
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inhibition of [FeFe]-hydrogenase by formaldehyde: proposed mechanism and reactivity of FeFe alkyl complexes†
Fanjun
Zhang
 *,
Toby J.
Woods
,
Lingyang
Zhu
and
Thomas B.
Rauchfuss
*
*,
Toby J.
Woods
,
Lingyang
Zhu
and
Thomas B.
Rauchfuss
*
School of Chemical Sciences, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. E-mail: rauchfuz@illinois.edu
First published on 16th November 2021
Abstract
The mechanism for inhibition of [FeFe]-hydrogenases by formaldehyde is examined with model complexes. Key findings: (i) CH2 donated by formaldehyde covalently link Fe and the amine cofactor, blocking the active site and (ii) the resulting Fe-alkyl is a versatile electrophilic alkylating agent. Solutions of Fe2[(μ-SCH2)2NH](CO)4(PMe3)2 (1) react with a mixture of HBF4 and CH2O to give three isomers of [Fe2[(μ-SCH2)2NCH2](CO)4(PMe3)2]+ ([2]+). X-ray crystallography verified the NCH2Fe linkage to an octahedral Fe(II) site. Although [2]+ is stereochemically rigid on the NMR timescale, spin-saturation transfer experiments implicate reversible dissociation of the Fe–CH2 bond, allowing interchange of all three diastereoisomers. Using 13CH2O, the methylenation begins with formation of [Fe2[(μ-SCH2)2N13CH2OH](CO)4(PMe3)2]+. Protonation converts this hydroxymethyl derivative to [2]+, concomitant with 13C-labelling of all three methylene groups. The Fe–CH2N bond in [2]+ is electrophilic: PPh3, hydroxide, and hydride give, respectively, the phosphonium [Fe2[(μ-SCH2)2NCH2PPh3](CO)4(PMe3)2]+, 1, and the methylamine Fe2[(μ-SCH2)2NCH3](CO)4(PMe3)2. The reaction of [Fe2[(μ-SCH2)2NH](CN)2(CO)4]2− with CH2O/HBF4 gave [Fe2[(μ-SCH2)2NCH2CN](CN)(CO)5]− ([4]−), the result of reductive elimination from [Fe2[(μ-SCH2)2NCH2](CN)2(CO)4]−. The phosphine derivative [Fe2[(μ-SCH2)2NCH2CN](CN)(CO)4(PPh3)]− ([5]−) was characterized crystallographically.
Introduction
The hydrogenase enzymes attract attention because they are extremely efficient catalysts for the production and utilization of hydrogen.1–4 These enzymes operate by an orchestration of protonations and electron transfers with the substrates being bound in a pocket that, at least in its Hox state, is a Frustrated Lewis Pair (FLP). These components are illustrated in Fig. 1.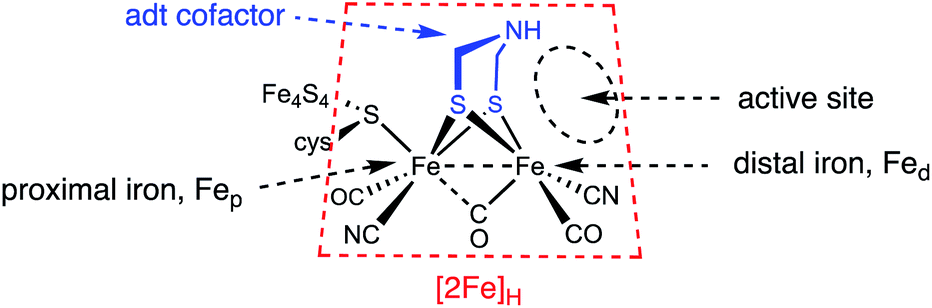 | ||
| Fig. 1 Nomenclature for the [2Fe]H active site of the [FeFe]-hydrogenases. | ||
Since these catalysts are based on iron, the most earth-abundant transition metal,5 Nature's designs promise to inspire to new synthetic catalysts that exhibit the enzyme-like activity but with more convenient molecular weight and air-sensitivity.
Given the significance of the [FeFe]-hydrogenases, many methods have been applied to elucidating their mechanism.6–10 One powerful mechanistic probe involves the use of inhibitors. Researchers from Bochum and Oxford described the reversible inhibition of the [FeFe]-hydrogenase from Clostridium acetobutylicum and Desulfovibrio desulfuricans with formaldehyde.11,12 The inhibited state was subsequently characterized for the spectroscopically simpler enzyme from Chlamydomonas reinhardtii.13 Spin resonance measurements were enabled by the presence of the S = ½ [4Fe–4S]+ center and augmented by the use of 13CH2O. Reversible inhibition is unequivocal, the molecular details of the inhibition remain uncertain. One important clue is that inhibition occurs for the reduced states of the enzyme, the oxidized states are less affected.
Scheme 1 summarizes some scenarios for the binding of CH2O at [2Fe]H.
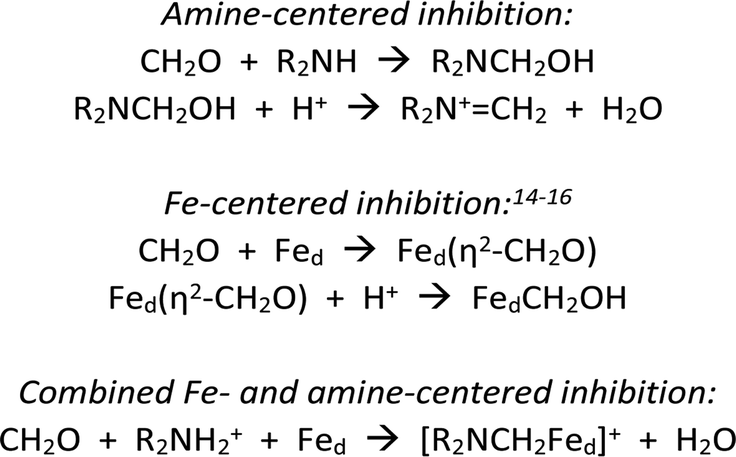 | ||
| Scheme 1 Hypotheses for inhibition of [FeFe]-hydrogenases by CH2O. | ||
The amine pathways, which involve standard organic reactions, might be relevant to what Bachmeier et al. refer to as “matrix” formaldehyde, i.e., unselective binding of formaldehyde in the vicinity of the active site. The Fe-centered reactions are precedented in organometallic chemistry, although not necessarily with iron. Bachmeier et al. favor this hypothesis. The third pathway, for which we provide evidence, involves covalent linking the amine and distal iron with a methylene bridge, locking up the [2Fe]H active site.
The model complexes used in this paper are Fe2[(μ-SCH2)2NH](CO)4L2, where L = PMe3 and CN−. These models feature the authentic azadithiolate cofactor bound to a pair of Fe(CO)2L centers. Such complexes are functional models in that they undergo protonation to give hydrides and are redox-active.17
Results and discussion
[Fe2[(μ-SCH2)2NCH2](CO)4(PMe3)2]+
Solutions of Fe2[(μ-SCH2)2NH](CO)4(PMe3)2 (1) were found to react with a mixture of HBF4 and paraformaldehyde to give [Fe2[(μ-SCH2)2NCH2](CO)4(PMe3)2]+ ([2]+). Using stoichiometric amounts of the three reagents, the conversion proceeds rapidly and in good yields at room temperature. These conditions are compatible with those reported for the enzyme. The formula of [2]+ was initially determined by ESI-MS, which showed a strong parent ion (eqn (1)).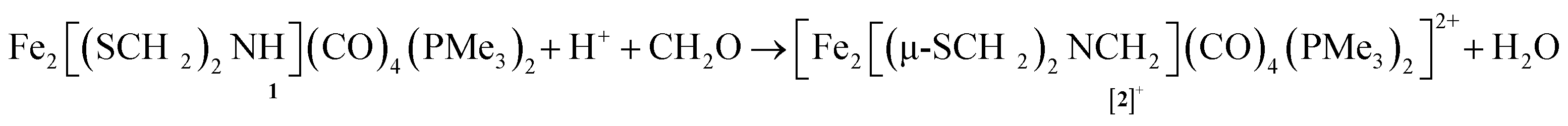 | (1) |
In a control experiment, the reaction of the propanedithiolate Fe2(μ-S2C3H6)(CO)4(PMe3)2 with HBF4 and paraformaldehyde afforded only the well-known hydride [HFe2(μ-S2C3H6)(CO)4(PMe3)2]+;18 the formaldehyde had no effect.
