
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A photoexcited halogen-bonded EDA complex of the thiophenolate anion with iodobenzene for C(sp3)–H activation and thiolation†
Tao
Li
,
Kangjiang
Liang
 ,
Jiaying
Tang
,
Yuzhen
Ding
,
Xiaogang
Tong
and
Chengfeng
Xia
*
,
Jiaying
Tang
,
Yuzhen
Ding
,
Xiaogang
Tong
and
Chengfeng
Xia
*
Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Chemical Science and Technology, Yunnan University, Kunming 650091, China. E-mail: xiacf@ynu.edu.cn
First published on 21st October 2021
Abstract
Thiophenol was discovered to form an EDA complex with iodobenzene through halogen bonding interactions upon treatment with KOH. A direct photochemical thiolation of C(sp3)–H bond-containing etheric, allylic, and benzylic substrates with thiophenol was developed. The reaction proceeded on the basis of the in situ generation of a thiyl radical and aryl radical through single electron transfer between the photoexcited thiophenolate anion and aryl iodide EDA complex. Then a C(sp3) centred-radical was formed by aryl radical-mediated hydrogen atom transfer and the thiolation products were delivered via a radical–radical cross-coupling with the thiyl radical.
1. Introduction
A visible light-induced photoreaction generates electronically excited open-shell species which expand novel and unique organic chemistry. Since most organic molecules do not possess the ability to absorb visible light, an external photocatalyst (e.g., ruthenium and iridium salts with pyridine-derived ligands, or organic dyes) is often required for desired chemical transformation.1 As an alternative, an electron donor–acceptor (EDA) complex, which is produced by ground-state association between an electron-rich donor and an electron-deficient acceptor through electrostatic interactions, can sometimes absorb light in the visible region and an electron-transfer event can occur without the need for any photocatalysts.2 The EDA complex can be formed by several recognition interactions, such as σ-type interactions, anion–π interactions, hydrogen bonding, and halogen bonding.3 Halogen bonding is a type of noncovalent interaction between a halogen atom (halogen bonding donor) and a negative site (halogen bonding acceptor) in two different molecules.4 The noncovalent interactions could be effectively analyzed and predicted from the electrostatic potential.5 It is discovered that there is a region of positive electrostatic potential surrounded by negative electrostatic potential, termed a “σ-hole”, on the outermost portions of the halogen atom surface.6 This electron deficiency area is responsible for the formation of halogen bonding interactions with a nucleophilic entity. Meanwhile, the halogen bonding interactions feature a near-linear structure with angles primarily between 160° and 180° because the “σ-hole” is centered on the C–X axis.7 Therefore, greater directionality in halogen bonding interactions is the advantage compared with other intermolecular non-covalent interactions, such as hydrogen bonding. In particular, the iodine atom participates in noncovalent interactions with electron-rich atoms or groups to form more efficient halogen bonding in the order I > Br > Cl.6b The halogen bonding EDA complexes have been exploited as efficient protocol for the photochemical transformation.8Organosulfur compounds have received great attention in the fields of pharmaceutical chemistry, chemical biology, and advanced functional materials.9 Among those established synthetic protocols, the methods for formation of C–S bonds via functionalization of the C–H bond mainly focused on the C(sp2)–H bonds.10 The thiolation of C(sp3)–H bonds normally relied on the cross-coupling of oxidatively generated radicals with disulfides11 or in situ generated disulfides.12 The disulfides were also applied to the nickel-catalyzed thiolation of β-methyl C(sp3)–H bonds of aliphatic carboxamides.13 Very recently, Wu reported the first direct use of thiophenol for the allylic C(sp3)–H thiolation.14 Under visible light irradiation, both the allylic radical and thiyl radical were generated on the surface of photocatalyst quantum dots via hydrogen evolution followed by direct radical–radical cross-coupling to afford the allylic C(sp3)–H thiolation products.
