
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural resolution and mechanistic insight into hydrogen adsorption in flexible ZIF-7†
Ryan A.
Klein
 ab,
Sarah
Shulda
a,
Philip A.
Parilla
a,
Pierre
Le Magueres
c,
Rachelle K.
Richardson
d,
William
Morris
d,
Craig M.
Brown
be and
C. Michael
McGuirk
*f
ab,
Sarah
Shulda
a,
Philip A.
Parilla
a,
Pierre
Le Magueres
c,
Rachelle K.
Richardson
d,
William
Morris
d,
Craig M.
Brown
be and
C. Michael
McGuirk
*f
aMaterial, Chemical, and Computational Sciences Directorate, National Renewable Energy Laboratory, Golden, Colorado 80401, USA
bCenter for Neutron Research, National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA
cRigaku Americas Corporation, 9009 New Trails Drive, The Woodlands, TX 77381, USA
dNuMat Technologies, 8025 Lamon Avenue, Skokie, Illinois 60077, USA
eDepartment of Chemical and Biomolecular Engineering, University of Delaware, Newark, Delaware 19716, USA
fDepartment of Chemistry, Colorado School of Mines, Golden, Colorado 80401, USA. E-mail: cmmcguirk@mines.edu
First published on 24th November 2021
Abstract
Flexible metal–organic frameworks offer a route towards high useable hydrogen storage capacities with minimal swings in pressure and temperature via step-shaped adsorption and desorption profiles. Yet, the understanding of hydrogen-induced flexibility in candidate storage materials remains incomplete. Here, we investigate the hydrogen storage properties of a quintessential flexible metal–organic framework, ZIF-7. We use high-pressure isothermal hydrogen adsorption measurements to identify the pressure–temperature conditions of the hydrogen-induced structural transition in ZIF-7. The material displays narrow hysteresis and has a shallow adsorption slope between 100 K and 125 K. To gain mechanistic insight into the cause of the phase transition correlating with stepped adsorption and desorption, we conduct powder neutron diffraction measurements of the D2 gas-dosed structures at conditions across the phase change. Rietveld refinements of the powder neutron diffraction patterns yield the structures of activated ZIF-7 and of the gas-dosed material in the dense and open phases. The structure of the activated phase of ZIF-7 is corroborated by the structure of the activated phase of the Cd congener, CdIF-13, which we report here for the first time based on single crystal X-ray diffraction measurements. Subsequent Rietveld refinements of the powder patterns for the gas-dosed structure reveal that the primary D2 adsorption sites in the dense phase form D2–arene interactions between adjacent ligands in a sandwich-like adsorption motif. These sites are prevalent in both the dense and the open structure for ZIF-7, and we hypothesize that they play an important role in templating the structure of the open phase. We discuss the implications of our findings for future approaches to rationally tune step-shaped adsorption in ZIF-7, its congeners, and flexible porous adsorbents in general. Lastly, important to the application of flexible frameworks, we show that pelletization of ZIF-7 produces minimal variation in performance.
Introduction
Hydrogen is emerging as a globally viable renewable fuel source. However, the widespread adaptation of hydrogen as a fuel is hindered by the intensive and expensive conditions required for transport, storage, and delivery. These processes currently rely on compression (700 bar) or liquification (20 K) to achieve appreciable volumetric energy capacities.1 One alternative method for storage is the use of porous adsorbates. Among porous adsorbates, metal–organic frameworks (MOFs) stand out as extremely promising for hydrogen storage applications.2–8 MOFs are porous crystalline materials composed of polytopic organic linkers bridging inorganic nodes. Some MOFs display framework flexibility.9 These are known as soft porous crystals, or flexible MOFs.10 Flexible MOFs undergo temperature-influenced pressure-induced reversible phase transitions between an activated, dense phase with reduced accessible porosity and a gas-dosed, open phase with greater accessible porosity.11–15 This behavior may manifest as a “step” in gas adsorption at the phase transition pressure (Fig. 1, blue curve).16,17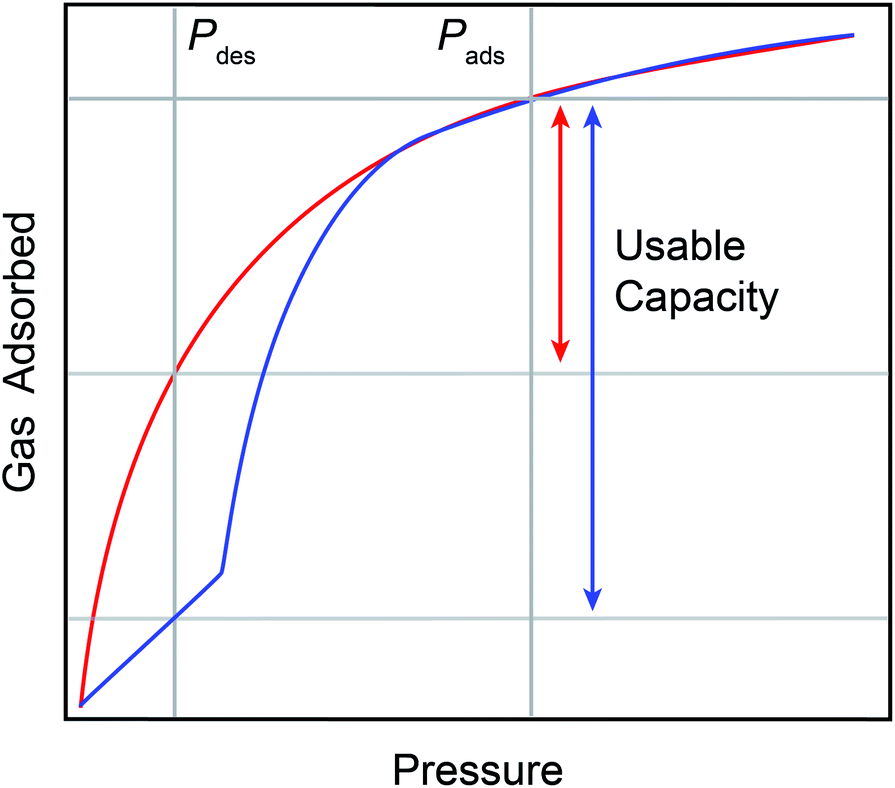 | ||
| Fig. 1 An idealized gas adsorption isotherm plot showing the difference between a type I isotherm (red trace) and a type V isotherm (blue trace). Type V isotherms promote larger usable capacities (blue arrow) versus type I isotherms (red arrow) over the same pressure interval in a pressure swing process, as indicated by Pads and Pdes in the figure. | ||
For storage and delivery applications, the stepped nature of both adsorption and desorption in some flexible materials presents key advantages over rigid materials with type I sorption (Fig. 1, red curve). First, the delivery of gas adsorbed across the step can be achieved with small changes in pressure or temperature, accessing enhanced useable capacities with minimal energetic input (Fig. 1). Second, framework flexibility obviates the need for precise tailoring of isosteric heats of adsorption via synthetic tuning of the gas adsorption sites needed to optimize the usable capacity for a material with type I sorption.18 Third, flexible MOFs can leverage the thermodynamics of the pressure-induced phase change to assist with thermal management during exothermic adsorption and endothermic desorption.19–21 Lastly, the pressure–temperature conditions at which the adsorption and desorption step occurs is synthetically tunable, presenting a path for minimizing energy consumption during pressure and temperature swing processes for a given application.22,23 However, the precise synthetic handles which can be used to rationally tune the step pressure are not yet fully understood, precluding informed adaptation of known flexible frameworks for storage applications. This is of particular importance as the discovery of flexible MOFs remains serendipitous. As such, in situ structural studies are needed to help clarify the mechanisms of hydrogen-induced phase changes in flexible MOFs.
The tunability of the step conditions in flexible MOFs has been investigated in several systems, including MIL-53 and its derivatives, Co(1,4-benzenedipyrazolate) (Co(bdp)), DUT-49,24 and in zeolitic imidazolate frameworks (ZIFs) for a variety of gasses. Among these, the best understood flexible MOF may be MIL-53(M) and its derivatives (M = Al3+, Cr3+, Fe3+).25–27 The breathing modes in MIL-53 have been thoroughly investigated, and the ‘accordion’ type mechanism for framework flexibility has been shown to depend on the identities of the metal centers,28–30 substituents on the ligands,31 and the adsorbate species.25,32,33 However, our understanding of the mechanism underlying flexibility remains incomplete, especially when the guest molecule is H2 or D2.34–36 Moreover, there are only a handful of examples of materials which display stepped hydrogen adsorption and desorption in the pressure regime relevant for storage applications-yet these operate at 87 K or colder.26,37–41 The most notable of these is Co(bdp),22,42–46 which also flexes following an ‘accordion’ type mechanism. Co(bdp) displays stepped hydrogen adsorption at cryogenic temperatures with large hysteresis.46 Indeed, no existing flexible framework is ideally suited for hydrogen storage and delivery applications at application-relevant temperature–pressure conditions. Therefore, an enhanced understanding of the structural causes of hydrogen-induced phase changes is needed to enable rational synthetic tuning of the pressure and temperature step conditions in candidate storage materials.8,47,48
To enhance our structural understanding of hydrogen-induced phase changes in porous frameworks we turned to another archetypal flexible material, ZIF-7 (Zn(benzimidazolate)2, SOD, ZnC14H10N4), in which ditopic benzimidazolate linkers bridge tetrahedrally coordinated Zn(II) sites.23,49,50 The water-solvated phase of ZIF-7 crystallizes in the trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) space group (Fig. 2).49,51 The high symmetry water-solvated phase contains three unique rings (Fig. 2b–d), which connect to form a sodalite-like topology (Fig. 2a). Upon activation (i.e. solvent removal) the framework undergoes a structural transition to a presumed dense phase, as evidenced by powder X-ray diffraction (PXRD) studies.23,52 Subsequent gas dosing of the activated phase reverts the framework to a higher symmetry, more porous structure, with step-shaped adsorption–desorption reported for CO2, CH4, and C2–C3 olefins and paraffins.23,52,53 The observed phase change is hypothesized to occur through a complex combination of distortions of the coordination skeleton (Fig. 2a) and ligand rotations about the Zn–N⋯N–Zn axes, producing a pore-gating mechanism with a relatively small change in volume.