The structure of [2]+ was determined by an X-ray crystallographic study of its BArF4− salt (ArF = C6H3-3,5-(CF3)2) (Fig. 2). The two PMe3 ligands are trans-dibasal. Fe1 has an S2(CO)2(PMe3)(alkyl) coordination sphere. The ligand–Fe1–ligand angles are suitable for octahedral coordination, as appropriate for Fe(II). The proximal Fe center Fe2 occupies a S2(CO)2(PMe3) coordination sphere. Its coordination number is ambiguous because one CO, primarily bound to Fe1, is semi-bridging: Fe1–CO = 1.796(2) and Fe2–CO = 2.559(2) Å. The Fe1–C5–O2 angle for the semi-bridging CO ligand is 167.8(2)°, suggesting that Fe2 is weakly Lewis acidic. Analogous to terminal hydride derivatives of Fe2II(μ-SR)2 complexes,19,20 the ligand trans to alkyl is CO. In a related thioaldehyde complexes21 Fe2(μ-SR)(μ-η2-SCHR′)(diphosphine)(CO)4, CO is also trans to alkyl.22
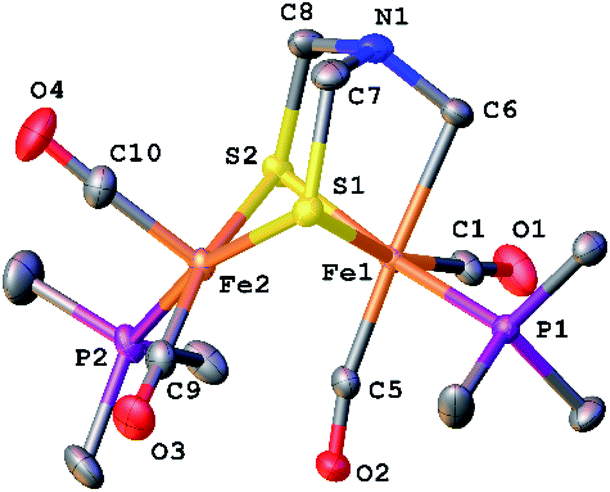 | ||
| Fig. 2 Structure of [Fe2[(μ-SCH2)2NCH2](CO)4(PMe3)2]BArF4 ([2]BArF4) with thermal ellipsoids shown at 50% probability. H atoms and BArF4− have been omitted for clarity. Selected distances and angles (Å and °): Fe1–Fe2, 2.5934(3); Fe1–C5, 1.796(2); Fe2–C5, 2.559(2); Fe1–C6, 2.168(2); N1–C6, 1.441(2); N1–C7, 1.431(3); N1–C8, 1.432(2); Fe1–C5–O2, 167.8(2); Fe1–C1–O1, 177.9(2). | ||
The NMR data for [2]+ are consistent with a stereo-rigid, chiral structure. For example, the 13C NMR spectrum shows four CO signals, three signals in the δ 211.45–210.70 region assigned to terminal CO groups, and one signal at δ 201.8 assigned to the semi-bridging CO. These 13CO signals are all coupled to 31P (JPC = 19.7 Hz), characteristic of CO cis to PMe3.23 The 13C NMR signal for the formaldehyde-derived methylene appears at δ 75.77. Its 1H NMR spectrum shows multiplets at δ 5.52 and 4.79, assigned to the diastereotopic CH2 protons. The four SCH2N protons are nonequivalent, also consistent with the low symmetry of the complex. Spin-saturation transfer experiments, which probes the exchange of signals at rates faster than 1/T1, were conducted on [2]+. Saturation of one of the SCH2 signals centered at δ 3.84 (T1 = 1.17 s for SCH2) or one of the NCH2Fe signals at δ 4.79 (T1 = 1.31 s for NCH2Fe) revealed that these sites do not exchange on the seconds time scale (Fig. S14 and S15†). As discussed below, 13C-labeling reveals that all three methylene groups do in fact exchange over the course of several minutes.
The 31P NMR data for [2]+ reveal the presence of a mixture of three (chiral) diastereoisomers in a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio. The minor diastereomers are not evident in the above-discussed 13C NMR data. If we assume that Fe1 center has CO trans to the alkyl ligand, as mentioned above, the three isomers result from the three diastereomeric sites on Fe2:
:1:1 ratio. The minor diastereomers are not evident in the above-discussed 13C NMR data. If we assume that Fe1 center has CO trans to the alkyl ligand, as mentioned above, the three isomers result from the three diastereomeric sites on Fe2:
We assume that the main isomer (A) has trans-dibasal phosphine ligands as established by X-ray crystallography. This dominant and one minor isomer (B) both show 31P–31P coupling (respectively, J = 7.4, 7.6 Hz). The two 31P NMR signals for the third isomer (minor, C) show no 31P, 31P coupling. Its unique (non)coupling is consistent with a unique structure, i.e., apical–basal disposition of the PMe3 ligands.
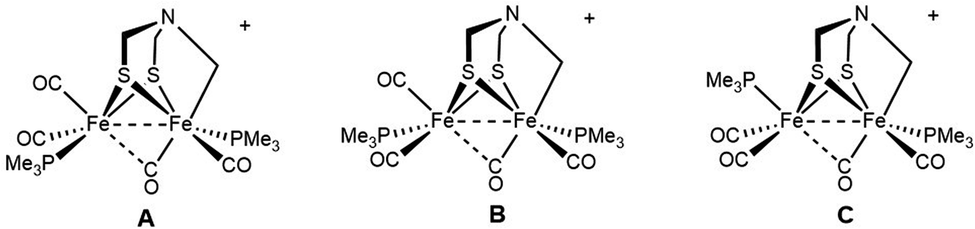
The entirety of the NMR data is accommodated by an exchange process involving reversible scission of the Fe–C bond, concomitant with regeneration of an Fe(I)Fe(I) species. Scission of the CH2–Fe bond introduces an effective plane of symmetry such that the two Fe(I) centers become equivalent (Scheme 2). Further relevant to stereodynamics, the exchange for the FeL3 sites is rapid in Fe(I)Fe(I) complexes, whereas bioctahedral Fe(II)Fe(II) complexes are more rigid.24
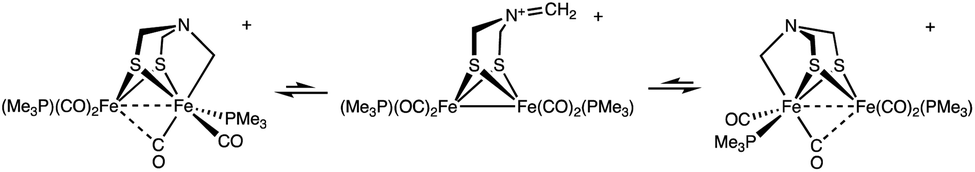 | ||
| Scheme 2 Proposed stereodynamics for [2]+. The process would proceed with racemization. | ||
Evidence of the process shown in Scheme 2 is provided by 31P NMR spin saturation experiments. The T1 of the signal at δ 9.45 was determined to be 8.2 s, and the exchange rate was k = 0.85 s−1. Saturation of either of the signals at δ 22.45 or 9.45 resulted in collapse of the other five 31P NMR signals (Fig. 3).
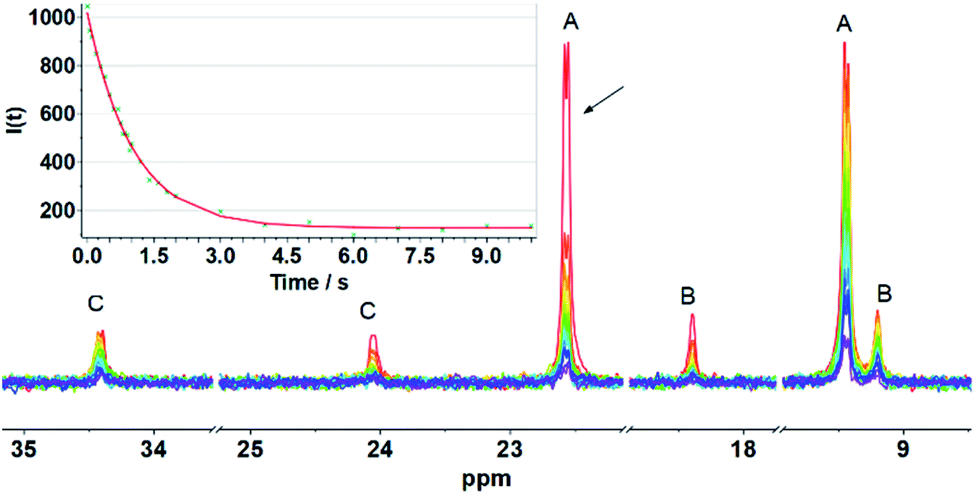 | ||
| Fig. 3 31P NMR spin saturation transfer spectra of [2]BArF4 at 298 K in CD2Cl2. Irradiation of the signal at δ 22.59. T1 = 8.2 s for the resonance at δ 9.45. Inset: graph of intensity (I) of the δ 9.45 peak vs. irradiation time at δ 22.59. Fitting: It = I0 × {1/(1 + τ/8.2) × exp[−t × (1/8.2 + 1/τ)] + 1/(1 + 8.2/τ)}, where τ = 1.178 s, k = 1/τ = 0.85 s−1. | ||
Mechanistic studies
Since Brønsted acids are required for the conversion of 1 to [2]+, we examined the ammonium complex [Fe2[(μ-SCH2)2NH2](CO)4(PMe3)2]+ ([1H]+). These results, which overlap with those reported earlier by Pickett,25 are rather fundamental and merit thorough analysis. 1H and 31P NMR spectra confirm that [1H]+ is stable in solution for days. Treating a CH2Cl2 solution of [1H]BF4 with NaBArF4 induced tautomerization to the hydride [HFe2[(μ-SCH2)2NH](CO)4(PMe3)2]+ ([H1]+). According to IR measurements, the tautomerization is complete after 3 h at room temperature (eqn (2)).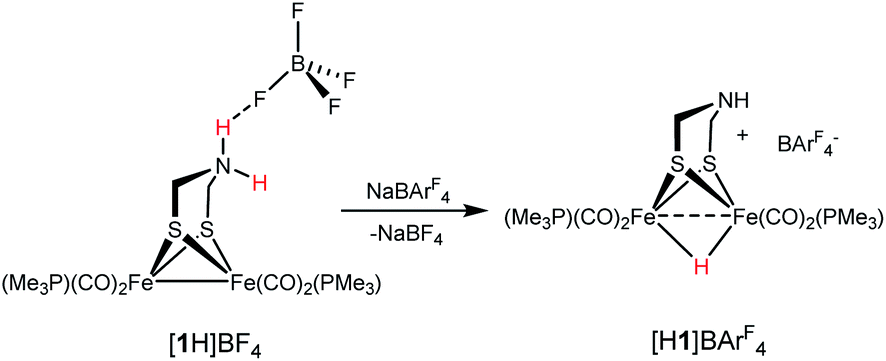 | (2) |
The 1H NMR spectrum for [H1]+ matches published data for related salts.18 The anion-dependent tautomerization reflects the stabilization of ammonium centers by hydrogen-bonding to BF4−, which persists in solution.26 One consequence of the ion-pairing (or its absence in the case of BArF4−) is that the proton-induced reaction of 1 with CH2O is sensitive to the identity of the acid: H(OEt2)BF4 cleanly gives [2]+ but H(OEt2)2BArF4, depending on the specific conditions, can afford significant quantities of the hydride [H1]+.
X-ray crystallography verified the extensive hydrogen-bonding in solid [1H]+ (Fig. 4). The asymmetric unit consists of three ion pairs; two cations have trans-dibasal phosphine ligands, one is apical–basal. All NH centers are hydrogen bonded to BF4−. The F⋯N distances range from 1.97–2.54 Å with the average distance of 2.22 Å.27
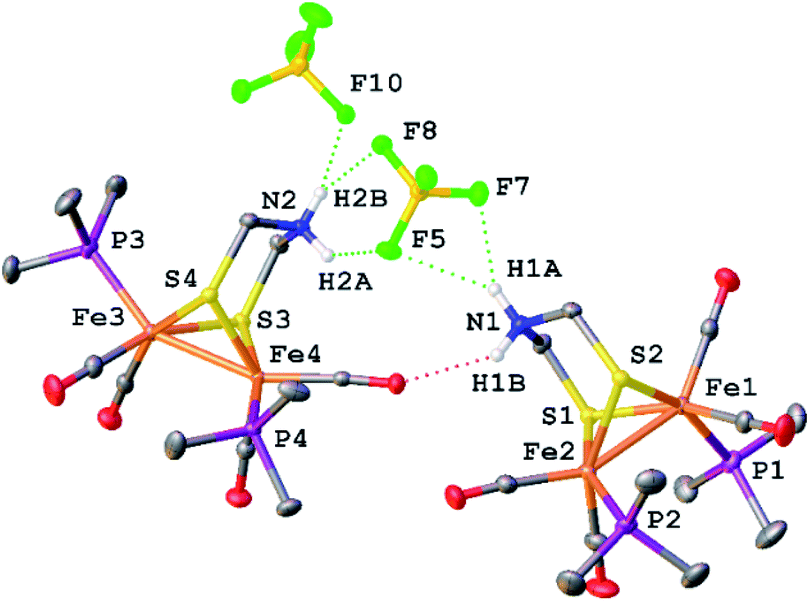 | ||
| Fig. 4 Structure of [Fe2[(μ-SCH2)2NH2](CO)4(PMe3)2]BF4 with thermal ellipsoids shown at 50% probability. H atoms except for the NH2 centres have been omitted for clarity. Two of the three ion pairs in the asymmetric unit are shown. Notice the presence of stereoisomers of [1H]+. Selected distances (Å): Fe1–Fe2, 2.5689(3); Fe3–Fe4 2.5444(3); H1A–F5, 2.54(2); H1A–F7, 2.10(2); H2A–F5, 2.30(2); H2B–F8, 2.41(2); H2B–F10, 2.00(2). | ||
The hydroxymethylation of secondary amines by formaldehyde is well studied.28 When a solution of 1 was treated with CH2O in the absence of acid, only subtle shifts (<5 cm−1) were observed in the IR spectrum in the νCO region. It is known, however, that νCO is relatively insensitive to substituents on nitrogen of the amine. For example, in this work we found that the νCO bands for Fe2[(μ-SCH2)2NR](CO)4(PMe3)2 are almost identical for R = H (1983, 1943, 1899 cm−1) and R = Me (1983, 1945, 1909, 1894 cm−1). 1H NMR spectroscopy proved to be a more sensitive indicator of the interaction of 1 and CH2O. A 1:0.25 mixture of these reactants generates ∼25% of a new species that we assign to the hydroxymethyl derivative Fe2[(μ-SCH2)2NCH2OH](CO)4(PMe3)2. The same species is observed with 13CH2O under otherwise identical conditions. In that experiment, the SCH2N groups did not show any enrichment. When 1 and CH2O were mixed in a 1:1 ratio, several species are observed, Fe2[(μ-SCH2)2NCH2OH](CO)4(PMe3)2, some unreacted 1, and what appears to be Fe2[(μ-SCH2)2N(CH2O)nCH2OH](CO)4(PMe3)2. Equilibration of these species is rapid, since the mixture reacts with 1 equiv. of HBF4 to cleanly give [2]+. No reaction was evident when 1 was treated with PhCHO, in the presence or absence of HBF4.