2. Results and discussion
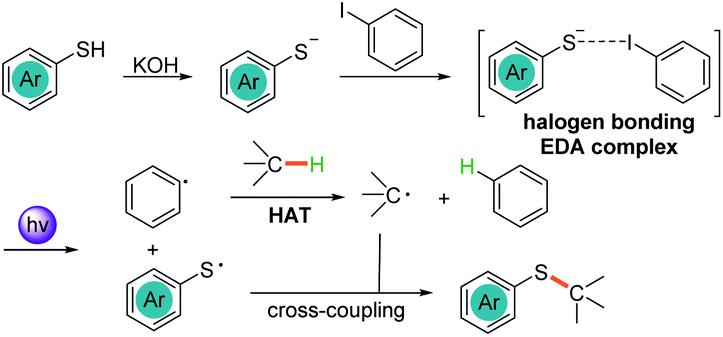
Herein, we report the photochemical etheric, allylic, benzylic, and even cycloalkyl C(sp3)–H thiolation via direct coupling with thiophenol under mild conditions. Miyake reported that the thiophenolate anion formed an electron donor–acceptor (EDA) complex via π–π interactions with aryl halides in the presence of Cs2CO3 and was excited under visible light irradiation (Scheme 1a).15 We discovered that when thiophenol 1 was treated with KOH instead of Cs2CO3 in THF, the spectrum of the thiophenolate anion showed a significant bathochromic shift with absorption tailing to the 400–450 nm region (Fig. 1a, red line). No significant shift was observed after addition of iodobenzene 2 (Fig. 1a, violet line). Similar results were obtained when the UV-visible absorption spectra were recorded in DMSO (see ESI Note 2†), and they were evidently different from Miyake's spectra of EDA complex formation. However, we observed the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between the thiophenolate anion and 2 with a binding constant Ka of 1.13 M−1 in DMSO-d6 using a 1H NMR titration method and a Job's plot analysis (Fig. 1b). We postulated that the thiophenolate anion and PhI formed an EDA complex through halogen bonding interactions8c,16 instead of π–π interactions2b,25 in Miyake's case.15 Therefore, the photo-excited thiophenolate anion–PhI EDA complex might generate the respective radicals via a single electron transfer (SET) process. We proposed that the photoexcited thiophenolate anion–PhI complex would be ideal for the C(sp3)–H thiolation by converting the aryl halide to an aryl radical, which could abstract a hydrogen from C(sp3)–H to give an alkyl radical.17 Then the alkyl radical would cross-couple with a thiyl radical to afford the C(sp3)–H thiolation products (Scheme 1b). Furthermore, we reasoned that a bulky aryl halide, such as the 4-tert-butyl-2,6-dimethyl-1-iodobenzene 2,17j would efficiently prevent the undesired coupling between the aryl radical and thiyl radical, thus avoiding the formation of C(sp2)–H thiolation products.15
:1 complex between the thiophenolate anion and 2 with a binding constant Ka of 1.13 M−1 in DMSO-d6 using a 1H NMR titration method and a Job's plot analysis (Fig. 1b). We postulated that the thiophenolate anion and PhI formed an EDA complex through halogen bonding interactions8c,16 instead of π–π interactions2b,25 in Miyake's case.15 Therefore, the photo-excited thiophenolate anion–PhI EDA complex might generate the respective radicals via a single electron transfer (SET) process. We proposed that the photoexcited thiophenolate anion–PhI complex would be ideal for the C(sp3)–H thiolation by converting the aryl halide to an aryl radical, which could abstract a hydrogen from C(sp3)–H to give an alkyl radical.17 Then the alkyl radical would cross-couple with a thiyl radical to afford the C(sp3)–H thiolation products (Scheme 1b). Furthermore, we reasoned that a bulky aryl halide, such as the 4-tert-butyl-2,6-dimethyl-1-iodobenzene 2,17j would efficiently prevent the undesired coupling between the aryl radical and thiyl radical, thus avoiding the formation of C(sp2)–H thiolation products.15
 | ||
| Scheme 1 Thiophenolate anions involved in photochemical C(sp2) thiolation (a) and C(sp3) thiolation (b). | ||
 | ||
| Fig. 1 UV-vis absorption spectrum of the thiophenolate anion (left) and 1H NMR determination of the binding stoichiometry of the thiophenolate anion–PhI complex (right). | ||
To test the feasibility of our hypothesis, we initiated an investigation by reacting thiophenol 1 and iodobenzene 2 in THF as a model photoreaction. After carefully screening the reaction conditions, we found that upon irradiating the reaction mixture with two 7 W purple LEDs (λ = 405 nm) at room temperature for 24 hours with KOH as a base, the thiolation product 3 was obtained in 84% yield with 5% yield of a C(sp2)–H thiolation byproduct (Table 1, entry 1). We also performed a 2.0 mmol scale reaction to prove the practicality of the thiolation reaction and product 3 was harvested in 72% yield after 36 hours. The formation of the thiolate anion formed was essential for this transformation because the reaction was completely inhibited in the absence of a base (entry 2). Controlled experiments revealed that no thiolation product formation was detected without light (entry 3), indicating the photochemical nature of this thiolation. The bulky phenyl iodide 2 was found to be the ideal electron acceptor while iodobenzene or 4-iodoanisole afforded lower yields with generation of the C(sp2)–H thiolation byproducts (8% and 12% yields of the byproduct formed, respectively, entries 4 and 5). Other bases, such as NaOH, CsOH·xH2O, KH, Cs2CO3, K2CO3, DBU, and TMG, were also evaluated (entries 6–12). It was found that NaOH was also an efficient base for the photochemical thiolation, albeit with a little lower yield. CsOH·xH2O only delivered the product in 28% yield. When KH was used instead of KOH, similar yield was obtained. As shown in Fig. S5 of ESI Note 2,† no significant bathochromic shift was observed in the presence of K2CO3. And we did not identify any thiolation products when the reaction mixture containing K2CO3 was irradiated under purple light (entry 10). The organic bases resulted in low yields too.
|
|
||
|---|---|---|
| Entrya | Variation from the reaction conditions | Yieldb [%] |
| a Reaction conditions: a mixture of thiophenol 1 (0.1 mmol), 4-(tert-butyl)-iodo-2,6-dimethyliodobenzene 2 (0.2 mmol), and KOH (0.2 mmol) in 1.0 mL of THF was irradiated with two 7 W purple LEDs for 24 h at room temperature. b Isolated yield. n.r. = no reaction. | ||
| 1 | None | 84 |
| 2 | Without base | n.r. |
| 3 | Without light | n.r. |
| 4 | Iodobenzene | 66 |
| 5 | 4-Iodoanisole | 78 |
| 6 | NaOH | 75 |
| 7 | CsOH·xH2O | 28 |
| 8 | KH | 81 |
| 9 | Cs2CO3 | 43 |
| 10 | K2CO3 | n.r. |
| 11 | DBU | 34 |
| 12 | TMG | 28 |
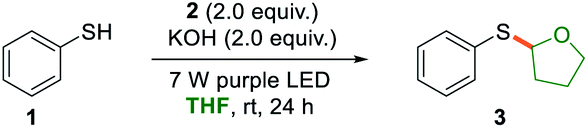
With the optimized reaction conditions in hand, we sought to evaluate the substrate scope of the thiophenolate anion induced photochemical thiolation. Different arylthiols were first examined as shown in Scheme 2. The thiophenols with different electronic nature of substitution patterns were found to be compatible and afforded the corresponding products in moderate to good yields. Under the standard conditions, arylthiols with electron-donating groups, such as alkyl and alkoxy substituted thiophenols, gave the related products in good yields (4–6). Halogenated thiophenols were also tolerated and afforded thiolation products in 51–78% yields (7–9). In addition, moderate yield was obtained when there is a phenyl substitution (10). It was discovered that the cyano group was not compatible for this thiolation and only 38% yield was delivered (11). Subsequently, several heteroarylthiols were exploited for the photochemical thiolation and gave the corresponding products in 41–72% yields (12–15). We next evaluated polycyclic thiophenols and found that naphthalene (16) and fluorene (17) were efficient coupling partners. In addition, the ortho-steric substitutions on the thiophenols did not affect the cross-coupling reaction (18 and 19).
 | ||
| Scheme 2 Substrate scope of thiophenols. Reaction conditions: a mixture of thiophenol (0.1 mmol), 2 (0.2 mmol), and KOH (0.2 mmol) in 1.0 mL of THF was irradiated with two 7 W purple LEDs for 24 h at room temperature. | ||
To explore the scope of the photochemical thiolation in cross-coupling with different C(sp3)–H bond-containing substrates, alternative solvents instead of THF should be exploited. We then performed the photoreaction in DMSO, DMF, MeCN, and acetone with 10 eq. of THF as a substrate. The experimental results showed that 65% yield was delivered in DMSO, while 13–27% yields were obtained in other solvents. As shown in Scheme 3, a series of ether compounds were first evaluated for the thiolation (20–26). Moderate yields were obtained for most of the ether compounds except the oxepane-derived product (21). We observed excellent regioselectivity for benzyl methyl ether (24) where the bond dissociation energies (BDEs) of two different C(sp3)–H bonds are significantly different. However, both of the two thiolation products 25 and 26 were generated when the 1,2-dimethoxyethane was subjected to photoreaction. Next, pyrrolidine and tetrahydrothiophene were also tested for the reaction, and thiolation products were obtained in moderate yields (27 and 28).