space group (Fig. 2).49,51 The high symmetry water-solvated phase contains three unique rings (Fig. 2b–d), which connect to form a sodalite-like topology (Fig. 2a). Upon activation (i.e. solvent removal) the framework undergoes a structural transition to a presumed dense phase, as evidenced by powder X-ray diffraction (PXRD) studies.23,52 Subsequent gas dosing of the activated phase reverts the framework to a higher symmetry, more porous structure, with step-shaped adsorption–desorption reported for CO2, CH4, and C2–C3 olefins and paraffins.23,52,53 The observed phase change is hypothesized to occur through a complex combination of distortions of the coordination skeleton (Fig. 2a) and ligand rotations about the Zn–N⋯N–Zn axes, producing a pore-gating mechanism with a relatively small change in volume.
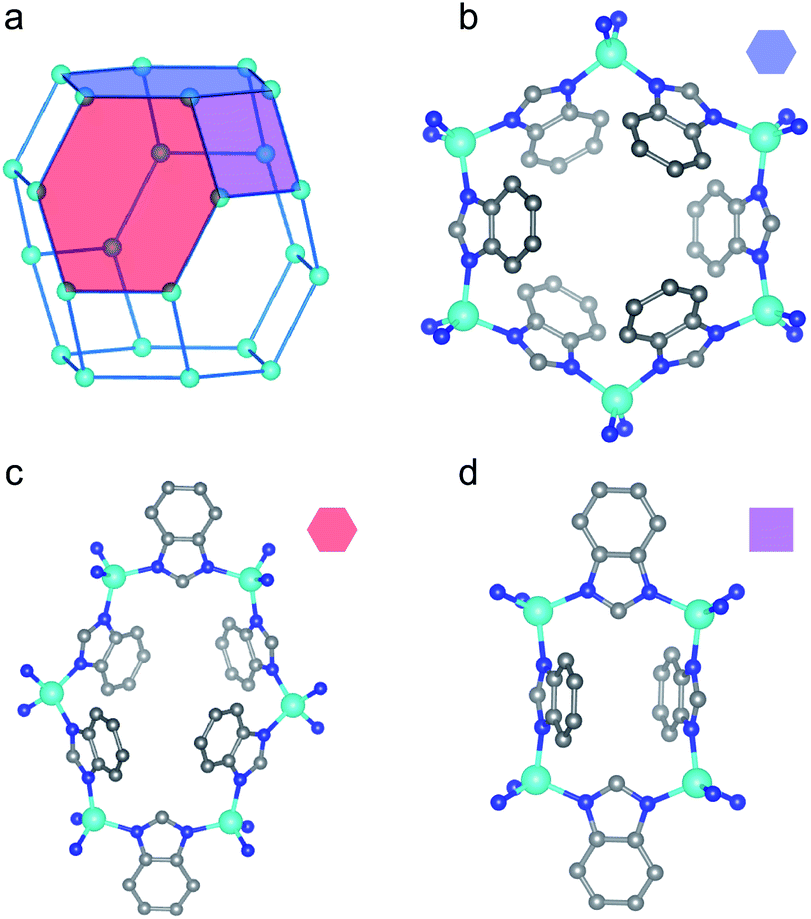 | ||
| Fig. 2 Portions of the water-solvated structure in the R space group, adapted from ref. 49, are shown here. (a) The 4- and 6-membered rings comprising the skeleton of the material connect to form a sodalite-like topology. The distinct rings in the structure are identified with colored polygons. (b) The first 6-membered ring is denoted by the blue hexagon. (c) The second 6-membered ring is denoted by the red hexagon. (d) The 4-membered ring is denoted by the purple rectangle. Teal, blue, and gray spheres depict Zn, N, and C atoms, respectively, while H atoms are omitted for clarity. | ||
While ZIF-7 is known to be flexible,23,52,54–57 stepped adsorption–desorption of hydrogen has not been observed, possibly due to the expected combination of high pressures and low temperatures needed to induce the phase change.58 Theoretical calculations predict relatively strong adsorption of hydrogen in ZIF-7 (ca. −10 kJ mol−1) for multiple calculated adsorption sites,59 although these calculations only investigate adsorption in the high-symmetry open phase and do not shed light on possible adsorption sites in the dense phase or on the phase change itself. This is largely because there are open questions surrounding the structure of the activated phase, which has been reported to possess a lower density than the water-solvated, open phase in the sodalite-like topology.60,61 This counterintuitive change in density contrasts with theoretical predictions, suggesting that the reported structure may be incorrect.23,62,63 A consequence of this possibly incorrect structure in the literature is that subsequent studies based on this structure may also lead to erroneous conclusions. As such, corroborating the existent activated structure—or correctly structurally characterizing the activated phase—and identifying the adsorption sites in the dense and open phases of ZIF-7 are critical for understanding the mechanism driving hydrogen-induced phase changes in this flexible MOF.
Here, we report the step-shaped adsorption and minimally hysteretic desorption of hydrogen in ZIF-7 using high pressure gas adsorption isothermal measurements conducted on phase-pure polycrystalline powder samples of ZIF-7 synthesized following established procedures22 (see ESI†). To the best of our knowledge, these measurements represent the first observation of step-shaped hydrogen adsorption–desorption in a flexible porous adsorbate above 87 K at operationally-relevant pressures. We then employ powder neutron diffraction (PND) measurements to investigate the structure of gas-dosed ZIF-7 across the phase transition. To investigate the structure of the activated phase of ZIF-7, we determined the crystal structure of the Cd congener of ZIF-7, CdIF-13, based on single crystal X-ray diffraction (SCXRD) measurements. The unit cell parameters found from the single crystal analysis of this material enabled successful Pawley fitting of the PND pattern for the activated phase of ZIF-7. Subsequent Rietveld refinements of the PND pattern yielded the crystal structure of activated ZIF-7. We show that the updated structural model of activated ZIF-7 is distinct from the existing model in the literature, and in doing so we resolve the apparent discrepancy in the densities of the activated and solvated phases. Using the sorption measurements as a guide, we then traverse the pressure–temperature conditions of the phase transition by conducting isobaric, variable-temperature PND measurements at ≈30 bar D2 between ≈77 K and ≈200 K. Based on Rietveld refinements of these patterns, we identify the adsorption sites before and after the phase change. By combining isothermal gas adsorption measurements, which provide a macroscopic understanding of the phase change, with the crystallographic probe of PND measurements, we thoroughly characterize the structural phase transition in ZIF-7 that occurs in response to increasing hydrogen pressure. The structural insights gained from our measurements help clarify the mechanism for structural flexibility in ZIF-7, which can be extended to give general insight into framework flexibility in MOFs.
