
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral, sequence-definable foldamer-derived macrocycles†
Toyah M. C.
Warnock
 a,
Sundaram
Rajkumar
b,
Matthew P.
Fitzpatrick
a,
Christopher J.
Serpell
c,
Paul
Dingwall
a and
Peter C.
Knipe
*a
a,
Sundaram
Rajkumar
b,
Matthew P.
Fitzpatrick
a,
Christopher J.
Serpell
c,
Paul
Dingwall
a and
Peter C.
Knipe
*a
aSchool of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Belfast, BT9 5AG, UK. E-mail: p.knipe@qub.ac.uk
bAlmac Group Ltd., 20 Seagoe Industrial Estate, Craigavon, BT63 5QD, UK
cSchool of Physical Sciences, University of Kent, Ingram Building, Canterbury, Kent CT2 7NH, UK
First published on 10th November 2021
Abstract
Nature's oligomeric macromolecules have been a long-standing source of inspiration for chemists producing foldamers. Natural systems are frequently conformationally stabilised by macrocyclisation, yet this approach has been rarely adopted in the field of foldamer chemistry. Here we present a new class of chiral cyclic trimers and tetramers formed by macrocyclisation of open-chain foldamer precursors. Symmetrical products are obtained via a [2 + 2] self-assembly approach, while full sequence control is demonstrated through linear synthesis and cyclisation of an unsymmetrical trimer. Structural characterisation is achieved through a combined X-ray and DFT approach, which indicates the tetramers adopt a near-planar conformation, while the trimers adopt a shallow bowl-like shape. Finally, a proof-of-concept experiment is conducted to demonstrate the macrocycles' capacity for cation binding.
Introduction
Nature frequently exploits macrocycles as functional molecules within living systems,1–3 and many of these and their derivatives have been exploited as therapeutics.4–6 Some of the most important drugs for human health are naturally-occurring macrocycles, including vancomycin and cyclosporin. The relative success of macrocyclic peptide drugs can be attributed to several factors including stability to degradation, cyclic constraint favouring the active conformation, and high membrane permeability. Inspired by macrocycles' use in Nature and potency in the clinic, chemists have developed numerous cyclisation strategies to stabilise the conformation of small bioactive peptides, including helix “stapling”7 and cross-linking to stabilise β-hairpin structures.8 Peptides have also been replaced entirely, with artificial folded molecules dubbed “foldamers”.9 Entirely abiotic macrocycles have also been used in applications outside biology, particularly in host-guest chemistry.10 Here, the pre-organisation of functionality within the macrocycle enables large binding constants and high guest-specificity.Foldamers have been developed to mimic peptide secondary11–14 and tertiary structures,15–17 and have shown promise as therapeutic agents in their own right.9 However, the field of foldamer-derived macrocycles – analogous to macrocyclic peptides – remains in its infancy. Extant foldamer-derived macrocycles typically display a large degree of symmetry,18–32 probably due to synthetic convenience (a notable exception is in mixed peptide/foldamer33–37 and peptoid38,39 systems). However, Nature demonstrates that diversity (rather than uniformity) of structure is crucial in enabling the use of macrocycles across a range of functions. In the past year the first two examples of sequence-defined abiotic foldamer-derived macrocycles have emerged from the laboratories of Flood,40 who reported cyclic carbazole-triazole trimers and Meisel and Hamilton41 who synthesised cavitand-like molecular containers (Fig. 1). Here, we present our synthesis of semi and fully sequence-definable abiotic foldamer-derived macrocycles possessing a homochiral backbone, allowing the defined positioning of sidechains in three-dimensions.
