
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed enantioselective carbonylation toward α-chiral secondary amides†
Yang
Yuan
a,
Fengqian
Zhao
a and
Xiao-Feng
Wu
 *ab
*ab
aLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: xiao-feng.wu@catalysis.de
bDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023, Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 25th August 2021
Abstract
Secondary amides are omnipresent structural motifs in peptides, natural products, pharmaceuticals, and agrochemicals. The copper-catalyzed enantioselective hydroaminocarbonylation of alkenes described in this study provides a direct and practical approach for the construction of α-chiral secondary amides. An electrophilic amine transfer reagent possessing a 4-(dimethylamino)benzoate group was the key to the success. This method also features broad functional group tolerance and proceeds under very mild conditions, affording a set of α-chiral secondary amides in high yields (up to 96% yield) with unprecedented levels of enantioselectivity (up to >99% ee). α,β-Unsaturated secondary amides can also be produced though the method by using alkynes as the substrate.
Introduction
Transition-metal-catalyzed hydrocarbonylations are one of the most fundamental and ideal reactions for the synthesis of numerous value-added carbonyl containing compounds from readily available alkenes.1 Within this class of reactions, hydroaminocarbonylation, also called hydroamidation, represents a straightforward route for the conversion of alkenes into amides.2 The original catalytic systems for hydroaminocarbonylations based on cobalt,3 nickel,4 iron,5 and ruthenium complexes6 require severe conditions such as high temperatures and high pressures and are often accompanied by aminoformylation side reactions, thus leading to poor chemoselectivity and limited substrate scope. