
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical chain monoalkylation of pyridines†
Samuel
Rieder‡
 a,
Camilo
Meléndez‡
a,
Fabrice
Dénès
a,
Harish
Jangra
b,
Kleni
Mulliri
a,
Hendrik
Zipse
*b and
Philippe
Renaud
*a
a,
Camilo
Meléndez‡
a,
Fabrice
Dénès
a,
Harish
Jangra
b,
Kleni
Mulliri
a,
Hendrik
Zipse
*b and
Philippe
Renaud
*a
aDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland. E-mail: philippe.renaud@unibe.ch
bDepartment of Chemistry, LMU München, Butenandtstrasse 5-13, 81377 München, Germany. E-mail: zipse@cup.uni-muenchen.de
First published on 3rd November 2021
Abstract
The monoalkylation of N-methoxypyridinium salts with alkyl radicals generated from alkenes (via hydroboration with catecholborane), alkyl iodides (via iodine atom transfer) and xanthates is reported. The reaction proceeds under neutral conditions since no acid is needed to activate the heterocycle and no external oxidant is required. A rate constant for the addition of a primary radical to N-methoxylepidinium >107 M−1 s−1 was experimentally determined. This rate constant is more than one order of magnitude larger than the one measured for the addition of primary alkyl radicals to protonated lepidine demonstrating the remarkable reactivity of methoxypyridinium salts towards radicals. The reaction has been used for the preparation of unique pyridinylated terpenoids and was extended to a three-component carbopyridinylation of electron-rich alkenes including enol esters, enol ethers and enamides.
Introduction
Aromatic heterocyclic compounds, especially nitrogen-containing rings, are core elements of vitamins, amino acids, nucleic acids, and alkaloids and have thus attracted the attention of the synthetic community and the pharmaceutical industry for many years.1 A recent study has shown that 59% of U.S. FDA approved small-molecule drugs contain nitrogen heterocycles.2 Their late-stage functionalization is of great interest to modify and tune their pharmaceutical properties. For instance, introduction of carbon substituents under mild conditions has great potential and remains a privileged goal.3 Homolytic aromatic substitution (SHAr) is a long-known reaction and early examples date back to over a century ago.4,5 The Minisci reaction, which involves the addition of a nucleophilic carbon-centered alkyl or acyl radical onto a protonated heteroaromatic compound, is of particular importance due to its broad scope.6,7 The classical method uses alkyl and acyl radicals generated by hydrogen atom abstraction with persulfate and a silver(I) salt.6,8 Over the years, many variations of this transformation have been reported with different sources of alkyl radicals such as alkyl trifluoroborates,9,10 boronic acids,11,12 alcohols,13 zinc sulfinates,14,15N-(acyloxy)-phthalimides,16 and simple alkanes and ethers17–20 to name some of the leading work. The efficacy of the Minisci reaction is highlighted in a recent review of Proctor and Phipps.21 All the above-mentioned examples involve a rearomatization process via single-electron oxidation using either a stoichiometric oxidant or photoredox catalysis. Non-acidic activation of pyridine derivatives has also been reported. Recently, Baran and co-workers have reported that easily prepared N-alkylpyridinium salts can be used to direct regioselective functionalization at C4.22 The use of pyridine-N-oxides,23,24N-iminopyridine ylides,18 and N-methoxypyridinium salts25–29 has been examined. These substrates are particularly interesting since the aromatization step does not require any external oxidant and the reaction affords simple pyridines that are less prone to further alkylation. In pioneer work, Mitchell and co-workers have developed a Minisci-type procedure for the hydroxymethylation of pyridines involving N-methoxypyridinium derivatives (Scheme 1A).25 Three different mechanisms were proposed for the rearomatization step, among them the one involving the fragmentation of a methoxyl radical that sustains a chain process via hydrogen atom transfer from methanol being the most plausible. The role of the oxidant (ammonium persulfate) used in a substoichiometric amount is to initiate the reaction. The scope of this reaction was limited to methanol and ethanol (one example) used as the solvent, but this study nicely demonstrated the usefulness of methoxypyridinium salts to perform monoalkylation of pyridine derivatives. This work was extended by Baik, Hong and co-workers who developed a site selective photocatalyzed functionalization of N-methoxypyridinium salts with phosphinoyl and carbamoyl radicals (Scheme 1B).30,31 Recently, related processes involving radical generation via hydrogen atom transfer were reported by Shen et al.,32 Lakhdar and co-workers33 and by Alfonzo and Hande.34 These examples extended the scope of Mitchell's strategy, and the desired alkyl radicals were generated by hydrogen atom transfer to the highly reactive alkoxyl radical released in the rearomatization process. Chemo- and regioselective control of this approach is very challenging and is usually limited to precursors presenting electron-rich weak C–H bonds positioned alpha to a heteroatom (O, N, S). The lack of selectivity between aliphatic C–H bonds presenting similar bond dissociation energies is usually circumvented by the choice of non-substituted cycloalkanes, used in excess to prevent polyalkylation. Herzon and co-workers extended considerably the scope of the Mitchell variant to the use of secondary and tertiary alkyl radicals generated from alkenes under cobalt-mediated hydrogen atom transfer conditions, and to a broad range of heterocycles such as pyridine, imidazole and pyridazine (Scheme 1C).28,29 This reaction requires a stoichiometric amount of cobalt and it was proposed that aromatization took place via reduction of the intermediate radical cation by a Co(II) species followed by methanol elimination. Interestingly, the reaction could be extended to a borono-Minisci reaction under oxidative conditions (potassium persulfate and silver(I)). In a related manner, three-component coupling reactions involving N-methoxypyridinium salts were developed by Baik, Hong and co-workers35 using a Mn(III)/Ag(I) oxidizing system and by Nagib and co-workers using an iridium based photocatalyst.36,37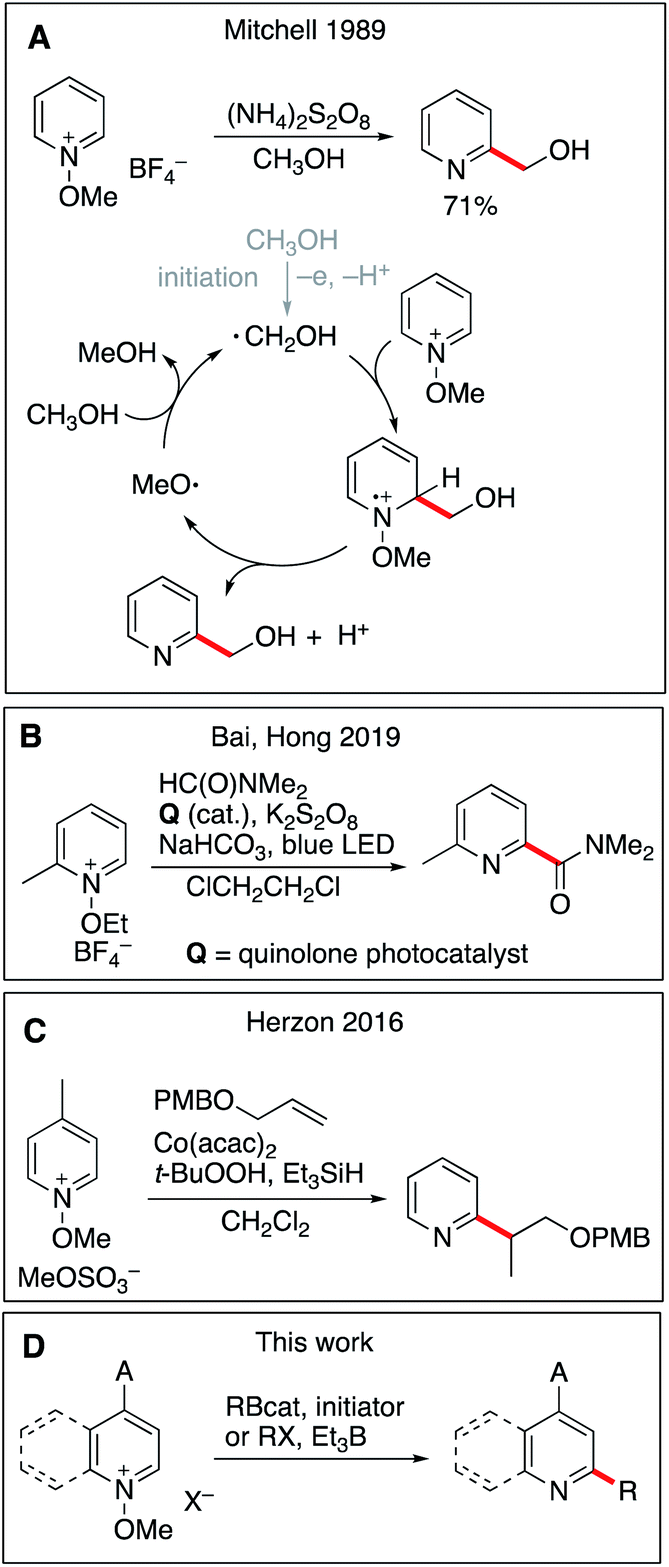 | ||
| Scheme 1 Radical addition to N-alkoxypyridinium salts. Previous leading work in the field (A–C) and this work (D). | ||
Based on these precedents, we hypothesized that the methoxyl radical proposed in Mitchell's mechanism could be used to sustain a radical chain process involving organoboranes. We report here a general method for a radical mediated monoalkylation of pyridines and related heterocycles using easily available B-alkylcatecholboranes (RBcat), alkyl iodides, and xanthates under radical chain reaction conditions without the need for any stoichiometric oxidant (Scheme 1D).