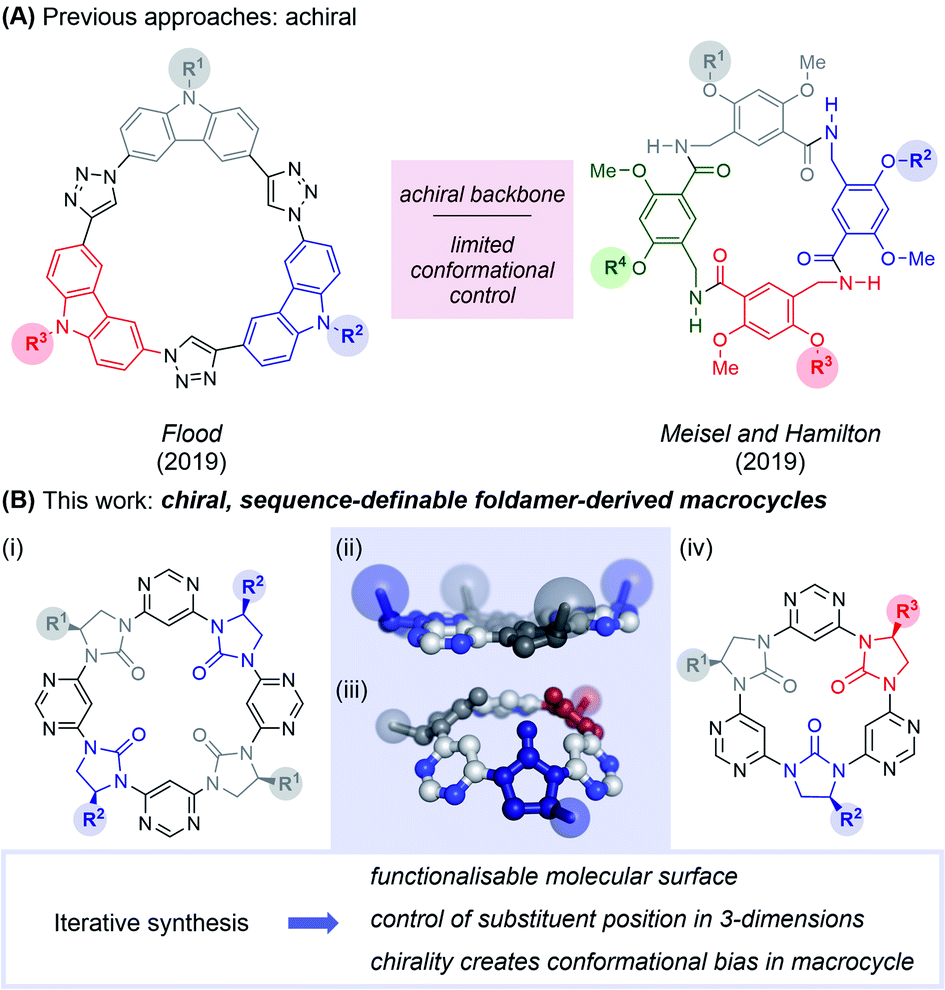 | ||
| Fig. 1 Sequence-defined chiral macrocyclic oligomers. (A) Recent examples of sequence-defined macrocyclic foldamers reported by Flood, and Hamilton and Meisel. The Flood macrocycles adopt an entirely planar conformation, forming 2-dimensional crystalline arrays, while those of Meisel and Hamilton adopt enantiomeric cavitand-like conformations under rapid exchange. (B) This study describes the synthesis, structure and cation-binding properties of a novel chiral foldamer-derived macrocycle. (i) General structure of tetrameric macrocycles; (ii) X-ray structure (top) and computed energy minimum (bottom) of tetrameric and trimeric macrocycles respectively; (iii) general structure of trimeric macrocycles. Sidechains are highlighted as coloured spheres. | ||
Results and discussion
Synthesis of macrocycles
We have previously described a foldamer architecture where dipolar repulsion between adjacent pyrimidine and imidazolidin-2-one components leads to structures with a turn per monomer of ∼86°.15 The attempted synthesis of related pyridine-linked macrocycles by Meth-Cohn failed entirely, generating only oligomeric material,42 but we hypothesised that the dipole-mediated pre-organisation in our putative linear precursor could favour macrocyclisation over oligomerisation. Similar preorganisation towards macrocyclisation has previously been exploited by Gong in the self-assembly of oligobenzamide foldamers.26,43 We tested this hypothesis by first synthesising linear dimer 2a by iterative deprotection/Buchwald–Hartwig coupling steps from a chiral, amino alcohol-derived monomeric precursor (see ESI† for procedures). Removal of the N-tert-butyl protecting group on 1a under acidic conditions afforded dimer 2a in 81% yield. Both the N-terminally protected and tert-butyl deprotected intermediates 1a and 2a were crystalline solids, with the latter purified conveniently by trituration with hot hexanes.Examination of their single crystal X-ray structures revealed that the expected dipole-opposed conformation was adopted, giving both molecules an overall crescent shape (Scheme 1B). We were therefore confident that treatment of dimer 2a under Buchwald–Hartwig cross-coupling conditions would lead directly to the C2-symmetrical macrocycle 3a, since the initial tetrameric product of homo-coupling would be pre-organised via dipolar repulsion for a second coupling reaction to generate the macrocycle. Thus, treatment of 2a with Pd2(dba)3, Xantphos and Cs2CO3 under reflux in toluene afforded macrocycle 3a in 56% isolated yield, with no evidence of the formation of larger macrocycles or polymeric material. With this synthetic strategy validated we proceeded to synthesise a further three dimeric precursor molecules 2b–d.44 Upon treatment under cross-coupling conditions all were converted to the corresponding C4-symmetrical macrocycles 3b–d in isolated yields of 61–85%.
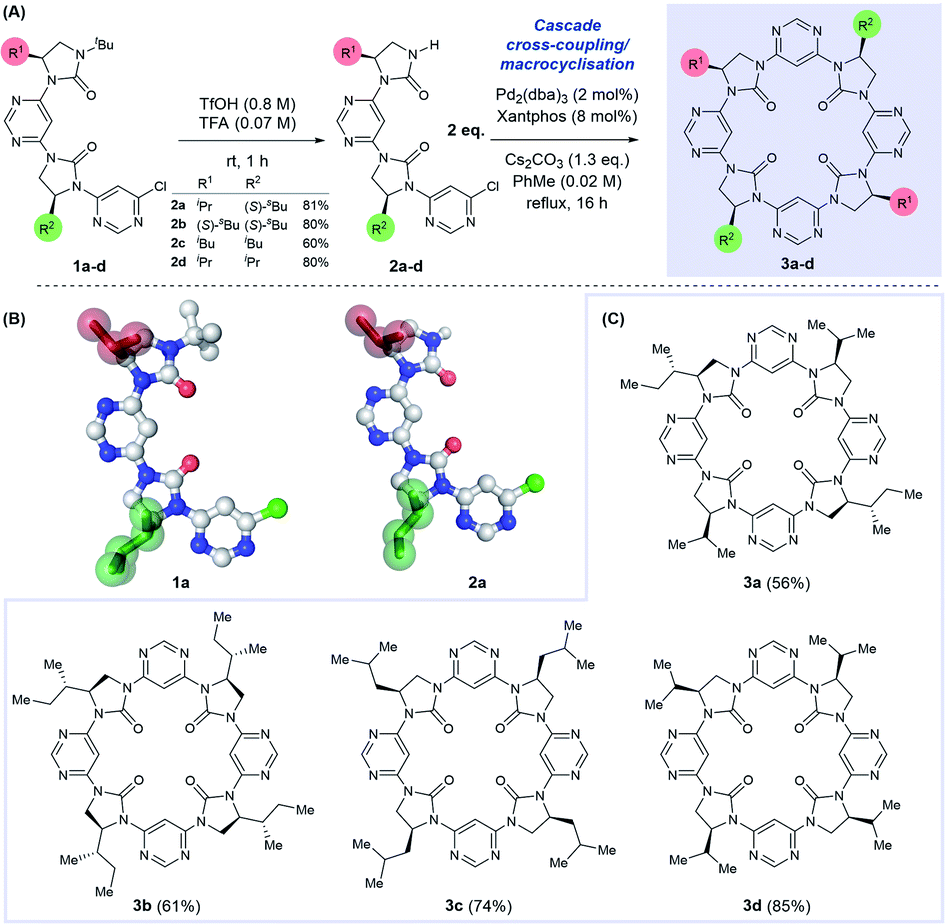 | ||
| Scheme 1 (A) Synthesis of tetrameric macrocycles via N-tBu deprotection of dimer 1a–d to afford 2a–d, and cascade dimerisation-cyclisation under Buchwald–Hartwig conditions. (B) Single crystal X-ray structures of 1a and 2a, sidechains are highlighted in green and red. (C) Synthetic scope of tetrameric macrocycle formation. | ||
To further test the limits of the macrocyclisation approach we synthesised linear trimers 4a and 4b (Scheme 2). While the cyclic tetramers were expected to be approximately planar and free from torsional strain, we anticipated that the trimers would be more strained, with the backbone forced out of the ideal planar conformation. To obtain the open-chain trimers it was necessary to add an additional pyrimidine and imidazolidin-2-one sub-unit to linear dimers 1c and 1e, which was achieved in four steps. In accordance with greater conformational strain, yields of the corresponding trimeric macrocycles were low relative to the tetrameric homologues, with 5a and 5b obtained in 6% and 29% yields respectively. Macrocycle 5b is especially noteworthy as it is entirely “sequence-defined”, with the cyclic ordering of the monomers predetermined by the order of monomer addition in the preparation of the linear precursor.
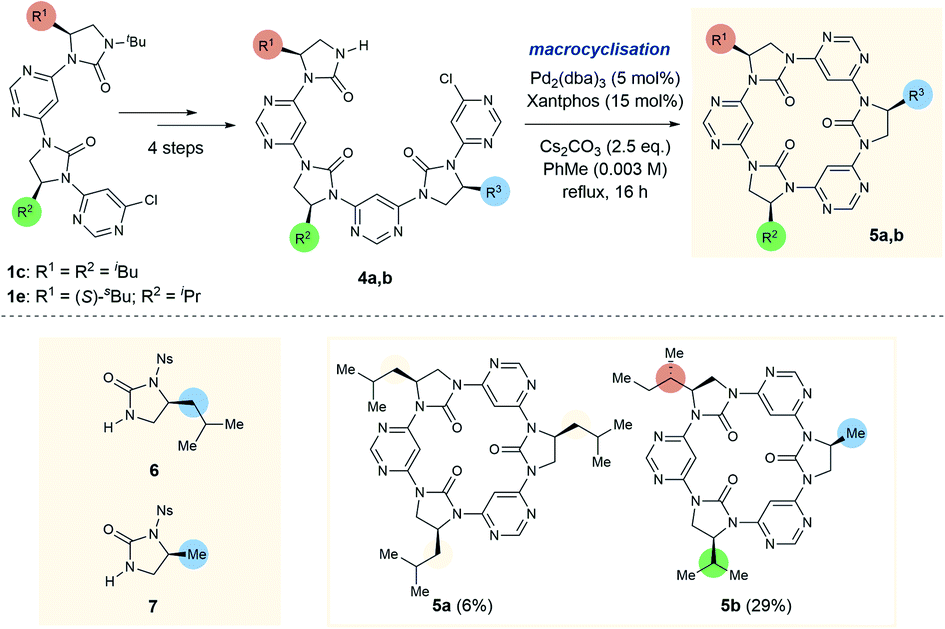 | ||
| Scheme 2 Synthesis of C3- (5a) and C1-symmetrical (5b) trimeric macrocycles. Synthesis of4a: (1) 6 (1.3 eq.), Pd2(dba)3 (5 mol%), Xantphos (15 mol%), K2CO3 (2.5 eq.), PhMe, reflux, 16 h, 73% brsm; (2) PhSH (1.5 eq.), K2CO3 (3 eq.), N,N-DMF, rt, 3 h, 90%; (3) 4,6-dichloropyrimidine (9 eq.), Pd2(dba)3 (5 mol%), Xantphos (15 mol%), K2CO3 (2.5 eq.), PhMe, reflux, 16 h, 62%; (4) TfOH:TFA 4:1 v:v, rt, 1 h, >99%. Synthesis of4b: (5) 7 (1.5 eq.), Pd2(dba)3 (5 mol%), Xantphos (15 mol%), K2CO3 (2.5 eq.), reflux, 16 h, 95%; (6) as step 2, 52%; (7) as step 3, 90%; (8) as step 4, >99%. 5b is an example of an entirely sequence-defined chiral foldamer-derived macrocycle. | ||
Conformational studies
In an effort to understand its conformational behavior, single crystals of macrocycle 3a were grown by vapour diffusion (Fig. 2A). Attempts to generate diffraction-quality crystals of the remaining macrocycles were unsuccessful. As expected, all four sidechains of 3a are projected from a single face of the macrocycle in a highly controlled manner. Inspection of the crystal packing reveals a layered, back-to-back stacking arrangement between planes of macrocycles (Fig. 2B–D), with the relative orientation of macrocycles between layers controlled in part by a dipole-opposed arrangement between adjacent imidazolidin-2-ones (Fig. 2C). Due to disorder and weak diffraction the X-ray data were insufficient to gain further structural insights, so we proceeded to explore the macrocycles' conformational behavior by DFT (Fig. 3).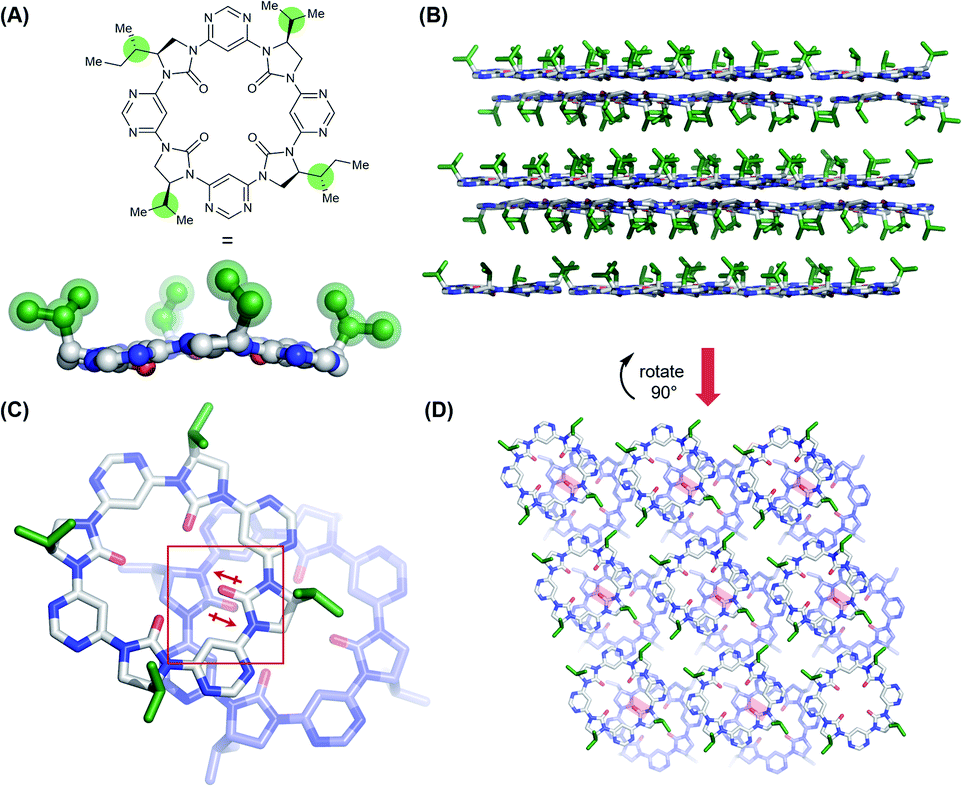 | ||
| Fig. 2 Single crystal X-ray structure of C2-symmetrical macrocycle 3a. Sidechains are highlighted in green. Sidechains cannot be identified beyond the γ-position due to disorder, so all are truncated to isopropyl. Solvent molecules and hydrogen atoms are omitted for clarity. (A) Single molecule view, showing controlled projection of side-chains from a single face; (B) crystal packing arrangement (viewed along crystalline b-axis) showing planes of macrocycles arranged back-to-back, with side-chains projected into the interplanar void; (C) stacking arrangement between macrocycles in adjacent planes (top plane – white; bottom plane – purple) displaying a dipole-opposed orientation (dipoles indicated in red); (D) crystal packing arrangement (viewed along the crystalline a-axis) showing the offset arrangement between adjacent planes of macrocycles (top plane – white; bottom plane –purple). Dipole-opposed interactions are indicated by red boxes. | ||
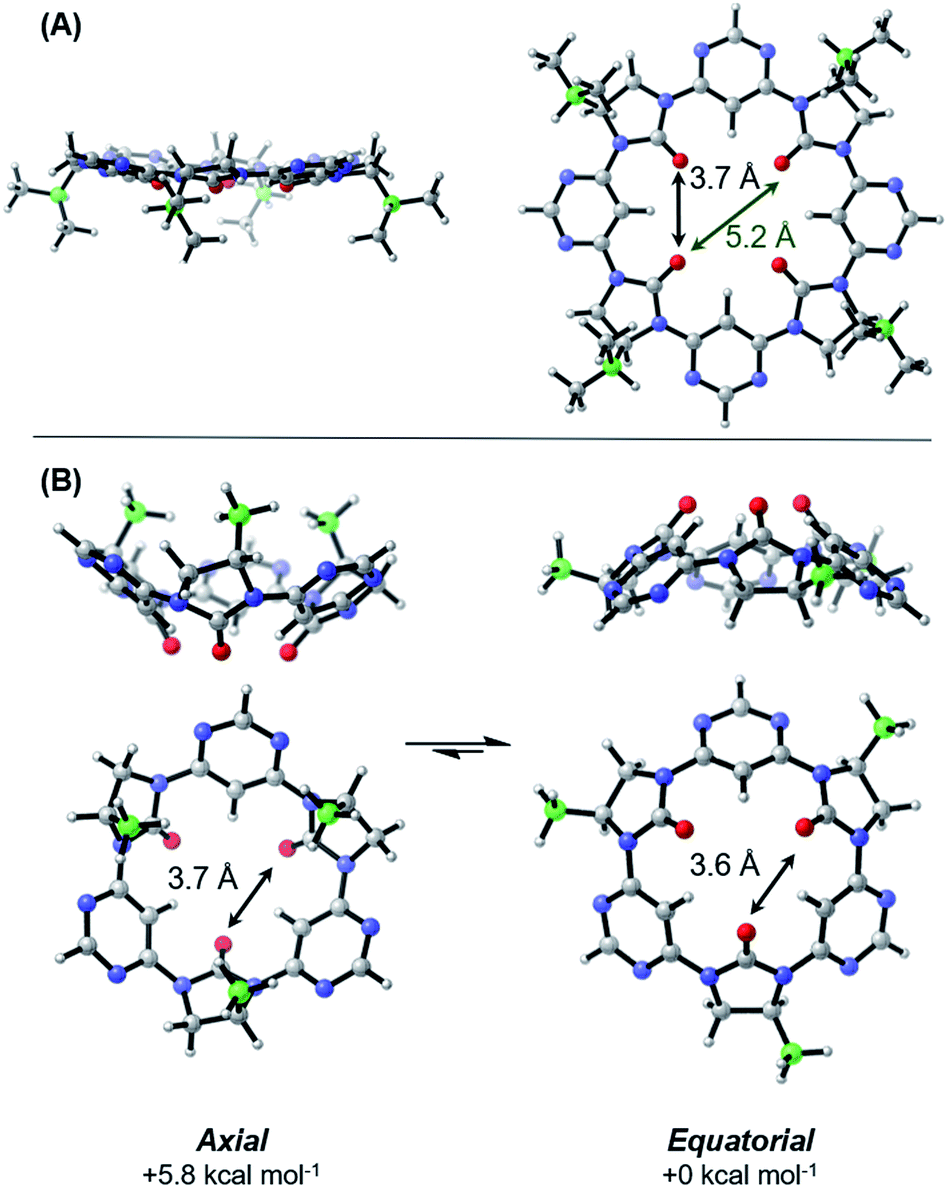 | ||
| Fig. 3 Computed lowest energy conformations of tetrameric (A) and trimeric (B) macrocycles. Averaged O–O interatomic distances are indicated. Level of theory: B3LYP/6-311+G(d,p)/GD3BJ/PCMToluene//B3LYP/6-31G(d,p)/GD3BJ. Isopropyl and methyl sidechains are shown for the tetramer and trimer respectively. | ||
The lowest energy conformation of the tetramer was found to be a shallow bowl, with all C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups puckered outwards from the same face of the macrocycle, and a transverse O–O distance of 5.2 Å. Two low energy conformers were identified for the trimer, corresponding to a macromolecular “ring-flip”, in which the sidechain substituents are placed in pseudo-axial or pseudo-equatorial positions (Fig. 3B). The equatorial conformer is lower in energy by 5.8 kcal mol−1 and represents the global energy minimum. No “mixed” conformers (in which one imidazolidin-2-one is puckered in an opposing direction to the others) are identified as minima. The less-planar structure of the trimer relative to the tetramer is also supported by 1H NMR data: the inward-pointing pyrimidine hydrogens in tetramers 3a–3d appear far downfield (∼9.9 ppm), likely due to the deshielding effect of the proximal oxygen lone-pairs, whereas the equivalent peaks in the trimers 5a and 5b appear at ∼8.7 ppm indicating that these hydrogens are subject to the deshielding effect of the lone pairs to a much lesser degree.
O groups puckered outwards from the same face of the macrocycle, and a transverse O–O distance of 5.2 Å. Two low energy conformers were identified for the trimer, corresponding to a macromolecular “ring-flip”, in which the sidechain substituents are placed in pseudo-axial or pseudo-equatorial positions (Fig. 3B). The equatorial conformer is lower in energy by 5.8 kcal mol−1 and represents the global energy minimum. No “mixed” conformers (in which one imidazolidin-2-one is puckered in an opposing direction to the others) are identified as minima. The less-planar structure of the trimer relative to the tetramer is also supported by 1H NMR data: the inward-pointing pyrimidine hydrogens in tetramers 3a–3d appear far downfield (∼9.9 ppm), likely due to the deshielding effect of the proximal oxygen lone-pairs, whereas the equivalent peaks in the trimers 5a and 5b appear at ∼8.7 ppm indicating that these hydrogens are subject to the deshielding effect of the lone pairs to a much lesser degree.
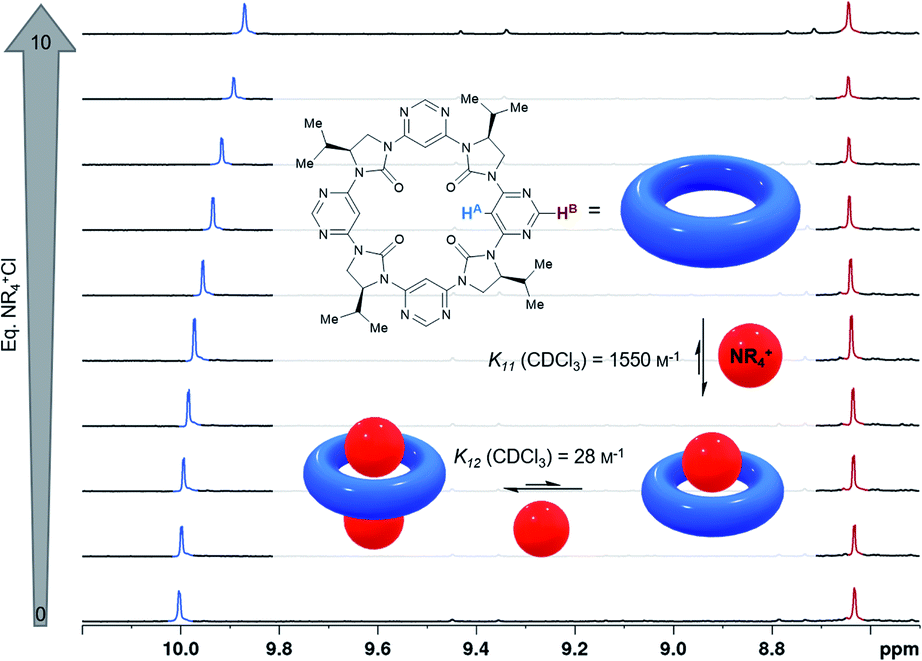 | ||
| Fig. 4 Binding of hexadecyltrimethylammonium chloride (depicted as a red sphere) by C4-symmetrical macrocycle 3d (blue torus) determined by 1H NMR titration (CDCl3, 600 MHz). Atoms HA and HB are highlighted and the corresponding 1H NMR signals are indicated in blue and red respectively. For full details refer to the ESI †. | ||
Lastly, an attempt was made to achieve diasteroselective recognition of a chiral guest using dibenzoyl tartaric acid, chosen for its solubility in CDCl3, availability in both enantiomeric forms, and ability to form hydrogen bonds to the acceptor-rich environment of the macrocycle interior. However, no difference in binding was observed between enantiomers (see ESI†). We attribute this to the ability of guests to bind the unsubstituted face of the macrocycles, where little stereo-differentiation is feasible; studies are ongoing to synthesise macrocycles incorporating substituents on both faces to address this issue, either by use of C2-symmetrical 1,2-disubstituted diamines, or through the use of alternating (R)- and (S)-configured imidazolidine-2-ones in adjacent monomers.
Conclusions
To summarise, we have synthesised and examined the properties of six chiral foldamer-derived macrocycles. The strategy enables their synthesis in entirely sequence-defined manner when required, through synthesis of a linear precursor, or in a convenient semi-defined manner through generation of linear dimers which undergo spontaneous cyclisation upon coupling to form the tetramer. DFT indicates the trimeric macrocycles adopt a bowl-like shape with substituents in a pseudo-equatorial position, while tetramers adopt a planar conformation, as demonstrated by single crystal X-ray diffraction, and bind metal and ammonium cations. The high level of conformational control means these macrocycles are an excellent platform for the controlled positioning of sidechain groups. Current work is ongoing to examine applications of the macrocycles in molecular recognition and catalysis, and to develop a solid-supported second-generation synthetic approach.Data availability
Crystallographic data for 1a, 2a, 2d and 3a has been deposited at the CCDC under accession numbers 2057484, 2057483, 2057482 and 2057486 respectively, and can be obtained from http://www.ccdc.cam.ac.uk. DFT data for this paper can be found at https://pure.qub.ac.uk with DOI: 10.17034/8953afcf-c4b8-41f4-8b4b-176fd07a8956.Author contributions
TMCW: data curation; formal analysis; investigation; methodology; validation; visualization; writing – original draft; writing – review & editing. SR: data curation; formal analysis; investigation; methodology; writing – review & editing. MPF: investigation; writing – review & editing. CJS: formal analysis (XRD); writing – review & editing; PD: formal analysis; investigation (DFT); visualization; writing – review & editing. PCK: conceptualization; data curation; formal analysis; funding acquisition; methodology; project administration; resources; supervision; visualization; writing – original draft; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from Queen’s University Belfast (PCK, TW), the Northern Ireland Department for the Economy (TW) and the EPSRC (EP/R021481/1, PCK, SR). LCMS facilities were funded through an EPSRC block equipment grant for early career researchers (EP/S018077/1). We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data for 3a.52Notes and references
- L. A. Wessjohann, R. Bartelt and W. Brandt, in Practical Medicinal Chemistry with Macrocycles, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2017, pp. 77–100 Search PubMed.
- D. J. Newman and G. M. Cragg, in Macrocycles in Drug Discovery, ed. J. Levin, The Royal Society of Chemistry, 2015, pp. 1–36 Search PubMed.
- C. T. Walsh, ACS Infect. Dis., 2018, 4, 1283–1299 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS PubMed.
- A. K. Yudin, Chem. Sci., 2015, 6, 30–49 RSC.
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- L. K. Henchey, A. L. Jochim and P. S. Arora, Curr. Opin. Chem. Biol., 2008, 12, 692–697 CrossRef CAS PubMed.
- N. Sawyer and P. S. Arora, ACS Chem. Biol., 2018, 13, 2027–2032 CrossRef CAS PubMed.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- M. K. P. P. Jayatunga, S. Thompson and A. D. Hamilton, Bioorg. Med. Chem. Lett., 2014, 24, 717–724 CrossRef CAS PubMed.
- W. A. Loughlin, J. D. A. Tyndall, M. P. Glenn and D. P. Fairlie, Chem. Rev., 2004, 104, 6085–6118 CrossRef CAS PubMed.
- T. Yamashita, P. C. Knipe, N. Busschaert, S. Thompson and A. D. Hamilton, Chem.–Eur. J., 2015, 21, 14699–14702 CrossRef CAS PubMed.
- E. A. German, J. E. Ross, P. C. Knipe, M. F. Don, S. Thompson and A. D. Hamilton, Angew. Chem., Int. Ed., 2015, 54, 2649–2652 CrossRef CAS PubMed.
- Z. Lockhart and P. C. Knipe, Angew. Chem., Int. Ed., 2018, 57, 8478–8482 CrossRef CAS PubMed.
- Z. E. Reinert, G. A. Lengyel and W. S. Horne, J. Am. Chem. Soc., 2013, 135, 12528–12531 CrossRef CAS PubMed.
- T. W. Craven, M.-K. Cho, N. J. Traaseth, R. Bonneau and K. Kirshenbaum, J. Am. Chem. Soc., 2016, 138, 1543–1550 CrossRef CAS PubMed.
- F. J. Carver, C. A. Hunter and R. J. Shannon, J. Chem. Soc., Chem. Commun., 1994, 1277–1280 RSC.
- L. He, Y. An, L. Yuan, K. Yamato, W. Feng, O. Gerlitz, C. Zheng and B. Gong, Chem. Commun., 2005, 1011, 3788–3790 RSC.
- L. Xing, U. Ziener, T. C. Sutherland and L. A. Cuccia, Chem. Commun., 2005, 5751–5753 RSC.
- J. K. H. Hui and M. J. MacLachlan, Chem. Commun., 2006, 1, 2480–2482 RSC.
- Z. J. Kinney and C. S. Hartley, J. Am. Chem. Soc., 2017, 139, 4821–4827 CrossRef CAS PubMed.
- Z. J. Kinney, V. C. Kirinda and C. S. Hartley, Chem. Sci., 2019, 10, 9057–9068 RSC.
- H. Juwarker, J. M. Suk and K. S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC.
- V. C. Kirinda, B. R. Schrage, C. J. Ziegler and C. S. Hartley, Eur. J. Org. Chem., 2020, 2020, 5620–5625 CrossRef CAS.
- L. Yuan, W. Feng, K. Yamato, A. R. Sanford, D. Xu, H. Guo and B. Gong, J. Am. Chem. Soc., 2004, 126, 11120–11121 CrossRef CAS PubMed.
- A. Gube, H. Komber, K. Sahre, P. Friedel, B. Voit and F. Böhme, J. Org. Chem., 2012, 77, 9620–9627 CrossRef CAS PubMed.
- F. Böhme, M. Rillich and H. Komber, Macromol. Chem. Phys., 1995, 196, 3209–3216 CrossRef.
- F. Böhme, C. Kunert, H. Komber, D. Voigt, P. Friedel, M. Khodja and H. Wilde, Macromolecules, 2002, 35, 4233–4237 CrossRef.
- H. Jiang, J. M. Léger, P. Guionneau and I. Huc, Org. Lett., 2004, 6, 2985–2988 CrossRef CAS PubMed.
- A. Zhang, Y. Han, K. Yamato, X. C. Zeng and B. Gong, Org. Lett., 2006, 8, 803–806 CrossRef CAS PubMed.
- S. Akine, T. Taniguchi and T. Nabeshima, Tetrahedron Lett., 2001, 42, 8861–8864 CrossRef CAS.
- R. Spencer, K. H. Chen, G. Manuel and J. S. Nowick, Eur. J. Org. Chem., 2013, 3523–3528 CrossRef CAS.
- C. A. Olsen, A. Montero, L. J. Leman and M. R. Ghadiri, ACS Med. Chem. Lett., 2012, 3, 749–753 CrossRef CAS PubMed.
- J. M. Rogers, S. Kwon, S. J. Dawson, P. K. Mandal, H. Suga and I. Huc, Nat. Chem., 2018, 10, 405–412 CrossRef CAS PubMed.
- J. D. Northrup, G. Mancini, C. R. Purcell and C. E. Schafmeister, J. Org. Chem., 2017, 82, 13020–13033 CrossRef CAS PubMed.
- A. Hennig, L. Fischer, G. Guichard and S. Matile, J. Am. Chem. Soc., 2009, 131, 16889–16895 CrossRef CAS PubMed.
- J. A. Schneider, T. W. Craven, A. C. Kasper, C. Yun, M. Haugbro, E. M. Briggs, V. Svetlov, E. Nudler, H. Knaut, R. Bonneau, M. J. Garabedian, K. Kirshenbaum and S. K. Logan, Nat. Commun., 2018, 9, 4396 CrossRef PubMed.
- A. M. Webster and S. L. Cobb, Chem.–Eur. J., 2018, 24, 7560–7573 CrossRef CAS PubMed.
- J. R. Dobscha, H. D. Castillo, Y. Li, R. E. Fadler, R. D. Taylor, A. A. Brown, C. Q. Trainor, S. L. Tait and A. H. Flood, J. Am. Chem. Soc., 2019, 141, 17588–17600 CrossRef CAS PubMed.
- J. W. Meisel, C. T. Hu and A. D. Hamilton, Org. Lett., 2019, 21, 7763–7767 CrossRef CAS PubMed.
- O. Meth-Cohn and Z. Yan, J. Chem. Soc., Perkin Trans., 1998, 1, 423–436 RSC.
- V. Martí-Centelles, M. D. Pandey, M. I. Burguete and S. V. Luis, Chem. Rev., 2015, 115, 8736–8834 CrossRef PubMed.
- The purification of 3a was initially hampered by the presence of an additional impurity (after trituration) which by 1H NMR appeared to have the symmetry characteristic of the macrocyclic product. ESI LC-MS suggested this impurity may be the macrocycle-Cs+ complex, and it was cleanly converted to the neutral “vacant” macrocycle by extensive washing with water. This raises the prospect that Cs+ plays a role in templating the overall cyclization. Meth-Cohn attempted the intramolecular SNAr cyclization of an analogous pyridine-imidazolidinone oligomer (where dipole effects would favour the extended conformation in which the termini are not in close proximity), and this reaction failed in the presence of CsF.42 In our case, the dipole-opposed conformation and any metal templation would act in unison (both favouring the compact conformation with the termini in close proximity), whilst in the work of Meth-Cohn the effects are in opposition. The failure of their system to cyclise suggests that any metal templation by coordination to the imidazolidinones is insufficient in strength to overturn the dipole-enforced innate conformational preference of the linear precursor.
- H. J. Buschmann, L. Mutihac and K. Jansen, J. Inclusion Phenom., 2001, 39, 1–11 CrossRef CAS.
- S. Lambert, K. Bartik and I. Jabin, J. Org. Chem., 2020, 85, 10062–10071 CrossRef CAS PubMed.
- M. A. Gamal-Eldin and D. H. Macartney, Org. Biomol. Chem., 2013, 11, 488–495 RSC.
- This guest was selected since it displays good solubility in chloroform, and the host macrocycles are poorly soluble in non-chlorinated solvents.
- D. J. Cram, Science, 1988, 240, 760–767 CrossRef CAS PubMed.
- C. E. Cannizzaro and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 7163–7169 CrossRef CAS PubMed.
- S. Kubik, J. Am. Chem. Soc., 1999, 121, 5846–5855 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2057482, 2057483, 2057484 and 2057486. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05021d |
| This journal is © The Royal Society of Chemistry 2021 |