
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentifying the true origins of selectivity in chiral phosphoric acid catalyzed N-acyl-azetidine desymmetrizations†
Pier Alexandre
Champagne
 *
*
Department of Chemistry and Environmental Science, New Jersey Institute of Technology, Newark, NJ, USA. E-mail: pier.a.champagne@njit.edu
First published on 23rd November 2021
Abstract
The first catalytic intermolecular desymmetrization of azetidines was reported by Sun and coworkers in 2015 using a BINOL-derived phosphoric acid catalyst (J. Am. Chem. Soc. 2015, 137, 5895–5898). To uncover the mechanism of the reaction and the origins of the high enantioselectivity, Density Functional Theory (DFT) calculations were performed at the B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G(d,p)/CPCM(toluene) level of theory. Comparison of four possible activation modes confirms that this reaction proceeds through the bifunctional activation of the azetidine nitrogen and the thione tautomer of the 2-mercaptobenzothiazole nucleophile. Upon thorough conformational sampling of the enantiodetermining transition structures (TSs), a free energy difference of 2.0 kcal mol−1 is obtained, accurately reproducing the experimentally measured 88% e.e. at 80 °C. This energy difference is due to both decreased distortion and increased non-covalent interactions in the pro-(S) TS. To uncover the true origins of selectivity, the TSs optimized with the full catalyst were compared to those optimized with a model catalyst through steric maps. It is found that the arrangements displayed by the substrates are controlled by strict primary orbital interaction requirements at the transition complex, and their ability to fit into the catalyst pocket drives the selectivity. A general model of selectivity for phosphoric acid-catalyzed azetidine desymmetrizations is proposed, which is based on the preference of the nucleophile and benzoyl group to occupy empty quadrants of the chiral catalyst pocket.
Introduction
The importance of chirality in all spheres of chemistry explains the ongoing extraordinary efforts toward developing stereoselective organic transformations.1,2 As new catalytic systems were discovered for this purpose throughout the years, models of selectivity were also developed to satisfy the practitioners' urge to rationalize and predict experimental results, as well as inform future improvements. These qualitative “chemically-intuitive” models, which were at first derived purely from empirical data and then increasingly supported by computational chemistry,3 focused on describing the principal factors affecting the energy of competing transition structures (TS) leading to stereoisomers. Only recently, with the rise of electronic structure calculations such as Density Functional Theory (DFT), from which chemically-accurate geometries and energies can be obtained at a reasonable computational cost,4 can such TSs of asymmetric transformations be routinely computed as the first step in the development of selectivity models.5 Qualitative modeling can be computationally expensive due to the requirement of optimizing multiple diastereomeric TSs which can be complicated by the large conformational space of the catalysts, but the resulting visualizations have been shown to provide intricate insights about the structures that can be distilled into chemically-intuitive explanations for the observed selectivity.6 A different approach to model stereoselective reactions without explicitly computing TSs is the statistical modeling, as popularized by Sigman.7–16 Such multivariate analyses allow for excellent quantitative predictions of enantioselectivity based on fitting experimental results to a set of physical organic chemistry descriptors, but their mechanistic interpretation is obfuscated behind complex equations often comprising cross-terms of parameters that have to be redefined for most regressions. Depending on the situation, chemically-intuitive or statistical modeling approaches can each be useful and should be considered as complementary instead of competing tools toward understanding and improving stereoselective transformations.In the field of enantioselective organocatalysis,17 chiral phosphoric acids (CPAs) enjoy a strong reputation as versatile and powerful catalysts that are applicable to a variety of chemical reactions.18 As such, there has been significant effort deployed toward the computational study of asymmetric CPA-catalyzed reactions over the last decades.19 Reactions involving imines and other sp2-hybridized carbon electrophiles are particularly well-studied experimentally and computationally (including multivariate analysis),10,20–32 yet few qualitative models have been proposed. Notably, Goodman and coworkers compiled an impressive breadth of quantitative computational data to develop their comprehensive qualitative model for imine additions, which focuses on the features making the TS leading to the major product favorable and explains why this arrangement is unfavorable for the minor enantiomer. The resulting eponymous Goodman model22,33 is the gold standard in CPA catalysis, and similar models for other reaction types are highly desirable. One class of reactions that is understudied and that would benefit from improved understanding is the desymmetrization of aziridines,34–39 epoxides,40–42 azetidines,43 and oxetanes44–49 by ring-opening with nucleophiles. For such sp3-hybridized carbon electrophiles, fewer experimental reports are known. Computational studies on these systems48,50–54 have relied on the optimization of stereodetermining TSs to identify if distortions or non-covalent interactions are the major contributors to selectivity, but few general models have been proposed.51,54
We were intrigued that DFT studies of organocatalyzed reactions often provide clear insights about the selectivity-determining TSs, but rarely yield general models of selectivity. We hypothesized that the focus of recent computational studies and statistical analyses on comparing only the stereodetermining TSs prevents the identification of the true origins of selectivity that are necessary to develop chemically-intuitive models. To demonstrate this effect, we present a DFT study of the first catalytic intermolecular desymmetrization of meso-azetidines, which was reported in 2015 by Sun and coworkers.43 Although the desymmetrization of aziridines and azetidines is an important transformation giving access to chiral amines,55,56 Sun's report is still the only example in this reaction class. Their system is limited to benzoyl-protected azetidines as electrophiles and mercaptobenzothiazoles as nucleophiles, and there are no reported extensions of this methodology. These limitations are not well understood, therefore studying this reaction presented a unique opportunity to uncover how its mechanism is linked to its selectivity, expand our understanding of such reactions, and allow for new developments to occur.
Methods
Fig. 1 reproduces the best optimization result from Sun et al., whereas meso N-acylazetidine 1, bearing a benzyloxy substituent at the 3-position, is reacted at 80 °C with 2-mercaptobenzothiazole 2a under catalysis from BINOL-derived (R)-3a, forming (S)-4a in 94% yield and 95% e.e. (system A). To reduce the computational cost associated with this study, we chose instead the example where the benzothiazole is unsubstituted (2b) and the catalyst bears a tris-(isopropyl)phenyl aryl substituent (3b), forming product 4b in 99% yield and 88% e.e. (system B). Sun's results indicate that the benzoyl group on the azetidine's nitrogen plays a key role in both reactivity and stereoinduction. Indeed, electron-poor benzamides were unreactive, while the 3,4,5-trimethoxy substitution pattern is crucial for high enantioselectivity. Electron-rich benzothiazoles were required for reactivity, but substitution was only tolerated at the 6-position for high selectivity. The reaction can be catalyzed by a range of BINOL-derived phosphoric acid catalysts, but only those with 2,6-iPr-4-R-C6H2 (R = iPr or Ad) groups provided high selectivity. Finally, a wide scope of substituents at the 3-position of the azetidine were tolerated without any impact on the high enantioselectivity, including alkyl or aryl ethers, alkanes, and arenes. Considering this experimental evidence, system B is a representative example of the transformation that did not require any approximations.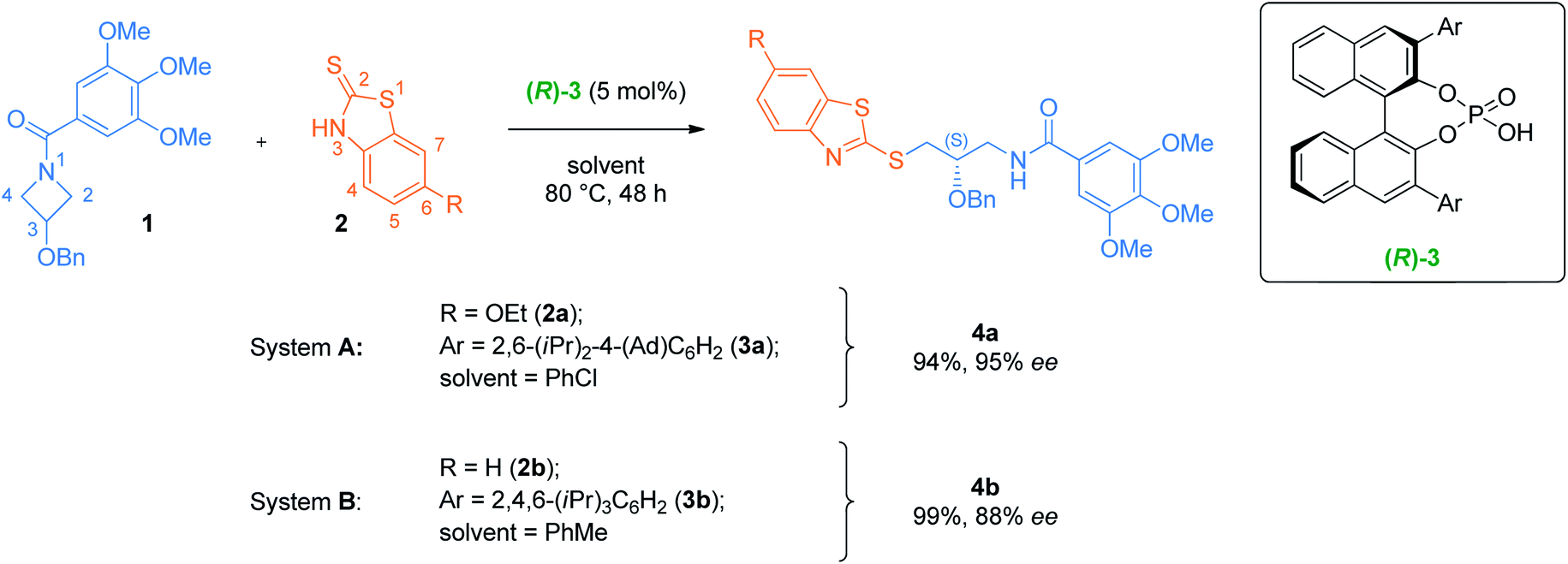 | ||
| Fig. 1 Selected examples of the enantioselective azetidine desymmetrizations reported by Sun. System A is the best reported system in the experimental study. System B is chosen as a computational target in this study. Ad = adamantyl. | ||
DFT geometry optimizations were performed with Gaussian 16 at the B97D3/6-31G(d,p) level of theory,57 with the CPCM implicit solvation58,59 model for toluene. This method has been shown to provide good results in previous DFT studies of CPA-catalyzed reactions52–54,60–62 and accounts for both dispersive interactions and solvation issues. Conformational sampling of each stationary point was performed using Grimme's CREST algorithm63 as implemented in the XTB code.64 Single-point energy (SPE) refinements were obtained with various methods, solvation models and basis sets, which all agreed on the relative free energies of the key stereodetermining TSs; the values from B97D3/6-311+G(2d,2p) with the SMD solvation model65 for toluene are presented in the main text as they are the most accurate. Visualizations of the structures were generated using CYLview.66 Full computational details can be found in the ESI.†
Results
Initially, we considered the tautomeric forms of 2-mercaptobenzothiazole 2b (more aptly named benzo[d]thiazole-2(3H)-thione) (Fig. 2A). Our results serve as a reminder that its thiol tautomer (2b-SH) is 8.3 kcal mol−1 less stable than its thione tautomer (2b-NH), thus its ground-state structure is not consistent with its usual drawing in the literature. That the thione tautomer is the major species in solution and the active nucleophile in such reactions has been known from other computational studies involving this nucleophile,52,53 and confirmed via13C NMR experiments by Pericàs and coworkers.67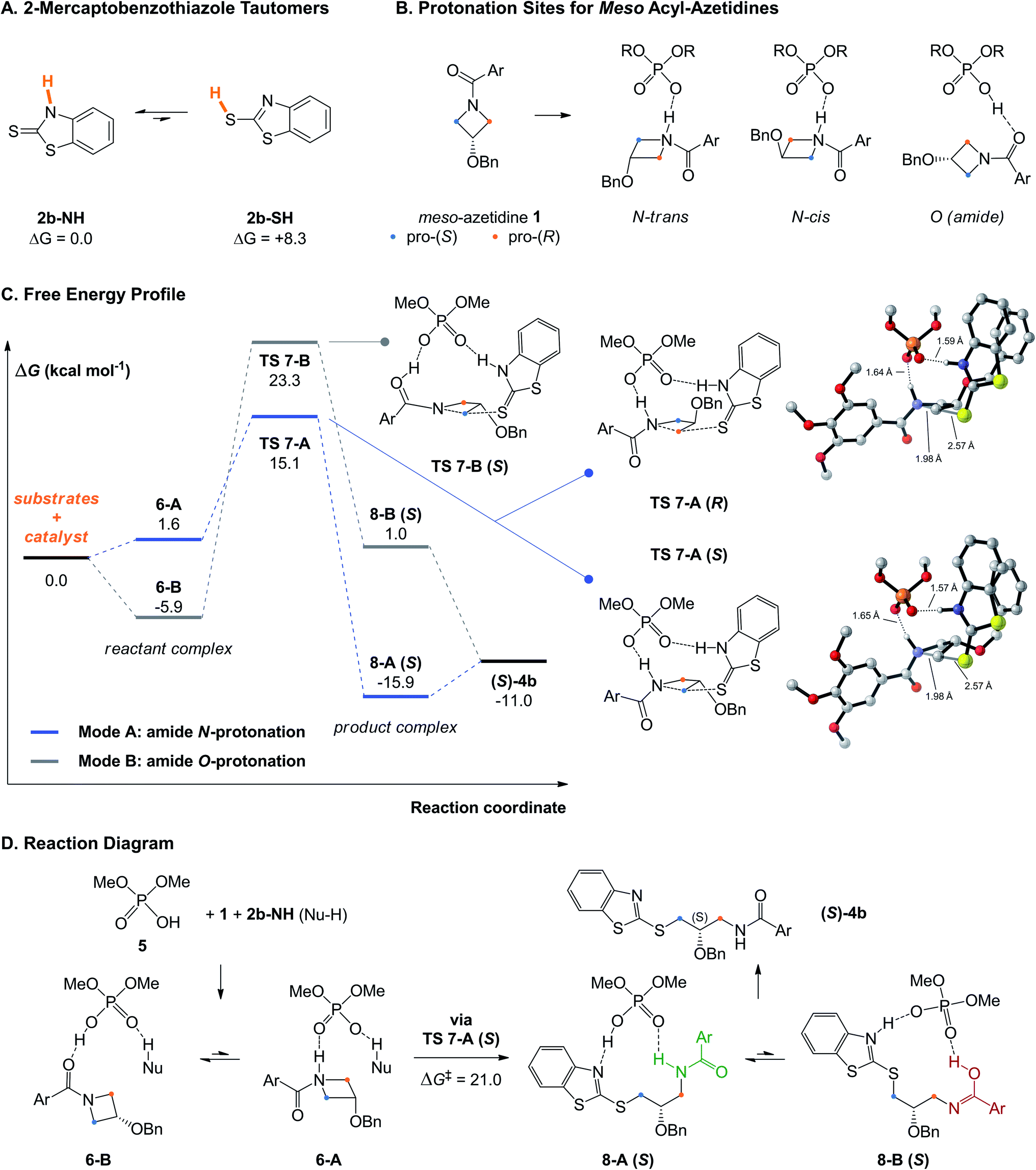 | ||
| Fig. 2 (A) Free energy difference (in kcal mol−1) between the thiol and thione tautomers of 2b. (B) Various protonation sites of meso acyl-azetidines by phosphoric acids. Ar = 3,4,5-(OMe)3C6H2. (C) Free energy profile for the reaction of 1 with 2b catalyzed by model catalyst 5, using modes A and B. Non-critical hydrogen atoms are hidden for improved clarity on the visualized structures. (D) Reaction diagram. | ||
Then, to study the mechanism of the transformation and the preferred conformations of the transition structures (TS) leading to the (R)- and (S)-products, we used dimethoxyphosphoric acid (5) as a model truncated catalyst, keeping a fixed conformation in line with the axial chirality of (R)-3. This approach has been used successfully in previous work,51,54,62 and the lowest-energy TSs located with the small catalyst often mimicked those obtained with the large BINOL- (or SPINOL-) derived catalyst, indicating that low-energy TS arrangements are independent of the catalyst's cavity shape (see below). In agreement with the known catalytic behavior of CPAs which predominantly activate both nucleophile and electrophile simultaneously via bifunctional activation,18 four possible activation modes were explored computationally. These possibilities arise as the acyl-azetidine substrate 1 can be activated by protonation of the nitrogen or oxygen atoms of its amide moiety (Fig. 2B), while nucleophile 2b can be activated from its two tautomeric forms (thiol or thione). Of those possibilities, mode A, where the azetidine ring nitrogen and the thione tautomer are activated, has the lowest activation free energy, followed by mode B where the azetidine is instead activated via the carbonyl.
The PES and rate-determining TSs for these two pathways en route to the major (S)-product are shown in Fig. 2C. In mode A, the reactant complex (RC) exhibits a complete transfer of the phosphoric acid proton to the azetidine nitrogen (6-A, Fig. 2D). The most stable RC located for this system uses mode B (6-B, −5.9 kcal mol−1), in line with the increased basicity of amide carbonyls versus nitrogens. However, the TS for nucleophilic attack has a lower free energy in mode A (TS 7-A, ΔG‡ = 21.0 kcal mol−1 from 6-B), while mode B entails an 8.2 kcal mol−1 penalty (TS 7-B, ΔG‡ = 29.2 kcal mol−1). The high energy of the TS using mode B is likely related to the unfavorable imidic acid tautomer of the amide which is initially formed using this pathway (8-B (S), 1.0 kcal mol−1). The reaction is under kinetic control (irreversible), due to the stability of the product-complex (PC) 8-A (S) (−15.9 kcal mol−1). TSs using the thiol tautomer of the nucleophile (2b-SH) were significantly higher in energy; thiol and amide nitrogen activation (mode C) is 14.5 kcal mol−1 higher than mode A, while the combination of thiol and oxygen activation (mode D) is 19.9 kcal mol−1 higher (see Fig. S2† for the structures of modes B–D). Overall, these results agree with the preliminary work from Sun43 and confirm that mode A is the only reaction pathway that is operative in this system.
Bifunctional activation, especially with displaced nucleophiles like 2b-NH (i.e. where deprotonation occurs from an atom conjugated with the nucleophilic sulfur), have very strict orbital requirements, or primary orbital interactions. For TS 7-A, nucleophilic attack must occur from the px lone pair of sulfur (orthogonal to the aromatic π system) into the azetidine's σ*C–N, following the ∼180° angle prescribed for SN2 reactions. Simultaneously, the azetidinium's sp3 N–H and the benzothiazole's sp2 N–H bonds must form hydrogen bonds with two of the phosphate's oxygen sp2 lone pairs (at a roughly 120° P–O–H angle). These requirements, along with the extent of bond breaking and bond forming due to the position of the TS along the reaction coordinate, make for a highly rigid transition complex (TC, the substrates in their TS geometry). This TC can, however, adopt different arrangements relative to the phosphoric acid's core structure, in addition to conformations arising from rotation around single bonds.
The arrangements located for TS 7-A (R and S) are depicted in Fig. 2C. For the pro-(S) TS, the best arrangement involves protonation of the azetidine trans from the benzyloxy substituent (see Fig. 2B), with the nucleophile's benzothiazole ring perpendicular to the phosphoric acid's structure. For the pro-(R) TS, the best arrangement is similar to the pro-(S) TS, except protonation occurs cis to the substituent now directed toward the catalyst structure, allowing the nucleophile to attack the opposite azetidine carbon. Eight other arrangements of the pro-(R) TC were located (Fig. 3), all of which displaying trans protonation yet at least 1.8 kcal mol−1 less favorable than TS 7-A (R). Two higher-energy arrangements were also located for TS 7-A (S). For each arrangement, many conformers were located, some with free energy differences as small as 0.2 kcal mol−1 (for other arrangements of TS 7-A (S) and examples of conformers, see Fig. S3–S5†). Overall, despite the varied tridimensional shapes of the arrangements, the primary orbital interactions kept the forming and breaking bond lengths in a narrow range (S⋯C bond between 2.53 and 2.61 Å and C⋯N bond between 1.96 and 2.02 Å). This indicates that even without the large groups on the BINOL-derived catalyst (R)-3b, the main difference between the pro-(R) and pro-(S) TSs is the orientation of the azetidine during the protonation.
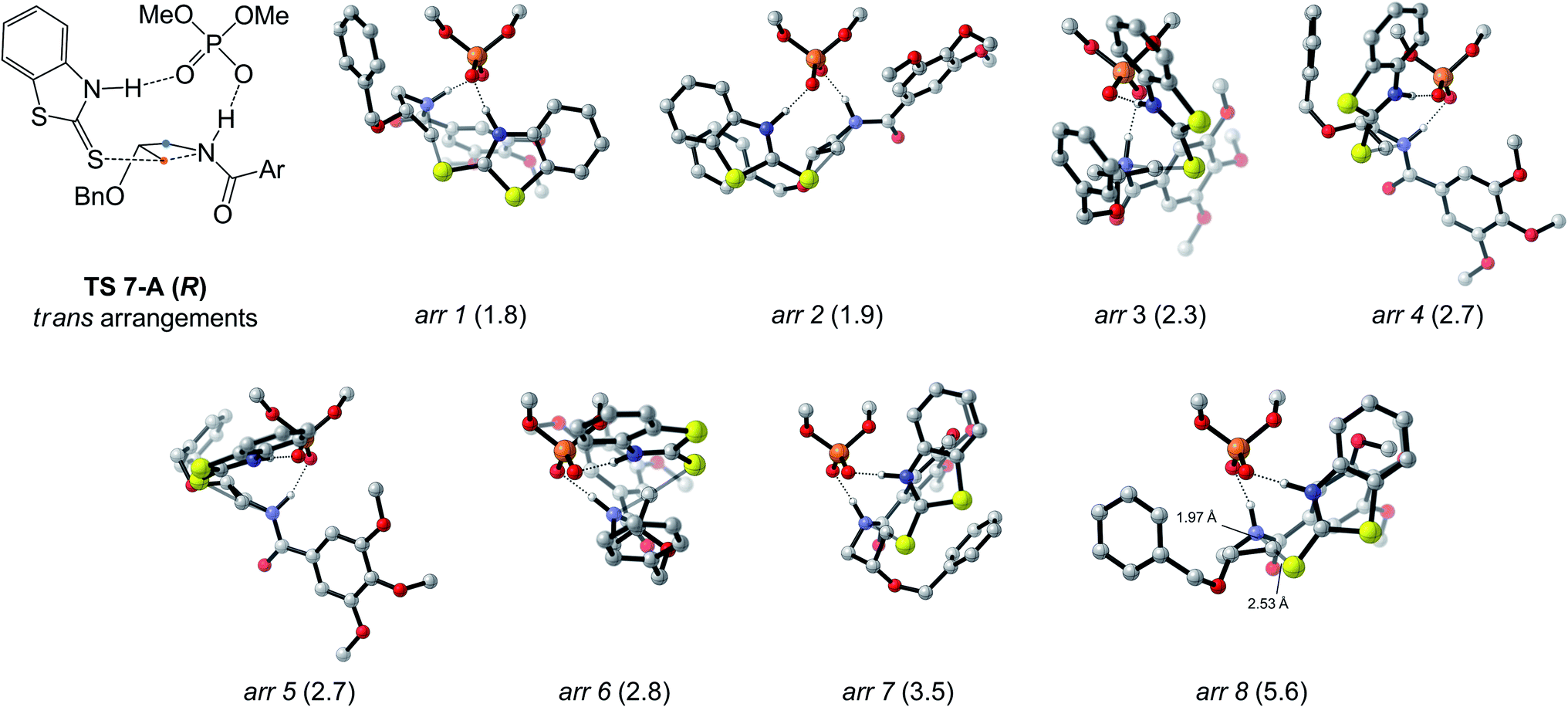 | ||
| Fig. 3 Other arrangements of the pro-(R) transition complex relative to the phosphoric acid catalyst, shown in the Goodman projection. Free energies relative to TS 7-A (R) (in kcal mol−1) are listed in parenthesis. For all computed structures, non-critical hydrogen atoms are hidden for improved clarity. Ar = 3,4,5-(OMe)3C6H2. | ||
With the mechanism confirmed as operating via amide N-protonation (mode A) and under Curtin-Hammett control, the origins of selectivity were investigated by computing the stereodetermining TSs for the reaction using the full catalyst (R)-3b. For the TSs leading to the experimentally preferred (S)-product, 19 unique conformers were located within a free energy span of 4.6 kcal mol−1. Of those, only 5 structures were within 1.2 kcal mol−1 of the lowest in energy (all within 0.6 kcal mol−1) and showed only minor conformational changes to the methoxy groups of the substrate or the isopropyl groups at the 4- and 4′-positions of the catalyst walls (see Fig. S1†). For the pro-(R) TSs, 45 unique conformers were located in a range of 5.3 kcal mol−1 and there were 13 structures within 1.2 kcal mol−1 with various conformations of the flexible benzyloxy substituent at the 3-position of the azetidine. Of note, the substrates, transition structures, and products are strongly stabilized within the pocket of (R)-3b, as displayed in the PES for the full catalyst (Fig. S6†). In particular, the computations predict that the products of the reaction (especially of the minor (R)-enantiomer) bind more strongly than the substrates to the catalyst (the ΔGbinding of the (R)-product is −15.3 kcal mol−1, while it is −10.3 kcal mol−1 for the best reactant complex). Therefore, the product complexes are the turnover-determining intermediates68 in this system and significant product inhibition is expected. This is in line with the long reaction times (48 h) at 80 °C that were required for complete conversion.43 Mode B is around 19 kcal mol−1 higher in free energy than mode A within (R)-3b, further confirming that only mode A is operative within the BINOL-derived catalyst. This is likely because protonation on the azetidine nitrogen ensures the benzoyl protecting group faces away from the catalyst backbone, while protonation of the carbonyl forces the benzoyl group to interfere with the catalyst (Fig. S6†) in addition to generating a worse imidic acid leaving group.
The lowest-energy transition structures leading to the (S)- and (R)-enantiomers of the product (TS 9 (S) and TS 9 (R), respectively) are shown in Fig. 4 as their Goodman33 (top row) and Terada-Himo69,70 “quadrant” (bottom row) projections. As protonation of the azetidine occurs at a roughly 109° angle from the plane of its ring, it is best positioned directly under the phosphoric acid moiety (Fig. 4B and C, top). The nucleophile adopts an orientation parallel to the C2-symmetric axis of the catalyst in one of its empty quadrants, while the benzamide leaving group occupies the opposite empty quadrant (Fig. 4B and C, bottom). Interestingly, this arrangement of the substrates under the phosphoric acid catalyst is almost identical to what was observed with the model catalyst 5 (compare Fig. 4 to Fig. 2C) and is reminiscent of oxetane, oxirane and aziridine openings.48,50–54 As was identified with the model catalyst, protonation happens trans to the azetidine substituent in TS 9 (S) and cis in TS 9 (R) (Fig. 4B and C, top). The cis approach in TS 9 (R) is accompanied by significant puckering of the C3 position in the ring (dihedral angle of 14° in TS 9 (R), only 5° in TS 9 (S)). In addition, the benzyloxy side chain adopts a partially eclipsed conformation vs. the C3 hydrogen in TS 9 (R) (dihedral angle of 40°), while a perfect staggered conformation is obtained in TS 9 (S) (dihedral angle of 58°). Both of these conformational changes likely occur to prevent clashes between the substituent and the catalyst wall, which are otherwise in close proximity. Overall, the calculations agree that the (S)-product is favored in this reaction, as TS 9 (S) is lower in free energy than TS 9 (R) by 2.0 kcal mol−1. This ΔΔG‡ value accounts for 89% e.e. at 80 °C, in excellent agreement with the 88% e.e. measured experimentally.
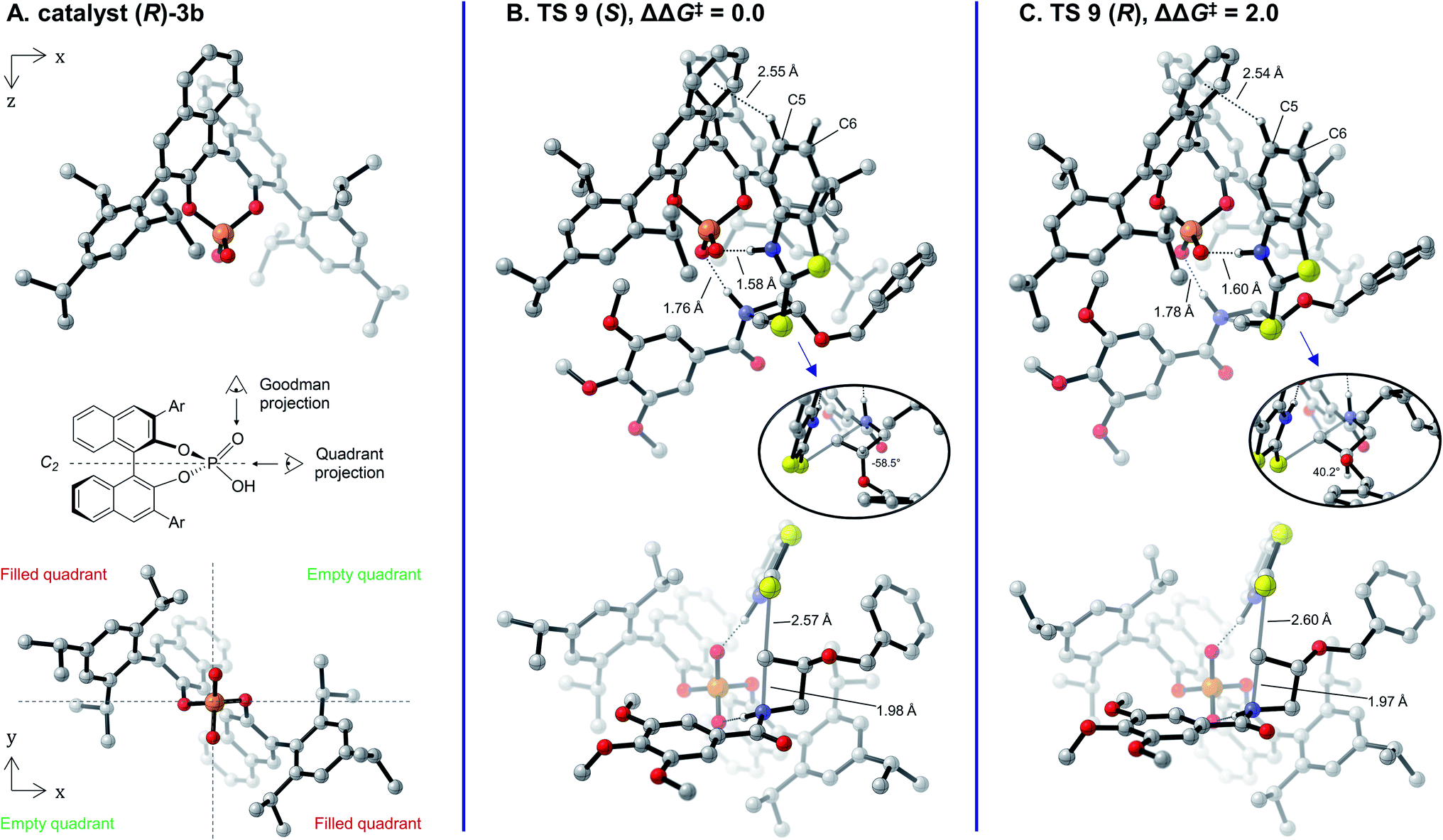 | ||
| Fig. 4 (A) Catalyst (R)-3b. (B) and (C) Lowest-energy TSs Leading to the major (S) and minor (R) products for the reaction of 1 and 2b catalyzed by (R)-3b. Top row: Goodman projection. Bottom row: quadrant projection. Insets: visualization of the H–C(3)–O–C dihedral angle of the azetidine substrate. Free energies are in kcal mol−1 and were obtained at the B97D3/6-311+G(2d,2p)/SMD(toluene)//B97D3/6-31G(d,p)/CPCM(toluene) level of theory. Non-critical hydrogen atoms are hidden in the visualized structures to improve clarity. Ar = 2,4,6-(iPr)3C6H2. | ||
Distortion/interaction (D/I) analysis71 of the TSs was performed by treating the structures as binary complexes between an anionic phosphate catalyst, and a cationic transition complex (azetidine + nucleophile, Fig. S8†). Due to the charged nature of the fragments, D/I analysis performed under various levels of theory gave different values for each component, although all methods agreed that TS 9 (S) exhibits both lower distortion and stronger interactions than TS 9 (R). The B97D3/6-311+G(2d,2p) level without the solvation model was identified as the best method (Table S2†) for this analysis. At that level of theory, the energy difference (ΔΔE‡ = ΔE(R) – ΔE(S)) of 3.4 kcal mol−1 is split into the distortion energy of the transition complex (azetidinium + benzothiazole, ΔΔE‡dist-complex = 1.2 kcal mol−1), the interaction energy of the anionic catalyst with that complex (ΔΔE‡int = 1.7 kcal mol−1), and the distortion of the catalyst (ΔΔE‡dist-catalyst = 0.5 kcal mol−1). Therefore, the free energy difference between the two stereodetermining structures is due to a combination of distortion and interactions. The large difference in distortion energy of the transition complex can be tracked down to the above-mentioned conformational changes in the azetidinium ring, since the distortion energy difference for the benzothiazole fragment is 0.0. The large difference in interaction energy is due to unequal non-covalent interactions (NCI) as visualized through NCI plots72 using the Multiwfn software.73 In both structures, favorable van der Waals (dispersive) interaction zones are found between many regions of the catalyst structure and the benzothiazole, methoxy substituents of the benzamide, azetidine ring methylenes, or benzyloxy group of the substrates (Fig. S9†). For TS 9 (S), larger interaction zones can be seen between the aromatic ring of the benzyloxy group and isopropyl substituent of the catalyst. However, the distortion and non-covalent interactions listed above are specific to substrate 1 and thus cannot be generalized to all substrates that are compatible with this reaction. Although they explain how the stereodetermining TSs studied here have different free energies, they are not the true origins of selectivity.
Identifying the true origins of selectivity
The above analysis would indicate that the size of the substituent on the azetidine substrate is driving the selectivity, since that substituent induces distortions raising the free energy of TS 9 (R). However, the scope of the Sun transformation shows that the catalytic system tolerates a wide variety of substituents on the substrate with minimal changes to the enantioselectivity. Similarly, as the trimethoxybenzoyl protecting group on the azetidine rests in the same position in TS 9 (S) and TS 9 (R) (Fig. 4), an obvious conclusion would be that it has no effect on the selectivity. Again, that is incorrect, as less crowded protecting groups on the azetidine lead to lower experimental enantioselectivity. The major issue is that D/I analysis, NCI plots and other conventional approaches focus on the symptoms of the selectivity, namely differences in geometry and energy between the two lowest-energy enantiodetermining TSs. Thus, these approaches do not explain why the two TSs are different. Considering that TS 9 (S) exhibits the best arrangement, with the protonation occurring trans from the substituent, why is the best TS 9 (R) (2.0 kcal mol−1 less favorable than the pro-(S) TS) occurring through an arrangement with the 3-substituent cis to the catalyst instead of any of the available trans arrangements (Fig. 3)? To identify the true origins of selectivity, we believe that we must uncover why TS 9 (S) and TS 9 (R) adopt geometries that are energetically different, and not simply compare their optimized structures.To illustrate this idea that comparing the optimized TSs leading to the major and minor products only displays the consequences of the true origin of selectivity, we turned to visualizations based on the steric maps pioneered by Cavallo.74–79 Such visualizations have been applied successfully for transition metal-catalyzed reactions, but not to organocatalyzed transformations.28,80–84 Using the SambVca 2.1 tool,85 the arrangements displayed with the full catalyst (R)-3b were compared to those using the model dimethoxyphosphoric acid catalyst 5. As mentioned above, for the pro-(S) TSs, the lowest-energy structure using the model catalyst has the same arrangement than with the full catalyst, barring minor conformational changes around the benzyloxy group. When the best model TS (TS 7-A (S), Fig. 5B) is superimposed with the catalyst (Fig. 5A, see SI for details), both the benzothiazole and the trimethoxybenzoyl groups align with empty quadrants of the catalyst pocket (Fig. 5C). Comparing this superimposition to the optimized TS 9 (S) (Fig. 5D) shows that only a minor reorganization of the flexible substituent is required, so the best structure for the (S)-leading TS fits easily in catalyst (R)-3b. This is a clear indication that the arrangements of substrates and catalysts in such TSs are governed by primary orbital interactions that dictate key bond lengths and angles. Fitting of this rigid transition complex in the catalyst pocket needs to happen before conformational changes occur to lower the distortion energy, or favorable non-covalent interactions between the substrates and catalyst are revealed.
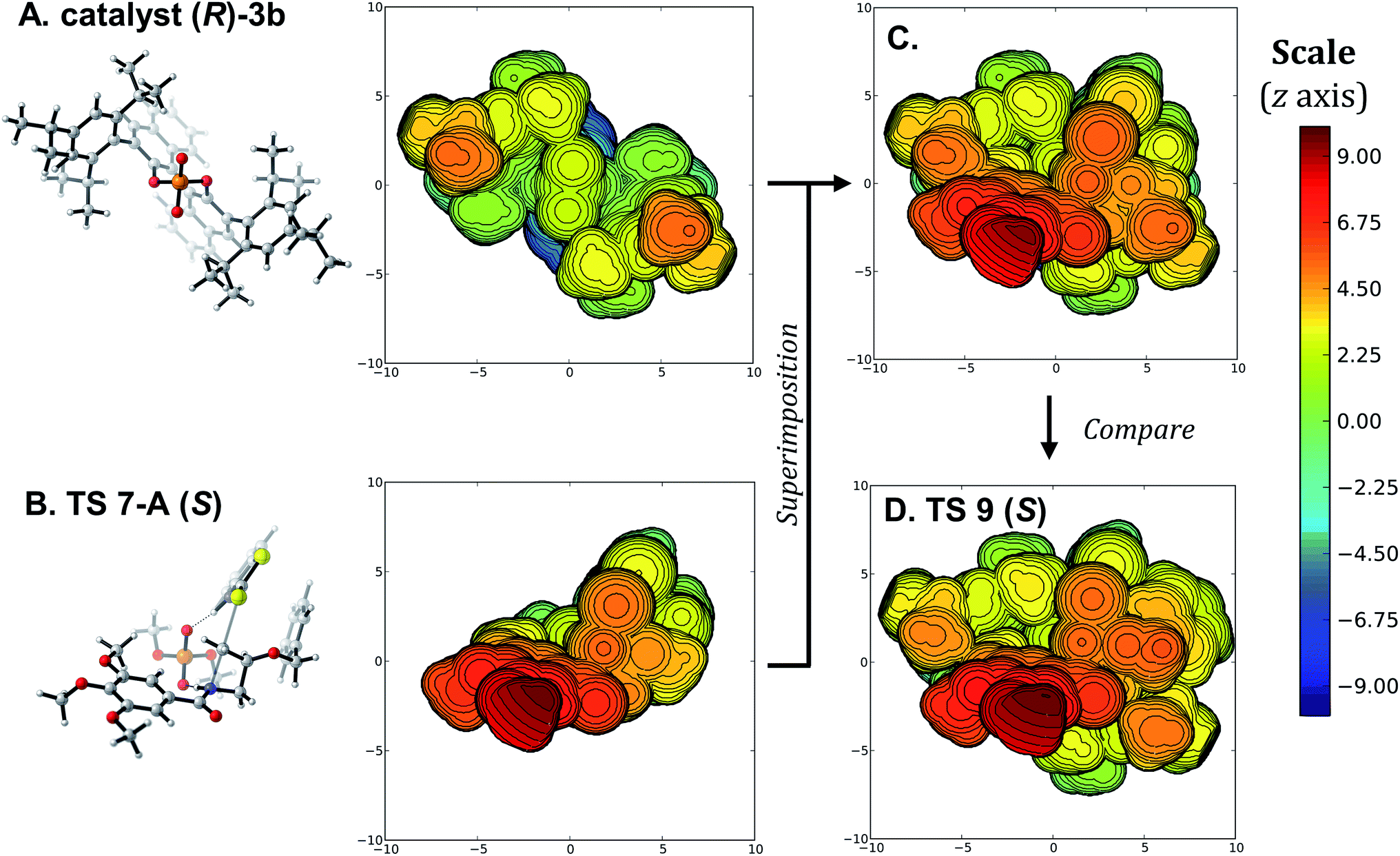 | ||
| Fig. 5 (A) and (B) Steric map visualizations for catalyst (R)-3b and TS 7-A (S), respectively. (C) Superimposition of TS 7-A (S) with catalyst (R)-3b. (D) Steric map visualization of TS 9 (S). See Fig. 4B for the regular visualization of TS 9 (S). Steric maps show the structures with their quadrant projection aligned with the z axis, visualized as a sphere of 10 Å radius with the phosphorus atom as the origin. Distances are in Å, and the distance along the z axis is color-coded from blue (−10 Å) to red (10 Å). | ||
To generate the (R)-enantiomer of the product, two main approaches are possible. First, the arrangement with cis protonation can be used, projecting the substituent toward the catalyst as was favored with the model catalyst (TS 7-A (R), Fig. 6A). Superimposition of this model structure into the full catalyst pocket shows a mostly favorable fit, placing all large and rigid groups in empty quadrants of the catalyst structure. TS 9 (R) (Fig. 6B) is very similar to that superimposition, except for conformational changes that alleviate unfavorable contacts between the substituent and crowded catalyst backbone. We then wondered why the trans arrangements did not compete favorably although they would de facto position the substituent away from the catalyst, so we optimized the located trans arrangements shown in Fig. 3 within the full catalyst (R)-3b. The resulting structures are heavily distorted and energetically unfavorable (see Fig. S7† for all structures). The best structure is derived from arrangement 3 (Fig. 6C), rotating the reaction axis (RA, green arrow) away from the phosphoric acid's C2-symmetric axis (PAA). When this arrangement is superimposed with the catalyst, the nucleophile occupies an empty quadrant (blue arrow #1), but the benzoyl group clashes with the catalyst wall (orange arrow #2). Full optimization of this structure induces large reorganizations, especially in the benzoyl group, and the final structure (Fig. 6D) is 5.1 kcal mol−1 higher in free energy than TS 9 (R). Another approach is derived from arrangement 8, bringing the RA closer to the PAA (Fig. 6E). This positions the nucleophile in an empty quadrant (blue arrow #1), but results in clashes between both the benzyloxy substituent (orange arrow #2) and the benzoyl group (orange arrow #3) with the two walls of the catalyst. The resulting fully-optimized structure (Fig. 6F) is 7.8 kcal mol−1 less stable than TS 9 (R), as it requires significant distortion of the catalyst structure. Therefore, none of the trans arrangements, which should in principle be preferable, actually fit in the catalyst pocket due to the large benzoyl substituent.
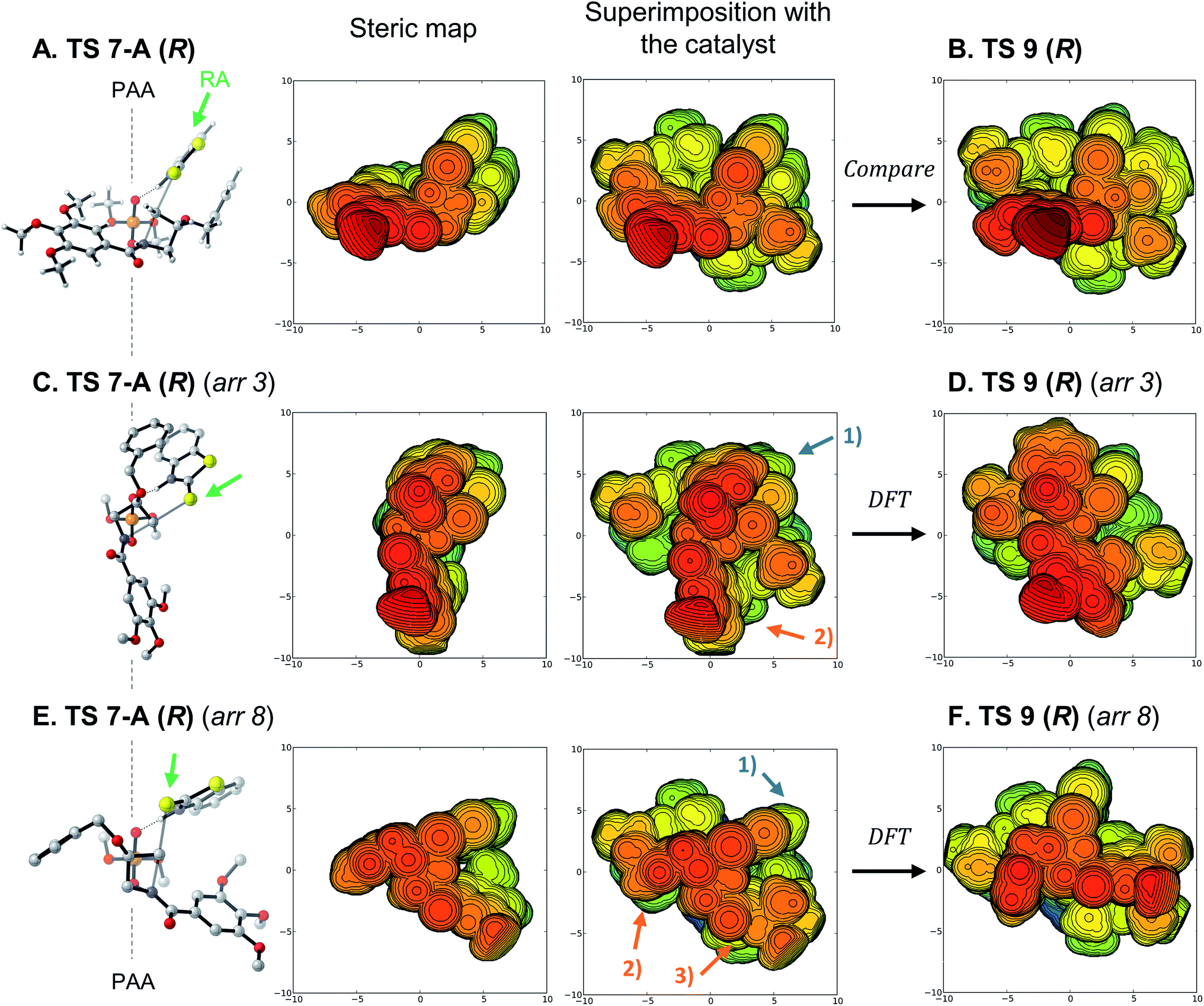 | ||
| Fig. 6 (A), (C) and (E) From left to right: quadrant projection and steric map of TS 7-A (R) arrangements, superimposition of the structures on catalyst (R)-3b. (B), (D) and (F) DFT-optimized TS arrangements in the full catalyst. PAA: phosphoric acid C2-symmetric axis; RA: reaction axis (green arrows); DFT: DFT optimization of the structure. Steric maps show the structures with the same orientation, scale and color code as Fig. 5. See Fig. 4B and S7† for the regular visualizations of TS 9 (R) and arrangements. | ||
Model of selectivity
To fit into the catalyst pocket, the important features from the transformation's strict primary orbital requirements are the following: (A) positioning of the TS as to align the SN2 axis with the empty quadrants. The alignment of the reaction axis with the empty quadrant is general to intermolecular openings of strained sp3-hybridized electrophiles, regardless of the nature of the nucleophile.39,50–53 (B) Positioning the benzothiazole nucleophile perpendicular to the reaction axis, where it can engage in favorable C–H⋯π interactions86 with the catalyst's binaphthyl backbone (see Fig. 4B and C, top). Interestingly, this would explain why substituents at the 4- or 5-positions of the benzothiazole were not well tolerated in this reaction.43 (C) Positioning of the azetidine's benzoyl group in an empty quadrant. (D) Positioning of the 3-substituent of the meso-azetidine trans from the catalyst structure. DFT studies of similar systems have consistently shown that ring substituents preferentially point away from the catalyst.39,50–53 As explained above, these features could only be identified by considering all of the available arrangements of the TS leading to the minor product (Fig. 6), and not from direct comparison of the lowest-energy structures (Fig. 4).Based on these features, a general model of selectivity for this reaction is proposed in Fig. 7. With the (R)-BINOL- (or (S)-SPINOL-)18,87 derived catalysts, the pro-(S) TS meets all criteria and is predicted to be major (Fig. 7B-I). The pro-(R) TS cannot simply mimic the arrangement of TS (S), since major steric repulsions would occur (Fig. 7B-II). This TS is thus disfavored, and will find a new arrangement allowing formation of the (R)-product, albeit at some energetic cost. Fulfilling criteria (A), (B) and (D) (Fig. 7B-IV) is also disfavored due to the important repulsions between the benzoyl group and the catalyst wall. In the specific case we studied here, the best arrangement for the minor TS involves fulfilling criteria (A), (B) and (C), while positioning the 3-substituent of the azetidine toward the catalyst (Fig. 7B-III).