Results and discussion
(a) Reaction with organoboranes generated via hydroboration of alkenes
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Solvent | CyBcat (equiv.) | 2,4,6-Collidine (equiv.) | Yieldb | |
| 2 | 2′ | ||||
| a Reagents and conditions (entries 3 and 5–7: 1a·BF4 (1 mmol), alkene (1.5 equiv.), catecholborane (3.0 equiv.), DMA (0.32 mol%), DTBHN (10 mol%), 40 °C, 18 h). b Yields are determined by GC unless otherwise stated. c Isolated yield. d Using 1a·PF6. e Air as an initiator instead of DTBHN. f No added initiator. | |||||
| 1 | CH2Cl2 | 3.0 | — | 36%c | 14%c |
| 2 | CH2Cl2 | 3.0 | 5 | 46% | — |
| 3 | CH2Cl2 | 1.5 | — | 55% | 5% |
| 4 | CH2Cl2 | 1.5 | 3 | 54% | 8% |
| 5 | EtOAc | 1.5 | — | 45%c | 17% |
| 6 | C6H6 | 1.5 | — | 10% | — |
| 7 | DCE | 1.5 | — | 63% | 5% |
| 8 | DCE | 1.5 | 3 | 63% | — |
| 9 | DCE | 1.5 | 3 | 63% | — |
| 10 | DCE | 1.5 | — | 55%e | 4% |
| 11 | DCE | 1.5 | — | 28%f | 2% |
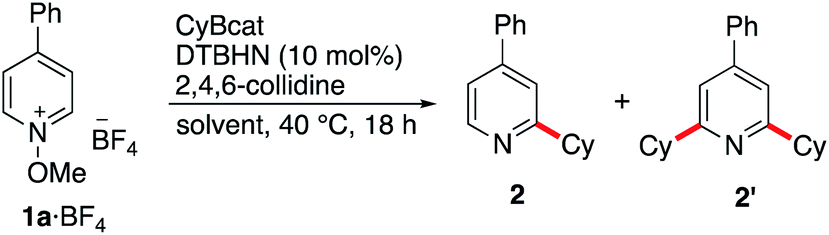
In dichloromethane and in the absence of any additive, the desired mono-alkylated product 2 was obtained in 36% yield along with 14% of the disubstituted product 2′. The formation of the bisalkylated product was attributed to acidic activation of pyridine 2 by protons released during the rearomatization process (see the mechanism in Scheme 4). The bis-alkylation could be suppressed by adding 2,4,6-trimethylpyridine (sym-collidine, 5 equiv.) as a base (Table 1, entry 2). Slightly higher yields were obtained by using a smaller excess (1.5 equivalents) of CyBcat with or without sym-collidine (Table 1, entries 3 and 4). Different solvents were tested next. Ethyl acetate (EtOAc) and benzene proved to be less efficient than dichloromethane (Table 1, entries 5 and 6) due to limited solubility of the methoxypyridinium 1a·BF4 in these solvents. The use of 1,2-dichloroethane (DCE) allows 1a·BF4 to be fully solubilized at 40 °C and the reaction afforded 2 in 63% yield, together with 5% of the dialkylated pyridine 2′ (Table 1, entry 7). When the reaction was run in DCE with 2,4,6-collidine as an additive (3 equiv.), the reaction exclusively afforded 2 in 63% isolated yield (Table 1, entry 8). Changing the counter-anion from tetrafluoroborate (BF4−) of 1a to hexafluorophosphate (PF6−) increased the solubility of the substrate but it had no influence on the outcome of the reaction (Table 1, compare entries 8 and 9). The reaction was also run using air initiation (open system) leading to a slight decrease of the yield (55%, Table 1, entry 10). In the absence of any added initiator, traces of oxygen proved to be sufficient to trigger the formation of 2 albeit in lower yield (28%, Table 1, entry 11) suggesting that an efficient chain process is taking place.
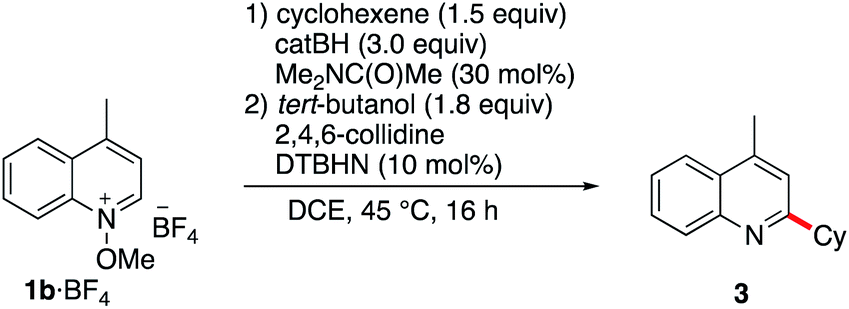
Since pyridine derivatives are challenging to purify, the reaction was further optimized with lepidine (Table 2). Reaction of the N-methoxylepidinium 1b·BF4 substrate furnished the alkylated lepidine 3 in 93% yield (GC analysis) (Table 2, entry 1). In this reaction, the use of 2,4,6-collidine was not necessary since no di- or polyalkylation was observed and the yield remains identical in the absence of collidine (Table 2, entry 2). An experiment with air initiation (open reaction vessel) gave the product in 95% yield (Table 2, entry 3). To guarantee an optimal reproducibility, all reactions were performed with DTBHN, air initiation being more influenced by the exact experimental setup. The nature of the alkoxy group (methoxy vs. ethoxy) was tested then (Table 2, entry 4) and did not affect the outcome of the reaction. Product isolation is often problematic with pyridine derivatives. Best results were obtained by filtration of the crude mixture through basic aluminum oxide to remove residual acid impurities and catechol byproducts followed by column chromatography on silica gel. With this method, the isolated yield closely matched the yield determined by GC analysis (Table 2, entry 5).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The reaction was also found to be efficient with alkenes containing a free hydroxy group. Starting from undec-10-en-1-ol, the linear hydroxyalkylated lepidine 10 was obtained in 65% yield. In this case too, a small amount (9%) of the branched isomer was also isolated. The silylether 11 was obtained in 79% yield from silylated 2-methallyl alcohol. Interestingly, in this first screening with 1b, products such as 9–11 are particularly easily accessible from alkenes with good to excellent regioselectivity control that cannot easily be matched using other Minisci type processes. A second series of experiments was performed with different N-methoxyheteroarenium salts. N-Methoxy-4-chloroquinolinium 1c and N-methoxyphenanthridin-5-ium 1d afforded upon reaction with cyclohexene the monoalkylated products 12 and 13 in 85% and 70% yield, respectively. Reaction of cyclohexene with N-methoxy-3-bromoquinolinium 1e gave product 14 in modest yield (38%). N-Methoxyquinaldinium 1f was not alkylated under the same reaction conditions. However, in the presence of 2,4,5-collidine (conditions A) the 4-cyclohexyquinaldine 15 was obtained in 38% yield together with a significant amount of quinaldine. A marginally higher yield was obtained with the N-ethoxyquinaldinium salt 1f′ (42%). A possible pathways leading to the formation of quinaldine involves deprotonation at the methyl position followed by a homolytic fragmentation of the N–OMe bond according to the work of Shen et al. liberating a methoxyl radical and a benzylic radical that can abstract a hydrogen from the reaction mixture.32 Reactions with substituted N-methoxypyridinium salts were also investigated. For these substrates, the use of 2,4,6-collidine had a very positive effect on the outcome of the reactions (compare conditions A with 2,4,6-collidine and B without a base). Reaction of 1a with cyclohexene and 1-dodecene under conditions A afforded 2 and 16 in 63% and 54% yields. Reaction with tetramethylethylene gave 17 in much lower yield. The 4-tert-butyl- and 4-ethoxycarbonyl-1-methoxypyridinium salts 1g and 1h were both monoalkylated with cyclohexene to afford 18 and 19 in 65% and 46% yields, respectively.
:3). The reaction was also found to be efficient with alkenes containing a free hydroxy group. Starting from undec-10-en-1-ol, the linear hydroxyalkylated lepidine 10 was obtained in 65% yield. In this case too, a small amount (9%) of the branched isomer was also isolated. The silylether 11 was obtained in 79% yield from silylated 2-methallyl alcohol. Interestingly, in this first screening with 1b, products such as 9–11 are particularly easily accessible from alkenes with good to excellent regioselectivity control that cannot easily be matched using other Minisci type processes. A second series of experiments was performed with different N-methoxyheteroarenium salts. N-Methoxy-4-chloroquinolinium 1c and N-methoxyphenanthridin-5-ium 1d afforded upon reaction with cyclohexene the monoalkylated products 12 and 13 in 85% and 70% yield, respectively. Reaction of cyclohexene with N-methoxy-3-bromoquinolinium 1e gave product 14 in modest yield (38%). N-Methoxyquinaldinium 1f was not alkylated under the same reaction conditions. However, in the presence of 2,4,5-collidine (conditions A) the 4-cyclohexyquinaldine 15 was obtained in 38% yield together with a significant amount of quinaldine. A marginally higher yield was obtained with the N-ethoxyquinaldinium salt 1f′ (42%). A possible pathways leading to the formation of quinaldine involves deprotonation at the methyl position followed by a homolytic fragmentation of the N–OMe bond according to the work of Shen et al. liberating a methoxyl radical and a benzylic radical that can abstract a hydrogen from the reaction mixture.32 Reactions with substituted N-methoxypyridinium salts were also investigated. For these substrates, the use of 2,4,6-collidine had a very positive effect on the outcome of the reactions (compare conditions A with 2,4,6-collidine and B without a base). Reaction of 1a with cyclohexene and 1-dodecene under conditions A afforded 2 and 16 in 63% and 54% yields. Reaction with tetramethylethylene gave 17 in much lower yield. The 4-tert-butyl- and 4-ethoxycarbonyl-1-methoxypyridinium salts 1g and 1h were both monoalkylated with cyclohexene to afford 18 and 19 in 65% and 46% yields, respectively.
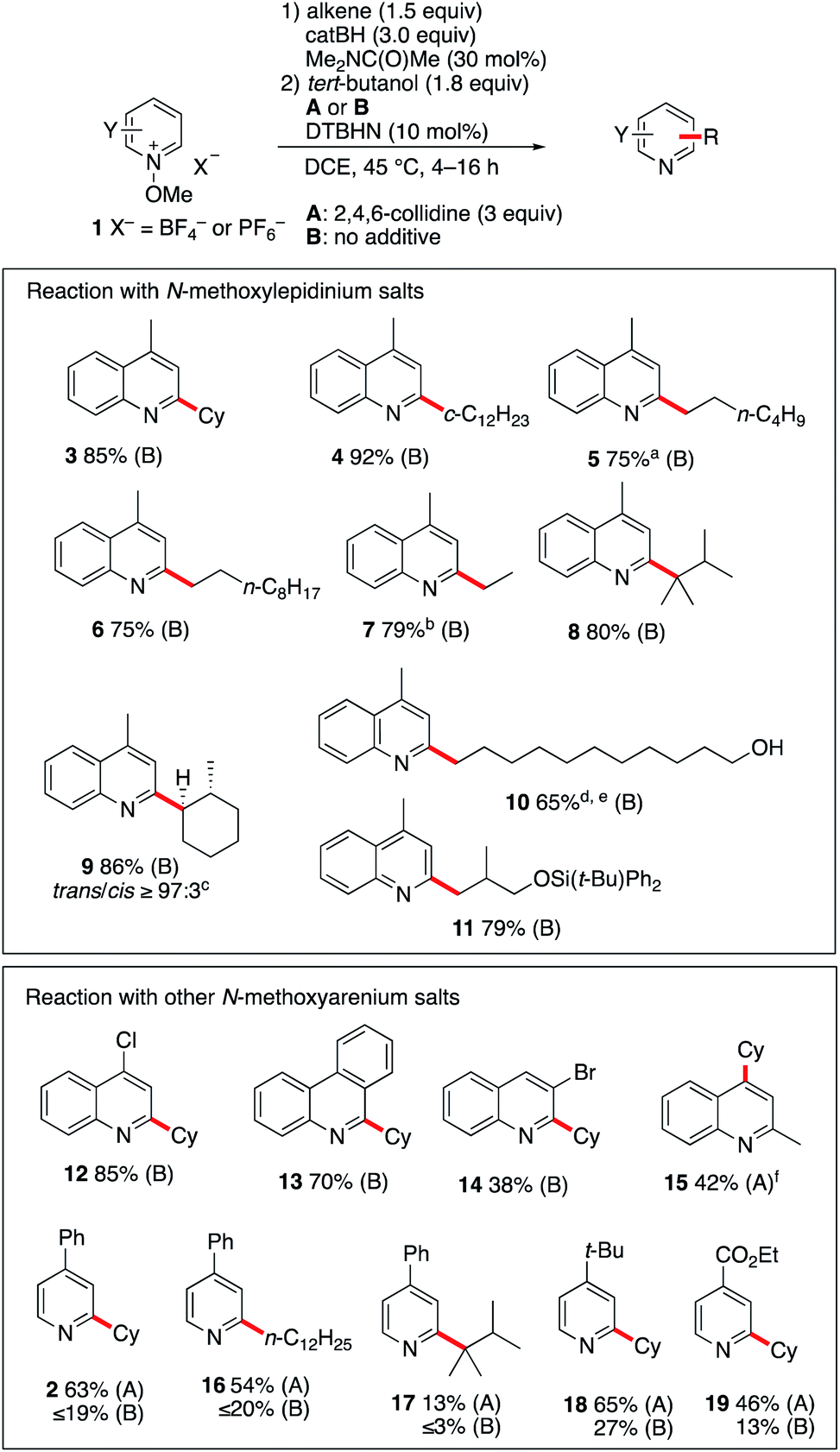 | ||
| Scheme 2 Reaction of N-methoxypyridinium salts 1 with a radical generated from B-alkylcatecholboranes. Yields refer to isolated products. (a) contains 11% of the branched 2-hexyl isomer. (b) Reaction run with Et3B (1.5 equiv.). (c) Diastereoselectivity determined by GC-analysis, only the major diastereoisomer is depicted. (d) Reaction run with catBH (4.5 equiv.). (e) The branched isomer (9%) was also isolated. (f) Using N-ethoxylepidinium tetrafluoroborate. | ||
In order to demonstrate further the utility of the hydroboration approach, the pyridinylation of a series of terpenoid natural products was investigated (Scheme 3). The reaction of α- and β-pinene provided the alkylated lepidines 20 and 21 in high yields and excellent stereocontrol.42 The reaction of O-benzyl-nopol was investigated next. It afforded the arylated quinoline and pyridines 22–26 in moderate to high yields. Satisfactory regiocontrol was observed, excepted for the 2-trifluromethylated pyridine 26 that was obtained as a nearly 1:1 mixture of C(2) and C(4) regioisomers. Non-protected nopol afforded the lepidinated product 27 in an excellent 75% yield. The reaction of camphene with 1b afforded the desired product 28 in 84% yield as a 3:1 mixture of diastereomers. Cholesteryl benzoate was converted to 29 in 58% yield with satisfactory stereocontrol (dr 92:8). Finally, the radical nature of the reaction was unambiguously demonstrated by the cyclopropane ring opening process observed when (+)-2-carene43,44 was used as a radical precursor. Reaction with 1b afforded the substituted lepidine 30 in 76% yield and high stereoselectivity (Scheme 3).
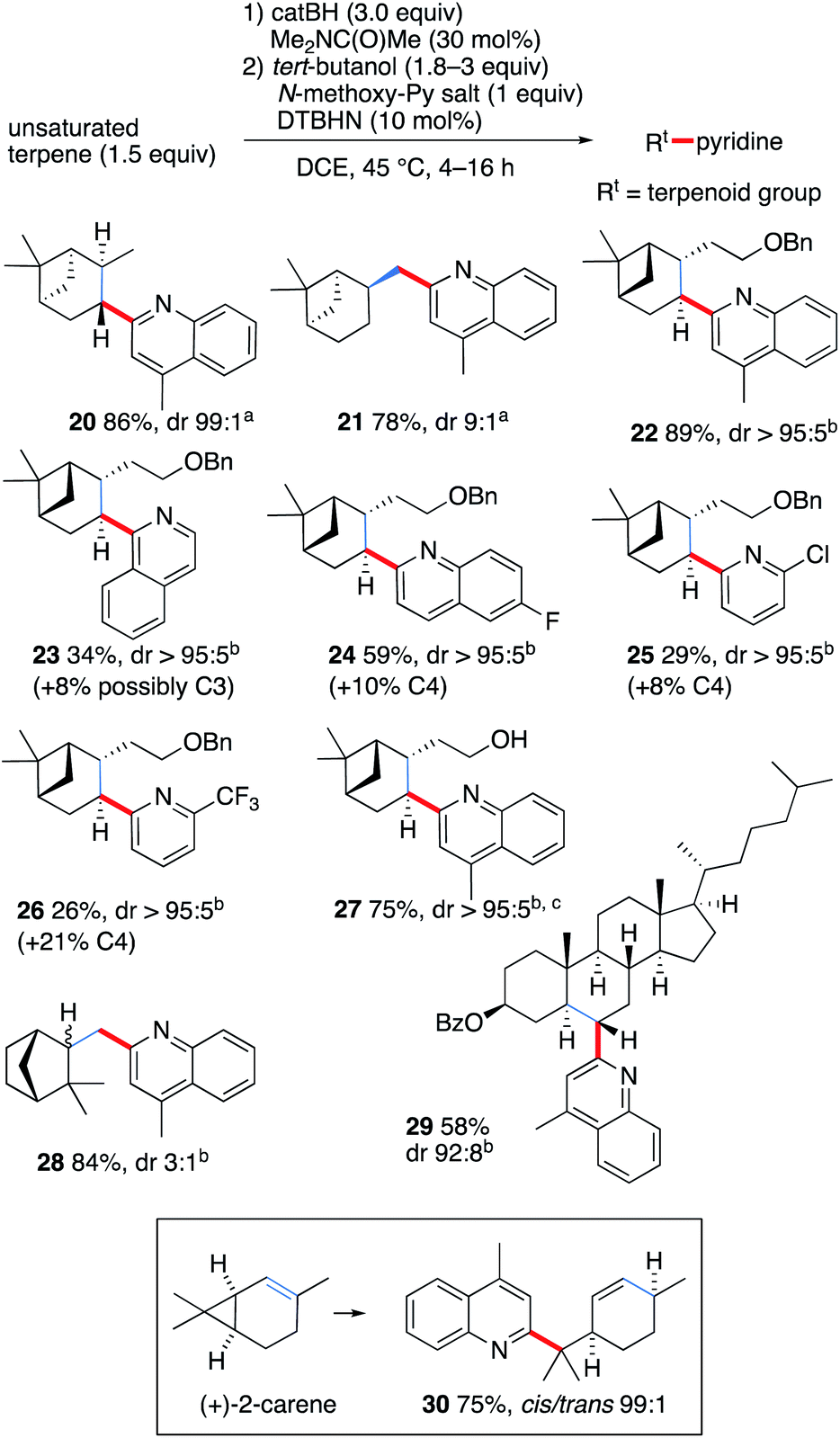 | ||
| Scheme 3 Pyridinylation of terpenoids. Radical probe experiment with (+)-2-carene. The position of the initial double bond is indicated in blue. Yields refer to isolated products. (a) Diastereoselectivity determined by GC-analysis. (b) Diastereoselectivity determined by 1H NMR. (c) Reaction run with catBH (4.5 equiv.). | ||
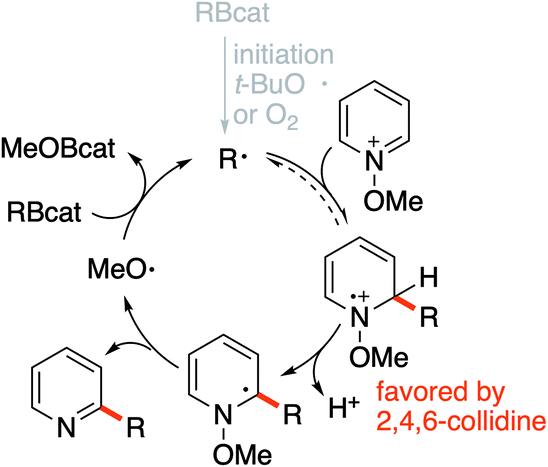 | ||
| Scheme 4 Proposed chain mechanism for the alkylation of N-methoxypyridinium salts. | ||
The formation of MeO–Bcat was confirmed by analysis of the crude product before purification. Although 1.5 equivalents of t-BuOBcat are formed during the treatment of excess catBH with tert-butanol, the presence of nearly one equivalent of MeOBcat can be detected by 11B NMR. Indeed, the two borate esters give distinct signals at +22.4 ppm (t-BuBcat) and +23.5 ppm (MeOBcat).53
(b) Reactions with alkyl iodides
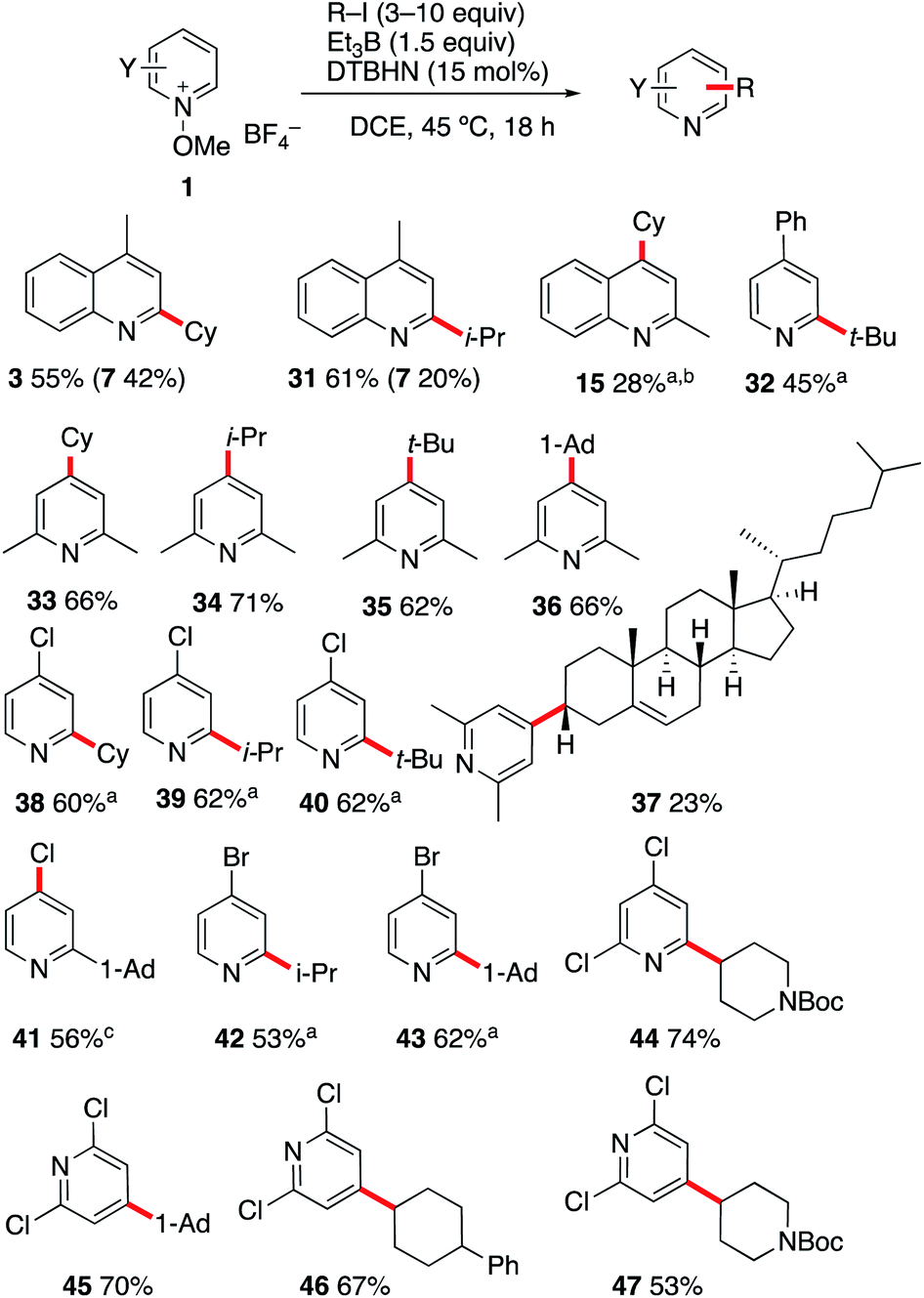 | ||