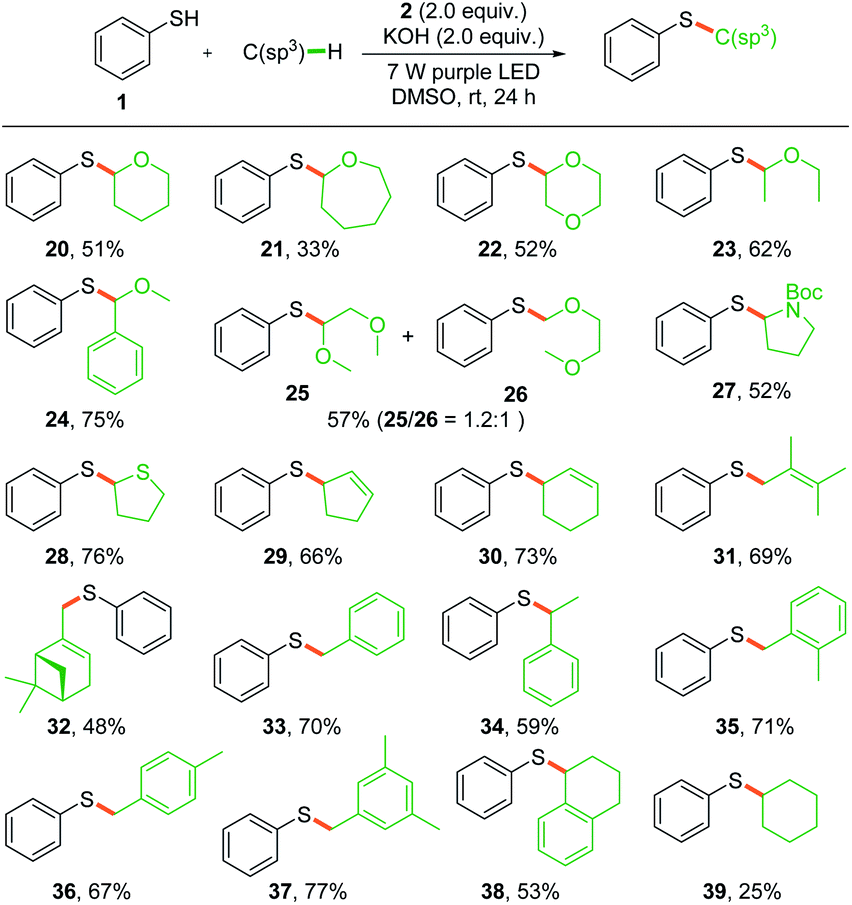 | ||
| Scheme 3 Substrate scope for C(sp3)–H bond activation. Reaction conditions: a mixture of thiophenol 1 (0.1 mmol), 2 (0.2 mmol), C(sp3)–H bond-containing substrate (1.0 mmol), and KOH (0.2 mmol) in 1.0 mL of DMSO was irradiated with two 7 W purple LEDs for 24 h at room temperature. | ||
The direct allylic C(sp3)–H thiolation is found to be difficult for the transition-metal catalyzed π-allyl metal pathway.18 Wu's pioneering photochemical radical–radical cross-coupling pathway provided an efficient method for the direct allylic C–H thiolation.14 We conjectured that the in situ generated phenyl radical would also be efficient in abstracting a hydrogen from allylic C(sp3)–H to give the allylic radical and enable direct allylic thiolation. Several substrates with allylic C(sp3)–H, such as cyclopentene, cyclohexene, 2,3-dimethyl-2-butene, and β-pinene, were tested under the standard conditions and the corresponding thiolation products were obtained in moderate yields (29–32). Moreover, the toluene derivatives were also proved to be suitable substrates for the radical–radical cross-coupling thiolation (33–38). Owing to the high BDEs, this method was found to be not effective in the thiolation of alkanes and only 25% yield was delivered for the cyclohexane (39).
The generated thiolation products, such as compound 3, are widely used in allylation, ketonylation, and other chemical conversions. To show the utility of the photochemical coupling, we also synthesized thiobenzimidazole, a selective CRTh2 receptor antagonist.19 As shown in Scheme 4, irradiation of the complex of thiol 40 with toluene in the presence of iodide 2 under basic conditions for 24 h efficiently delivered the target product 41 in 61% yield in one step.
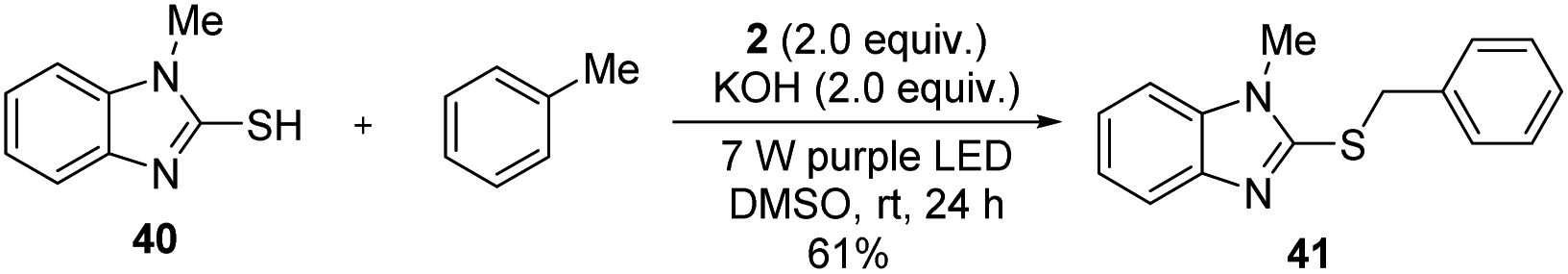 | ||
| Scheme 4 The photochemical synthesis of thiobenzimidazole. | ||
The radical pathway of the photochemical thiolation was confirmed by the controlled experiments (Scheme 4). When the radical scavenger TEMPO was added to the reaction mixture under standard reaction conditions, the thiolation was completely inhibited. Instead, the TEMPO-trapped compounds 42–44 were detected from the reaction (Scheme 5a), indicating the formation of the phenyl radical, thiophenolic radical, and the etheric radical. To verify that the etheric radical was generated through phenyl radical-mediated hydrogen atom transfer (HAT), a deuteration experiment was performed. The isolation of deuterated arene 46 undeniably validated this process (Scheme 5b). The quantum yield for the model reaction was determined to be Φ = 0.23 (see ESI Note 7†), indicating that the reported thiolation reaction was a non-chain process20 or an inefficient chain process.21
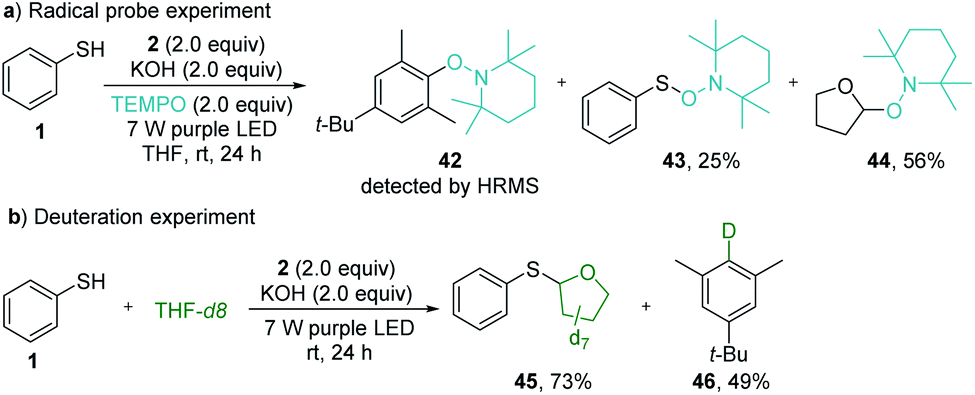 | ||
| Scheme 5 Controlled experiments. (a) Trapping radical intermediates with TEMPO. (b) Deuteration experiment for verifying the hydrogen atom transfer process. | ||
To better visualize the interactions of the thiophenolate anion and aryl iodide complex, we carried out DFT calculations as shown in Fig. 2. The electrostatic potential surfaces (ESPs) of the iodine atom contained a positive area along the extension of the Ar–I bond that was surrounded by negative electrostatic potential with a distance of 2.19 Å as mapped in GaussView 6.0.16 (ref. 22) (ESI Fig. 10†), indicating the existence of a “σ-hole” (Fig. 2a and b).4a This region was necessary for the formation of halogen bonding with negative parts of the thiophenolate anion. We noticed that the most positive value of electrostatic potential (Vs,max, 14.33 kcal mol−1) of the “σ-hole”23 was a reliable indicator for analyzing and predicting the character of a non-covalent bond.24 Its sign determined whether the positive contribution of nuclei or the negative contribution of electrons was dominant in any area of space. Since there were three parameters, d, θ, and φ, which were essential in the properties of halogen bonding, we subsequently conducted geometry optimization and interaction energy calculations of the complex intermediate. In the equilibrium structure shown in Fig. 2c, the S−⋯I interactions had a distance d of 3.46 Å which was shorter than the sum of van der Waals radii of the two interacting atoms (3.78 Å).25 Thus the S−⋯I interactions should be responsible for the production of the EDA complex. The calculated angle θ of Ar–I⋯S was 178.7°, indicating that the interactions were highly directional along the extension of the covalent Ar–I bond and were in good agreement with the nature of halogen bonding.4a The binding energy calculated was −8.61 kcal mol−1 (Table S1 in the ESI†) which demonstrated that the interaction of the thiophenolate anion and aryl iodide complex was relatively stable. The lower HOMO–LUMO energy gaps (0.22319 eV) revealed that the electron transfer process occurred favourably under visible light irradiation (Fig. 2d).
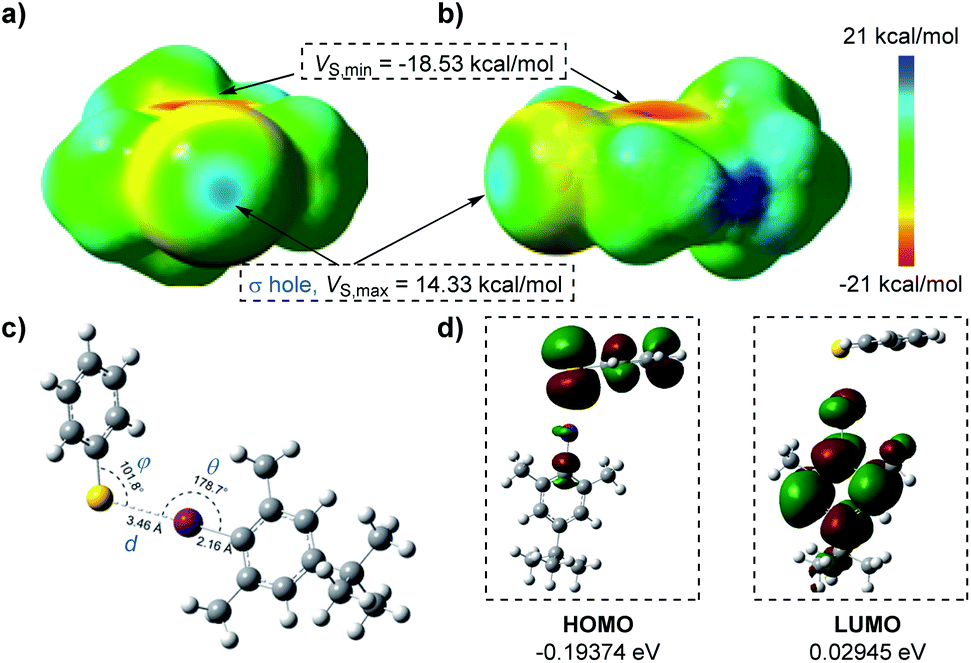 | ||
| Fig. 2 (a) and (b) Electrostatic potentials mapped on the molecular surface (electron density ρ = 0.001 electrons per bohr3, kcal mol−1) calculated at the M06-2X/def2-TZVP level; the iodine atom is pointing out of the page (a) and to the left (b). (c) Calculated geometries, bond distances, and bond angles of the thiophenolate anion and aryl iodide complex. (d) Calculated highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of the EDA complex. | ||
At the same time, the electrostatic potential over the aromatic ring was found to be extremely negative (Vs,min = −18.53 kcal mol−1) (Fig. 2a and b). These results implied that the iodobenzene 2 was not suited for establishing interactions with the thiophenolate anion (Vs,min = −124.17 kcal mol−1) (Fig. S10 in the ESI†) via a π–π system where the existence of both electron-rich and electron-deficient π systems was necessary,2b,15,26 or for establishing anion–π interactions.27 Moreover, the possible hydrogen bonding was also excluded due to the fact that no reactions were observed with the weak base Cs2CO3 (ESI Note 1†). Meanwhile, the attempted optimization for conformations of possible π–π interactions, anion–π interactions, or σ-type interactions indeed led to dissociation of the complex due to the mutual electrostatic repulsion of the two negative systems.
In order to elaborate on the interactions more clearly, we performed a Quantum Theory of Atoms in Molecules (QTAIM) analysis,28 which has been widely utilized to determine the bond critical points (BCPs) of halogen bonding interactions.27a,29 The properties at the BCPs provide a lot of information that can be used to characterize the chemical structure and nature of chemical bonds. From QTAIM calculations performed using the Multiwfn 3.8 package,30 we found the existence of BCPs that had two negative and one positive eigenvalues of Hessian (Table S2 in the ESI†). Based on the relatively low value of electron densities ρ and the positive value of Laplacian of electron density ∇2ρ at BCPs (Table 2), this kind of halogen bond can be classified as a “closed-shell” bond. Furthermore, the positive sign of energy density H meant that the interactions were not covalent but dominantly electrostatic. Additionally, the absolute value for the ratio of eigenvalues of Hessian λ1 and λ3, and that of potential energy density V and Lagrangian kinetic energy G were less than 0.25 and 1, respectively. These results illuminated explicitly the fact that the interactions of the thiophenolate anion and aryl iodide complex were closed-shell non-covalent interactions.
| Interactions | ρ | ∇2ρ | V | H | G |
|---|---|---|---|---|---|
| a BCPs = bond critical points. ρ = electron density. ∇2ρ = Laplacian of electron density. V = potential energy density. H = energy density. G = Lagrangian kinetic energy. ρ, ∇2ρ, H, V, and G are in atomic units. | |||||
| S−⋯I | 0.0134 | 0.0326 | −0.0067 | 0.0007 | 0.0074 |
On the basis of the above results, we proposed the mechanism of photoexcited C(sp3)–H bond activation and thiolation as depicted in Scheme 6. By deprotonation with KOH, the generated thiophenolate anion formed an EDA complex with iodobenzene 2via halogen bonding interactions. The photoexcited EDA complex underwent a SET process to afford two radicals, a thiyl radical and aryl radical, as shown in Scheme 6. The aryl radical then abstracted a hydrogen atom from the C(sp3)–H in the HAT pathway to deliver the alkyl radical. Finally, the radical–radical cross-coupling between the thiyl radical and the alkyl radical achieved the C(sp3)–H thiolation products.
 | ||
| Scheme 6 Proposed photochemical mechanism. | ||
3. Conclusions
In summary, we discovered that the thiophenolate anion and iodobenzene could form an EDA complex through halogen bonding interactions and be excited under visible light irradiation. A transition-metal-free photochemical thiolation of C(sp3)–H bond-containing compounds with thiophenol was successfully developed. Mechanistic studies indicated that the excited thiolate anion–PhI EDA complex underwent SET to generate a thiyl radical and aryl radical followed by a HAT between the aryl radical and C(sp3)–H bond-containing substrates to give a C(sp3) centred-radical. The radical–radical cross-coupling between the thiyl radical and C(sp3)-centred radical delivered thiolation products with high structural diversity.Data availability
All relevant experimental and computational details are provided in the ESI.†Author contributions
T. Li conducted most of the experiments and wrote the initial manuscript draft. K. Liang, J. Tang, and X. Tong performed some of the experiments. Y. Ding performed the DFT calculations. C. Xia conceptualized and directed the project and finalized the manuscript draft. All authors contributed to discussions. We are grateful for the computational resources provided by the High-Performance Computing Platform of Yunnan University.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21871228 and 22061044), the Natural Science Foundation of Yunnan Province (2018FY001015), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT_17R94).Notes and references
- For selected reviews, see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (c) M. D. Kärkäs, J. A. Porco and C. R. J. Stephenson, Chem. Rev., 2016, 116, 9683 CrossRef PubMed; (d) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41 CrossRef CAS PubMed; (e) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911 CrossRef CAS PubMed; (f) M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261 CrossRef CAS PubMed; (g) D. Staveness, I. Bosque and C. R. J. Stephenson, Acc. Chem. Res., 2016, 49, 2295 CrossRef CAS PubMed; (h) X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833 CrossRef CAS PubMed; (i) X. Lang, J. Zhao and X. Chen, Chem. Soc. Rev., 2016, 45, 3026 RSC; (j) S. Protti, S. Garbarino, D. Ravelli and A. Basso, Angew. Chem., Int. Ed., 2016, 55, 15476 CrossRef CAS PubMed; (k) J. Xie, H. Jin and A. S. K. Hashmi, Chem. Soc. Rev., 2017, 46, 5193 RSC; (l) M. Schmalzbauer, M. Marcon and B. König, Angew. Chem., Int. Ed., 2021, 60, 6270 CrossRef CAS PubMed; (m) D.-M. Yan, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 378 CrossRef CAS PubMed.
- (a) J. K. Kochi, in Advances in Physical Organic Chemistry, ed. D. Bethell, Academic Press, 1994, vol. 29, p. 185 Search PubMed; (b) G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461 CrossRef CAS PubMed; (c) C. G. S. Lima, T. d. M. Lima, M. Duarte, I. D. Jurberg and M. W. Paixão, ACS Catal., 2016, 6, 1389 CrossRef CAS; (d) L. Zheng, L. Cai, K. Tao, Z. Xie, Y.-L. Lai and W. Guo, Asian J. Org. Chem., 2021, 10, 711 CrossRef CAS; (e) Y.-q. Yuan, S. Majumder, M.-h. Yang and S.-r. Guo, Tetrahedron Lett., 2020, 61, 151506 CrossRef CAS.
- O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2007, 129, 48 CrossRef CAS PubMed.
- (a) P. Politzer, P. Lane, M. C. Concha, Y. Ma and J. S. Murray, J. Mol. Model., 2007, 13, 305 CrossRef CAS PubMed; (b) M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281 CrossRef CAS PubMed; (c) R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004 CrossRef CAS PubMed; (d) S. Scheiner, Struct. Chem., 2019, 30, 1119 CrossRef CAS.
- (a) J. S. Murray and P. Politzer, J. Mol. Struct.: THEOCHEM, 1998, 425, 107 CrossRef CAS; (b) P. Politzer, J. S. Murray and Z. Peralta-Inga, Int. J. Quantum Chem., 2001, 85, 676 CrossRef CAS.
- (a) H. Wang, W. Wang and W. J. Jin, Chem. Rev., 2016, 116, 5072 CrossRef CAS PubMed; (b) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS PubMed.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308 CrossRef CAS.
- (a) A. Postigo, Eur. J. Org. Chem., 2018, 2018, 6391 CrossRef CAS; (b) M. Schmalzbauer, T. D. Svejstrup, F. Fricke, P. Brandt, M. J. Johansson, G. Bergonzini and B. König, Chem, 2020, 6, 2658 CrossRef CAS; (c) Y. Wang, J. Wang, G.-X. Li, G. He and G. Chen, Org. Lett., 2017, 19, 1442 CrossRef CAS PubMed; (d) B. Dereka, I. Fureraj, A. Rosspeintner and E. Vauthey, Molecules, 2019, 24, 4361 CrossRef PubMed; (e) M. Bracker, L. Helmecke, M. Kleinschmidt, C. Czekelius and C. M. Marian, Molecules, 2020, 25, 1606 CrossRef CAS PubMed; (f) A. Nandy, I. Kazi, S. Guha and G. Sekar, J. Org. Chem., 2021, 86, 2570 CrossRef CAS PubMed; (g) N. Sundaravelu, A. Nandy and G. Sekar, Org. Lett., 2021, 23, 3115 CrossRef CAS PubMed.
- (a) G. Liu, J. T. Link, Z. Pei, E. B. Reilly, S. Leitza, B. Nguyen, K. C. Marsh, G. F. Okasinski, T. W. von Geldern, M. Ormes, K. Fowler and M. Gallatin, J. Med. Chem., 2000, 43, 4025 CrossRef CAS PubMed; (b) M. C. Carreno, Chem. Rev., 1995, 95, 1717 CrossRef CAS; (c) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (d) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- (a) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (b) D.-Q. Dong, S.-H. Hao, D.-S. Yang, L.-X. Li and Z.-L. Wang, Eur. J. Org. Chem., 2017, 2017, 6576 CrossRef CAS; (c) M. Pramanik, K. Choudhuri and P. Mal, Org. Biomol. Chem., 2020, 18, 8771 RSC.
- (a) R.-Y. Tang, Y.-X. Xie, Y.-L. Xie, J.-N. Xiang and J.-H. Li, Chem. Commun., 2011, 47, 12867 RSC; (b) S.-r. Guo, Y.-q. Yuan and J.-n. Xiang, Org. Lett., 2013, 15, 4654 CrossRef CAS PubMed; (c) B. Du, B. Jin and P. Sun, Org. Lett., 2014, 16, 3032 CrossRef CAS PubMed; (d) X. Zhu, X. Xie, P. Li, J. Guo and L. Wang, Org. Lett., 2016, 18, 1546 CrossRef CAS PubMed; (e) J. Kim, B. Kang and S. H. Hong, ACS Catal., 2020, 10, 6013 CrossRef CAS; (f) L. I. Panferova, M. O. Zubkov, V. A. Kokorekin, V. V. Levin and A. D. Dilman, Angew. Chem., Int. Ed., 2021, 60, 2849 CrossRef CAS PubMed.
- (a) J. Yuan, X. Ma, H. Yi, C. Liu and A. Lei, Chem. Commun., 2014, 50, 14386 RSC; (b) Q. Chen, G. Yu, X. Wang, Y. Ou and Y. Huo, Green Chem., 2019, 21, 798 RSC.
- (a) X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970 CrossRef CAS PubMed; (b) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341 RSC.
- C. Huang, R.-N. Ci, J. Qiao, X.-Z. Wang, K. Feng, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 11779 CrossRef CAS PubMed.
- B. Liu, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 13616 CrossRef CAS PubMed.
- (a) P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed; (b) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC; (c) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS PubMed; (d) X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638 CrossRef CAS PubMed.
- (a) S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974 RSC; (b) A. Clerici, R. Cannella, W. Panzeri, N. Pastori, E. Regolini and O. Porta, Tetrahedron Lett., 2005, 46, 8351 CrossRef CAS; (c) D. P. Curran and J. Xu, J. Am. Chem. Soc., 1996, 118, 3142 CrossRef CAS; (d) F.-Q. Huang, X. Dong, L.-W. Qi and B. Zhang, Tetrahedron Lett., 2016, 57, 1600 CrossRef CAS; (e) S. Du, E. A. Kimball and J. R. Ragains, Org. Lett., 2017, 19, 5553 CrossRef CAS PubMed; (f) A.-F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629 CrossRef CAS PubMed; (g) K. A. Hollister, E. S. Conner, M. L. Spell, K. Deveaux, L. Maneval, M. W. Beal and J. R. Ragains, Angew. Chem., Int. Ed., 2015, 54, 7837 CrossRef CAS PubMed; (h) F. W. Friese, C. Mück-Lichtenfeld and A. Studer, Nat. Commun., 2018, 9, 2808 CrossRef PubMed; (i) B. Ye, J. Zhao, K. Zhao, J. M. McKenna and F. D. Toste, J. Am. Chem. Soc., 2018, 140, 8350 CrossRef CAS PubMed; (j) Z. Liu, M. Li, G. Deng, W. Wei, P. Feng, Q. Zi, T. Li, H. Zhang, X. Yang and P. J. Walsh, Chem. Sci., 2020, 11, 7619 RSC.
- (a) T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205 CrossRef CAS PubMed; (b) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516 CrossRef CAS PubMed.
- J. Pothier, M. A. Riederer, O. Peter, X. Leroy, A. Valdenaire, C. Gnerre and H. Fretz, Bioorg. Med. Chem. Lett., 2012, 22, 4660 CrossRef CAS PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- (a) J. E. Argüello, A. B. Peñéñory and R. A. Rossi, J. Org. Chem., 2000, 65, 7175 CrossRef PubMed; (b) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730 CrossRef CAS PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 6.1.1, Semichem Inc., Shawnee Mission, KS, 2019 Search PubMed.
- (a) G. Wang, Z. Chen, Z. Xu, J. Wang, Y. Yang, T. Cai, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 610 CrossRef CAS PubMed; (b) Z. Yang, Z. Xu, Y. Liu, J. Wang, J. Shi, K. Chen and W. Zhu, J. Phys. Chem. B, 2014, 118, 14223 CrossRef CAS PubMed; (c) Z. Chen, G. Wang, Z. Xu, J. Wang, Y. Yu, T. Cai, Q. Shao, J. Shi and W. Zhu, J. Phys. Chem. B, 2016, 120, 8784 CrossRef CAS PubMed.
- (a) P. Politzer and J. S. Murray, Fluid Phase Equilib., 2001, 185, 129 CrossRef CAS; (b) J. S. Murray and P. Politzer, J. Mol. Struct., 1998, 425, 107 CrossRef CAS.
- F. Zhou, J. Han, R. Liu, P. Li and H. Zhang, Comput. Theor. Chem., 2014, 1044, 80 CrossRef CAS.
- (a) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC; (b) S. R. Kandukuri, A. Bahamonde, I. Chatterjee, I. D. Jurberg, E. C. Escudero-Adán and P. Melchiorre, Angew. Chem., Int. Ed., 2015, 54, 1485 CrossRef CAS PubMed.
- (a) Y. Chen, J. Phys. Chem. A, 2013, 117, 8081 CrossRef CAS PubMed; (b) L. Buglioni, M. M. Mastandrea, A. Frontera and M. A. Pericàs, Chem.–Eur. J., 2019, 25, 11785 CrossRef CAS PubMed.
- U. Koch and P. L. A. Popelier, J. Phys. Chem., 1995, 99, 9747 CrossRef CAS.
- (a) F. Zhou, R. Liu, P. Li and H. Zhang, New J. Chem., 2015, 39, 1611 RSC; (b) O. A. Syzgantseva, V. Tognetti and L. Joubert, J. Phys. Chem. A, 2013, 117, 8969 CrossRef CAS PubMed; (c) M. Jabłoński and M. Palusiak, J. Phys. Chem. A, 2012, 116, 2322 CrossRef PubMed; (d) S. Sarkar, P. K. Sruthi, N. Ramanathan and K. Sundararajan, New J. Chem., 2020, 44, 7116 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, DFT calculations, and characterisation for all compounds. See DOI: 10.1039/d1sc03667j |
| This journal is © The Royal Society of Chemistry 2021 |