Results and discussion
Hydrogen adsorption measurements
We conducted gas adsorption–desorption isothermal measurements to identify the precise pressure–temperature conditions at which hydrogen induces a phase change in ZIF-7. Isothermal measurements collected between 77 K to 303 K are provided in Fig. 3 and S2 in the ESI† document. A step, or change in slope, in the isothermal adsorption and desorption at 100 K, 110 K, and 125 K is readily observable below 105 bar, indicating a pressure-induced phase change to a phase with more accessible porosity during adsorption and less accessible porosity during desorption. The closed hysteresis loop near the onset of the adsorption step provides evidence for complete reversibility. At higher temperatures, 150–303 K, no step is observed below the accessible upper pressure limit, indicating that a phase change has not occurred under these specific pressure–temperature conditions, following the trend of increasing step pressure threshold with increasing temperature observed between 100 K and 125 K. At 77 K, the pressure threshold for the onset of the phase change occurs below 3 bar, precluding observation of the pre-step regime in the dense, low porosity phase at this temperature.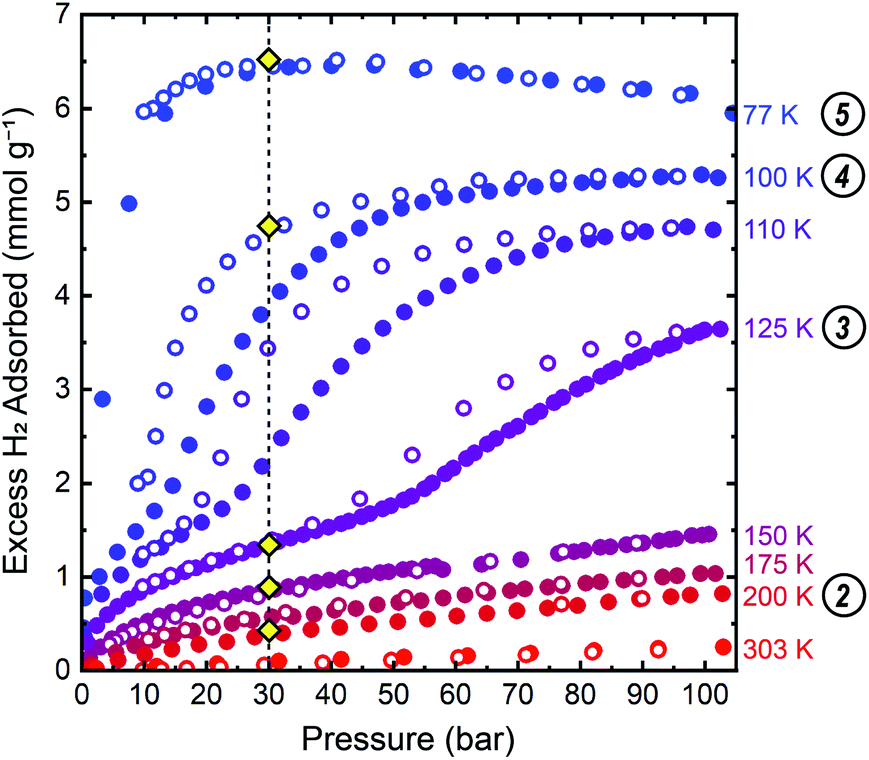 | ||
| Fig. 3 Isothermal hydrogen adsorption–desorption data up to 105 bar for ZIF-7 show stepped adsorption and desorption with minimal hysteresis between 100 K and 125 K. Closed and open symbols represent data collected upon adsorption and desorption, respectively. The vertical dashed line at 30 bar is a guide for the eye denoting the target pressure used in the PND measurements. Gold diamond symbols mark the approximate conditions at which PND patterns were collected. Circled numbers to the right of the temperatures correspond to the structure numbering used in Fig. 3–5, and throughout the text. | ||
A single-site Freundlich–Langmuir model was used to fit the data at 100 K, 110 K, and 125 K for both the low-pressure and high-pressure phases. A discretized version of the Clausius–Clapeyron equation (eqn (S2)†) was then used to extract the isoexcess enthalpies of adsorption, −ΔHads, for H2 in ZIF-7. The fits are shown in Fig. S3–S5† and the fit parameters are tabulated in Table S1.† In the pre-step regime, −ΔHads ≈ 8 kJ mol−1 at a loading concentration of 0.5 mmol g−1, in good agreement with the previous theoretical calculations.58 In the post step regime, −ΔHads ≈ 5.5 kJ mol−1 at a loading concentration of 3.0 mmol g−1, indicating weaker adsorption interactions in the high-pressure phase.
Diffraction measurements of activated CdIF-13 and ZIF-7
We then sought to thoroughly characterize the structure of ZIF-7 across the phase change identified by the step-shaped isothermal gas adsorption measurements. To start, we collected a PND pattern for the activated phase at ≈7 K (Fig. 4(1)). The crystallinity of the material suffers somewhat upon activation such that sharp features in the pattern are only observed out to a scattering vector value of Q ≈ 2 Å−1. The results reported here are consistent with the resolution of the data. Successful indexing of this phase was very difficult due to the low symmetry and large unit cell size. Indeed, difficulty in indexing the structure is one of the factors that rendered this crystal structure elusive for over a decade. To circumvent this issue, we attempted to isolate single crystals of the activated phase of ZIF-7, and, because it is a structural analog to ZIF-7, single crystals of activated CdIF-13. While efforts with ZIF-7 were unsuccessful, we successfully isolated a crystal of activated CdIF-13 suitable for SCXRD measurements, from which we obtained the single crystal structure for activated CdIF-13 (Fig. S14†). We then used the unit cell parameters of the activated phase of CdIF-13 as a starting point for indexing the PND pattern for activated ZIF-7. Based on these cell parameters we were able to perform a successful Pawley fit of the PND pattern for activated ZIF-7.64 For the activated phases of both CdIF-13 and ZIF-7, we find unit cells with significantly lower volume compared to the previously reported structure for activated ZIF-7 (Table S2†).63 Based on the successful Pawley fit, we were able to perform a subsequent Rietveld refinement of the PND pattern to obtain the structure of the activated phase of ZIF-7 (Fig. 5 (activated ZIF-7), and Fig. S7†).65 Only the unit cell parameters from the activated CdIF-13 structure were used to inform the structure solution of the Zn framework. Importantly, the density for the activated structure is higher than that of the parent water-solvated structure (≈1.390 g cm−3 in the open R phase, including solvent molecules, ≈1.511 g cm−3 in the activated P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) phase, Table S4†).49 This change in density corresponds to an ≈18.3% decrease in volume upon activation (calculated as the density of the activated phase divided by the framework density of the water-solvated phase, Table S4†). The obtained density quantitatively agrees with previous theoretical calculations62 and is chemically intuitive. Moreover, the structure obtained from the Rietveld refinement of the PND pattern is similar to the structure obtained for activated CdIF-13 (Fig. S14†), even though only the unit cell parameters for the latter were used to inform the structure solution for the former. Thus, we believe this to be the most accurate determination of the activated ZIF-7 structure to date.