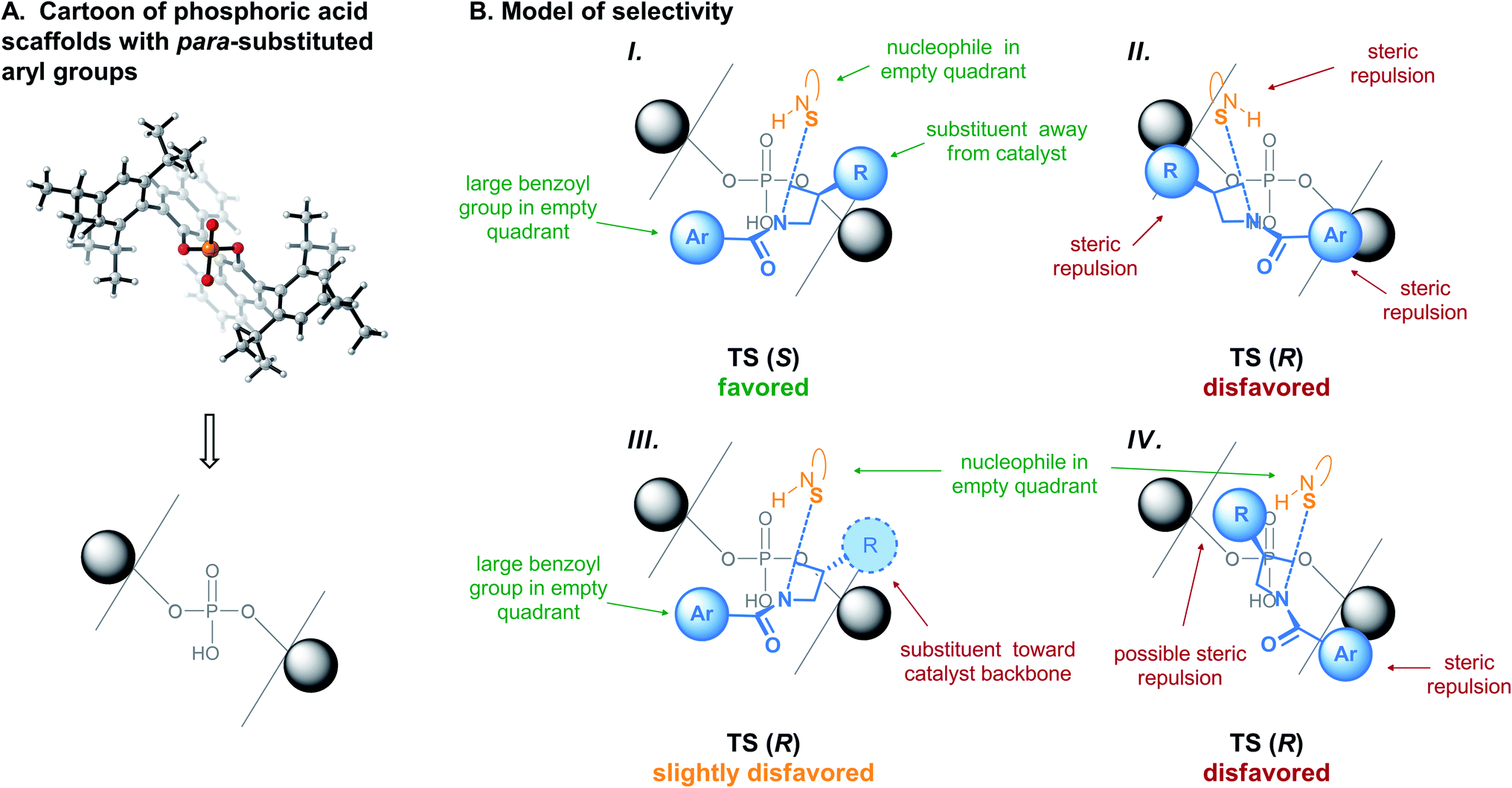 | ||
| Fig. 7 (A) Cartoon of chiral phosphoric acids seen from the quadrant projection. (B) General model of selectivity for (R)-BINOL- or (S)-SPINOL-derived phosphoric acid-catalyzed azetidine desymmetrizations. | ||
That the ideal arrangement is not possible for TS (R) does not imply that the lowest-energy (R)-yielding TS will always involve the exact arrangement as shown in Fig. 7B-III. In fact, depending on the nature of the substrates and catalyst, another arrangement could become more stable or new distortions/interactions could impact the energy. To illustrate this point, we have re-optimized TS 9 (S), TS 9 (R), and all other arrangements optimized with (R)-3b, replacing the three methoxy substituents on the azetidine's benzoyl group with hydrogens. This substrate had been tested by Sun and provided lower selectivity than 1.43 Upon re-optimization, the pro-(R) TS derived from arrangement 3 is only 2.2 kcal mol−1 less stable than the one derived from TS 9 (R), itself only 1.5 kcal mol−1 higher than the one derived from TS 9 (S) (Fig. S10 and Table S3†). This matches the experimental observation of lower selectivity, but also indicates that alternative arrangements are much closer in energy than with the trimethoxybenzoyl group of 1. Thus, although the identity of the lowest-energy structures will depend on each substrate/catalyst combination, the origin of the selectivity remains the same for this reaction: the minor TS cannot accommodate all three bulky groups in their preferred positions, due to the chiral nature of the catalyst pocket. Therefore, the above model can predict which enantiomer is favored and provide a rationale for this behavior, but cannot predict which geometry the minor TS will employ. It also cannot predict a magnitude of selectivity. This is in line with other intuitive organic chemistry models for chiral phosphoric acids,33,51,54 or famous qualitative examples such as the Felkin-Anh88–90 or Corey-Bakshi-Shibata models.91–94
Conclusions
In conclusion, we have studied the chiral phosphoric acid-catalyzed intermolecular desymmetrization of meso-acyl-azetidines reported by Sun, using state-of-the-art DFT calculations. Our results indicate that the reaction is kinetically-controlled and proceeds through bifunctional activation of the azetidine nitrogen and of the thione tautomer of the mercaptobenzothiazole nucleophile. Significant catalyst poisoning explains the low reactivity observed in this system. The calculations accurately reproduce experimental selectivity and the difference in energy between the major pro-(S) and the minor pro-(R) TSs is attributed to a combination of distortions and interactions. We have shown that the true origins of the selectivity cannot be assigned simply by comparing the two stereodetermining TSs to each other since they showcase different arrangements of the substrates relative to the catalyst structure. Instead, a complete analysis of the possible arrangements and conformations was necessary. Our results showed that the transition complexes in this reaction are highly-organized, following strict primary orbital interaction requirements. Using steric maps, we have shown that only the pro-(S) arrangements can fit easily in the chiral pocket of the catalyst. Considering the preferred orientation of the reaction axis in CPA-catalyzed desymmetrizations, the bulky trimethoxybenzoyl group on the azetidine is the main functional group discriminating between potential arrangements of the minor TS, and as such is the main origin of selectivity. Our approach of comparing TSs optimized with a model catalyst to the structures optimized with the full catalyst allowed for the development of a general model for this reaction. We anticipate that this approach will be helpful in the study of other types of enantioselective transformations, and work in that direction in ongoing in our group.Data availability
The Gaussian 16 output files of all optimizations and frequency calculations presented in this article are archived on Zenodo at https://doi.org/10.5281/zenodo.4923662.Author contributions
P. A. C. conceived the project, performed and analyzed the calculations, and prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Startup funds from NJIT are gratefully acknowledged. Calculations for this research were performed on the Kong and Lochness clusters at NJIT and on the Extreme Science and Engineering Discovery Environment (XSEDE, allocation number TG-CHE190061), which is supported by the National Science Foundation (grant number ACI-1548562). P. A. C thanks Dr Floris Buttard (NJIT) and Prof. Shuming Chen (Oberlin College) for their critical comments and suggestions during the preparation of this manuscript.References
- B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5348–5355 CrossRef CAS.
- I. Ojima, Catalytic asymmetric synthesis, John Wiley & Sons, Hoboken, NJ, 2010 Search PubMed.
- K. Houk, M. Paddon-Row, N. Rondan, Y. Wu, F. Brown, D. Spellmeyer, J. Metz, Y. Li and R. Loncharich, Science, 1986, 231, 1108–1117 CrossRef CAS.
- L. Simon and J. M. Goodman, Org. Biomol. Chem., 2011, 9, 689–700 RSC.
- Y.-h. Lam, M. N. Grayson, M. C. Holland, A. Simon and K. N. Houk, Acc. Chem. Res., 2016, 49, 750–762 CrossRef CAS PubMed.
- A. J. Sterling, S. Zavitsanou, J. Ford and F. Duarte, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2021, e1518, DOI:10.1002/wcms.1518.
- A. Milo, A. J. Neel, F. D. Toste and M. S. Sigman, Science, 2015, 347, 737–743 CrossRef CAS.
- A. J. Neel, A. Milo, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 3863–3875 CrossRef CAS PubMed.
- J. P. Reid, R. S. J. Proctor, M. S. Sigman and R. J. Phipps, J. Am. Chem. Soc., 2019, 141, 19178–19185 CrossRef CAS PubMed.
- J. P. Reid and M. S. Sigman, Nature, 2019, 571, 343–348 CrossRef CAS.
- M. D. Levin, J. M. Ovian, J. A. Read, M. S. Sigman and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 14831–14837 CrossRef CAS.
- J. Miró, T. Gensch, M. Ellwart, S.-J. Han, H.-H. Lin, M. S. Sigman and F. D. Toste, J. Am. Chem. Soc., 2020, 142, 6390–6399 CrossRef.
- S. G. Robinson, X. Wu, B. Jiang, M. S. Sigman and S. Lin, J. Am. Chem. Soc., 2020, 142, 18471–18482 CrossRef CAS PubMed.
- C.-C. Tsai, C. Sandford, T. Wu, B. Chen, M. S. Sigman and F. D. Toste, Angew. Chem., Int. Ed., 2020, 59, 14647–14655 CrossRef CAS.
- J. Werth and M. S. Sigman, J. Am. Chem. Soc., 2020, 142, 16382–16391 CrossRef CAS.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed.
- K. N. Houk and B. List, Acc. Chem. Res., 2004, 37, 487 CrossRef CAS.
- D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- R. Maji, S. C. Mallojjala and S. E. Wheeler, Chem. Soc. Rev., 2018, 47, 1142–1158 RSC.
- L. Simón and J. M. Goodman, J. Org. Chem., 2010, 75, 589–597 CrossRef.
- T. Hirata and M. Yamanaka, Chem.–Asian J., 2011, 6, 510–516 CrossRef CAS PubMed.
- L. Simón and J. M. Goodman, J. Org. Chem., 2011, 76, 1775–1788 CrossRef.
- M. N. Grayson, S. C. Pellegrinet and J. M. Goodman, J. Am. Chem. Soc., 2012, 134, 2716–2722 CrossRef CAS.
- M. N. Grayson and J. M. Goodman, J. Am. Chem. Soc., 2013, 135, 6142–6148 CrossRef CAS.
- J. P. Reid and J. M. Goodman, J. Am. Chem. Soc., 2016, 138, 7910–7917 CrossRef CAS PubMed.
- L. Simon and R. S. Paton, Org. Biomol. Chem., 2016, 14, 3031–3039 RSC.
- J. P. Reid and J. M. Goodman, Org. Biomol. Chem., 2017, 15, 6943–6947 RSC.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS.
- K. Ermanis, A. C. Colgan, R. S. J. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 21091–21101 CrossRef CAS PubMed.
- C. J. Laconsay, T. J. Seguin and S. E. Wheeler, ACS Catal., 2020, 10, 12292–12299 CrossRef.
- M. A. Maskeri, A. C. Brueckner, T. Feoktistova, M. J. O'Connor, D. M. Walden, P. H.-Y. Cheong and K. A. Scheidt, Chem. Sci., 2020, 11, 8736–8743 RSC.
- B. N. Falcone and M. N. Grayson, Org. Biomol. Chem., 2021, 19, 3656–3664 RSC.
- J. P. Reid, L. Simón and J. M. Goodman, Acc. Chem. Res., 2016, 49, 1029–1041 CrossRef CAS.
- E. B. Rowland, G. B. Rowland, E. Rivera-Otero and J. C. Antilla, J. Am. Chem. Soc., 2007, 129, 12084–12085 CrossRef CAS.
- S. E. Larson, J. C. Baso, G. Li and J. C. Antilla, Org. Lett., 2009, 11, 5186–5189 CrossRef CAS PubMed.
- G. D. Sala and A. Lattanzi, Org. Lett., 2009, 11, 3330–3333 CrossRef.
- M. Senatore, A. Lattanzi, S. Santoro, C. Santi and G. D. Sala, Org. Biomol. Chem., 2011, 9, 6205–6207 RSC.
- M. R. Monaco, B. Poladura, M. Diaz de Los Bernardos, M. Leutzsch, R. Goddard and B. List, Angew. Chem., Int. Ed., 2014, 53, 7063–7067 CrossRef CAS PubMed.
- S. Sun, Z. Wang, S. Li, C. Zhou, L. Song, H. Huang and J. Sun, Org. Lett., 2021, 23, 554–558 CrossRef CAS PubMed.
- Z. Wang, W. K. Law and J. Sun, Org. Lett., 2013, 15, 5964–5966 CrossRef CAS PubMed.
- M. R. Monaco, S. Prévost and B. List, J. Am. Chem. Soc., 2014, 136, 16982–16985 CrossRef CAS PubMed.
- S. Liao, M. Leutzsch, M. R. Monaco and B. List, J. Am. Chem. Soc., 2016, 138, 5230–5233 CrossRef CAS PubMed.
- Z. Wang, F. K. Sheong, H. H. Y. Sung, I. D. Williams, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 5895–5898 CrossRef CAS PubMed.
- Z. Chen, Z. Wang and J. Sun, Chem.–Eur. J., 2013, 19, 8426–8430 CrossRef CAS PubMed.
- Z. Wang, Z. Chen and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 6685–6688 CrossRef CAS PubMed.
- W. Yang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 1868–1871 CrossRef CAS.
- W. Yang, Z. Wang and J. Sun, Angew. Chem., Int. Ed., 2016, 55, 6954–6958 CrossRef CAS PubMed.
- R. Zhang, W. Guo, M. Duan, K. N. Houk and J. Sun, Angew. Chem., Int. Ed., 2019, 58, 18055–18060 CrossRef CAS.
- X. Zou, G. Sun, H. Huang, J. Wang, W. Yang and J. Sun, Org. Lett., 2020, 22, 249–252 CrossRef CAS.
- M. J. Ajitha and K.-W. Huang, Org. Biomol. Chem., 2015, 13, 10981–10985 RSC.
- P. A. Champagne and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 12356–12359 CrossRef CAS PubMed.
- T. J. Seguin and S. E. Wheeler, ACS Catal., 2016, 6, 2681–2688 CrossRef CAS.
- T. J. Seguin and S. E. Wheeler, ACS Catal., 2016, 6, 7222–7228 CrossRef CAS.
- R. Maji, P. A. Champagne, K. N. Houk and S. E. Wheeler, ACS Catal., 2017, 7, 7332–7339 CrossRef CAS.
- A. Borissov, T. Q. Davies, S. R. Ellis, T. A. Fleming, M. S. W. Richardson and D. J. Dixon, Chem. Soc. Rev., 2016, 45, 5474–5540 RSC.
- H. Mughal and M. Szostak, Org. Biomol. Chem., 2021, 19, 3274–3286 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- F. Li, T. Korenaga, T. Nakanishi, J. Kikuchi and M. Terada, J. Am. Chem. Soc., 2018, 140, 2629–2642 CrossRef CAS.
- L. Zhu, H. Yuan and J. Zhang, J. Catal., 2020, 383, 230–238 CrossRef CAS.
- S. Wang, A. J. Arguelles, J.-H. Tay, M. Hotta, P. M. Zimmerman and P. Nagorny, Chem.–Eur. J., 2020, 26, 4583–4591 CrossRef CAS.
- S. Grimme, C. Bannwarth, S. Dohm, A. Hansen, J. Pisarek, P. Pracht, J. Seibert and F. Neese, Angew. Chem., Int. Ed., 2017, 56, 14763–14769 CrossRef CAS PubMed.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009, http://www.cylview.org Search PubMed.
- J. Lai, M. Fianchini and M. A. Pericàs, ACS Catal., 2020, 10, 14971–14983 CrossRef CAS.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS.
- I. D. Gridnev, M. Kouchi, K. Sorimachi and M. Terada, Tetrahedron Lett., 2007, 48, 497–500 CrossRef CAS.
- T. Marcelli, P. Hammar and F. Himo, Chem.–Eur. J., 2008, 14, 8562–8571 CrossRef CAS PubMed.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 2009, 1759–1766 CrossRef.
- A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem.–Eur. J., 2010, 16, 14348–14353 CrossRef CAS.
- F. Ragone, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2010, 132, 4249–4258 CrossRef CAS PubMed.
- P. Wucher, L. Caporaso, P. Roesle, F. Ragone, L. Cavallo, S. Mecking and I. Göttker-Schnetmann, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8955–8959 CrossRef CAS.
- A. Poater, L. Falivene, C. A. Urbina-Blanco, S. Manzini, S. P. Nolan and L. Cavallo, Procedia Comput. Sci., 2013, 18, 845–854 CrossRef.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- P. Liu, J. Montgomery and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 6956–6959 CrossRef CAS PubMed.
- X. Hong, P. Liu and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 1456–1462 CrossRef CAS.
- G. Lu, C. Fang, T. Xu, G. Dong and P. Liu, J. Am. Chem. Soc., 2015, 137, 8274–8283 CrossRef CAS.
- G. Huang and P. Liu, ACS Catal., 2016, 6, 809–820 CrossRef CAS.
- A. Gomez-Suarez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html .
- E. H. Krenske and K. N. Houk, Acc. Chem. Res., 2013, 46, 979–989 CrossRef CAS.
- (R)-BINOL and (+)-SPINOL catalysts have the same orientation of their axial chirality, hence why the model is applicable to both.
- O. E. Nguyen Trong Anh, Nouv. J. Chim., 1977, 1, 61–70 CAS.
- Y. D. Wu and K. N. Houk, J. Am. Chem. Soc., 1987, 109, 908–910 CrossRef CAS.
- R. J. Smith, M. Trzoss, M. Bühl and S. Bienz, Eur. J. Org. Chem., 2002, 2002, 2770–2775 CrossRef.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551–5553 CrossRef CAS.
- V. Nevalainen, Tetrahedron: Asymmetry, 1994, 5, 289–296 CrossRef CAS.
- G. J. Quallich, J. F. Blake and T. M. Woodall, J. Am. Chem. Soc., 1994, 116, 8516–8525 CrossRef CAS.
- G. Alagona, C. Ghio, M. Persico and S. Tomasi, J. Am. Chem. Soc., 2003, 125, 10027–10039 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full computational details, additional figures and discussion, energies and XYZ coordinates of all computed structures. See DOI: 10.1039/d1sc04969k |
| This journal is © The Royal Society of Chemistry 2021 |