| Scheme 5 Alkylation of quinolines and pyridines with alkyl iodides. (a) Using 2,4,6-collidine (3 equiv.) as a base. (b) Using 20 equiv. of CyI. (c) Using K2CO3 (3 equiv.) as a base. | ||
(c) Reaction with xanthates
The generation of more functionalized radicals such α-oxygenated and β-silylated radicals from iodide radical precursors cannot be performed due to the instability of the required iodides. This issue could be efficiently circumvented by using the more stable O-ethyl xanthate radical precursors (type I).55–58 Theses xanthates 48a–d were prepared by adding ethyl 2-((ethoxycarbonothioyl)thio)acetate to 1-nonene, allyltrimethylsilane, vinyl acetate, and vinyl butyl ether. Reaction of these three radical precursors with different N-methoxypyridinium salts 1i–k have been examined and results are reported in Scheme 6. The products 49–54 were isolated in moderate to excellent yields and the reaction tolerates the presence of a trimethylsilyl group at position 2 as well as 1-acetoxy and 1-butoxy groups. Extension of the reaction to S-methyl xanthates (type II), the well-established intermediates of the Barton–McCombie deoxygenation process,59 was examined next. The simple secondary S-methyl xanthate 55 was engaged in a reaction with N-methoxy-2,6-lutidinium 1i. Satisfyingly, it provided the desired 4-alkylated lutidine 56 in moderate 53% yield. Extending the reaction to the S-methyl xanthate 57 derived from isopinocampheol and 1i provided the dithiocarbonate rearrangement product 58 in 13% yield as the only identified product.60,61 This result is not surprising since xanthates of type II are good radical traps, and, in contrast to the type I xanthates, their reaction with the liberated alkyl radical is not a degenerative reaction liberating the original xanthate, but it leads to the formation of radical inactive dithiocarbonates such as 58.55,62 Finally, reaction of type II xanthate derived for cholesterol with N-methoxylepidinium 1b was attempted. In this case however, the only isolated product was 7 indicating that 1b was reacting much faster with the ethyl radical than the type II xanthate (see the ESI† for details). The results summarized in Scheme 6 indicate that type I xanthates are of synthetic interest for the alkylation of pyridines under Et3B-mediated conditions, while type II xanthates have a much more limited scope.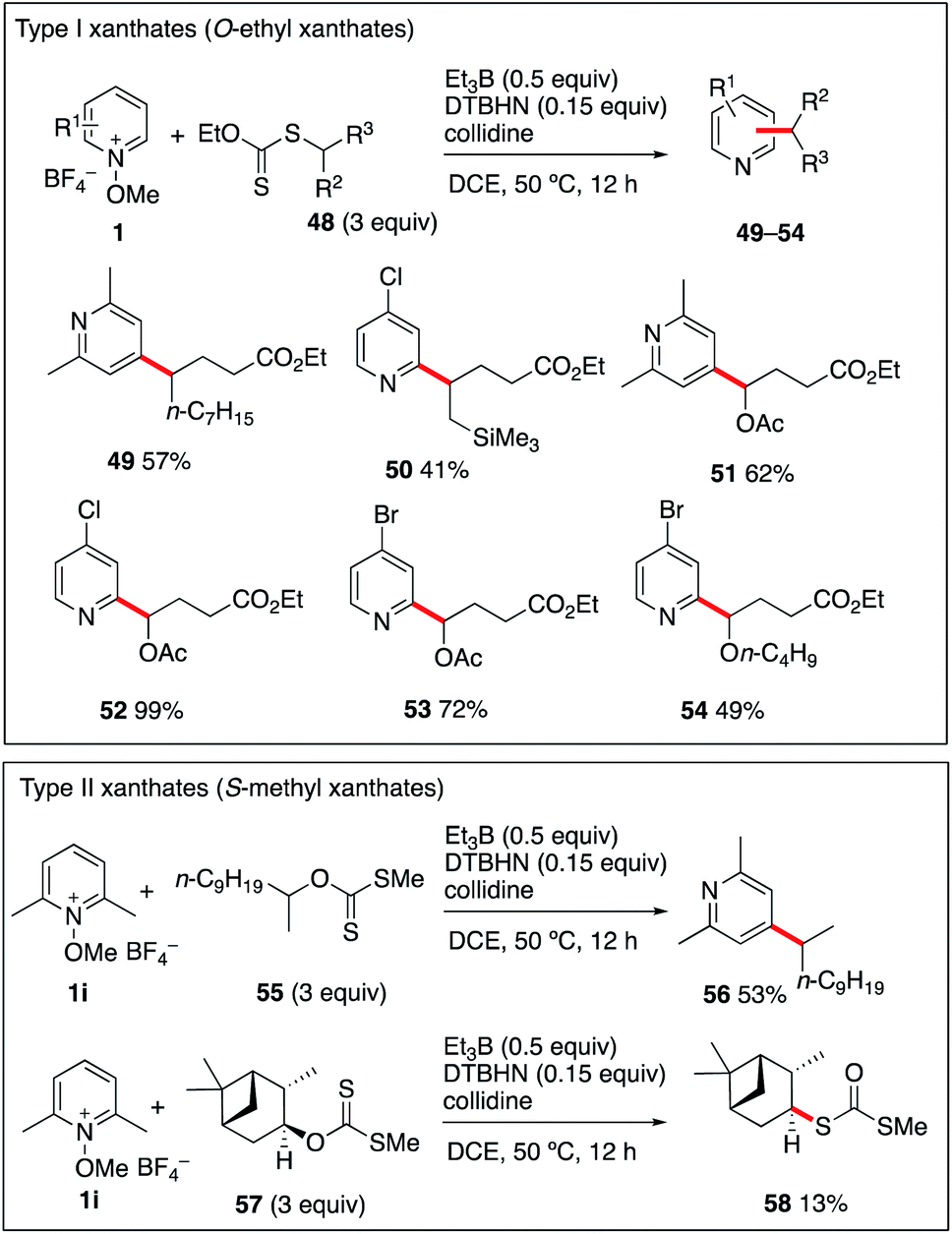 | ||
| Scheme 6 Alkylation of pyridines with xanthates. | ||
(d) One-pot three-component alkylation of N-methoxy-pyridinium salts
The reaction involving iodides and xanthates described above opens the possibility to develop a three-component coupling process involving a radical precursor, an alkene, and an N-methoxypyridinium salt. This approach is expected to complement the related photoredox catalyzed approaches involving N-methoxypyridinium salts,35–37 lepidinium and quinaldinium trifluoroacetate,63 as well as quinoxalin-2(1H)-ones.64 As the xanthates 48a–d are prepared by radical mediated xanthate transfer addition to the corresponding alkenes,55–58 all products presented in Scheme 6 result formally from a two-step carbopyridinylation reaction. Since electrophilic radicals are not expected to react with the N-methoxypyridinium salts 1, a one-pot process appears feasible. Gratifyingly, the Et3B-mediated three-component coupling process involving N-methoxypyridinium salts 1, electron-rich alkenes and ethyl α-iodo- or α-((ethoxycarbonothioyl)thio)acetate afforded the desired products in moderate yields (Scheme 7). Reaction of 1-nonene with ethyl iodoacetate and the N-methoxypyridinium salts 1b, 1j and 1d led to the formation of 59–61 in 28–52% yield. Interestingly, even the non-protected allyl alcohol could be used in this process giving 62 in 26% yield. For 2,2-disubstituted alkenes such as 1-phenyl-1-methylenecyclohexene, the best results for the formation of 63 were obtained with the xanthate radical precursor. Similar results were obtained with n-vinylpyrrolidinone and 2-acetoxypropene. In these last examples, the one-pot three-component approach is the only way to perform the transformation since isolation of the intermediate tertiary xanthate proved to be impossible. Moderate yields for the formation of 64–66 were obtained when the reactions were performed under these conditions.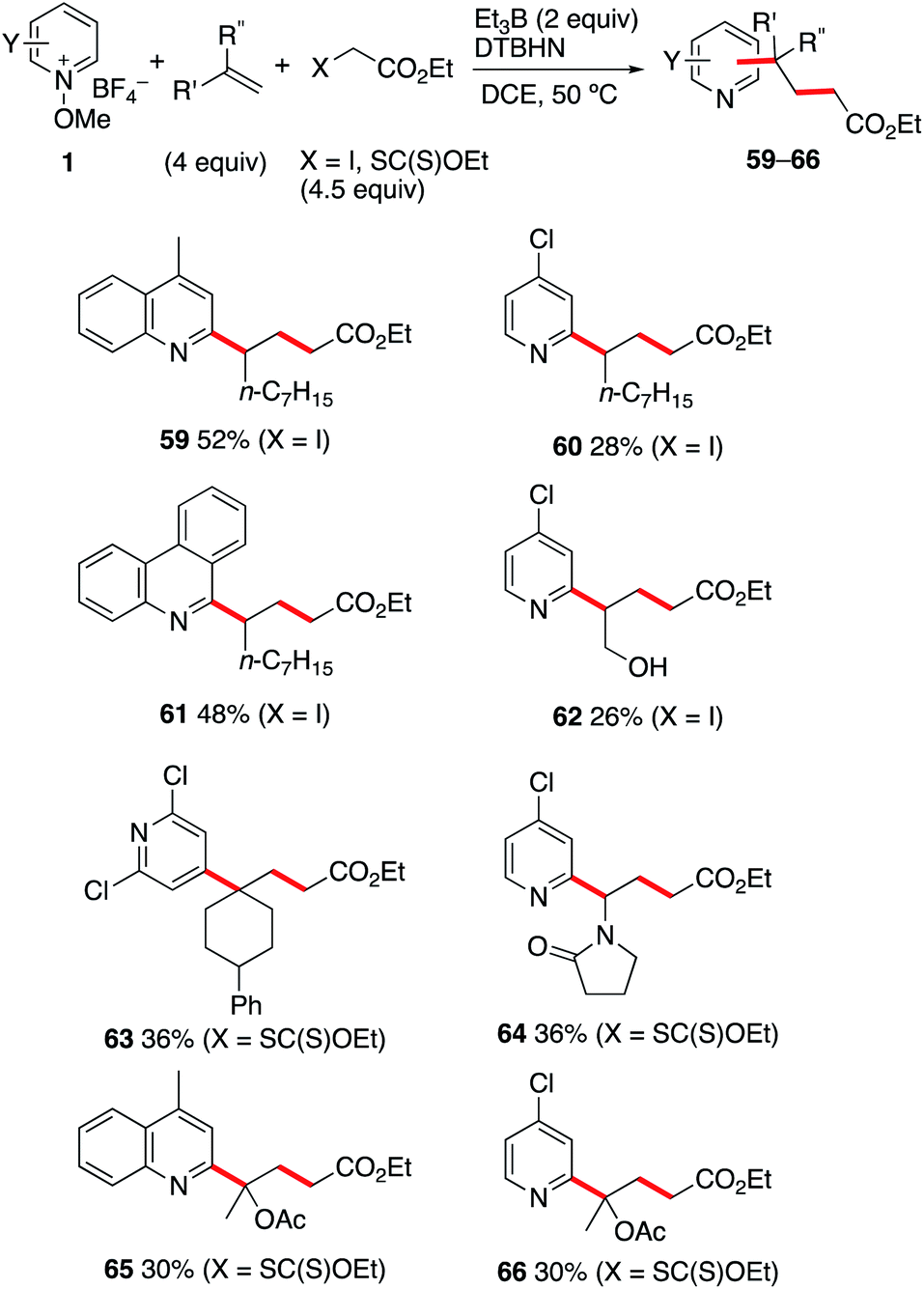 | ||
| Scheme 7 Carbopyridinylation of alkenes (three-component coupling process). | ||
(e) Mechanistic investigations
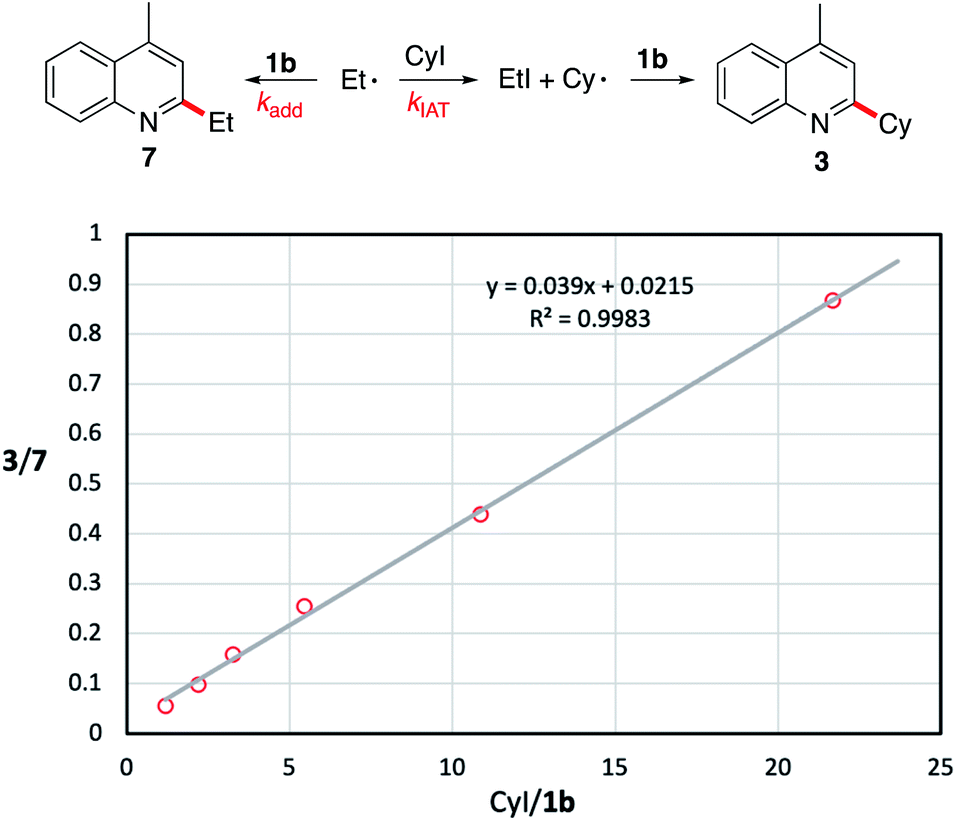 | ||
| Fig. 1 Estimation of the rate constant kadd for the addition of the ethyl radical to 1b at 50 °C. Reported kIAT = 5.4 ± 0.9 ×105 M−1 s−1 at 50 °C.54kadd = kIAT/slope. Using the experimental slope of 0.039, kadd (50 °C) = 1.4 ± 1 × 107 M−1 s−1. | ||
:1 mixture of 4- and 6-isopropyl regioisomers (Scheme 8A). By running the reaction in the presence of K2CO3, a similar yield was obtained for 67 but the formation of the 6-isopropyl-2-chloropyridine became the major process. A similar trend, albeit less pronounced was observed when 2,4,6-collidine was used as a base. This influence of the base on the regioselectivity is attributed to a faster deprotonation of the intermediate radical cation that favors formation of the 6-isopropyl addition product.