phase, Table S4†).49 This change in density corresponds to an ≈18.3% decrease in volume upon activation (calculated as the density of the activated phase divided by the framework density of the water-solvated phase, Table S4†). The obtained density quantitatively agrees with previous theoretical calculations62 and is chemically intuitive. Moreover, the structure obtained from the Rietveld refinement of the PND pattern is similar to the structure obtained for activated CdIF-13 (Fig. S14†), even though only the unit cell parameters for the latter were used to inform the structure solution for the former. Thus, we believe this to be the most accurate determination of the activated ZIF-7 structure to date.
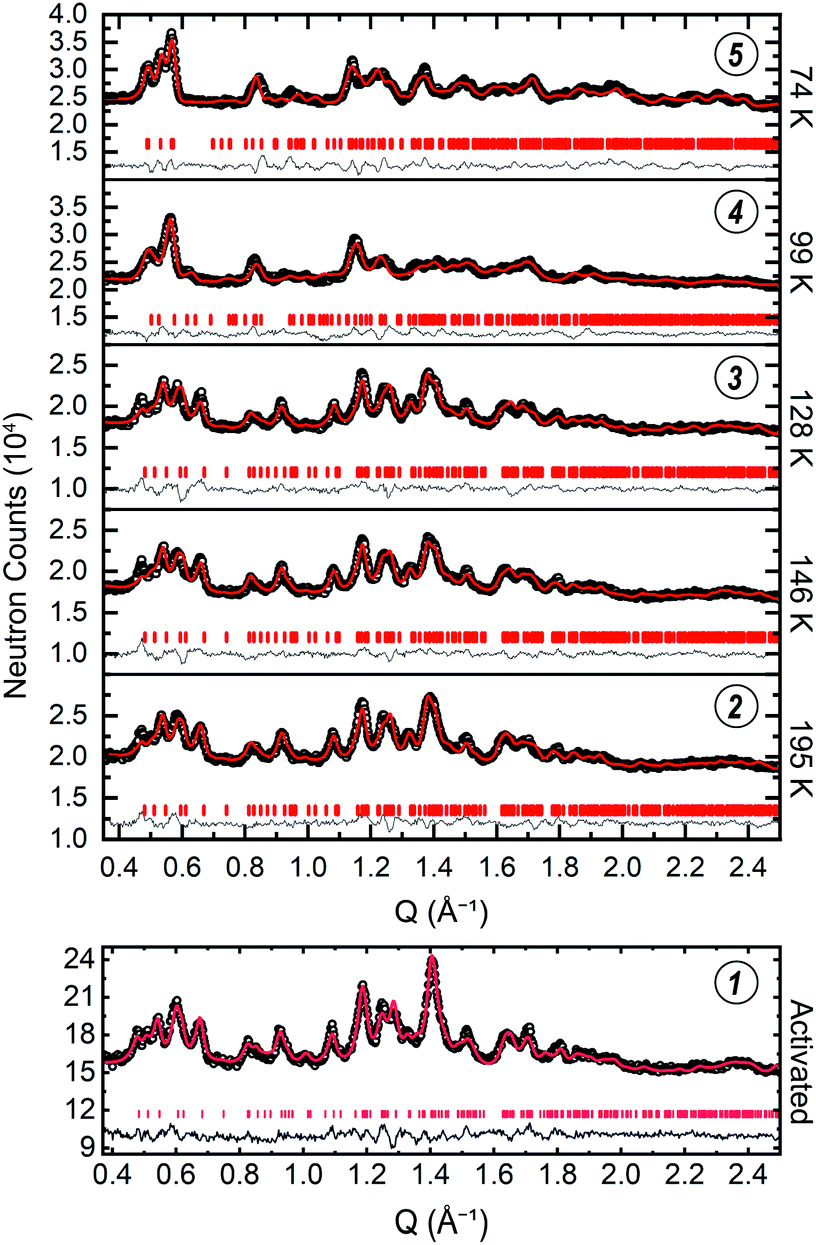 | ||
| Fig. 4 The low-Q regions of the PND patterns for the data collected at ≈30 bar D2 are stacked in the y-axis direction based on the temperature at which the data were collected, as denoted to the right of the plots of the data (top). The patterns collected at 74 K and 99 K index to the P21/n space group while the patterns collected at 128 K, 146 K, and 195 K index in the space group. Below, the data for the activated structure are shown (bottom). The black circles, red curve, grey curve, and vertical tick marks denote the raw data, the Rietveld refinement curve, the difference curve, and the hkl positions, respectively. The full Q range for the patterns is shown in Fig. S7, S9–S13.† Circled numbers above the plots of the data correspond to the structure numbering used in Fig. 3–5, and throughout the text. | ||
The structures of the activated phases of CdIF-13 and ZIF-7 are qualitatively similar (Fig. S14†). Here we focus on the structure of activated ZIF-7, which is distorted compared to the high symmetry, water-solvated phase. The skeleton of ZIF-7 has four distinct rings (Fig. 5, activated ZIF-7, (1)) instead of three, all of which have lower symmetry than their counterparts in the water-solvated structure. Especially notable is the loss of planarity of the 6-membered ring 1 (Fig. 5). The {ZnN4} tetrahedra display bond and angle distortions in the activated phase of ZIF-7 compared to the trigonal, water-solvated phase (Fig. S16†).49 In the trigonal, water-solvated structure the shortest Zn–N distance is d(Zn–N) = 1.983(4) Å and the longest is d(Zn–N) = 2.001(6) Å (values in parentheses indicate 1 s). Likewise the N–Zn–N angles in the tetrahedra are almost ideal in the trigonal structure, ranging between ∠(N–Zn–N) = 105.3(2)° and ∠(N–Zn–N) = 115.2(2)°. By comparison, the shortest and longest Zn–N distances in the activated phase are d(Zn–N) = 1.93(7) Å and d(Zn–N) = 2.06(6) Å, respectively. Similarly, the bond angles in the {ZnN4} tetrahedra in the activated phase show an expanded range of ∠(N–Zn–N) = 100(3)° to ∠(N–Zn–N) = 119(4)°. The Zn–N bond metrics are statistically identical within the ±3σ regime between the water-solvated phase and the activated phase of ZIF-7 described here. Another difference between the high symmetry phase and the activated phase is that some of the ligands in the activated phase twist between the Zn ions (Fig. 5). While the torsion angles are negligible for most of the ligands, the most extreme case results in a torsion angle of 107(5)°, where the out-of-plane twist minimizes sterically repulsive ligand–ligand contacts in the dense phase. This type of torsion is observed in the activated phases of Co(bdp)22 and MIL-53(Al),25 and in the N,N-dimethylformamide- and dichloromethane-solvated phases of CdIF-13.23
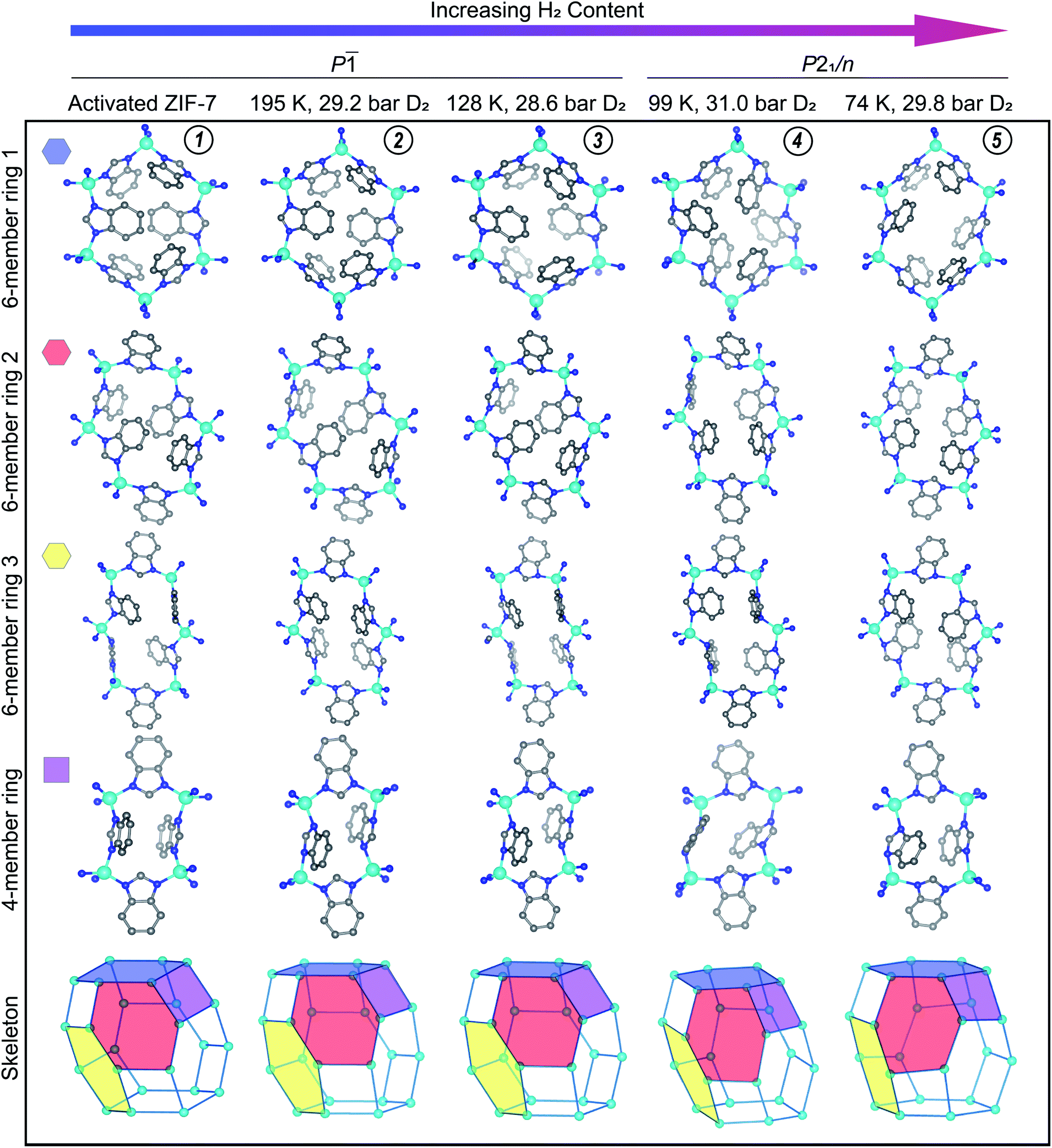 | ||
| Fig. 5 The activated structure for ZIF-7 is compared to the D2-dosed structures. The conditions under which each data set was measured are listed at the top of the figure. The approximate position of these conditions on the gas adsorption plot are represented in Fig. 3 with gold diamond symbols, with the vertical dashed line at ≈30 bar of D2. The amount of adsorbed D2 increases from left to right in this figure. D2 molecules and H atoms are omitted for clarity. Teal, blue, and gray spheres depict Zn, N, and C atoms, respectively. D2 adsorption sites are displayed in Fig. 6 and S18.† Circled numbers at the top of each column correspond to the structure numbering used in Fig. 3–5, and throughout the text. | ||
The gas adsorption data suggest that ZIF-7 exhibits low accessible porosity in the pre-step regime from a macroscopic point of view. This is corroborated at a microscopic level by the observed ligand-based pore gating in the crystal structure of activated ZIF-7 (Fig. 5(1)). The collapsed 4- and 6-member rings in the dense phase form face-to-face and edge-to-face π–π interactions which are absent in the high symmetry system. In Co(bdp), structural collapse to a ground state narrow pore structure is stabilized by edge-to-face π–π interactions.22 We hypothesize that the π–π interactions observed here similarly stabilize the dense activated phase of ZIF-7. Per unit cell, there is one unique offset face-to-face stacking interaction, in which the closest C–C distance is 3.149(7) Å. This offset stacking interaction occurs between the benzene moieties of the benzimidazolate ligands across the 6-membered ring 1 depicted in Fig. 4 (activated ZIF-7, top row). The apparent offset face-to-face interaction across 6-membered ring 2 is just outside of meaningful π–π stacking range, with the closest C–C contact between rings at 3.8(1) Å. Edge-to-face π–π interactions are much more prevalent in the activated phase. Given the close nature of the pores, each ligand participates in at least two unique edge-to-face π–π interactions per unit cell with distances ranging up to 3.3 Å in length.
In situ powder neutron diffraction measurements of D2-dosed ZIF-7
With a firm understanding of the structure of the activated phase established, we sought to investigate the hydrogen-induced phase change in ZIF-7 at varying dosing concentrations. To do so, we conducted in situ variable-temperature deuterium gas dosing PND measurements (Fig. 4). We use D2 instead of H2 gas because neutrons coherently scatter well from deuterium such that PND measurements can be used to structurally characterize the D2 adsorption sites in the structure and because of the incoherent scattering length of hydrogen which contributes to diffuse scattering in diffraction patterns.66 We acknowledge the recently observed differences in flexible framework responses between hydrogen and deuterium adsorption.34–36 Throughout the following sections, we consistently compare the structural changes inferred from the macroscopic probe of isothermal hydrogen gas adsorption measurements with the deuterium gas pressure-induced structural changes found from the microscopic probe of crystallography. To check that hydrogen and deuterium induce comparable structural changes in ZIF-7, we compare the quantity of gas adsorbed for the various conditions (Table S3†), the reversibility of the phase transition, and the pressure–temperature conditions for which the phase transition is observed for both gases. Using these checks, we find good agreement for the measurements conducted using hydrogen compared to the measurements conducted using deuterium gas. From these D2-dosed PND experiments, we aimed to identify: (1) the structure of the dense phase dosed with gas (corresponding to the pre-step regime of the isotherm plot), (2) the structure of the D2-opened phase after the phase change (the post-step regime), and (3) the adsorption sites in the two phases of ZIF-7 as a function of gas dosing pressure. We collected six additional PND patterns at various temperature and deuterium gas pressure conditions. The structure refinements, and the insights gained from these experiments, are discussed in detail below.Powder neutron diffraction measurements at 0.7 eq. D2 and 11 K
The first D2-dosed pattern was collected at 11 K with a stoichiometric dosing concentration of 0.7 molar equivalents D2 molecules per Zn2+ ion per formula unit. This experiment aimed to identify the primary D2 adsorption site upon initial adsorption from the activated phase of ZIF-7. The pattern collected at these conditions is depicted in Fig. S8† along with the corresponding Rietveld refinement curve. The Rietveld refinement analysis modeled the D2 molecules as super-atoms (see ESI†).67 The refinement yielded the crystal structure of the dense phase of ZIF-7 dosed with 0.7 eq. D2 gas. The primary adsorption site, site I, for D2 is sandwiched between the imidazolate ring of one ligand and the benzene ring of an adjacent ligand in 6-membered ring 3 (Fig. 6a, S17 and S18†). Of note, compared to rings 1 and 2, 6-membered ring 3 in the activated structure has a more accessible pore (Fig. 5 and S17†), which possibly explains why this is the initial adsorption site. At this loading concentration, the adsorption distances are 2.66(8) Å from the D2 molecule to the centroid of the imidazolate ring and 2.68(8) Å to the centroid of the benzene ring. These close adsorption distances indicate relatively strong D2–arene interactions.39 This strong interaction gives rise to the relatively high value for −ΔHads at low loading concentrations (Fig. S6†). Moderately strong van der Waals interactions with H atoms on separate adjacent ligands of 2.84(9) Å and 2.7(2) Å further stabilize the adsorbed D2 molecule. Compared to the fully activated phase, the framework skeleton and the ligands adjust very minimally to maximize the adsorption interaction at site I (Fig. S17† (0.7 eq. D2 dosed)). The sandwich adsorption motif observed here qualitatively agrees with the adsorption behavior previously predicted by theoretical calculations for H2 adsorption between ligands.59Data collection and Pawley fitting of 74–195 K, ≈30 bar D2 data sets
Armed with the structural information of the dense phase and the roadmap of the phase transition provided by the isotherm data, we next sought to structurally investigate the deuterium-induced phase change in ZIF-7. We conducted isobaric, variable-temperature PND measurements at ≈30 bar D2 at temperatures of 74 K (5), 99 K (4), 128 K (3), 146 K, and 195 K (2) (Fig. 5 and S7–S13†). By starting at 74 K and 29.8 bar D2, we measured the open phase of the material first. As we increased the measurement temperature, D2 desorbed from the structure such that we followed the desorption data from the isothermal measurements (open symbols, Fig. 3 approximate measurement conditions indicated by diamond symbols). According to the isotherm data, we expected to observe a phase transition in the PND patterns between 99 K and 128 K as the structure transitioned from the open phase to the dense phase. Indeed, the patterns are qualitatively distinct. Our initial Pawley fits of the patterns indicated a change in symmetry from the triclinic P space group at 128 K to the monoclinic P21/n space group at 99 K (Table S2†), marking the phase change. Incidentally, P21/n is the same space group assumed by the N,N-dimethylformamide- and dichloromethane-solvated CdIF-13 structures, the Cd-based congener of ZIF-7.23 Of note, even at 77 K, which shows complete saturation at 30 bar (Fig. 3), the D2-dosed open phase is lower in symmetry than the water-solvated structure, which we hypothesize is caused by the thermodynamic favorability of the sandwich-like adsorption sites with the small gas molecules enabled by the lower-symmetry structure (the kinetic diameter of H2 is just 2.9 Å).68 We find that, while both the 74 K and 99 K patterns can be indexed in the monoclinic structure, the change in the unit cell parameters is greater than can be expected from thermal expansion alone (Table S2†). We attribute the differences in unit cell parameters to the fact that the 74 K pattern was collected along the plateau of the isotherm, indicating completion of the phase transition, while the 99 K pattern was collected along the sloped portion of the step, indicating that at these conditions the phase transition was incomplete. We note that, during the experiment, long equilibration times for the pressure changes were given (ca. 1 to 3 h) and that the measurements were long (ca. 20 to 24 h). Therefore, we posit that the PND patterns reflect the equilibrated structures. As temperature was increased further, the patterns remained qualitatively similar (from 128 K up to 195 K) and the structures remain in the dense P state.
Rietveld refinement and crystal structure analysis of 74–195 K, ≈30 bar D2 data
We then performed Rietveld refinement analysis on these patterns and derived the crystal structures. Importantly, there is quantitative agreement between the amount of D2 adsorbed in the structures found from the PND measurements when compared with the amount of H2 adsorbed in ZIF-7 found from the isothermal measurements for all measurements (Table S3†).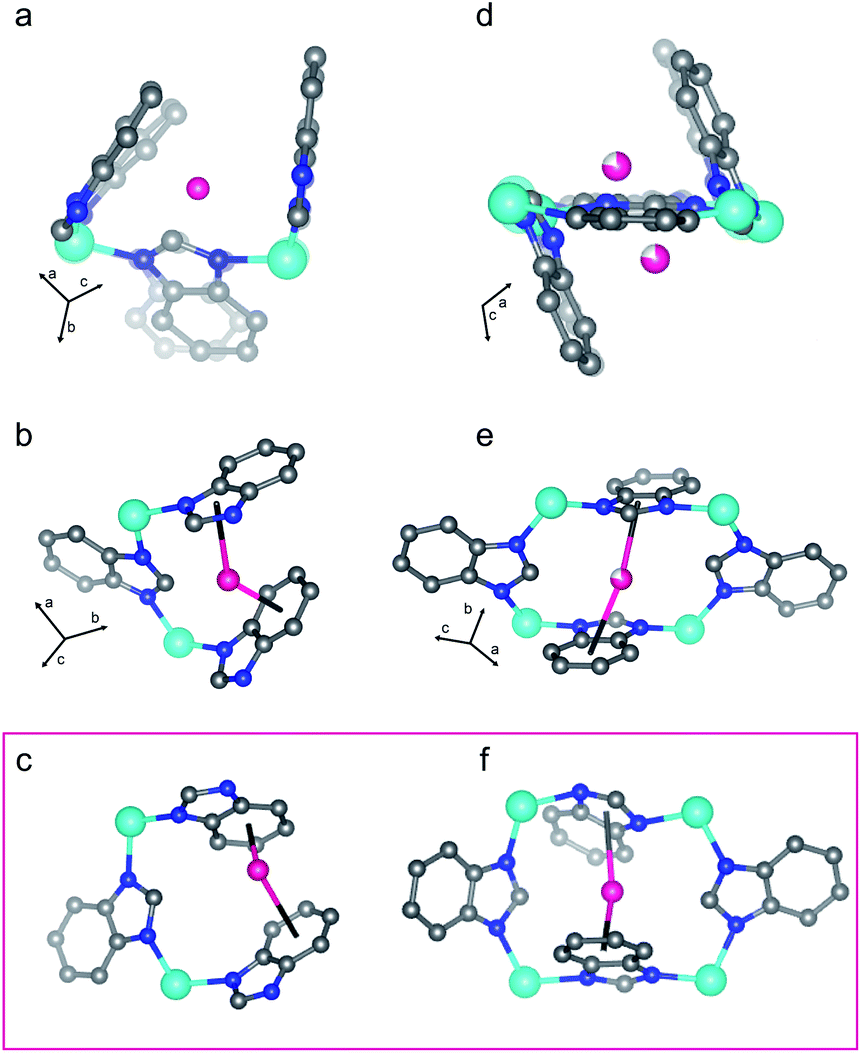 | ||
| Fig. 6 D2 adsorption sites I (a and b) and II (d and e) in the pre-step dense structure of ZIF-7 are shown. Additional representations are displayed in Fig. S18.† Both adsorption sites give a sandwich-like adsorption motif for D2 between an imidazole and a benzene ring. (a) and (d) show the structure derived from the Rietveld refinement of the pattern collected at 125 K and 28.6 bar D2 (pre-step conditions, Fig. 3) overlayed on top of the activated structure (faded) such that the unit cell origins overlap. The framework adjusts slightly to optimize the adsorption interactions. The boxed (c and f) illustrate similar adsorption sites to (a and b) and (d and e), respectively, in the post-step, fully open structure from the 74 K, 29.8 bar D2 data set (Fig. 3). Teal, blue, gray, and pink spheres depict Zn, N, and C atoms and D2 molecules, respectively, while H atoms are omitted for clarity. Incomplete shading denotes partial occupancy. | ||
The crystal structure of ZIF-7 at 99 K and 31.0 bar D2 (Fig. 5(4)) reveals that the ligands swing about the Zn–N⋯N–Zn axes in a fan-like motion (Fig. 5), that the {ZnN4} tetrahedra do not distort significantly more than in the activated phase (Fig. S14†), and that, at these conditions, there are some instances of significant ligand torsion. At these conditions, there is an ≈10% relative increase in the volume of the structure compared to the activated phase (Fig. S20†). The skeleton of ZIF-7 trends towards higher symmetry in this phase, and the 6-membered ring 1 becomes more planar than in the activated, triclinic phase (Fig. 5). The PND measurement conditions correspond to the sloped portion of the desorption step in the isotherm data. As such, the structure is captured in between the dense and the fully D2-opened states. Six total adsorption sites are observed in the structure (Fig. S18†). Sites I and II observed in the dense P phase at 128 K, shown in Fig. 5, are fully occupied in the open P21/n phase at 99 K, with the distinction that the D2 molecule adsorbs more strongly to the benzene ring than the imidazole ring in the D2-opened phase at 99 K. The site I D2–centroid distances are 3.21(4) Å and 2.27(5) Å for the imidazole and the benzene rings, respectively. For site II, the D2–centroid distances for the adsorption interactions with the imidazole and benzene rings are 3.95(6) Å and 2.38(8) Å, respectively. For site II, the D2–imidazole distance is very long, out of range of a significant D2–arene interaction.16 In addition to these D2–arene contacts, 6 D2–H van der Waals interactions stabilize site II. These van der Waals contacts range in distance between 2.48(8) Å and 3.33(7) Å (Fig. S18†). Notably, these are formed as additional adjacent ligands move closer to site II while the ligand containing the capping imidazole ring originally forming the sandwich-like adsorption motif moves farther away from site II. In addition to sites I and II, 4 new adsorption sites become occupied. Of these, 3 sites (sites III, IV, and V) adsorb to just one aryl ring each and are stabilized by additional van der Waals interactions with the ligands and by adsorbate–adsorbate interactions (Fig. S18†). These can be described as half-sandwich-like adsorption sites. The weaker adsorption at these sites gives rise to lower values of −ΔHads at higher loading concentrations (Fig. S6†). The remaining adsorption site (site VI) does not form a D2-arene contact and is instead adsorbed in the pores of the material, stabilized by adsorbate–adsorbate interactions and by non-D2–arene van der Waals contacts with the ligands. In the pores, the D2–D2 contacts have distances of 2.94(7) Å, 2.97(7) Å, 3.19(9) Å, and 3.2(1) Å, much shorter than the intermolecular distance observed in solid H2.70 This sequential filling of the small adsorption pockets yielding sandwich-like adsorption motifs followed by a liquid like filling of the pores agrees with the theory of micropore filling.16
Under 29.8 bar D2, the D2-induced phase change in ZIF-7 is complete at 74 K, and the structure is in the most open phase observable under accessible conditions with deuterium (or hydrogen) gas as the adsorbent (Fig. 5(5)). There is an ≈18.9% increase in the volume of the fully D2-opened phase compared to the activated material (Fig. S20†). Importantly, no additional step in adsorption is observed at 77 K up to 100 bar (Fig. 3), suggesting that no additional phase changes occur. In comparison to the 99 K structure, at 74 K, the ligands swing further about the Zn–N⋯N–Zn axes in a fan-like motion to open the pores more fully (Fig. 5(5)). This pore-gating motion is most clearly observed in the 6-member ring 1 (Fig. 5, top row), where the close C–C contact of 3.149(7) Å between the ligands bridging this ring in the activated phase expands to 7.81(1) Å in the open phase. In the fully D2-opened phase, the {ZnN4} tetrahedra remain slightly distorted compared to the water-solvated phase (Fig. S14†). The Zn–N distances are slightly elongated, with distances between 2.0(1) Å and 2.1(1) Å. The ∠(N–Zn–N) angles are close to ideal tetrahedral angles for two of the three unique Zn centers in the asymmetric unit. The third Zn tetrahedron is slightly more distorted, with angles ranging between 100(5)° and 117(5)°. As in the activated phase, ligand torsion occurs between the Zn centers (Fig. 5). The skeleton becomes more symmetric than in the activated phase, and the 6-membered ring 1 becomes planar. Yet the symmetry of the skeleton is still lower than in the water-solvated phase. In the D2-opened P21/c phase there are still 4 distinct rings, and the rings are distorted compared to their high-symmetry counterpart.
A total of 9 D2 adsorption sites are observed. Site I remains fully occupied in the fully D2-opened structure. The D2 molecule at site I shifts slightly and adsorbs between two benzene rings, rather than between a benzene and imidazole ring as observed at higher temperatures (Fig. 6c vs. b). The D2 molecule adsorbs much more strongly to the one of the benzene rings than the other, with D2-centroid distances of 2.04(4) Å and 3.04(5) Å. The occupancy of site II decreases slightly to 0.93(4) (Table S4†) and the D2 molecule remains relatively centered between the imidazole and benzene rings, with D2-centroid distances of 2.67(5) Å and 2.77(5) Å, respectively (Fig. 6f). Given the change in unit cell symmetry, this site is no longer mirrored across the 4-membered ring and the site is consolidated to one central position. In addition, four half-sandwich-like adsorption motifs are observed at sites III–VI (Fig. S18†). These adsorption sites sit between two ligands, however, the fan-like motion of the benzimidazolate linkers separates the rings such that the D2 molecules at these sites do not form D2-arene interactions with both rings. Additional non-arene D2–framework van der Waals interactions stabilize these sites. The occupancies for sites III–VI are 0.78(5), 1.00(5), 0.35(4), and 0.52(4) (Table S4†), respectively, indicating that the non-arene van der Waals interactions play an important role in determining the adsorption thermodynamics at these sites. The final 3 adsorption sites, sites VII, VIII, and IX, occur in the pores of the material in accordance with a liquid like filling of the pores and are stabilized by longer adsorbent–adsorbate and adsorbate–adsorbate interactions.
Structure–mechanism correlation
The mechanism underlying the hydrogen-induced phase change can be deduced based on analyzing the adsorption isotherm data in tandem with the structures derived from the PND measurements. The ligands in ZIF-7 can rotate about the Zn–N⋯N–Zn axes in a fan-like motion (Fig. 5), giving rise to the so-called pore gating mechanism, which alters the accessible porosity of the framework. In addition, there is some tolerance for out-of-plane ligand torsion between the Zn2+ ions. The combination of these two types of ligand motion, enabled by subtle distortions of the ZIF-7 skeleton, allows the framework to conform to the kinetic diameter of the D2 molecules to form optimized sandwich-like adsorption sites between arene rings on neighboring ligands for the primary adsorption sites. Adsorption to two electron-rich arene rings is energetically favorable,59 and indeed the adsorption sites I and II, common between the dense and the D2-opened structure, occupy such positions.ZIF-7 does not contain Lewis acidic coordinatively unsaturated metal ions as possible adsorption sites. Instead, the most thermodynamically favorable adsorption sites in ZIF-7 are formed by multiple Lewis basic organic ligands. Strong hydrogen adsorption is realized in ZIF-7 because of the material's ability to responsively adapt its structure to fit to the kinetic diameter of the adsorbate and form the sandwich-like adsorption sites. ZIF-7 does not simply undergo a binary phase transition between the P phase and the R phase upon gas dosing. Rather, the framework can access, and be stabilized in, a lower symmetry P21/n state which optimizes the adsorbate–adsorbent interactions while minimizing ligand–ligand repulsive interactions. As such, hydrogen adsorption in the dense phase templates and partially determines the ligand orientations as the structure opens into the P21/n phase. In contrast, strong adsorption sites in rigid adsorbents are achieved through strongly basic or acidic sites, or through exceedingly small pores with walls that can form multiple simultaneous contacts with hydrogen, which must be synthetically tailored on a case-by-case basis. As such, framework responsiveness is a possible path for achieving strong adsorption with chemically inert surface chemistries.39
Here we showed that the benzene rings of the benzimidazolate ligands play a key role in the stabilization of the dense activated phase and in the formation of the sandwich-like adsorption motifs that serve as the primary adsorption sites. Therefore, we hypothesize that the adsorption step pressure at a given temperature can be finely tuned by synthetically modifying the benzimidazolate ligand along the benzene backbone. In Co(bdp), modulation of the methane adsorption pressure step has been controlled through chemical substitution of the ligands, and substitution-induced weakening (or strengthening) of the edge-to-face π–π interactions effectively shifted the step temperature–pressure conditions.22 In ZIF-7, we hypothesize that substitutions that reduce attractive non-covalent ligand–ligand interactions in the activated phase and/or increase the Lewis basicity of the arene ring to increase adsorption strength at the sandwich-like adsorption sites should shift the step threshold to lower pressures at a given temperature. Alternatively, ligand substitutions that strengthen ligand–ligand interactions and/or weaken adsorption should shift the step threshold to higher pressures. With this enhanced mechanistic understanding and a suite of possible substitutions, shifting the step pressure threshold to operationally relevant temperatures and pressures in derivatized ZIF-7 appears achievable. Lastly, the Department of Energy Hydrogen and Fuel Cells Technologies Office set ambitious targets for the volumetric capacity in storage materials for use in light-duty hydrogen fuel cell electric vehicles (ultimately 50 g L−1).18,45 Currently, no materials come close to reaching this goal at operationally relevant conditions, in part due to discussed issues with type I adsorption–desorption. Towards this end, we hypothesize that the volumetric capacity in porous materials like ZIF-7 may be increased by moving to less dense topologies, such as RHO,49 by increasing the metal–ligand bond length via metal center substitution, or by increasing the metal–metal spacing by modifying the linkers.
Adsorption by pelletized ZIF-7
For a porous adsorbent to be commercially relevant for applications such as hydrogen storage and delivery it must be synthetically scalable and amenable to pelletization. With our current synthetic approach, ZIF-7 is scalable to at least the 10's of grams batch size (see ESI†) and the material is bench-top stable. Yet the application of external pressure and the process of pelletization has been known to negatively affect the gas adsorption properties of porous frameworks. High applied pressures can induce phase transitions in MOFs and modify the phase transition conditions in flexible frameworks.71–76 For example, the application of pressure to Co(bdp) reduces the flexible MOF's methane adsorption capacity by greater than 50% as the bulk density of the material increases from 0.2 g cm−3 to 0.75 g cm−3 and shifts the step in the adsorption isotherm to higher pressure.19 Moreover, the process of pelletization has been shown to reduce gas storage capacities.77–80 Pelletization of MIL-53(Al) with polyvinyl alcohol binder led to a reduction of the N2 adsorption capacity by 32%.81 On the other hand, interestingly, the hydroxy-functionalized MIL-53(Al)-OH and MIL-53(Al)-(OH)2 showed minimal reduction of methane storage capacity upon pelletization.82 Here, we generated several pellets of ZIF-7 using 5 mass% polyvinylpyrrolidone as a binder under the application of varying external pressure of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 psi (≈690 bar), 30000 psi (≈2070 bar), and 50000 psi (≈3450 bar). We characterized the resulting pellets using PXRD measurements (Fig. S19†). Analysis of the diffraction patterns revealed the open structure for all solvated pellets followed by a transition to the closed phase upon activation, correlating with our observations of the compound in powder form. Regardless of the applied pressure, a monolith density of ≈65% to 70% was achieved for the pellets in the open phase of the material. To investigate the effects of pelletization on the adsorption properties, we measured isothermal H2 and CO2 adsorption–desorption for pelletized ZIF-7 (Fig. S21 and S22†). For both gasses, when the polyvinylpyrrolidone binder is accounted for, we find minimal reduction of the gas adsorption properties for pelletized ZIF-7. Moreover, we find that the CO2 adsorption properties do not vary with the external pressure used in the pelletization process. These results indicate that the gas storage properties of ZIF-7 are robust to pelletization and suggest that, given the proper functionalization of the benzene ring, properly derivatized pelletized ZIF-7 may be a commercially viable candidate hydrogen storage material.
000 psi (≈690 bar), 30000 psi (≈2070 bar), and 50000 psi (≈3450 bar). We characterized the resulting pellets using PXRD measurements (Fig. S19†). Analysis of the diffraction patterns revealed the open structure for all solvated pellets followed by a transition to the closed phase upon activation, correlating with our observations of the compound in powder form. Regardless of the applied pressure, a monolith density of ≈65% to 70% was achieved for the pellets in the open phase of the material. To investigate the effects of pelletization on the adsorption properties, we measured isothermal H2 and CO2 adsorption–desorption for pelletized ZIF-7 (Fig. S21 and S22†). For both gasses, when the polyvinylpyrrolidone binder is accounted for, we find minimal reduction of the gas adsorption properties for pelletized ZIF-7. Moreover, we find that the CO2 adsorption properties do not vary with the external pressure used in the pelletization process. These results indicate that the gas storage properties of ZIF-7 are robust to pelletization and suggest that, given the proper functionalization of the benzene ring, properly derivatized pelletized ZIF-7 may be a commercially viable candidate hydrogen storage material.
Conclusions
In conclusion, we identified the hydrogen-induced phase change in ZIF-7 using variable temperature adsorption isothermal measurements. We then structurally characterized this phase change in ZIF-7 and identified the adsorption sites using in situ PND measurements. We conducted variable-temperature, isobaric measurements at ≈30 bar to traverse the phase space containing the phase transition as identified by the isothermal measurements. Rietveld refinements of the resulting patterns revealed that the structure crystalizes in the P space group in the activated phase and in the P21/n space group in the fully D2-opened phase. The D2 adsorption in ZIF-7 primarily occurs via a sandwich-like motif between the benzene ring of one ligand and the imidazole ring of an adjacent ligand, which creates an energetically favorable adsorption environment for the gas molecules. In the D2-opened structure, adsorption sites displaying only one D2-arene interaction in a half-sandwich-like motif are also present as is liquid-like filling of the pores. Based on our results, we hypothesize that synthetic substitution along the benzene ring in the benzimidazolate linkers will enable fine tuning of the phase change to achieve step-shaped adsorption–desorption under pressure-temperature conditions relevant to hydrogen storage, transport, and delivery, contributing to surmounting a key barrier in the hydrogen fuel economy.
Data availability
The experimental details and datasets supporting this article are available in the ESI.† The crystallographic information files can be found with the supplemental materials associated with this work, and on the Cambridge Crystallographic Data Center under deposition numbers 2101613–2101619.† The structure for activated CdIF-13 is deposited under CCDC deposition number 2114713.†Author contributions
C. M. M. synthesized the compounds and grew the single crystal of activated CdIF-13. P. L. M. solved the crystal structure for activated CdIF-13. R. A. K. and S. S. conducted the PND and isotherm measurements, respectively. R. A. K. wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Juscelino Leão for invaluable technical support. R. A. K. acknowledges funding by the U.S. Department of Energy (USDOE), Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office (HFTO) under contract no. DE-AC36-8GO28308 to the National Renewable Energy Laboratory (NREL). This material is based upon work supported by the USDOE EERE HFTO under Award Number DE-EE0008823. NuMat is grateful for the support from the USDOE EERE SBIR/STTR program and the HFTO (DE-SC0018532). Certain commercial equipment, instruments, or materials are identified in this document. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the products identified are necessarily the best available for the purpose. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- M. Hirscher, V. A. Yartys, M. Baricco, J. B. von Colbe, D. Blanchard, R. C. Bowman Jr, D. P. Broom, C. E. Buckley, F. Chang, P. Chen and others, J. Alloys Compd., 2020, 82, 7153548 Search PubMed.
- K. M. Thomas, Catal. Today, 2007, 120, 389–398 CrossRef CAS.
- D. Zhao, D. Yuan and H.-C. Zhou, Energy & Environ. Sci., 2008, 1, 222–235 RSC.
- J. Y. Lee, D. H. Olson, L. Pan, T. J. Emge and J. Li, Adv. Funct. Mater., 2007, 17, 1255–1262 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- M. T. Kapelewski, T. Runčevski, J. D. Tarver, H. Z. Jiang, K. E. Hurst, P. A. Parilla, A. Ayala, T. Gennett, S. A. FitzGerald, C. M. Brown and others, Chem. Mater., 2018, 30, 8179–8189 CrossRef CAS PubMed.
- D. E. Jaramillo, H. Z. Jiang, H. A. Evans, R. Chakraborty, H. Furukawa, C. M. Brown, M. Head-Gordon and J. R. Long, J. Am. Chem. Soc., 2021, 143, 6248–6256 CrossRef CAS PubMed.
- R. A. Klein, H. A. Evans, B. A. Trump, T. J. Udovic and C. M. Brown, Neutron scattering studies of materials for hydrogen storage, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2021, ISBN 9780124095472, DOI:10.1016/B978-0-12-823144-9.00028-5.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CrossRef CAS PubMed.
- S. Seth and S. Jhulki, Mater. Horiz., 2021, 8, 700–727 RSC.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS.
- Z. Chang, D.-H. Yang, J. Xu, T.-L. Hu and X.-H. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed.
- C. R. Murdock, B. C. Hughes, Z. Lu and D. M. Jenkins, Coord. Chem. Rev., 2014, 258, 119–136 CrossRef.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. Sing, Adsorption by powders and porous solids: principles, methodology and applications, Academic press, 2nd edn, 2013 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- M. D. Allendorf, Z. Hulvey, T. Gennett, A. Ahmed, T. Autrey, J. Camp, E. S. Cho, H. Furukawa, M. Haranczyk, M. Head-Gordon and others, Energy Environ. Sci., 2018, 11, 2784–2812 RSC.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown and others, Nature, 2015, 52, 7357–7361 Search PubMed.
- X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS PubMed.
- R. Numaguchi, H. Tanaka, S. Watanabe and M. T. Miyahara, J. Chem. Phys., 2013, 138, 54708 CrossRef PubMed.
- M. K. Taylor, T. Runčevski, J. Oktawiec, M. I. Gonzalez, R. L. Siegelman, J. A. Mason, J. Ye, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2016, 138, 15019–15026 CrossRef CAS PubMed.
- C. M. McGuirk, T. Runčevski, J. Oktawiec, A. Turkiewicz, M. K. Taylor and J. R. Long, J. Am. Chem. Soc., 2018, 140, 15924–15933 CrossRef CAS PubMed.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Toebbens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and others, Nature, 2016, 532, 348–352 CrossRef CAS PubMed.
- Y. Liu, J.-H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813–11818 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- F. M. Mulder, B. Assfour, J. Huot, T. J. Dingemans, M. Wagemaker and A. Ramirez-Cuesta, J. Phys. Chem. C, 2010, 114, 10648–10655 CrossRef CAS.
- F. Millange, N. Guillou, R. I. Walton, J.-M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2008, 39, 4732–4734 RSC.
- A. Boutin, D. Bousquet, A. U. Ortiz, F.-X. Coudert, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, J.-P. Bellat, G. Ortiz and others, J. Phys. Chem. C, 2013, 117, 8180–8188 CrossRef CAS.
- L. R. Parent, C. H. Pham, J. P. Patterson, M. S. Denny Jr, S. M. Cohen, N. C. Gianneschi and F. Paesani, J. Am. Chem. Soc., 2017, 139, 13973–13976 CrossRef CAS PubMed.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Greneche and others, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS.
- A. Boutin, M.-A. Springuel-Huet, A. Nossov, A. Gedeon, T. Loiseau, C. Volkringer, G. Férey, F.-X. Coudert and A. H. Fuchs, Angew. Chem., Int. Ed., 2009, 121, 8464–8467 CrossRef.
- A. V. Neimark, F.-X. Coudert, A. Boutin and A. H. Fuchs, J. Phys. Chem. Lett., 2010, 1, 445–449 CrossRef CAS PubMed.
- J. Y. Kim, L. Zhang, R. Balderas-Xicohténcatl, J. Park, M. Hirscher, H. R. Moon and H. Oh, J. Am. Chem. Soc., 2017, 139, 17743–17746 CrossRef CAS PubMed.
- J. Y. Kim, J. Park, J. Ha, M. Jung, D. Wallacher, A. Franz, R. Balderas-Xicohténcatl, M. Hirscher, S. G. Kang, J. T. Park and others, J. Am. Chem. Soc., 2020, 142, 13278–13282 CrossRef CAS PubMed.
- R. Muhammad, S. Jee, M. Jung, J. Park, S. G. Kang, K. M. Choi and H. Oh, J. Am. Chem. Soc., 2021, 143, 8232–8236 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC.
- A. Noguera-Díaz, N. Bimbo, L. T. Holyfield, I. Y. Ahmet, V. P. Ting and T. J. Mays, Colloids Surf., A, 2016, 496, 77–85 CrossRef.
- D. P. Halter, R. A. Klein, M. A. Boreen, B. A. Trump, C. M. Brown and J. R. Long, Chem. Sci., 2020, 11, 6709–6716 RSC.
- R. A. Pollock, J.-H. Her, C. M. Brown, Y. Liu and A. Dailly, J. Phys. Chem. C, 2014, 118, 18197–18206 CrossRef CAS.
- C. Yang, X. Wang and M. A. Omary, J. Am. Chem. Soc., 2007, 129, 15454–15455 CrossRef CAS PubMed.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and others, J. Am. Chem. Soc., 2010, 132, 13782–13788 CrossRef CAS PubMed.
- H. J. Choi, M. Dincă, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117–123 RSC.
- J. Kang, S.-H. Wei and Y.-H. Kim, J. Am. Chem. Soc., 2010, 132, 1510–1511 CrossRef CAS PubMed.
- U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, https://www.energy.gov/sites/prod/files/2017/05/f34/fcto_targets_onboard_hydro_storage_explanation.pdf, accessed July 2021 Search PubMed.
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 CrossRef CAS PubMed.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109–119 RSC.
- M. D. Allendorf, V. Stavila, M. Witman, C. K. Brozek and C. H. Hendon, J. Am. Chem. Soc., 2021, 143, 6705–6723 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- X. Huang, J. Zhang and X. Chen, Chin. Sci. Bull., 2003, 48, 1531–1534 CrossRef CAS.
- S. Aguado, G. Bergeret, M. P. Titus, V. Moizan, C. Nieto-Draghi, N. Bats and D. Farrusseng, New J. Chem., 2011, 35, 546–550 RSC.
- C. Gücüyener, J. Van Den Bergh, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2010, 132, 17704–17706 CrossRef PubMed.
- D.-L. Chen, N. Wang, F.-F. Wang, J. Xie, Y. Zhong, W. Zhu, J. K. Johnson and R. Krishna, J. Phys. Chem. C, 2014, 118, 17831–17837 CrossRef CAS.
- A. Arami-Niya, G. Birkett, Z. Zhu and T. E. Rufford, J. Mater. Chem. A, 2017, 5, 21389–21399 RSC.
- Y. Du, B. Wooler, M. Nines, P. Kortunov, C. S. Paur, J. Zengel, S. C. Weston and P. I. Ravikovitch, J. Am. Chem. Soc., 2015, 137, 13603–13611 CrossRef CAS PubMed.
- Y. Du, K. Mao, B. Wooler, A. K. Sharma, D. Colmyer, M. Nines and S. C. Weston, J. Phys. Chem. C, 2017, 121, 28090–28095 CrossRef CAS.
- A. Noguera-Díaz, N. Bimbo, L. T. Holyfield, I. Y. Ahmet, V. P. Ting and T. J. Mays, Colloids Surf., A, 2016, 496, 77–85 CrossRef.
- M. Dixit, D. T. Major and S. Pal, Chem. Phys. Lett., 2016, 651, 178–182 CrossRef CAS.
- P. Zhao, G. I. Lampronti, G. O. Lloyd, E. Suard and S. A. Redfern, J. Mater. Chem. A, 2014, 2, 620–623 RSC.
- P. Zhao, G. I. Lampronti, G. O. Lloyd, M. T. Wharmby, S. Facq, A. K. Cheetham and S. A. Redfern, Chem. Mater., 2014, 26, 1767–1769 CrossRef CAS PubMed.
- Y. Du, B. Wooler, M. Nines, P. Kortunov, C. S. Paur, J. Zengel, S. C. Weston and P. I. Ravikovitch, J. Am. Chem. Soc., 2015, 137, 13603–13611 CrossRef CAS PubMed.
- P. Zhao, H. Fang, S. Mukhopadhyay, A. Li, S. Rudić, I. J. McPherson, C. C. Tang, D. Fairen-Jimenez, S. E. Tsang and S. A. Redfern, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- G. Pawley, J. Appl. Crystallogr., 1981, 14, 357–361 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- T. Runčevski, M. T. Kapelewski, R. M. Torres-Gavosto, J. D. Tarver, C. M. Brown and J. R. Long, Chem. Commun., 2016, 52, 8251–8254 RSC.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- Y. G. Andreev, G. S. MacGlashan and P. G. Bruce, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, 12011 CrossRef CAS.
- J. V. Kranendonk and H. Gush, The Crystal Structure of Solid Hydrogen, Phys. Letters, 1962, 1, 22 CrossRef.
- L. R. Redfern, M. Ducamp, M. C. Wasson, L. Robison, F. A. Son, F.-X. Coudert and O. K. Farha, Chem. Mater., 2020, 32, 5864–5871 CrossRef CAS.
- I. Beurroies, M. Boulhout, P. L. Llewellyn, B. Kuchta, G. Férey, C. Serre and R. Denoyel, Angew. Chem., Int. Ed., 2010, 49, 7526–7529 CrossRef CAS PubMed.
- N. Chanut, A. Ghoufi, M.-V. Coulet, S. Bourrelly, B. Kuchta, G. Maurin and P. L. Llewellyn, Nature Commun, 2020, 11, 1–7 CrossRef PubMed.
- L. R. Redfern, L. Robison, M. C. Wasson, S. Goswami, J. Lyu, T. Islamoglu, K. W. Chapman and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 4365–4371 CrossRef CAS PubMed.
- L. Robison, R. J. Drout, L. R. Redfern, F. A. Son, M. C. Wasson, S. Goswami, Z. Chen, A. Olszewski, K. B. Idrees, T. Islamoglu and others, Chem. Mater., 2020, 32, 3545–3552 CrossRef CAS.
- M. Wahiduzzaman, N. Reimer, J.-P. Itié, N. Stock, G. Maurin and P. G. Yot, Polyhedron, 2018, 155, 144–148 CrossRef CAS.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- M. Tagliabue, C. Rizzo, R. Millini, P. D. Dietzel, R. Blom and S. Zanardi, J. Porous Mater., 2011, 18, 289–296 CrossRef CAS.
- E. Mahmoud, L. Ali, A. El Sayah, S. A. Alkhatib, H. Abdulsalam, M. Juma and A. H. Al-Muhtaseb, Crystals, 2019, 9, 406 CrossRef CAS.
- J. Cousin-Saint-Remi, A.-L. Finoulst, C. Jabbour, G. V. Baron and J. F. Denayer, Microporous Mesoporous Mater., 2020, 304, 109322 CrossRef CAS.
- V. Finsy, L. Ma, L. Alaerts, D. De Vos, G. Baron and J. Denayer, Microporous Mesoporous Mater., 2009, 120, 221–227 CrossRef CAS.
- T. Kundu, B. B. Shah, L. Bolinois and D. Zhao, Chem. Mater., 2019, 31, 2842–2847 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ESI figures, synthetic details, details of isotherm fitting, and tables of structural information. CCDC 2101613–2101619, 2114713. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04618g |
| This journal is © The Royal Society of Chemistry 2021 |