Treatment of a solution of 13CH2O and 1 with H(Et2O)2BArF4 gave [132]+ with selective formation of the Fe–13CH2 isotopomer. Interestingly, this label exchanges with the other methylene groups in the complex over the course of hours (Scheme 3). The kinetics of exchange are first order in [2]+ up to about 90% conversion, which points to an intramolecular process. The NMR data show that this 13CH2/12CH2 exchange affects the diastereotopic SCH2 groups equally. We suggest that exchange occurs for the Fe(I)Fe(I) species where the diastereomerization is rapid. The ESI-MS of the product of the labelling shows only singly labelled [2]+. Intermolecular processes would be expected to yield detectable levels of doubly labeled product.
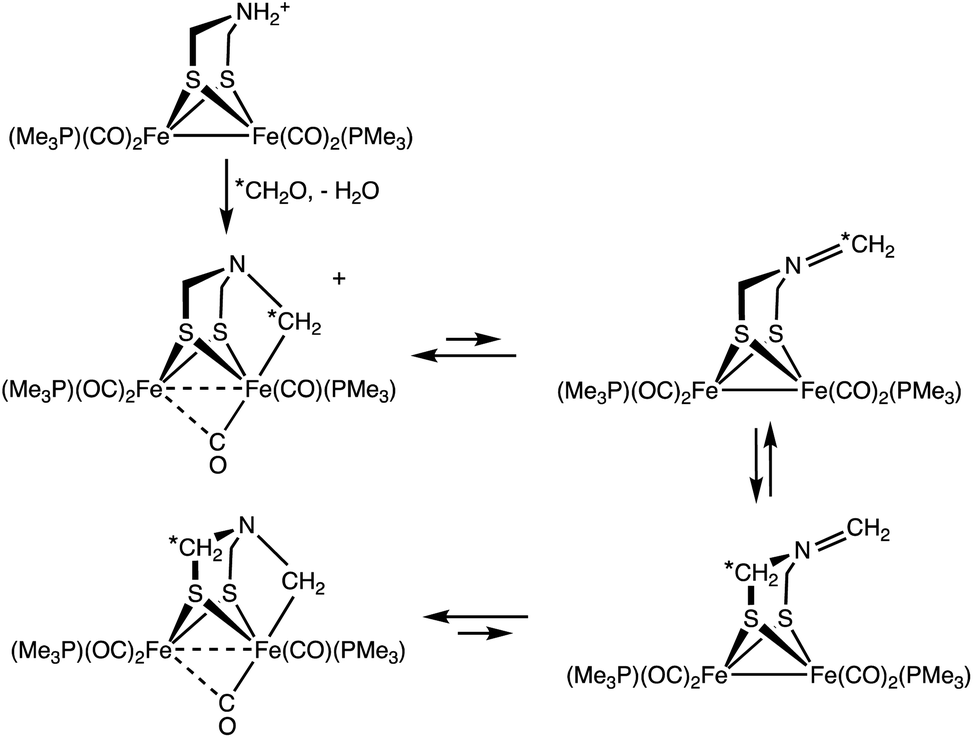 | ||
| Scheme 3 Synthesis of [2]+ using 13CH2O, showing site exchange. | ||
Reaction of [Fe2[(μ-SCH2)2NH](CN)2(CO)4]2− with CH2O/HBF4
The reaction of paraformaldehyde with [Fe2[(μ-SCH2)2NH](CN)2(CO)4]2− ([3]2−) was investigated because this complex resembles the [2Fe]H active site, which is also a dicyanide. In the presence of one equiv. of HBF4, [3]2− converts to the ammonium derivative, which is stable in MeCN solution for several minutes prior to irreversible tautomerization to the hydride (Fig. S39†). Addition of paraformaldehyde to [3H]− gives one main product, which we assign as the pentacarbonyl [Fe2[(μ-SCH2)2NCH2CN](CN)(CO)5]− ([4]−). This formula is supported by ESI-MS analysis. Attempted purification of [4]− was unsuccessful, however its FT-IR spectrum is very similar to that for [Fe2[(μ-S2C3H6)](CN)(CO)5]−. When 13CH2O was used, the singly labeled product was generated according to ESI-MS. We propose that [4]− arises by reductive elimination of the nitrile from [Fe2[(μ-SCH2)2NCH2](CN)2(CO)4]− followed by CO-scavenging (Scheme 4).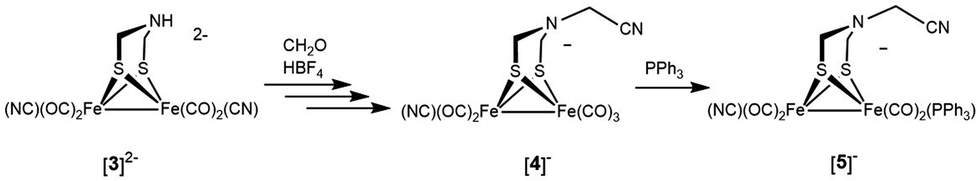 | ||
| Scheme 4 Pathway for formation of [Fe2[(μ-SCH2)2NCH2CN](CN)(CO)4(PPh3)]− ([5]−). | ||
The phosphine derivative of [4]− was obtained when the reaction of [3]2− with CH2O was conducted in the presence of PPh3 (Scheme 4). The 31P NMR signal of this product at δ 60.1 indicates coordination of PPh3, leading to the formula [Fe2[(μ-SCH2)2NCH2CN](CN)(CO)4(PPh3)]− ([5]−). In the FT-IR spectrum of [5]−, the νCO bands are shifted by 21 cm−1 toward high energy compared to dicyanide complex [3]2−. The structure of [5]− was verified by X-ray crystallography (Fig. 5), which confirms the presence of a conventional [Fe2(μ-SR)2(CN)(CO)4(PPh3)]− complex,29 and, most importantly, the presence of the cyanomethyl substituent.
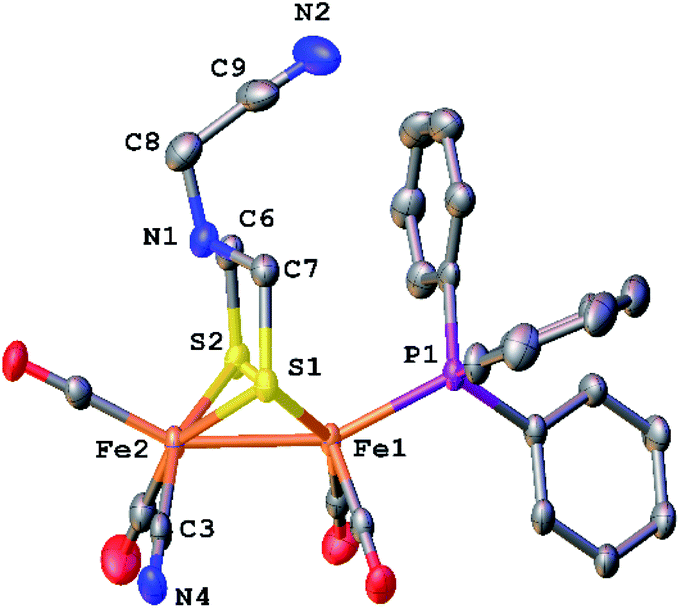 | ||
| Fig. 5 Structure of Et4N[Fe2[(μ-SCH2)2NCH2CN](CN)(CO)4(PPh3)] (Et4N[5]) with thermal ellipsoids shown at 50% probability. H atoms and the Et4N+ cation have been omitted for clarity. Selected distances (Å): Fe1–Fe2, 2.5263(8); Fe2–C3, 1.928(5); C3–N4, 1.148(6); C9–N2, 1.144(8). | ||
Reactions of methylenated FeFe complex with nucleophiles
The Fe–CH2N bond in [2]+ is electrophilic. For example, treating [2]+ with PPh3 cleanly gave [Fe2[(μ-SCH2)2NCH2PPh3](CO)4(PMe3)2]BF4 ([6]+), the result of C–P bond formation. Dealkylation of Fe involves reduction to an [Fe(I)]2 complex (eqn (3)).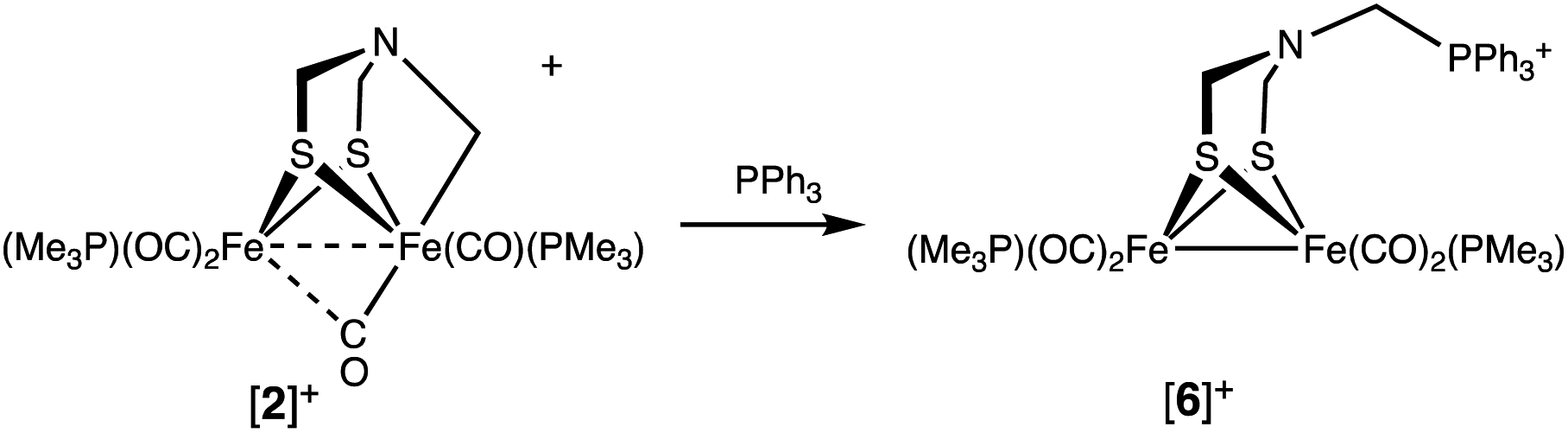 | (3) |
The PMe3 ligands in [6]+ appear equivalent, as is typical for related [Fe(I)]2 complexes. The presence of a phosphonium center is indicated by the 31P NMR singlet at δ 9.17, much higher field than δ 65.7 for Fe2(μ-S2C3H6)(CO)5(PPh3).30 In the region assigned to NCH2P, the 1H NMR spectrum features a broad signal at δ 4.90. The broadness is associated with the nonequivalent protons, each of which is coupled to 31P. The IR spectrum of [6]BF4 also agrees with reduction of Fe(II)Fe(II) to Fe(I)Fe(I): νCO shifts to lower frequency by 55 cm−1 (1978, 1948, 1903 cm−1). These frequencies are comparable to those in 1.
The structure of [6]BF4 was verified by X-ray crystallography (Fig. 6). The complex is a conventional Fe2(μ-SR)2(CO)6−xLx butterfly. The bulky phosphonium substituent is distant from the Fe2 core.
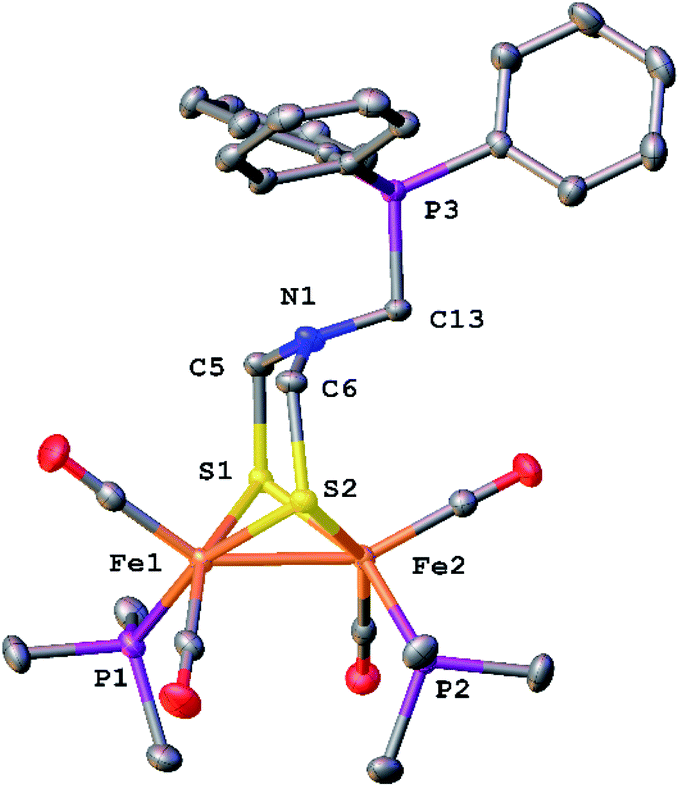 | ||
| Fig. 6 Structure of [Fe2[(μ-SCH2)2NCH2PPh3](CO)4(PMe3)2]BF4 ([6]BF4) with thermal ellipsoids shown at 50% probability. H atoms and BF4− anion have been omitted for clarity. Selected distances (Å): Fe1–Fe2, 2.5706(4); N1–C5, 1.436(3); N1–C6, 1.445(3); N1–C13, 1.449(3). | ||
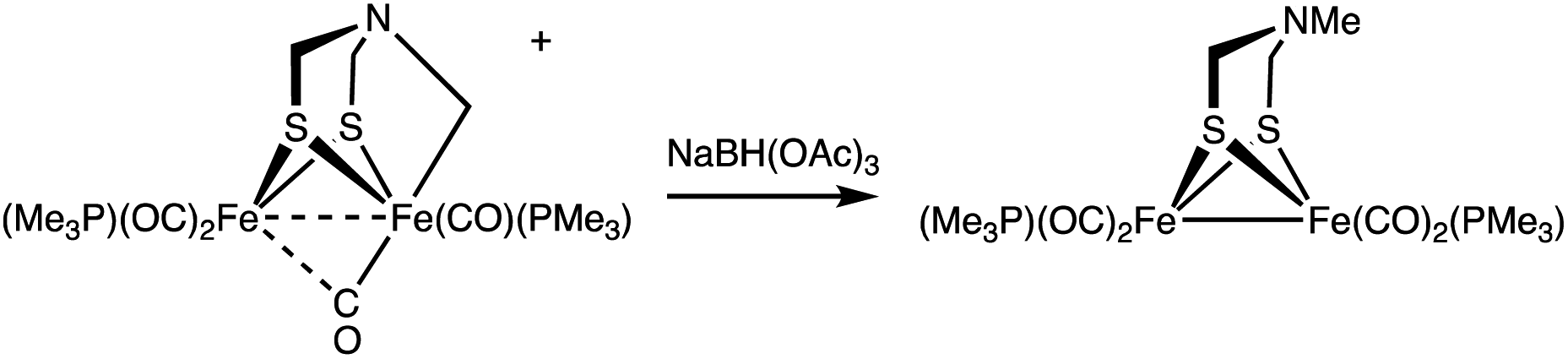
Conversion of [2]+ back to 1 was induced upon treatment with Et4NOH. From this reaction, 1 was recovered in 40% yield after purification by column chromatography. The electrophilic nature of the Fe–CH2 bond is also supported by the reaction of [2]+ with BH(OAc)3−, a mild hydride donor. In this case, Fe2[(μ-SCH2)2NMe](CO)4(PMe3)2 was obtained in good yield (eqn (4)).
 | (4) |
Conclusions
As a specific conclusion, this work provides a plausible model for the inhibition of [FeFe]-hydrogenases by formaldehyde. The methylene group donated by formaldehyde occupies both substrate binding sites, amine and the distal Fe. The methylenation proceeds by addition of the aldehyde to the secondary amine followed by generation of the iminium cation, which oxidatively adds to one of the Fe(I) centers, oxidizing the diiron site by 2e− (Scheme 5). Many examples exist for the addition of iminium cations to low-valent metals.31 The methylenation reaction does not proceed from the diiron μ-hydride. In accord with the results of Bachmeier et al.,13 the methylenation is selective for reduced state(s) of the diiron center, as required for oxidative addition.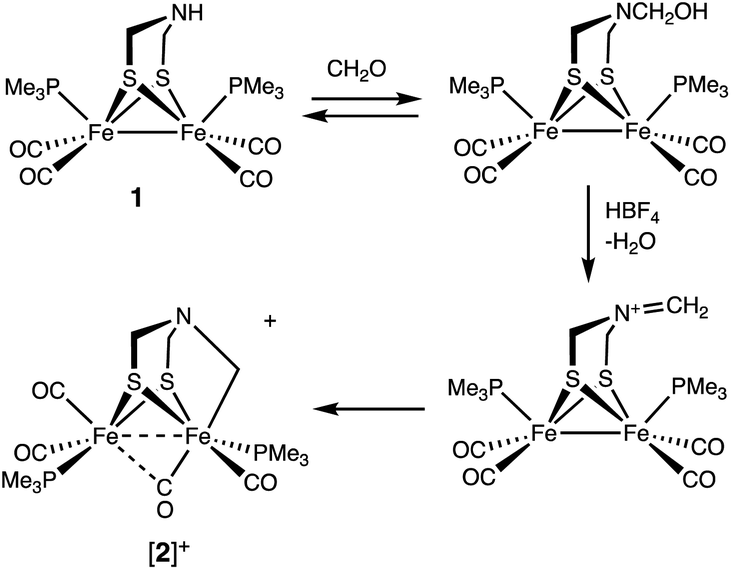 | ||
| Scheme 5 Proposed pathway for the methylenation of 1. | ||
Complex [2]+ represents a rare mimic of a terminal hydride for the [FeFe]-hydrogenases. Normally terminal hydrides of synthetic diiron dithiolates rapidly isomerize,20 which precludes extensive characterization of this key intermediate.32 In the Mulheim mechanism, the diferrous terminal hydride corresponds to Hhyd state, defined as [4Fe–4S]+–Fep(II)–amine–Fed(II)H. In this state, the hydride is protic, being reversibly deprotonated by the amine cofactor. Consistent with this model, the alkyl ligand in [2]+ is electrophilic. Furthermore, analogous to the reversible deprotonation of Hhyd, [2]+ reversibly dealkylates (see Scheme 2) to give a Fe(I)Fe(I) species as proposed for the HsredH+ state. Complex [2]+ exists as an equilibrium mixture of three isomers. The finding that these isomers are separated by less than ∼1 kcal mol−1, shows that stereochemistry of the other ligands on the diiron dithiolate has little influence on the Fe–alkyl bond (and by inference Fe–hydride bond). Also like Hhyd state, [2]+ has a highly unsymmetrical semi-bridging CO trans to R (= alkyl, hydride) on the distal Fe is persistent. In a future paper we plan to describe the redox chemistry of this electrophilic Fe(II)Fe(II) in our quest to further probe analogues of the elusive HhydH+ state.
As established by its reactions with a range of nucleophiles (Scheme 6), [2]+ presents opportunities for appending the Fe2(μ-SR)2 center to other scaffolds.
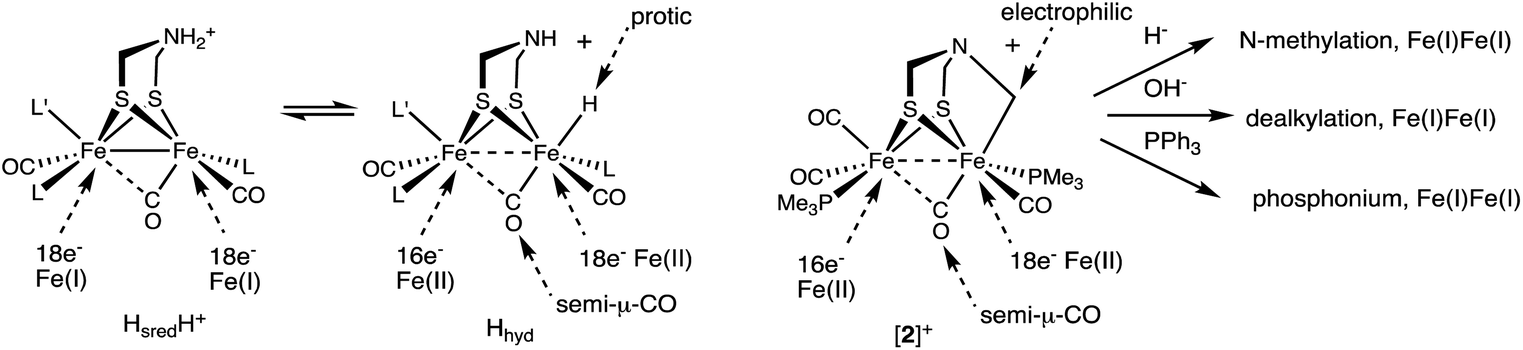 | ||
| Scheme 6 Comparison of the [2Fe]H centers in the HsredH+ and Hhyd states (L = CN−) and [2]+, including its reactions. | ||
The susceptibility of [2]+ to nucleophilic attack is reminiscent of Co(III)-alkyls as represented by vitamin B12 and its derivatives and models.33,34 Given the vast chemistry of B12 mimics, it is possible that a wide range of diiron alkyl chemistry awaits discovery and development.
Experimental
Materials and methods
Reactions were conducted in stirred solutions or slurries under nitrogen at room temperature unless otherwise indicated. Sample work-up routinely included rinsing solids with Et2O or pentane and storage under vacuum to remove traces of solvent. All reactions and purifications were conducted using standard Schlenk techniques or in an MBraun glovebox under N2. Solvents were purified using solvent purification system equipped with alumina filtration column. CD2Cl2 was degassed by freeze–pump–thaw cycles and dried using 4 Å molecular sieves. 1H, 31P{1H}, and 13C NMR spectra were recorded on Varian 500, Varian 600, Bruker 500, or Bruker Ascend 600 MHz spectrometers. Chemical shifts (δ/ppm) are referenced to residual solvent peak (5.32 ppm for 1H and 53.84 ppm for 13C in CD2Cl2). Chemical shifts (δ/ppm) for 31P{1H} NMR were calibrated using 85% H3PO4 as an external reference (0 ppm). Solution IR spectra were recorded on a PerkinElmer Spectrum 100 FTIR spectrometer. Elemental analysis was performed utilizing an Exeter CE-440 elemental Analyzer. A Waters Micromass Quattro II spectrometer was used to acquire ESI-MS data. Crystallographic data were collected on a Bruker D8 Venture kappa diffractometer equipped with a Photon II CPAD detector. An Iμs Microfocus Mo source (λ = 0.71073 Å) coupled with a multi-layer mirror monochromator provided the incident beam. Literature procedures were followed for the synthesis of (Et4N)2[Fe2[(μ-SCH2)2NH](CN)2(CO)4],35 Fe2[(μ-SCH2)2NH](CO)4(PMe3)2,25 and Fe2[(μ-SCH2)2NMe](CO)4(PMe3)2.23,25 Other chemicals were purchased from commercial sources and used without further purification.Et4N[4]. A solution of (Et4N)2[Fe2[(μ-SCH2)2NH](CN)2(CO)4] (50 mg, 0.078 mmol) in MeCN was treated with paraformaldehyde (4.7 mg, 0.016 mmol). After stirring this mixture for 2 h, a solution of H(OEt2)BF4 (13 mg, 0.078 mmol) in 2 mL of MeCN was added dropwise. The color of the reaction mixture changed from deep red to dark brown immediately. FT-IR: νCO = 2038, 2000, 1980, 1945, 1935 (sh), 1912 cm−1; νCN = 2108 cm−1. ESI-MS: m/z calcd for [M−], 423.8. Found, 423.8. When the experiment was conducted in the presence of 13CH2O, the FT-IR spectrum was the same. ESI-MS: m/z calcd for [M−], 424.8. Found, 424.8.
Et4N[5]. The experiment above was repeated using [HPPh3]BF4 (27 mg, 0.078 mmol) in place of H(OEt2)BF4. A solution of (Et4N)2[Fe2[(μ-SCH2)2NH](CN)2(CO)4] (50 mg, 0.078 mmol) in MeCN was treated with paraformaldehyde (4.7 mg, 0.016 mmol). After 2 h, the IR spectrum showed no change in the CO region. A solution of [HPPh3]BF4 (27 mg, 0.078 mmol) in 2 mL of MeCN was then added dropwise. The color of the reaction mixture changed from red to dark brown immediately. After 12 h, the color turned to red again. The mixture was then concentrated to 2 mL, and the concentrate was filtered through Celite and layered with 20 mL of Et2O. After 2 days at −30 °C, the layered solution yielded a red solid. Yield: 75% (45 mg). 1H NMR (600 MHz, CD2Cl2): δ 7.68–7.66 (m, 6H, ArH) 7.40–7.39 (m, 9H, ArH), 3.17–3.15 (q, 8H, +N(CH2CH3)4), 2.57–2.52 (br, 4H, SCH2), 2.43 (s, 2H, NCH2CN), 1.25 (t, 12H, +N(CH2CH3)4). 31P{1H} NMR (203 MHz, CD2Cl2): δ 60.06. 13C NMR (151 MHz, CD2Cl2): δ 218.28 (CO), 138.69 (d, CN), 133.71 (d, JPC = 11.5 Hz, P(C6H5)3), 129.67 (d, JPC = 2 Hz, P(C6H5)3), 128.44 (d, JPC = 9.0 Hz, P(C6H5)3), 114.82 (NCH2CN), 53.23 (+N(CH2CH3)4), 50.64 (SCH2), 46.77 (NCH2CN), 8.00 (+N(CH2CH3)4). IR (CH2Cl2): νCO = 1988, 1950, 1916 cm−1, νCN = 2081 cm−1. Anal. calcd for C35H41Fe2N4O4PS2·0.2CH2Cl2: C, 52.49; H, 5.18; N, 6.96. Found, C, 52.48; H, 5.42; N, 7.15. ESI-MS: m/z calcd for [M−], 657.9. Found, 657.9. Single crystals were grown by diffusion of Et2O into a CH2Cl2 solution at room temperature.
Data availability
All experimental and crystallographic data are available in the ESI.†Author contributions
Methodology, investigation, writing: F. Z.; conceptualisation and writing: T. B. R. Investigation: L. Z. and T. J. W. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by GM-61153 from the National Institutes of Health.Notes and references
- H. Land, M. Senger, G. Berggren and S. T. Stripp, Current State of [FeFe]-Hydrogenase Research: Biodiversity and Spectroscopic Investigations, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS
.
- J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, [FeFe]-Hydrogenases: Maturation and Reactivity of Enzymatic Systems and Overview of Biomimetic Models, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC
- A. Dutta, A. M. Appel and W. J. Shaw, Designing Electrochemically Reversible H2 Oxidation and Production Catalysts, Nat. Rev. Chem., 2018, 2, 244–252 CrossRef CAS
-
Bioinspired Catalysis, ed. Schollhammer, P. and Weigand, W., Wiley-VCH, Weinheim, 2015 Search PubMed
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Using Nature's Blueprint to Expand Catalysis with Earth-Abundant Metals, Science, 2020, 369, eabc3183 CrossRef CAS PubMed
- E. J. Reijerse, C. C. Pham, V. Pelmenschikov, R. Gilbert-Wilson, A. Adamska-Venkatesh, J. F. Siebel, L. B. Gee, Y. Yoda, K. Tamasaku, W. Lubitz, T. B. Rauchfuss and S. P. Cramer, Direct Observation of an Iron-Bound Terminal Hydride in [FeFe]-Hydrogenase by Nuclear Resonance Vibrational Spectroscopy, J. Am. Chem. Soc., 2017, 139, 4306–4309 CrossRef CAS PubMed
- V. Pelmenschikov, J. A. Birrell, C. C. Pham, N. Mishra, H. Wang, C. Sommer, E. Reijerse, C. P. Richers, K. Tamasaku, Y. Yoda, T. B. Rauchfuss, W. Lubitz and S. P. Cramer, Reaction Coordinate Leading to H2 Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory, J. Am. Chem. Soc., 2017, 139, 16894–16902 CrossRef CAS PubMed
- B. L. Greene, G. E. Vansuch, B. C. Chica, M. W. W. Adams and R. B. Dyer, Applications of Photogating and Time Resolved Spectroscopy to Mechanistic Studies of Hydrogenases, Acc. Chem. Res., 2017, 50, 2718–2726 CrossRef CAS PubMed
- V. Fourmond, N. Plumeré and C. Léger, Reversible Catalysis, Nat. Rev. Chem., 2021, 5, 348–360 CrossRef CAS
- V. Artero, G. Berggren, M. Atta, G. Caserta, S. Roy, L. Pecqueur and M. Fontecave, From Enzyme Maturation to Synthetic Chemistry: The Case of Hydrogenases, Acc. Chem. Res., 2015, 48, 2380–2387 CrossRef CAS PubMed
- C. E. Foster, T. Kramer, A. F. Wait, A. Parkin, D. P. Jennings, T. Happe, J. E. McGrady and F. A. Armstrong, Inhibition of [FeFe]-Hydrogenases by Formaldehyde and Wider Mechanistic Implications for Biohydrogen Activation, J. Am. Chem. Soc., 2012, 134, 7553–7557 CrossRef CAS PubMed
- A. F. Wait, C. Brandmayr, S. T. Stripp, C. Cavazza, J. C. Fontecilla-Camps, T. Happe and F. A. Armstrong, Formaldehyde—A Rapid and Reversible Inhibitor of Hydrogen Production by [FeFe]-Hydrogenases, J. Am. Chem. Soc., 2011, 133, 1282–1285 CrossRef CAS PubMed
- A. Bachmeier, J. Esselborn, S. V. Hexter, T. Kramer, K. Klein, T. Happe, J. E. McGrady, W. K. Myers and F. A. Armstrong, How Formaldehyde Inhibits Hydrogen Evolution by [FeFe]-Hydrogenases: Determination by 13C ENDOR of Direct Fe-C Coordination and Order of Electron and Proton Transfers, J. Am. Chem. Soc., 2015, 137, 5381–5389 CrossRef CAS PubMed
- H. Berke, G. Huttner, G. Weiler and L. Zsolnai, Struktur und Reaktivität eines Formaldehydeisen-Komplexes, J. Organomet. Chem., 1981, 219, 353–362 CrossRef CAS
- C. P. Casey, M. W. Meszaros, S. M. Neumann, I. G. Cesa and K. J. Haller, Synthesis and X-Ray Crystal Structures of an Analog Pair of Iron Formyl and Iron Acetyl Complexes, Organometallics, 1985, 4, 143–149 CrossRef CAS
- K. Toyohara, K. Tsuge and K. Tanaka, Comparison of Ru-C Bond Characters Involved in Successive Reduction of Ru-CO2 to Ru-CH2OH, Organometallics, 1995, 14, 5099–5103 CrossRef CAS
- D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS PubMed
- X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, H/D Exchange Reactions in Dinuclear Iron Thiolates as Activity Assay Models of Fe-H2ase, J. Am. Chem. Soc., 2001, 123, 9710–9711 CrossRef CAS PubMed
- J. I. van der Vlugt, T. B. Rauchfuss, C. M. Whaley and S. R. Wilson, Characterization of a Diferrous Terminal Hydride Mechanistically Relevant to the Fe-Only Hydrogenases, J. Am. Chem. Soc., 2005, 127, 16012–16013 CrossRef CAS PubMed
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, Synthetic Models for the Active Site of the [FeFe]-Hydrogenase: Catalytic Proton Reduction and the Structure of the Doubly Protonated Intermediate, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef CAS PubMed
- P. Mathur, B. Manimaran, C. V. V. Satyanarayana and B. Varghese, Synthesis, Spectroscopic and Structural Characterisation of (CO)6Fe2EE'{μ-C(H)(CH3)}2 and (CO)6Fe2{μ-EC(H)(CH3)E'} (E,E' = S, Se, Te), J. Organomet. Chem., 1997, 527, 83–91 CrossRef CAS
- D. Zheng, N. Wang, M. Wang, S. Ding, C. Ma, M. Y. Darensbourg, M. B. Hall and L. Sun, Intramolecular Iron-Mediated C-H Bond Heterolysis with an Assist of Pendant Base in a [FeFe]-Hydrogenase Model, J. Am. Chem. Soc., 2014, 136, 16817–16823 CrossRef CAS PubMed
- R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Gioia and G. Zampella, Terminal vs. Bridging Hydrides of Diiron Dithiolates: Protonation of Fe2(dithiolate)(CO)2(PMe3)4, J. Am. Chem. Soc., 2012, 134, 19260–19269 CrossRef CAS PubMed
- M. T. Olsen, T. B. Rauchfuss and S. R. Wilson, Role of the Azadithiolate Cofactor in Models for [FeFe]-Hydrogenase: Novel Structures and Catalytic Implications, J. Am. Chem. Soc., 2010, 132, 17733–17740 CrossRef CAS PubMed
- J. A. Wright, L. Webster, A. Jablonskyte, P. M. Woi, S. K. Ibrahim and C. J. Pickett, Protonation of [FeFe]-Hydrogenase Sub-Site Analogues: Revealing Mechanism Using FTIR Stopped-Flow Techniques, Faraday Discuss., 2011, 148, 359–371 RSC
- G. M. Chambers, S. I. Johnson, S. Raugei and R. M. Bullock, Anion Control of Tautomeric Equilibria: Fe–H vs. N–H Influenced by NH⋯F Hydrogen Bonding, Chem. Sci., 2019, 10, 1410–1418 RSC
- J. Emsley, Very Strong Hydrogen Bonds, Chem. Soc. Rev., 1980, 9, 91–124 RSC
- F. E. Rogers and R. J. Rapiejko, Thermochemistry of Carbonyl Addition Reactions. II. Enthalpy of Addition of Dimethylamine to Formaldehyde, J. Phys. Chem., 1974, 78, 599–603 CrossRef CAS
- F. Gloaguen, J. D. Lawrence, M. Schmidt, S. R. Wilson and T. B. Rauchfuss, Synthetic and Structural Studies on [Fe2(SR)2(CN)x(CO)6−x]x− as Active Site Models for Fe-Only Hydrogenases, J. Am. Chem. Soc., 2001, 123, 12518–12527 CrossRef CAS PubMed
- P. Li, M. Wang, C. He, G. Li, X. Liu, C. Chen, B. Åkermark and L. Sun, Influence of Tertiary Phosphanes on the Coordination Configurations and Electrochemical Properties of Iron Hydrogenase Model Complexes: Crystal Structures of [(μ-S2C3H6)Fe2(CO)6−nLn] (L = PMe2Ph, n = 1, 2; PPh3, P(OEt)3, n = 1), Eur. J. Inorg. Chem., 2005, 2005, 2506–2513 CrossRef
- W.-Y. Chu, C. P. Richers, E. R. Kahle, T. B. Rauchfuss, F. Arrigoni and G. Zampella, Imine-Centered Reactions in Imino-Phosphine Complexes of Iron Carbonyls, Organometallics, 2016, 35, 2782–2792 CrossRef CAS
- J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, Spectroscopic and Computational Evidence that [FeFe] Hydrogenases Operate Exclusively with CO-Bridged Intermediates, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed
- P. A. Butler and B. Kräutler, Biological Organometallic Chemistry of B12, Top. Organomet. Chem., 2006, 17, 1–55 CrossRef CAS
- J. Demarteau, A. Debuigne and C. Detrembleur, Organocobalt Complexes as Sources of Carbon-Centered Radicals for Organic and Polymer Chemistries, Chem. Rev., 2019, 119, 6906–6955 CrossRef CAS PubMed
- H. Li and T. B. Rauchfuss, Iron Carbonyl Sulfides, Formaldehyde, and Amines Condense To Give the Proposed Azadithiolate Cofactor of the Fe-Only Hydrogenases, J. Am. Chem. Soc., 2002, 124, 726–727 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data and crystallographic data. CCDC 2104104–2104107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05803g |
| This journal is © The Royal Society of Chemistry 2021 |