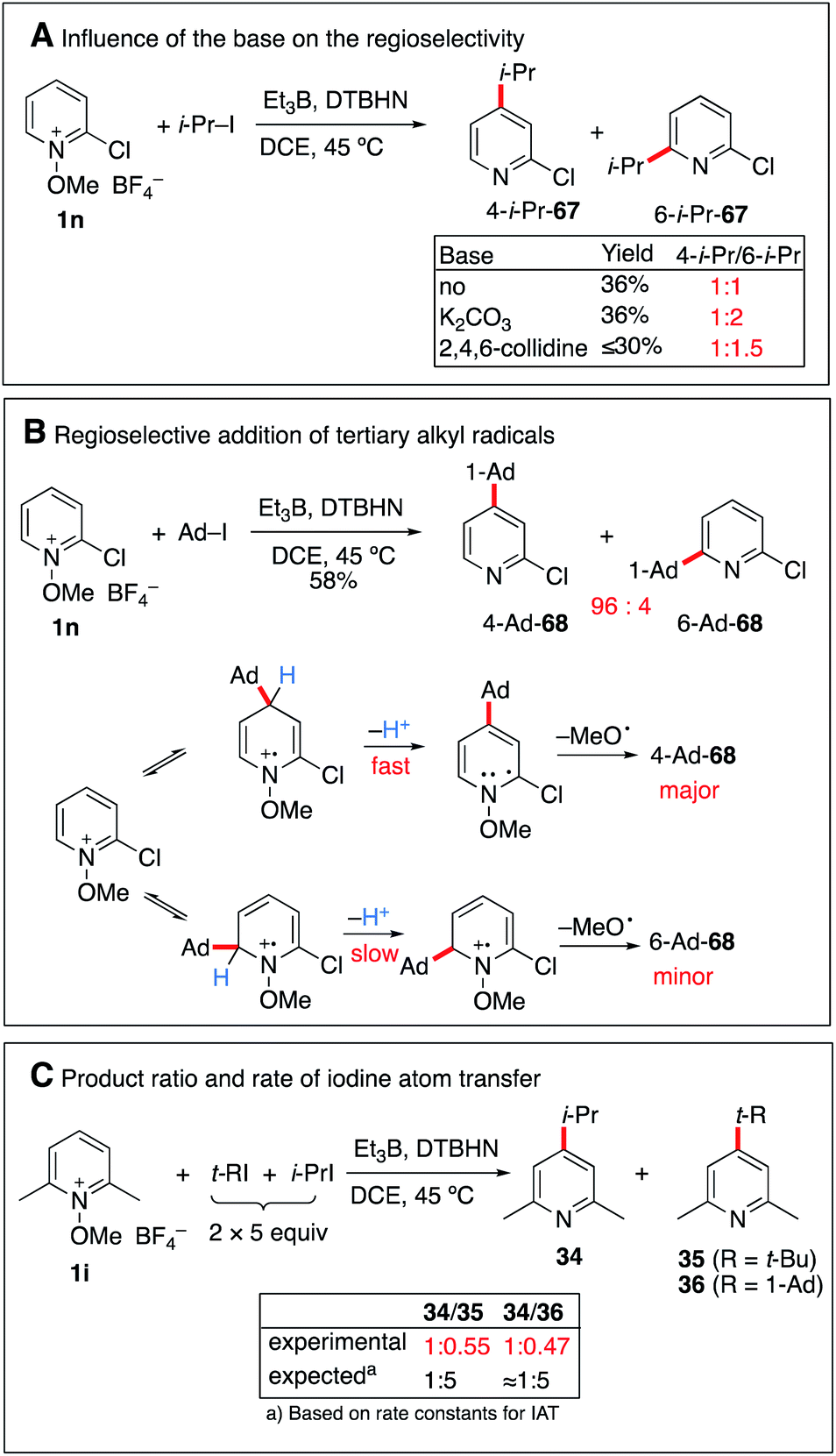 | ||
| Scheme 8 Regioselectivity and possible reversibility of the radical addition. | ||
Interestingly, the same reaction with 1-iodoadamantane afforded 68 in 58% yield as a 94:6 mixture of 4-Ad/6-Ad (Scheme 8B). Similar regioselectivities for tertiary alkyl radicals were already observed by Herzon and co-workers.29 The impressive difference of regioselectivity between a secondary and a tertiary radical may not strictly reflect the different rates of addition but it may also be a consequence of the possible reversibility of the reaction. Indeed, addition of tertiary radicals is expected to be fast and reversible (see the Calculations below) (Fig. 2) and the observed product ratio may be governed by the difference of deprotonation rates, the 4-adamantyl adduct being more difficult to deprotonate due to steric reasons (see Scheme 8B).
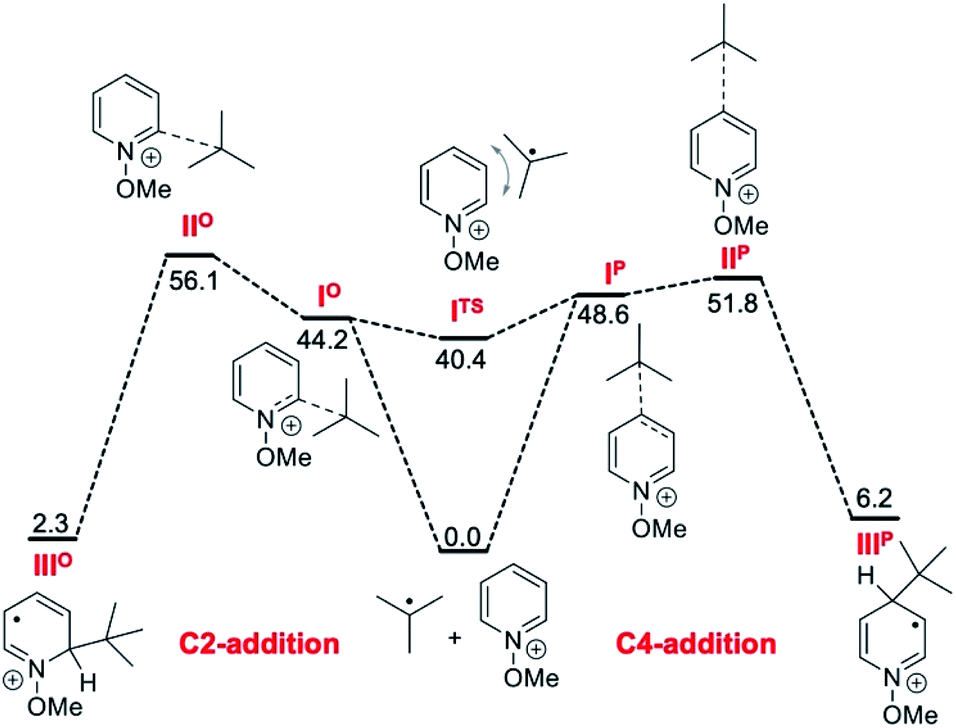 | ||
| Fig. 2 Reaction free energy profile (kJ mol−1) in dichloromethane for the reaction of the tert-butyl radical with the N-methoxypyridinium cation calculated at the SMD(DCM)/DLPNO-CCSD(T)/cc-pVTZ//M06-2X/def2-TZVP level of theory. Single point solvation energies are calculated at the SMD(DCM)/UB3LYP/6-31+G(d)//M06-2X/def2-TZVP level of theory. | ||
A further indication of reversibility was observed when the alkylation of N-methoxy-2,6-dimethylpyridine 1i was run with an equimolar excess of a tert-butyl or adamantyl iodide and isopropyl iodide (Scheme 8C). In both cases, the major product was the 4-isopropylpyridine 34 despite the fact that iodine atom abstractions involving tert-butyl iodide (kIAT = 3 × 106 M−1 s−1 at 50 °C)54 and presumably also adamantyl iodide are faster than the one involving isopropyl iodide (5.6 × 105 M−1 s−1 at 50 °C).54 This result is best explained by the higher reversibility of the addition of the more stable tert-butyl radical compared to the isopropyl radical.
The reactivities of various N-methoxypyridinium systems towards tert-butyl radical addition were investigated next by comparing the addition energy for different systems such as N-methoxypyridinium (C4-addition, ΔG‡ = +51.8 kJ mol−1, ΔG = +6.2 kJ mol−1), N-methoxy-2,6-lutidinium 1i (C4-addition, ΔG‡ = +60.2 kJ mol−1, ΔG = +16.2 kJ mol−1), N-methoxyquinaldinium 1f (C4-addition, ΔG‡ = +47.2 kJ mol−1, ΔG = −3.4 kJ mol−1, see ESI†) and N-methoxylepidinium 1b (C2-addition, ΔG‡ = +46.9 kJ mol−1, ΔG = −6.2 kJ mol−1) as well as protonated pyridine (C4-addition, ΔG‡ = +52.2 kJ mol−1, ΔG = +14.4 kJ mol−1). For the more reactive quinolinium salts 1f and 1b, the radical addition reactions are slightly exergonic (ΔG = −3.4 and −6.2 kJ mol−1, respectively). In the case of the less reactive N-methoxy-2,6-lutidinium 1i, the reaction is strikingly endergonic (ΔG = +16.2 kJ mol−1) and therefore reversibility is expected to occur (Fig. 3A) supporting the experimental results presented in Scheme 8C. The energy profiles of the addition of the tert-butyl radical to the methoxypyridinium salt (Fig. 3B) and the protonated pyridine (Fig. 3C) have similar activation energies, but differ noticeably in their thermochemistry, the addition to the protonated pyridine being 8 kJ mol−1 more endergonic. A similar trend was found for the addition of the ethyl radical to N-methoxylepidinium and protonated lepidine (see Fig. 4B and C). The difference of the addition rates observed for these two systems may result from stronger interactions of the protonated species with the counterions, an effect that was neglected in our calculations.
