. Conformations with the adenosine ring of the ATP exposed are much less populated. Dimerization can enhance ATP binding in the kinase domain by assembling the C-spine. The R-spine is the signature of the active ON-state conformation.55 The distances between H574 (H466 in C-Raf) and F595 (F487 in C-Raf) in the R-spine were also considerably reduced upon the dimerization (Fig. S3B†), which indicates that dimerization also facilitates the assembly of the R-spine.
Dimerization disrupts inhibitory hydrophobic interactions in the kinase domain
The OFF-state conformation of the Raf kinase domain is stabilized by the inhibitory hydrophobic interactions between the A-loop and the N-lobe hydrophobic surface. The OFF-to-ON transition requires the disruption of the inhibitory hydrophobic interactions, enabling the αC-helix and A-loop to become IN and extended56 (Fig. 2A). Located at the center of these inhibitory hydrophobic interactions, the V600 (V492 in C-Raf) residue plays a major role in stabilizing the OFF-state conformation (Fig. 2B). The hotspot B-Raf mutation, V600E, disrupts the inhibitory hydrophobic interactions, replacing hydrophobic valine with negatively charged glutamic acid, and activates Raf.57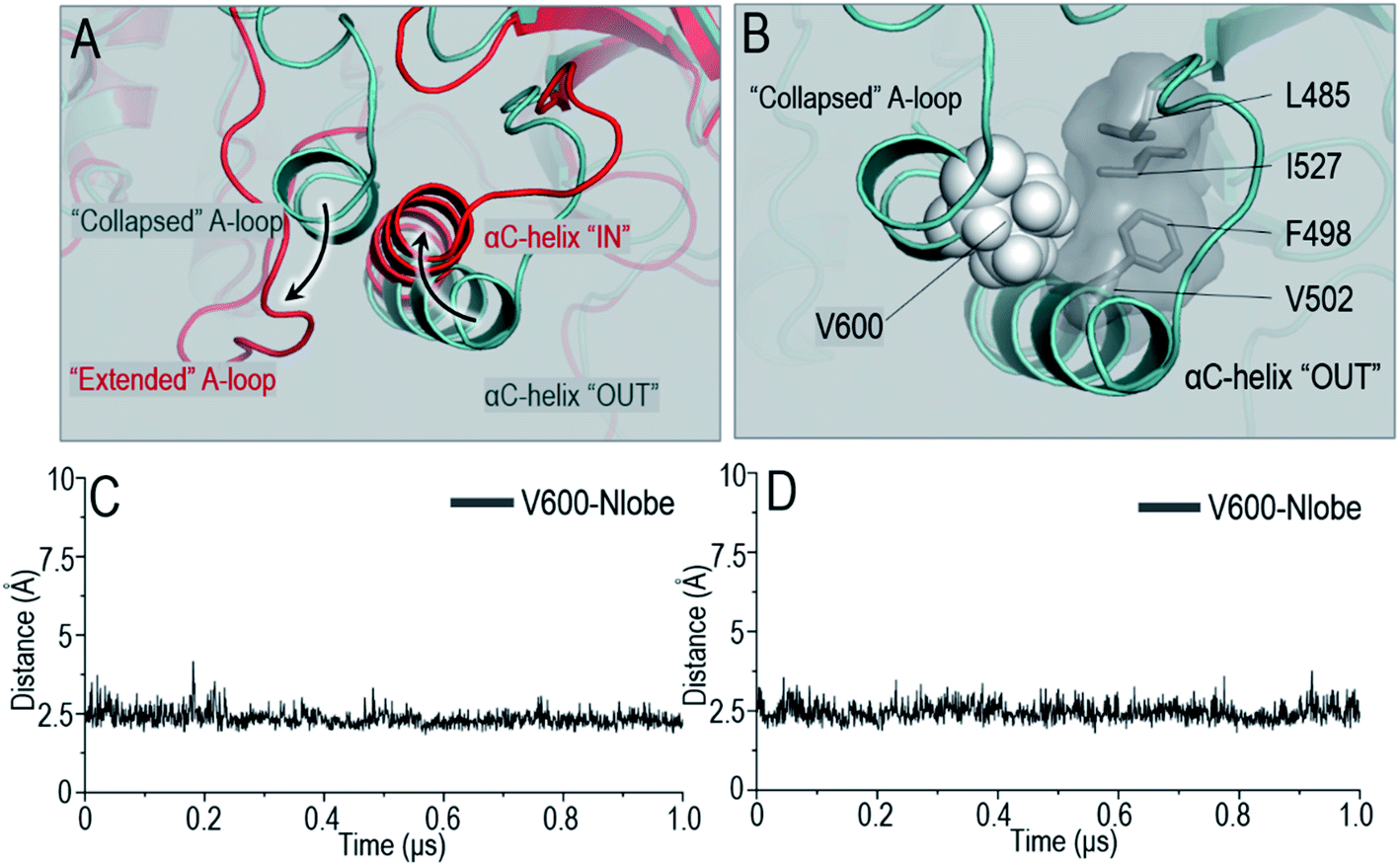 | ||
| Fig. 2 Hydrophobic interactions in Raf OFF-state conformation. (A) The OFF-state kinase domain [cyan] is stabilized by the inhibitory hydrophobic interactions between the collapsed A-loop and the hydrophobic surface in N-lobe. Upon dimerization, the inhibitory hydrophobic interactions are disrupted to enable the A-loop and αC-helix to become extended and IN [red]. (B) The V600 in the A-loop locates at the center and plays a major role in the OFF-state hydrophobic interactions. The V600/N-lobe inhibitory hydrophobic interactions are maintained in (C) B-Raf and (D) C-Raf monomers. | ||
The V600/N-lobe distance, defined as the minimal distance between any atom of V600 (V492 in C-Raf) and any atom of N-lobe's hydrophobic residues (L485, F498, V502 and I527 in B-Raf; L377, F390, V394, I419 in C-Raf), was calculated to characterize the hydrophobic interactions in OFF-state Raf. In Raf monomers, the inhibitory hydrophobic interactions appear to be well maintained with the V600/N-lobe distances less than ∼3.5 Å (Fig. 2C and D). However, upon dimerization, notable disruptions of the inhibitory hydrophobic interactions can be observed, particularly for two systems: BI in BI/BA and BI in BI/CI. In the dimeric systems, the V600/N-lobe distances can be up to ∼6 Å and ∼9 Å (Fig. 3). We observed that upon dimerization V600 moved out from the N-lobe hydrophobic surface (Fig. S4†), suggesting that the side-to-side dimerization can disrupt the inhibitory hydrophobic interactions in the kinase domain OFF-state.
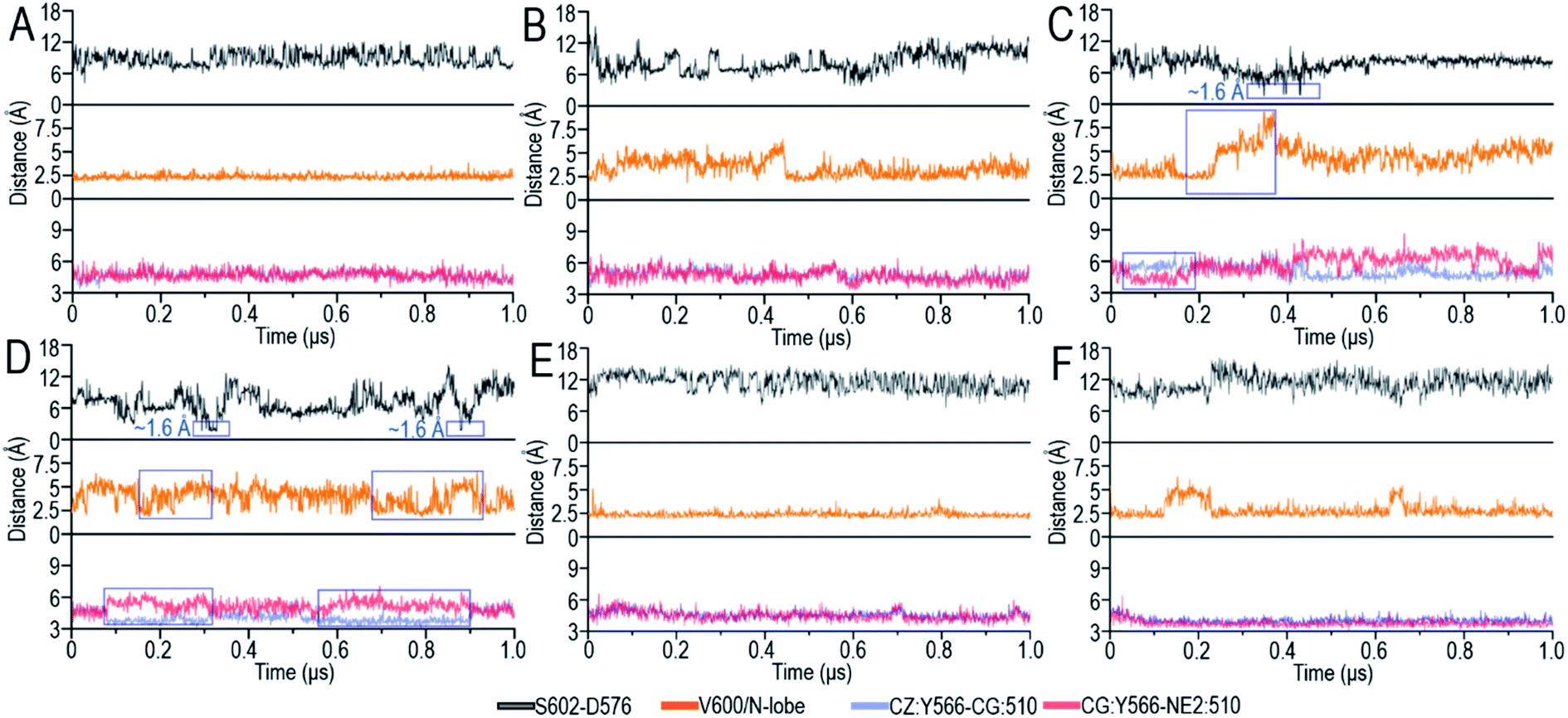 | ||
| Fig. 3 Time series of the S602–D576 distances [top panel], V600/N-lobe distances [middle panel] and intramolecular π–π stacking at the dimer interface [bottom panel] for (A) BI [chain A] in BI/BI, (B) BI [chain B] in BI/BI, (C) BI in BI/BA, (D) BI in BI/CI, (E) CI in BI/CI and (F) BI in BI/CA. The S602–D576 distance is defined as the minimal atom pair distance between the hydrogen atom (HG1) in the hydroxyl group of S602 (S494 in C-Raf) and the oxygen atom (OD1 and OD2) in the carboxyl group of D576 (D468 in C-Raf). The V600/N-lobe distance is defined as the minimal distances between any atom of V600 (V492 in C-Raf) and any atom of N-lobe's hydrophobic residues (L485, F498, V502 and I527 in B-Raf and L377, F390, V394, I419 in C-Raf). The intramolecular π–π stacking is characterized by the atom pair distances of CZ:Y566–CG:H510 and CG:Y566–NE2:H510. BI refers to B-Raf with inactive conformation, CI refers to C-Raf with inactive conformation, where B and C stand for B-Raf and C-Raf respectively. BA, CA refer to the respective active conformations. The blue boxes highlight the conformational changes upon dimerization. | ||
Dimerization promotes cis-autophosphorylation for OFF-to-ON transition
A-loop phosphorylation (cis-autophosphorylation) is a hallmark for full Raf activation.58,59 Based on the catalytic scenario of protein kinases, A-loop phosphorylation consists of two steps: (i) the deprotonation of the phosphorylation sites in the A-loop and (ii) the phosphorylation of the deprotonated residues in the A-loop by ATP.49,60 The deprotonation is executed by the catalytic HRD motif in the kinase domain. The aspartic acid, D576 (D468 in C-Raf) in the catalytic HRD motif removes the proton from the serine/threonine residues.49,60 In Raf, the phosphorylation sites in the A-loop, T599 and S602 (T491 and S494 in C-Raf), are conserved. Our results show that S602 (S494) was consistently closer to the catalytic HRD motif than T599 (T491) in all systems (Fig. S5A†), suggesting that S602 is more likely to be phosphorylated first, followed by T599.The time-dependent distances between the hydrogen atom (HG1) in the hydroxyl group of S602 (S494 in C-Raf) and the oxygen atoms (OD1 and OD2) in the carboxyl group of D576 (D468 in C-Raf) in the HRD motif, the key atomic contacts for the deprotonation, were calculated (Fig. 3). In the monomers, the distances between S602 and D576 of the HRD motif fluctuated by ∼4–17 Å, which are too far to allow for the deprotonation (Fig. S5B†). It is interesting to observe that in systems with disrupted inhibitory hydrophobic interactions (BI in BI/BA and BI in BI/CI), the S602–D576 distances went as low as ∼1.6 Å, at which the deprotonation can be executed (Fig. 3C and D). We observed that the A-loop was released from the structural constraint of the N-lobe in the kinase domain and rotated towards the catalytic HRD motif when the inhibitory hydrophobic interactions were disrupted (Fig. S4†). This effectively reduced the S602–D576 distances to allow for cis-autophosphorylation. These results suggest that the disruption of the inhibitory hydrophobic interactions upon dimerization can make the A-loop phosphorylation sites approach the catalytic HRD motif for cis-autophosphorylation, promoting the OFF-to-ON transition.
Dimerization activates Raf through the π–π stacking at the dimer interface
To understand the underlying mechanism of the dimerization-induced conformational changes, comprehensive comparisons were performed. The results point to the important role of π–π stacking at the dimer interface in promoting the OFF-to-ON transition. In Raf's kinase domain, Y566 (Y458 in C-Raf) in αE-helix and H510 (H402 in C-Raf) at the C-terminal end of the αC-helix form intramolecular π–π stacking. Upon the side-to-side dimerization, the Y566–H510 intramolecular π–π stacking is approached by another aromatic residue, H477 (H369 in C-Raf), from another protomer at the dimer interface (Fig. 4A). The Y566–H510 intramolecular π–π stacking competes with, and is disrupted by, the intermolecular Y566–H477 π–π stacking, facilitating the OFF-to-ON transition.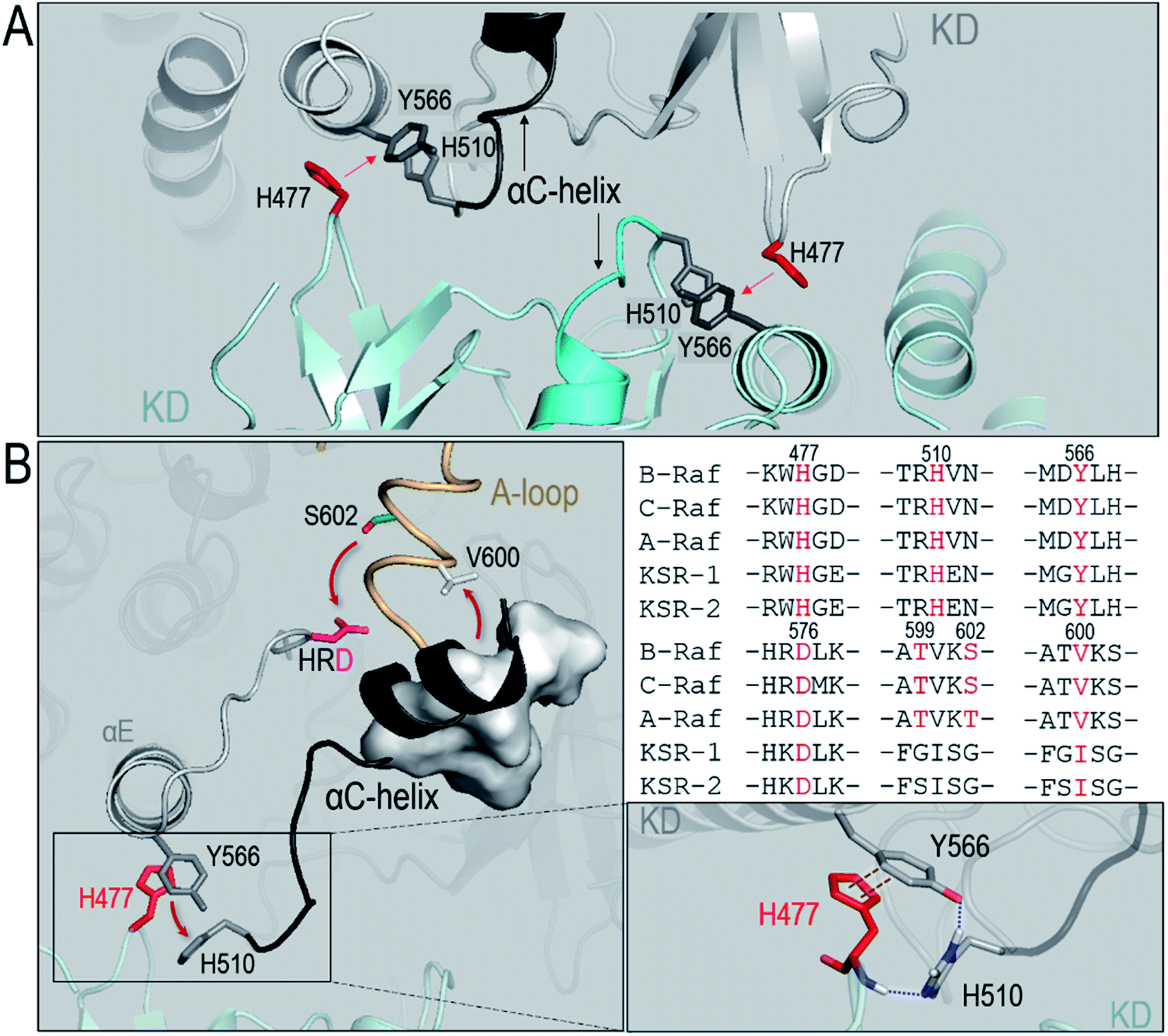 | ||
| Fig. 4 Collective conformational changes upon dimerization promoting the OFF-to-ON transition. (A) In B-Raf, the intramolecular π–π stacking between Y566 and H510 is surrounded by H477 from the partner kinase at the dimer interface. The H510 is at the C-terminal end of the αC-helix. Red letters in the vertical label denote the His residue from the partner kinase. (B) A snapshot of the collective conformational change shows that the disruption of the intramolecular π–π stacking by intermolecular π–π stacking at the dimer interface induces the conformational changes of the αC-helix and the A-loop, promoting the approach of the phosphorylation site, S602, in the A-loop to the HRD motif for cis-autophosphorylation [left panel]. The replaced H510 forms the hydrogen bonds with H477 and Y566 [bottom-right panel]. The key residues for the collective conformational changes are conserved in the Raf isoforms and KSR proteins [top-right panel]. | ||
The atom pair distances between the aromatic rings of Y566 and H510 were measured to quantify the intramolecular π–π stacking. While not all inactive systems disrupted this stacking, two systems (BI in BI/BA and BI in BI/CI) with disrupted inhibitory hydrophobic interactions and reduced S602–D576 distances exhibited more unstable and separated atom pair distance profiles than other systems, which confirmed the disruption of the intramolecular π–π stacking in the systems (Fig. 3). In the BI of the BI/BA dimer, the Y566–H510 intramolecular stacking can be completely broken and replaced by the Y566–H477 intermolecular π–π stacking at the dimer interface. The replacement of the intramolecular by the intermolecular π–π stacking leads to the favorable local residue contacts. The replaced H510 formed the hydrogen bonds with Y566 and H477 at the dimer interface (Fig. 4B), which indicates that the conformational changes upon dimerization may generate local energy minimum along the reaction coordinates for Raf's activation. Similar phenomena, including the intra-π–π disruption and replacement, can also be observed in the active Raf in the dimer and in the replicate systems, confirming the behaviors (Fig. S6†). The consequence of the disrupted intramolecular π–π stacking is the release of the H510 from the aromatic interactions. The time-dependent distance profiles in Fig. 3 show that the disruption of the intramolecular π–π stacking at the dimer interface may precede the V600/N-lobe disruption and the S602–D576 interactions. H510 is at the C-terminal end of the αC-helix (Fig. 4A). Its release from the intramolecular π–π stacking removed the structural constraint of the αC-helix at the C-terminal end, which promoted the conformational changes of the αC-helix in the kinase domain. This facilitated the movements of the V600 out from the N-lobe hydrophobic surface and the approach of S602 in the A-loop to the catalytic HRD motif in the Raf kinase domain. These actions collectively promote the structural transition of Raf's kinase domain towards the ON-state conformation, explaining how it is activated through the side-to-side dimerization (Fig. 4B). The sequence analysis shows that the key residues in the collective conformational changes are conserved in the Raf family and KSR proteins (Fig. 4B), which implies that the observed mechanism can be applied to diverse Raf homo- and hetero-dimerization scenarios.
ON-state conformation is stabilized by the NtB motif in the dimer
Another important question related to Raf dimerization is how the fully activated, ON-state kinase domain can be stabilized in the dimer for Raf signaling. Here, we show that the stabilization of the ON-state kinase domain can be achieved through the N-terminal basic (NtB) motif. The NtB motif is at the N-terminal end of the NtA motif. Sequence analysis shows that the highly basic NtB motifs in the Raf family are conserved (Fig. 5A). Extensive work has shown the significance of the NtA motif in Raf dimerization,22,50 but the nearby NtB motif is much less studied. As shown in Fig. S7,† the tandem NtB–NtA motif can cover another protomer in the dimer, indicating that the NtB motif holds the potential to interact with the kinase domain upon dimerization. In the OFF state, the NtA motif interacts with the basic residues (506RKTR509 in B-Raf) in the αC-helix of the partner Raf (Fig. 5B). In the ON state, the NtB motif interacts with the exposed pS602 in the extended A-loop of the partner Raf (Fig. 5C).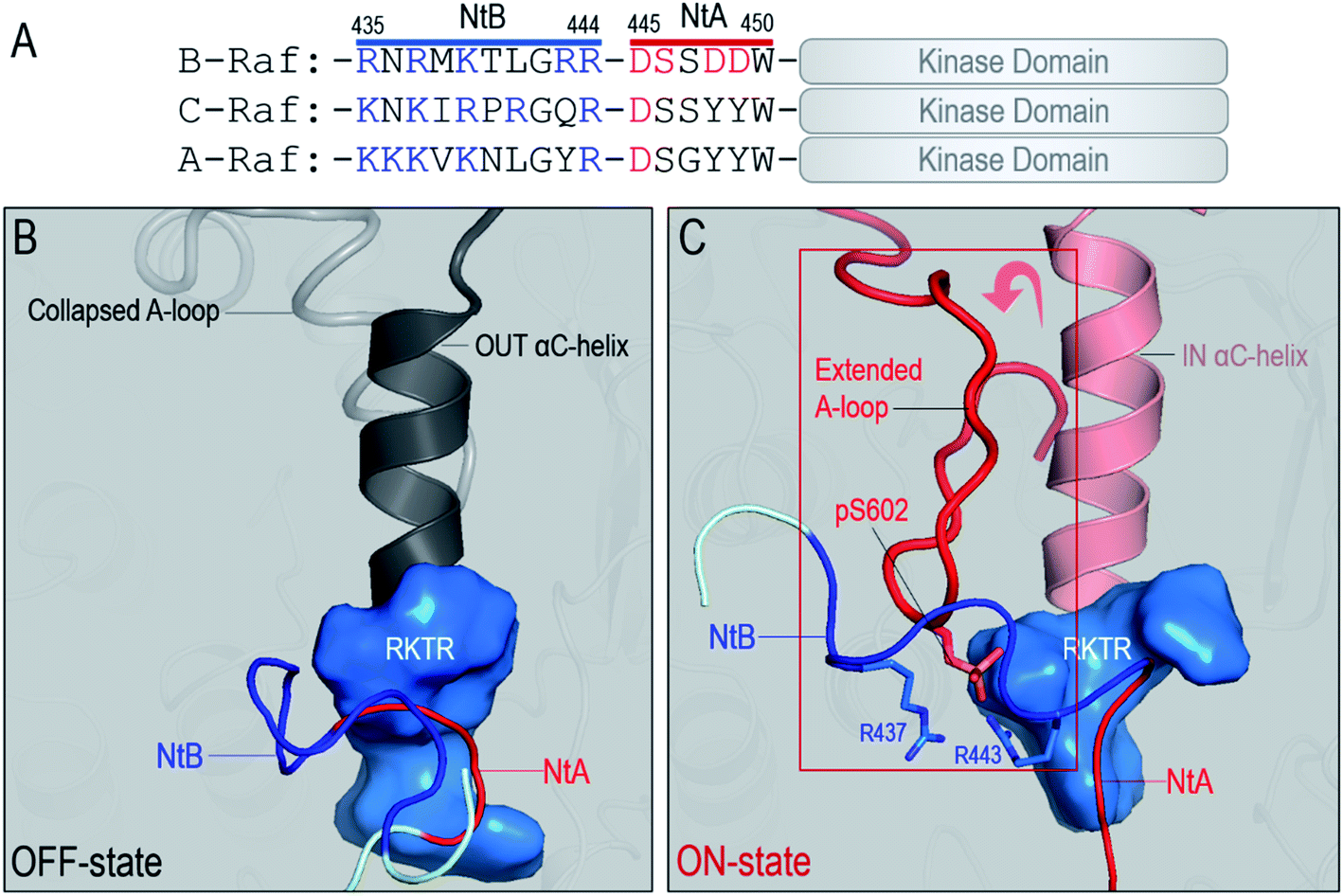 | ||
| Fig. 5 Interactions between the NtB motif and the kinase domain in the Raf dimers. (A) The N-terminal basic (NtB, 435–444 in B-Raf) motif is at the N-terminal end of the N-terminal acidic (NtA) motif. The NtB motifs are highly basic and conserved in Raf family. Snapshots representing (B) the interaction of the NtA motif with the kinase domain of partner Raf in the OFF state and (C) the interaction of the NtB motif with the kinase domain of partner Raf in the ON state. Key residues involved in the interactions are marked. | ||
The role of the NtB motif is to stabilize the fully activated ON-state kinase domain in the dimer for Raf signaling. Our results show that at the early stage of the dimerization, before the kinase domain was fully activated, the interactions of the NtB motif with the OFF-state kinase domain were dynamic. However, once the kinase domain is fully activated (BA in BI/BA, BA in BA/BA and CA in BI/CA), the NtB motif showed strong interactions with the extended A-loop, stabilizing the ON-state conformation of the kinase domain in the dimer (Fig. 6A–E). In the ON-state kinase domain, the A-loop is phosphorylated and extended. We observed that the extended A-loop exposed the pS602 to the dimer interface. The acidic pS602 can be captured by the basic NtB motif, generating strong electrostatic interactions. These interactions stabilized the ON-state conformation of the kinase domain in the dimer. The pS602–NtB interactions can be observed in all the ON-state kinase domains in the Raf dimers (Fig. 6A–E). Other residue contacts, including those between R603 (K492 in C-Raf) and NtA motif, and between pS602 and RKTR residues in αC-helix, also contributed to the stabilization of ON-state kinase domain in the dimer. The interaction energies at the dimer interface increased when more kinase domains are fully activated (Fig. 6F), which confirms that the NtB motif and nearby residues provided additional interactions to stabilize the ON-state kinase domain in the dimer.
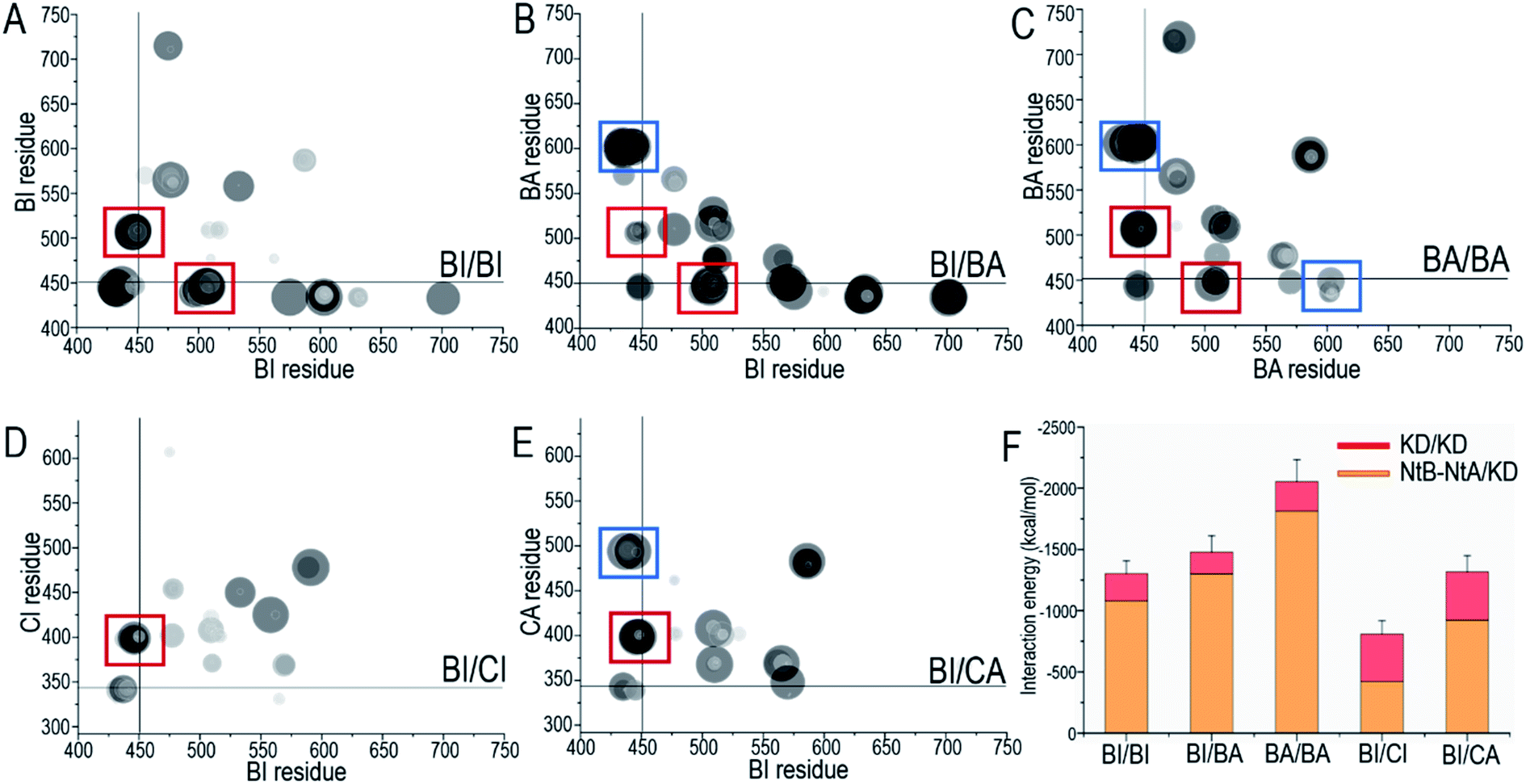 | ||
| Fig. 6 Key residue contacts at the dimer interface. The residue contacting maps for (A) the BI/BI homodimer, (B) the BI/BA homodimer, (C) the BA/BA homodimer, (D) the BI/CI heterodimer, and (E) the BI/CA heterodimer. The interactions of the NtA and NtB motifs at the dimer interface are highlighted by the red and blue boxes, respectively. (F) The total interaction energies show the contributions from the KD/KD and NtB–NtA/KD interfaces, where KD denotes kinase domain. BI refers to B-Raf with inactive conformation, CI refers to C-Raf with inactive conformation, where (B) and (C) stand for B-Raf and C-Raf respectively. BA, CA refer to the respective active conformations. | ||
Discussion
In this work, we showed that the N- and C-lobes are further apart in Raf monomers than in the dimers. The side-to-side dimerization leads to the CLOSED kinase domain conformation. The conformational changes upon dimerization help assemble the C-spine, leading to a more compact ATP pocket in the Raf dimers than in the monomers. The less compact ATP pocket in Raf monomers can be more readily outcompeted by inhibitors. These results are consistent with experimental observations, explaining the drug resistance in the constitutive Raf dimerization by p61 spliced B-Raf mutant.38,39,61We determine an atomic-level mechanism of Raf activation through dimerization, which reveals that the disruption of the intramolecular π–π stacking by the intermolecular stacking at the dimer interface can release the structural constraint of the αC-helix and initiate the OFF-to-ON transition in the kinase domain (Fig. 7). The released αC-helix promotes the disruption of the inhibitory hydrophobic interactions in the OFF state, making the phosphorylation sites in the A-loop approach the catalytic HRD motif for cis-autophosphorylation. These results show how the transverse side-to-side dimerization in Raf promotes cis-autophosphorylation for full activation. The dimerization makes the overall structure of the kinase domain more compact. In the compact dimer structure, the local conformational changes in kinase domain promotes the OFF-to-ON transition. Both are required for Raf's full activation (Fig. 7). These actions promote the phosphorylation of the A-loop, which further induces the conformational changes to allow α-C helix moving IN and the A-loop to become extended. The sequence analysis shows that the key residues in the mechanism, including the Y566, H510 and H477 for the π–π stacking at the dimer interface and S602 and D576 for cis-autophosphorylation, are fully conserved in Raf family and KSR proteins, which implies that the mechanism sampled in this work can be general for diverse dimerization scenarios in the Raf family. The Y–H intramolecular π–π stacking can be also found in many other protein kinases, including Src, JAK and ERK. This suggests that the intramolecular π–π stacking is an evolutionally conserved feature in protein kinases. However, whether other protein kinases also adopt a similar strategy to control αC-helix for the full activation requires further studies.
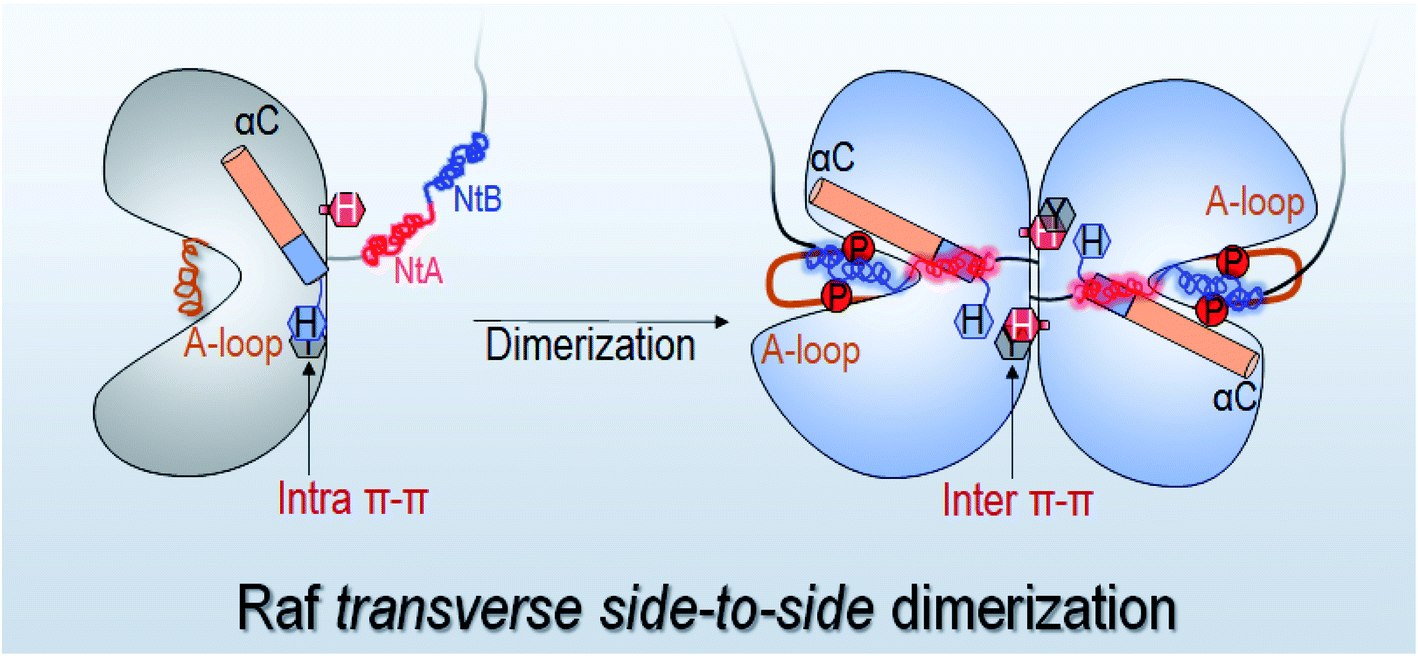 | ||
| Fig. 7 Schematic figure for the side-to-side dimerization induced Raf full activation through the destabilization of the intramolecular π–π stacking at the dimer interface. The H477 (H369 in C-Raf) may interfere with and replace the intramolecular π–π stacking for the Raf full activation. The NtB and NtA motifs form the electrostatic interactions at the dimer interface. | ||
The mechanism in this work is supported by the B-Raf Y566C oncogenic mutation. This missense mutation removes the aromatic ring of Y566 and disrupts the intramolecular π–π stacking, resulting in cutaneous melanoma.62 Since this mutation is located at the dimer interface, it could also interfere with dimerization, weakening its activation potential in cancer and making it less frequent. The disruption of the intramolecular π–π stacking at the dimer interface can be also observed in the Raf crystal and cryo-EM structures (Fig. S8†), however the disruptions observed there are less significant than those in the simulations. This is possibly because the conformational changes during the OFF-to-ON transition sampled in this work are intermediate states, which cannot fully represent the energy minimum in the full free energy landscape.
Different from previous computational works,50,63 we modeled the NtB motif based on structural analysis and included both OFF-state and ON-state kinase domains in the dimers. This protocol allows us not only to understand Raf activation by dimerization, but also to study how the fully activated ON-state kinase domain is stabilized in the dimer for Raf signaling. The results suggest that the NtB motif near to the NtA motif is functionally important for Raf dimerization. The NtB motif can capture the exposed pS602 in the extended A-loop, stabilizing the ON-state kinase domain in the dimer for Raf signaling. This is consistent with experimental observations that the removal of NtB–NtA motif attenuated Raf signalling.22
The dimer interface in Raf has been a major therapeutic target.43 The consensus has been reached that effective Raf inhibitors should target both the mutations and dimerization.35,43,64–66 The next-generation inhibitor, PLX8394, that evades the paradoxical MAPK activation by disrupting the Raf dimerization, has been in clinical trials.45,67 The insights gained from this work lead us to propose a drug–peptide conjugate. The NtB and NtA motifs are adjacent. The single conjugate with the drug that is specific to the Raf mutant and the conjugated peptide with charged residues that can disrupt the electrostatic forces at the dimer interface by the NtB and NtA motifs may destabilize the NtB-enhanced ON-state conformation and prevent NtA-induced dimerization.
Conclusions
Here we modeled and simulated B-Raf monomers, C-Raf monomers, B-Raf/B-Raf homodimers, and B-Raf/C-Raf heterodimers to decipher the detailed mechanism of Raf activation through the transverse side-to-side dimerization. The results show that Raf monomers are OPEN with the N- and C-lobes drifting further apart. The ATP pockets are less tightly assembled in the monomers than in the dimers, which can explain drug resistance to constitutive Raf dimerization. The unraveled atomic-level mechanism clarifies how the transverse side-to-side dimerization promotes the OFF-to-ON transition of Raf's kinase domain through the π–π stacking at the dimer interface, a long-standing enigma. Our simulations observe that the disruption of the intramolecular π–π stacking by the intermolecular π–π stacking at the dimer interface can release the structural constraint of the αC-helix in the kinase domain, facilitating the disruption of the inhibitory hydrophobic interactions in the OFF state and decreasing the distance of the phosphorylation sites in the A-loop to the HRD motif for the cis-autophosphorylation. Our results also suggest that the under-explored NtB motif can be functionally important. It stabilizes the fully activated ON-state kinase domain in the dimer for Raf signaling. Finally, it further leads us to suggest a new structural strategy for drug discovery targeting Raf dimerization.Methods and materials
Modeling of B-Raf monomers and C-Raf monomers
The inactive conformations of B-Raf and C-Raf kinase domains were modeled based on the crystal structure (PDB ID: 3SKC). In the inactive kinase domains, the A-loops were collapsed, and the αC-helices were OUT. The active conformation of B-Raf kinase domain was modeled based on the crystal structure (PDB ID: 4E26), and for C-Raf (PDB ID: 3OMV). In the active kinase domains, the A-loops were extended, and the αC-helices were IN. The N-terminal motifs, residues 432–450 in B-Raf and residues 324–342 in C-Raf, were modeled into the Raf monomers. The B-Raf NtA motif was phosphorylated at S446, while the C-Raf NtA motif was unphosphorylated. The modeled NtA motifs had no initial contact with the kinase domain. For B-Raf and C-Raf monomers in the active conformation, the A-loop residues were unphosphorylated. The ATP molecule with the magnesium ion (Mg2+) were included in all the systems.Modeling of B-Raf/B-Raf homodimers and B-Raf/C-Raf heterodimers
As summarized in Table S1,† we modeled the B-Raf/B-Raf homodimers and the B-Raf/C-Raf heterodimers. Here, BI and CI denoted inactive B-Raf and C-Raf, respectively. Since the dimer interfaces are identical, the modeled dimer systems with different isoforms and initial conformations were considered as the replicates to understand the Raf activation through dimerization. The replicate simulations for BI/BA and BI/CI were also performed for checking the behaviors. Likewise, BA and CA denote active B-Raf and C-Raf with the phosphorylated A-loop, respectively. By combining the inactive/active B-Raf and C-Raf, we obtained BI/BI, BI/BA, and BA/BA homodimers, and BI/CI and BI/CA heterodimers. The dimer interface in the modeled Raf dimers was identical to that in the crystal structures. The N-terminal motifs were included in all the systems.MD simulation protocol
The all-atom MD simulations were conducted with the NAMD package68 using the CHARMM all-atom additive forcefield (version C36).69 The systems were solvated by the explicit TIP3 water model. Water molecules within 2.4 Å of the proteins were removed to avoid unreasonable atom collapse. The ions, Na+ and Cl−, were used to neutralize the systems and generate a ∼0.15 mol L−1 ionic strength. The MD simulations were performed at 310 K. Covalent bonds involving hydrogen atoms were constrained. Short-range van der Waals (vdW) and long-rang electrostatic interactions were calculated by the switch function and the particle mesh Ewald (PME) algorithm. The time-step of 2 fs was used in the MD simulations. The VMD70 and NAMD68 programs were used for modeling and analyzing the systems. 1 μs simulation was performed for each generated system. The statistics of the NC distances, NC residue distance, the C-spine distances, the R-spine distances, the interaction energies, and the residue contacting maps were calculated based on the last 500 ns trajectories.Author contributions
MZ, HJ, and RN conceived and designed the study. MZ conducted most of the simulations and anlayzed the results. MZ, RM, HJ and RN wrote the paper.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261201500003I. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, CCR, LCIM. The calculations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (https://hpc.nih.gov/).References
- H. Lavoie and M. Therrien, Nat. Rev. Mol. Cell Biol., 2015, 16, 281–298 CrossRef CAS PubMed.
- C. J. Tsai and R. Nussinov, Curr. Opin. Struct. Biol., 2018, 53, 100–106 CrossRef CAS PubMed.
- S. Pfister, W. G. Janzarik, M. Remke, A. Ernst, W. Werft, N. Becker, G. Toedt, A. Wittmann, C. Kratz, H. Olbrich, R. Ahmadi, B. Thieme, S. Joos, B. Radlwimmer, A. Kulozik, T. Pietsch, C. Herold-Mende, A. Gnekow, G. Reifenberger, A. Korshunov, W. Scheurlen, H. Omran and P. Lichter, J. Clin. Invest., 2008, 118, 1739–1749 CrossRef CAS PubMed.
- U. Degirmenci, M. Wang and J. Hu, Cells, 2020, 9, 198 CrossRef CAS PubMed.
- G. Bollag, J. Tsai, J. Z. Zhang, C. Zhang, P. Ibrahim, K. Nolop and P. Hirth, Nat. Rev. Drug Discovery, 2012, 11, 873–886 CrossRef CAS PubMed.
- N. H. Tran, X. Wu and J. A. Frost, J. Biol. Chem., 2005, 280, 16244–16253 CrossRef CAS PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Nature, 2002, 417, 949–954 CrossRef CAS PubMed.
- P. T. Wan, M. J. Garnett, S. M. Roe, S. Lee, D. Niculescu-Duvaz, V. M. Good, C. M. Jones, C. J. Marshall, C. J. Springer, D. Barford, R. Marais and P. Cancer Genome, Cell, 2004, 116, 855–867 CrossRef CAS PubMed.
- C. A. Pratilas, B. S. Taylor, Q. Ye, A. Viale, C. Sander, D. B. Solit and N. Rosen, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4519–4524 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Front. Oncol., 2019, 9, 1231 CrossRef PubMed.
- T. Travers, C. A. Lopez, Q. N. Van, C. Neale, M. Tonelli, A. G. Stephen and S. Gnanakaran, Sci. Rep., 2018, 8, 8461 CrossRef PubMed.
- S. Lu, H. Jang, S. Muratcioglu, A. Gursoy, O. Keskin, R. Nussinov and J. Zhang, Chem. Rev., 2016, 116, 6607–6665 CrossRef CAS PubMed.
- T. Improta-Brears, S. Ghosh and R. M. Bell, Mol. Cell. Biochem., 1999, 198, 171–178 CrossRef CAS PubMed.
- S. Li, H. Jang, J. Zhang and R. Nussinov, Structure, 2018, 26, 513–525 CrossRef CAS PubMed.
- S. S. Taylor, M. M. Keshwani, J. M. Steichen and A. P. Kornev, Philos. Trans. R. Soc. London, Ser. B, 2012, 367, 2517–2528 CrossRef CAS PubMed.
- E. Park, S. Rawson, K. H. Li, B. W. Kim, S. B. Ficarro, G. Gonzalez-Del Pino, H. Sharif, J. A. Marto, H. Jeon and M. J. Eck, Nature, 2019, 575, 545–550 CrossRef CAS PubMed.
- Y. Kondo, J. Ognjenovic, S. Banerjee, D. Karandur, A. Merk, K. Kulhanek, K. Wong, J. P. Roose, S. Subramaniam and J. Kuriyan, Science, 2019, 366, 109–115 CrossRef CAS PubMed.
- M. Zhang, H. Jang, Z. Li, D. B. Sacks and R. Nussinov, Structure, 2021, 768–777 CrossRef CAS PubMed.
- E. M. Terrell and D. K. Morrison, Cold Spring Harbor Perspect. Med., 2019, 9, a033746 CrossRef CAS PubMed.
- T. Cookis and C. Mattos, Biomolecules, 2021, 11, 996 CrossRef CAS PubMed.
- C. K. Weber, J. R. Slupsky, H. A. Kalmes and U. R. Rapp, Cancer Res., 2001, 61, 3595–3598 CAS.
- J. Hu, E. C. Stites, H. Yu, E. A. Germino, H. S. Meharena, P. J. S. Stork, A. P. Kornev, S. S. Taylor and A. S. Shaw, Cell, 2013, 154, 1036–1046 CrossRef CAS PubMed.
- T. Pantsar, Comput. Struct. Biotechnol. J., 2020, 18, 189–198 CrossRef CAS PubMed.
- J. R. Haling, J. Sudhamsu, I. Yen, S. Sideris, W. Sandoval, W. Phung, B. J. Bravo, A. M. Giannetti, A. Peck, A. Masselot, T. Morales, D. Smith, B. J. Brandhuber, S. G. Hymowitz and S. Malek, Cancer Cell, 2014, 26, 402–413 CrossRef CAS PubMed.
- K. T. Flaherty, I. Puzanov, K. B. Kim, A. Ribas, G. A. McArthur, J. A. Sosman, P. J. O'Dwyer, R. J. Lee, J. F. Grippo, K. Nolop and P. B. Chapman, N. Engl. J. Med., 2010, 363, 809–819 CrossRef CAS PubMed.
- R. Garcia-Gomez, X. R. Bustelo and P. Crespo, Trends Cancer, 2018, 4, 616–633 CrossRef CAS PubMed.
- M. Holderfield, M. M. Deuker, F. McCormick and M. McMahon, Nat. Rev. Cancer, 2014, 14, 455–467 CrossRef CAS PubMed.
- L. K. Rushworth, A. D. Hindley, E. O'Neill and W. Kolch, Mol. Cell. Biol., 2006, 26, 2262–2272 CrossRef CAS PubMed.
- A. K. Freeman, D. A. Ritt and D. K. Morrison, Mol. Cell, 2013, 49, 751–758 CrossRef CAS PubMed.
- A. S. Shaw, A. P. Kornev, J. Hu, L. G. Ahuja and S. S. Taylor, Mol. Cell. Biol., 2014, 34, 1538–1546 CrossRef PubMed.
- H. Lavoie, M. Sahmi, P. Maisonneuve, S. A. Marullo, N. Thevakumaran, T. Jin, I. Kurinov, F. Sicheri and M. Therrien, Nature, 2018, 554, 549–553 CrossRef CAS PubMed.
- D. A. Ritt, D. M. Monson, S. I. Specht and D. K. Morrison, Mol. Cell. Biol., 2010, 30, 806–819 CrossRef CAS PubMed.
- M. K. Dougherty, J. Muller, D. A. Ritt, M. Zhou, X. Z. Zhou, T. D. Copeland, T. P. Conrads, T. D. Veenstra, K. P. Lu and D. K. Morrison, Mol. Cell, 2005, 17, 215–224 CrossRef CAS PubMed.
- P. I. Poulikakos, C. Zhang, G. Bollag, K. M. Shokat and N. Rosen, Nature, 2010, 464, 427–430 CrossRef CAS PubMed.
- D. E. Durrant and D. K. Morrison, Br. J. Cancer, 2018, 118, 3–8 CrossRef CAS PubMed.
- R. Yaeger, Z. Yao, D. M. Hyman, J. F. Hechtman, E. Vakiani, H. Zhao, W. J. Su, L. Wang, A. Joelson, A. Cercek, J. Baselga, E. de Stanchina, L. Saltz, M. F. Berger, D. B. Solit and N. Rosen, Cancer Res., 2017, 77, 6513–6523 CrossRef CAS PubMed.
- S. H. Chen, Y. Y. Zhang, R. D. van Horn, T. G. Yin, S. Buchanan, V. Yadav, I. Mochalkin, S. S. Wong, Y. G. Yue, L. Huber, I. Conti, J. R. Henry, J. J. Starling, G. D. Plowman and S. B. Peng, Cancer Discovery, 2016, 6, 300–315 CrossRef CAS PubMed.
- P. I. Poulikakos, Y. Persaud, M. Janakiraman, X. J. Kong, C. Ng, G. Moriceau, H. B. Shi, M. Atefi, B. Titz, M. T. Gabay, M. Salton, K. B. Dahlman, M. Tadi, J. A. Wargo, K. T. Flaherty, M. C. Kelley, T. Misteli, P. B. Chapman, J. A. Sosman, T. G. Graeber, A. Ribas, R. S. Lo, N. Rosen and D. B. Solit, Nature, 2011, 480, 387–390 CrossRef CAS PubMed.
- Z. Yao, N. M. Torres, A. Tao, Y. J. Gao, L. S. Luo, Q. Li, E. de Stanchina, O. Abdel-Wahab, D. B. Solit, P. I. Poulikakos and N. Rosen, Cancer Cell, 2015, 28, 370–383 CrossRef CAS PubMed.
- G. Hatzivassiliou, K. Song, I. Yen, B. J. Brandhuber, D. J. Anderson, R. Alvarado, M. J. Ludlam, D. Stokoe, S. L. Gloor, G. Vigers, T. Morales, I. Aliagas, B. Liu, S. Sideris, K. P. Hoeflich, B. S. Jaiswal, S. Seshagiri, H. Koeppen, M. Belvin, L. S. Friedman and S. Malek, Nature, 2010, 464, 431–435 CrossRef CAS PubMed.
- S. J. Heidorn, C. Milagre, S. Whittaker, A. Nourry, I. Niculescu-Duvas, N. Dhomen, J. Hussain, J. S. Reis-Filho, C. J. Springer, C. Pritchard and R. Marais, Cell, 2010, 140, 209–221 CrossRef CAS PubMed.
- A. B. Cotto-Rios XM, N. Gitego, E. Zacharioudakis, O. Giricz, Y. Wu, Y. Zou, A. Verma, P. I. Poulikakos and E. Gavathiotis, Nat. Commun., 2020, 11, 4370 CrossRef PubMed.
- T. Rajakulendran, M. Sahmi, M. Lefrancois, F. Sicheri and M. Therrien, Nature, 2009, 461, 542–545 CrossRef CAS PubMed.
- T. Huang, M. Karsy, J. Zhuge, M. Zhong and D. Liu, J. Hematol. Oncol., 2013, 6, 30 CrossRef CAS PubMed.
- C. Zhang, W. Spevak, Y. Zhang, E. A. Burton, Y. Ma, G. Habets, J. Z. Zhang, J. Lin, T. Ewing, B. Matusow, G. Tsang, A. Marimuthu, H. Cho, G. X. Wu, W. R. Wang, D. Fong, H. Nguyen, S. Y. Shi, P. Womack, M. Nespi, R. Shellooe, H. Carias, B. Powell, E. Light, L. Sanftner, J. Walters, J. Tsai, B. L. West, G. Visor, H. Rezaei, P. S. Lin, K. Nolop, P. N. Ibrahim, P. Hirth and G. Bollag, Nature, 2015, 526, 583–586 CrossRef CAS PubMed.
- V. Modi and R. L. Dunbrack Jr, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6818–6827 CrossRef CAS PubMed.
- H. S. Meharena, P. Chang, M. M. Keshwani, K. Oruganty, A. K. Nene, N. Kannan, S. S. Taylor and A. P. Kornev, PLoS Biol., 2013, 11, e1001680 CrossRef CAS.
- S. A. Jamieson, Z. Ruan, A. E. Burgess, J. R. Curry, H. D. Mcmilan, J. L. Brewster, A. K. Dunbier, A. D. Axtman, N. Kannan and P. D. Mace, Sci. Signaling, 2018, 11, eaau0597 CrossRef.
- R. Roskoski Jr, Pharmacol. Res., 2018, 135, 239–258 CrossRef.
- P. G. Jambrina, N. Rauch, R. Pilkington, K. Rybakova, L. K. Nguyen, B. N. Kholodenko, N. V. Buchete, W. Kolch and E. Rosta, Angew. Chem., Int. Ed., 2016, 55, 983–986 CrossRef CAS PubMed.
- D. K. Morrison and R. E. Cutler, Curr. Opin. Cell Biol., 1997, 9, 174–179 CrossRef CAS PubMed.
- C. Hyeon, P. A. Jennings, J. A. Adams and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3023–3028 CrossRef CAS PubMed.
- R. Mega, N. Nakagawa, S. Kuramitsu and R. Masui, PLoS One, 2020, 15, e0233689 CrossRef CAS PubMed.
- A. P. Kornev, S. S. Taylor and L. F. Ten Eyck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14377–14382 CrossRef CAS PubMed.
- A. P. Kornev, N. M. Haste, S. S. Taylor and L. F. Eyck, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17783–17788 CrossRef CAS PubMed.
- W. Yeung, Z. Ruan and N. Kannan, IUBMB Life, 2020, 72, 1189–1202 CrossRef CAS PubMed.
- R. C. Maloney, M. Zhang, H. Jang and R. Nussinov, Comput. Struct. Biotechnol. J., 2021, 19, 3349–3363 CrossRef CAS PubMed.
- M. Kohler and T. Brummer, Cell Cycle, 2016, 15, 1171–1173 CrossRef PubMed.
- D. K. Morrison, G. Heidecker, U. R. Rapp and T. D. Copeland, J. Biol. Chem., 1993, 268, 17309–17316 CrossRef CAS.
- R. Roskoski Jr, Biochem. Biophys. Res. Commun., 2010, 399, 313–317 CrossRef PubMed.
- H. Lavoie, N. Thevakumaran, G. Gavory, J. J. Li, A. Padeganeh, S. Guiral, J. Duchaine, D. Y. Mao, M. Bouvier, F. Sicheri and M. Therrien, Nat. Chem. Biol., 2013, 9, 428–436 CrossRef CAS PubMed.
- E. Cerami, J. Gao, U. Dogrusoz, B. E. Gross, S. O. Sumer, B. A. Aksoy, A. Jacobsen, C. J. Byrne, M. L. Heuer, E. Larsson, Y. Antipin, B. Reva, A. P. Goldberg, C. Sander and N. Schultz, Cancer Discovery, 2012, 2, 401–404 CrossRef PubMed.
- A. Tse and G. M. Verkhivker, PLoS One, 2016, 11, e0166583 CrossRef PubMed.
- Z. Karoulia, E. Gavathiotis and P. I. Poulikakos, Nat. Rev. Cancer, 2017, 17, 676–691 CrossRef CAS PubMed.
- G. Posternak, X. Tang, P. Maisonneuve, T. Jin, H. Lavoie, S. Daou, S. Orlicky, T. Goullet de Rugy, L. Caldwell, K. Chan, A. Aman, M. Prakesch, G. Poda, P. Mader, C. Wong, S. Maier, J. Kitaygorodsky, B. Larsen, K. Colwill, Z. Yin, D. F. Ceccarelli, R. A. Batey, M. Taipale, I. Kurinov, D. Uehling, J. Wrana, D. Durocher, A. C. Gingras, R. Al-Awar, M. Therrien and F. Sicheri, Nat. Chem. Biol., 2020, 16, 1170–1178 CrossRef CAS PubMed.
- Z. Yao, Y. J. Gao, W. J. Su, R. Yaeger, J. Tao, N. Na, Y. Zhang, C. Zhang, A. Rymar, A. Tao, N. M. Timaul, R. Mcgriskin, N. A. Outmezguine, H. Zhao, Q. Chang, B. Qeriqi, M. Barbacid, E. de Stanchina, D. M. Hyman, G. Bollag and N. Rosen, Nat. Med., 2019, 25, 284–291 CrossRef CAS PubMed.
- F. Janku, U. N. Vaishampayan, V. Khemka, M. Bhatty, E. J. Sherman, J. Tao, J. R. Whisenant, D. S. Hong, N. Buix, S. Kummar, L. G. Feun, A. R. Parikh, C. Zhang, G. Michelson, E. Martin, R. Shellooe, P. Severson, M. Pelayo, D. A. Karlin and S. Sharma, J. Clin. Oncol., 2018, 36, 2583 Search PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks 3rd, A. D. Mackerell Jr, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14(33–38), 27–38 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03444h |
| This journal is © The Royal Society of Chemistry 2021 |