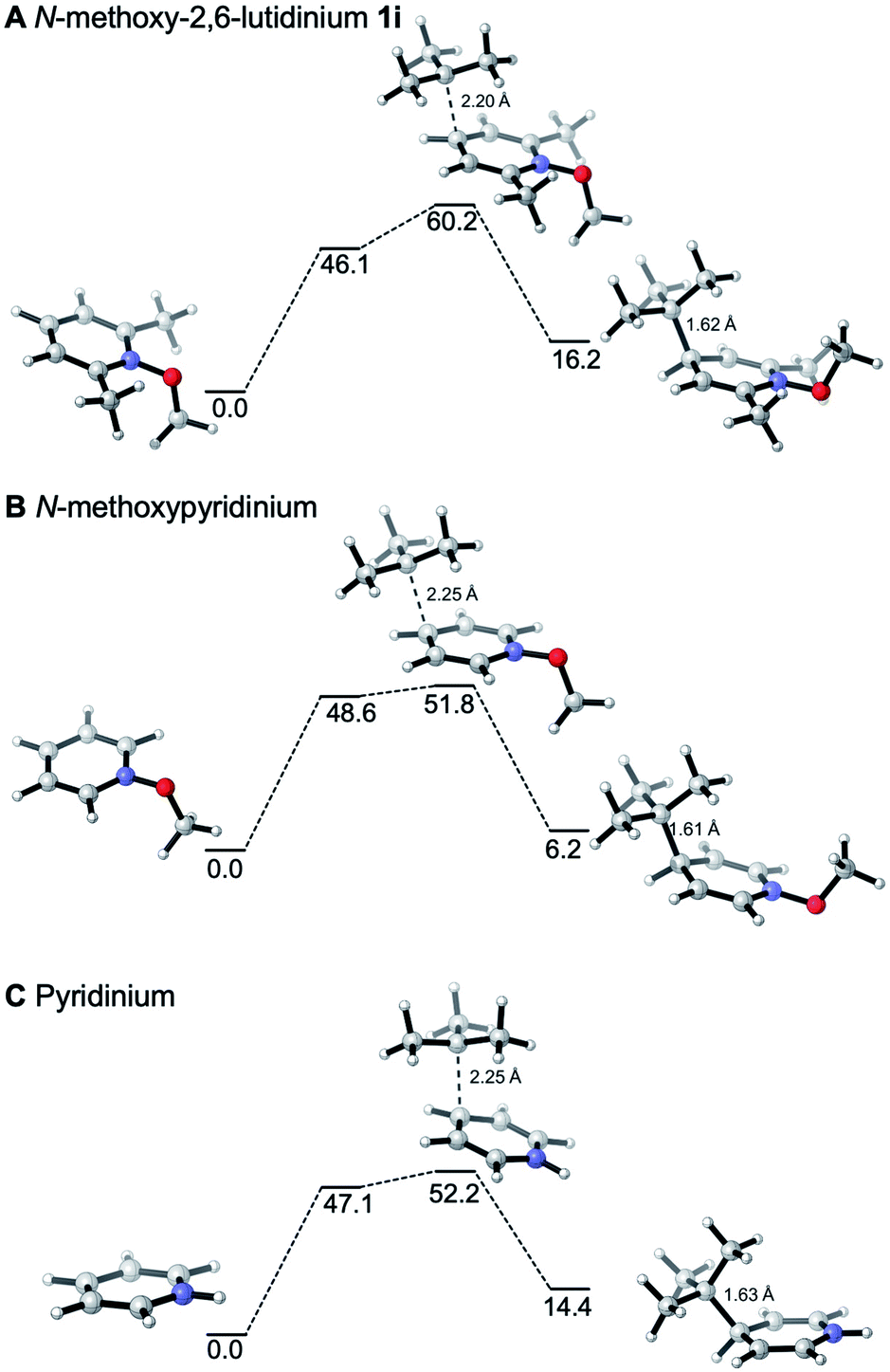 | ||
| Fig. 3 Reaction free energy profiles (kJ mol−1) for the addition of the tert-butyl radical to the N-methoxy-2,6-lutidinium salt (A), N-methoxypyridine (B) and protonated pyridine (C) at the SMD(DCM)/DLPNO-CCSD(T)/cc-pVTZ//M06-2X/def2-TZVP level of theory. Single point solvation energies are calculated at the SMD(DCM)/UB3LYP/6-31+G(d)//M06-2X/def2-TZVP level of theory. Energies are given in kJ mol−1. | ||
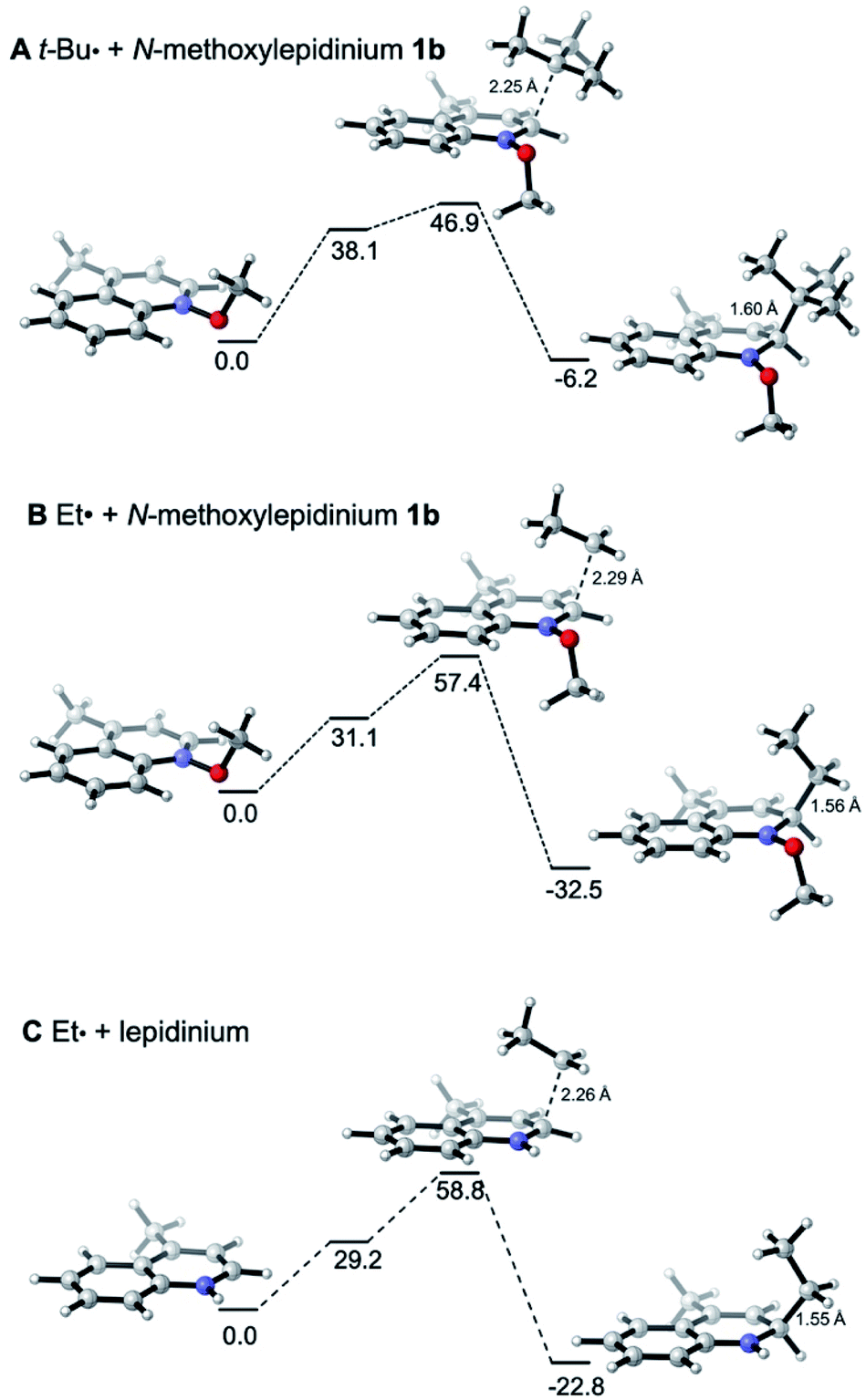 | ||
| Fig. 4 Reaction free energy profiles (ΔGsol, kJ mol−1) for the addition of the (A) tert-butyl and (B) ethyl radicals to N-methoxylepidinium and of the (C) ethyl radical to protonated lepidine calculated at the SMD(DCM)/DLPNO-CCSD(T)/cc-pVTZ//M06-2X/def2-TZVP level of theory. Single point solvation energies are calculated at the SMD(DCM)/UB3LYP/6-31+G(d)//M06-2X/def2-TZVP level of theory. | ||
In Fig. 4, the energy profiles for the addition of the tert-butyl and ethyl radicals to N-methoxylepidinium 1b (Fig. 4A and B) as well as the addition of the ethyl radical to the protonated lepidinium (Fig. 4C) are depicted. As anticipated based on the nucleophilic character of the radicals, the energy barrier for the addition of the ethyl radical (ΔG‡ = +57.4 kJ mol−1) is higher than the one calculated for the tert-butyl radical (ΔG‡ = +46.9 kJ mol−1). Also anticipated, the addition of the ethyl radical is more exothermic (ΔG = −32.5 kJ mol−1) than the addition of the tert-butyl radical (ΔG = −6.2 kJ mol−1). These results support the hypothesis made in our kinetic study that the addition of the ethyl radical to N-methoxylepidinium is irreversible under our reaction conditions. Finally, the addition of the ethyl radical to the protonated lepidine indicates, in agreement with the pyridine system discussed above (Fig. 3B and C), that the activation energies for the addition step are very similar but the thermochemistry is less favorable by about 10 kJ mol−1 for the protonated lepidine relative to the N-methoxylepidinium 1b.
The outcome of our calculations fits well with literature reports. Minisci and co-workers have reported experimental evidence for the reversibility of the addition of stabilized radicals such as benzyl,79tert-butyl80 and α-oxygenated81 radicals to protonated pyridines. This reversibility was recently invoked by Phipps and co-workers to rationalize the enantioselective Minisci reactions involving stabilized α-aminoalkyl radicals.82 Our calculations support these findings showing that the addition of a tert-butyl radical to protonated pyridine is an endergonic process. N-Methoxypyridinium salts behave similarly but the reaction is less endergonic and even slightly exergonic with quinoline derivatives. To get a full picture of the factors that govern the regioselectivity, the energy barrier for the fast subsequent deprotonation step should be calculated. However, we did not perform these calculations because of their arbitrary character due to the strong dependence on the solvation model used.
Conclusion
An efficient and experimentally simple method for monoalkylation of pyridine derivatives and related compounds has been developed. The transformation is achieved by reaction of N-methoxypyridinium salts, easily prepared by alkylation of N-oxides with trimethyloxonium tetrafluoroborate (Meerwein's salt). The generality of the process is demonstrated by using radicals generated either from alkenes via a hydroboration process, from alkyl iodides and from xanthates. In terms of regioselectivity control, the hydroboration approach complements nicely the cobalt-mediated pyridinylation developed by Herzon. The high selectivity observed for the formation of monoalkylated products is best explained by the exceptional reactivity of the N-methoxypyridinium salts towards radicals. Indeed, these pyridine salts were found to react faster with radicals than the corresponding protonated pyridines. All these reactions rely on an efficient chain reaction involving the fragmentation of a weak N–OMe bond leading to rearomatization and generation of a methoxyl radical that sustains the chain process by reaction with an organoboron species. Based on the strong favorable polar effects, a three-component coupling process leading to the carbopyridinylation of electron-rich alkenes could also be performed. This work is expected to find applications in natural product synthesis and this aspect of the chemistry is currently being further investigated.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
P. R. secured funding for the project and wrote the initial research proposal. P. R., S. R., C. M. and F. D. conceptualized the work and interpreted the results. S. R., C. M., K. M. and F. D. conceived and performed the experiments. H. J. and H. Z. performed the calculations. S. R. wrote the initial draft, P. R., C. R. and F. D. prepared the final version and all the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Swiss National Science Foundation (Project 200020_201092) is gratefully acknowledged for financial support. The authors gratefully acknowledge the computational and data resources provided by the Leibniz Supercomputing Centre (http://www.lrz.de)References
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- F. O'Hara, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2013, 135, 12122–12134 CrossRef PubMed.
- F. Minisci, in Synthetic and Mechanistic Organic Chemistry, ed. F. Minisci, J. B. Hendrickson and C. Wentrup, Springer, Berlin, Heidelberg, 1976, pp. 1–48 Search PubMed.
- W. R. Bowman and J. M. D. Storey, Chem. Soc. Rev., 2007, 36, 1803–1822 RSC.
- F. Minisci, R. Bernardi, F. Bertini, R. Galli and M. Perchinummo, Tetrahedron, 1971, 27, 3575–3579 CrossRef CAS.
- M. A. J. Duncton, MedChemComm, 2011, 2, 1135–1161 RSC.
- T. Caronna, G. Fronza, F. Minisci, O. Porta and G. P. Gardini, J. Chem. Soc., Perkin Trans. 2, 1972, 1477–1481 RSC.
- G. A. Molander, V. Colombel and V. A. Braz, Org. Lett., 2011, 13, 1852–1855 CrossRef CAS PubMed.
- M. Presset, N. Fleury-Bregeot, D. Oehlrich, F. Rombouts and G. A. Molander, J. Org. Chem., 2013, 78, 4615–4619 CrossRef CAS PubMed.
- G.-X. Li, C. A. Morales-Rivera, Y. Wang, F. Gao, G. He, P. Liu and G. Chen, Chem. Sci., 2016, 7, 6407–6412 RSC.
- I. B. Seiple, S. Su, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed.
- J. Jin and D. W. C. MacMillan, Nature, 2015, 525, 87–90 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, F. O'Hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99 CrossRef CAS PubMed.
- Y. Fujiwara, J. A. Dixon, R. A. Rodriguez, R. D. Baxter, D. D. Dixon, M. R. Collins, D. G. Blackmond and P. S. Baran, J. Am. Chem. Soc., 2012, 134, 1494–1497 CrossRef CAS PubMed.
- W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem.–Eur. J., 2017, 23, 2537–2541 CrossRef CAS PubMed.
- G. Deng and C.-J. Li, Org. Lett., 2009, 11, 1171–1174 CrossRef CAS PubMed.
- L. Fang, L. Chen, J. Yu and L. Wang, Eur. J. Org. Chem., 2015, 1910–1914 CrossRef CAS.
- A. P. Antonchick and L. Burgmann, Angew. Chem., Int. Ed., 2013, 52, 3267–3271 CrossRef CAS PubMed.
- X. Li, H.-Y. Wang and Z.-J. Shi, New J. Chem., 2013, 37, 1704–1706 RSC.
- R. S. J. Proctor and R. J. Phipps, Angew. Chem., Int. Ed., 2019, 58, 13666–13699 CrossRef CAS PubMed.
- J. Choi, G. Laudadio, E. Godineau and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 11927–11933 CrossRef CAS PubMed.
- G. Deng, K. Ueda, S. Yanagisawa, K. Itami and C.-J. Li, Chem.–Eur. J., 2009, 15, 333–337 CrossRef CAS PubMed.
- W. Sun, Z. Xie, J. Liu and L. Wang, Org. Biomol. Chem., 2015, 13, 4596–4604 RSC.
- R. B. Katz, J. Mistry and M. B. Mitchell, Synth. Commun., 1989, 19, 317–325 CrossRef CAS.
- G. A. Russell, L. Wang and C.-F. Yao, J. Org. Chem., 1995, 60, 5390–5395 CrossRef CAS.
- M. A. AmrollahiBiyouki, R. A. J. Smith, J. J. Bedford and J. P. Leader, Synth. Commun., 1998, 28, 3817–3825 CrossRef.
- X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2016, 138, 8718–8721 CrossRef CAS PubMed.
- X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 5998–6007 CrossRef CAS PubMed.
- I. Kim, G. Kang, K. Lee, B. Park, D. Kang, H. Jung, Y.-T. He, M.-H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 9239–9248 CrossRef PubMed.
- S. Jung, H. Lee, Y. Moon, H.-Y. Jung and S. Hong, ACS Catal., 2019, 9, 9891–9896 CrossRef CAS.
- L. Shen, X. Gao, N. Luan, Z. Liu, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Biomol. Chem., 2020, 18, 1738–1742 RSC.
- F. Rammal, D. Gao, S. Boujnah, A. Gaumont, A. A. Hussein and S. Lakhdar, Org. Lett., 2020, 22, 7671–7675 CrossRef CAS PubMed.
- E. Alfonzo and S. M. Hande, Org. Lett., 2021, 23, 6115–6120 CrossRef CAS PubMed.
- Y.-T. He, J. Won, J. Kim, B. Park, T. Kim, M.-H. Baik and S. Hong, Org. Chem. Front., 2018, 5, 2595–2603 RSC.
- J. Q. Buquoi, J. M. Lear, X. Gu and D. A. Nagib, ACS Catal., 2019, 9, 5330–5335 CrossRef CAS PubMed.
- J. M. Lear, J. Q. Buquoi, X. Gu, K. Pan, D. N. Mustafa and D. A. Nagib, Chem. Commun., 2019, 55, 8820–8823 RSC.
- A.-P. Schaffner and P. Renaud, Eur. J. Org. Chem., 2004, 2004, 2291–2298 CrossRef.
- M. R. Sabol, J. M. Owen and W. R. Erickson, Synth. Commun., 2000, 30, 427–432 CrossRef CAS.
- C. E. Garrett and G. C. Fu, J. Org. Chem., 1996, 61, 3224–3225 CrossRef CAS.
- J. Boukouvalas, S. Cren and P. Renaud, in Encyclopedia of Reagents for Organic Synthesis, American Cancer Society, 2007 Search PubMed.
- A.-P. Schaffner and P. Renaud, Angew. Chem., Int. Ed., 2003, 42, 2658–2660 CrossRef CAS PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- V. W. Bowry and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 4992–4996 CrossRef CAS.
- C. Carra and J. C. Scaiano, Eur. J. Org. Chem., 2008, 4454–4459 CrossRef CAS.
- G. Villa, G. Povie and P. Renaud, J. Am. Chem. Soc., 2011, 133, 5913–5920 CrossRef CAS PubMed.
- E. D. Lorance, W. H. Kramer and I. R. Gould, J. Am. Chem. Soc., 2004, 126, 14071–14078 CrossRef CAS PubMed.
- D. Shukla, W. G. Ahearn and S. Farid, J. Org. Chem., 2005, 70, 6809–6819 CrossRef CAS PubMed.
- E. D. Lorance, K. Hendrickson and I. R. Gould, J. Org. Chem., 2005, 70, 2014–2020 CrossRef CAS PubMed.
- E. D. Lorance and I. R. Gould, J. Phys. Chem. A, 2005, 109, 2912–2919 CrossRef CAS PubMed.
- R. Ueno, Y. Ikeda and E. Shirakawa, Eur. J. Org. Chem., 2017, 4188–4193 CrossRef CAS.
- R. Ueno and E. Shirakawa, Org. Biomol. Chem., 2014, 12, 7469–7473 RSC.
- G. Povie, G. Villa, L. Ford, D. Pozzi, C. H. Schiesser and P. Renaud, Chem. Commun., 2010, 46, 803–805 RSC.
- M. Newcomb, R. M. Sanchez and J. Kaplan, J. Am. Chem. Soc., 1987, 109, 1195–1199 CrossRef CAS.
- S. Z. Zard, in Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH, 2008, vol. 1, pp. 90–108 Search PubMed.
- S. W. McCombie, B. Quiclet-Sire and S. Z. Zard, Tetrahedron, 2018, 74, 4969–4979 CrossRef CAS.
- S. Z. Zard, Helv. Chim. Acta, 2019, 102, e1900134 CrossRef.
- S. Z. Zard, Chem.–Eur. J., 2020, 26, 12689–12705 CrossRef CAS PubMed.
- S. W. McCombie, W. B. Motherwell and M. J. Tozer, Organic Reactions, American Cancer Society, 2012, pp. 161–432 Search PubMed.
- J. Marco-Contelles, P. Ruiz-Fernandez and B. Sanchez, J. Org. Chem., 1993, 58, 2894–2898 CrossRef CAS.
- Q. Wan, A. Lubineau and M. Scherrmann, J. Carbohydr. Chem., 2004, 23, 83–93 CrossRef CAS.
- B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 1998, 39, 9435–9438 CrossRef CAS.
- T. McCallum and L. Barriault, Chem. Sci., 2016, 7, 4754–4758 RSC.
- D. Zheng and A. Studer, Org. Lett., 2019, 21, 325–329 CrossRef CAS PubMed.
- A. Citterio, F. Minisci, O. Porta and G. Sesana, J. Am. Chem. Soc., 1977, 99, 7960–7968 CrossRef CAS.
- T. Caronna, A. Citterio, M. Ghirardini and F. Minisci, Tetrahedron, 1977, 33, 793–796 CrossRef CAS.
- B. Giese and G. Kretzschmar, Chem. Ber., 1984, 117, 3160–3164 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Rammal, D. Gao, S. Boujnah, A. A. Hussein, J. Lalevée, A.-C. Gaumont, F. Morlet-Savary and S. Lakhdar, ACS Catal., 2020, 10, 13710–13717 CrossRef CAS.
- F. Neese and E. F. Valeev, J. Chem. Theory Comput., 2011, 7, 33–43 CrossRef CAS PubMed.
- M. Saitow, U. Becker, C. Riplinger, E. F. Valeev and F. Neese, J. Chem. Phys., 2017, 146, 164105 CrossRef PubMed.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- F. Minisci, E. Vismara, G. Morini, F. Fontana, S. Levi, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 476–479 CrossRef CAS.
- A. Citterio, F. Minisci and V. Franchi, J. Org. Chem., 1980, 45, 4752–4757 CrossRef CAS.
- F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1987, 52, 730–736 CrossRef CAS.
- K. Ermanis, A. C. Colgan, R. S. J. Proctor, B. W. Hadrys, R. J. Phipps and J. M. Goodman, J. Am. Chem. Soc., 2020, 142, 21091–21101 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterization of all new compounds including copies of 1H- and 13C-NMR spectra. See DOI: 10.1039/d1sc02748d |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |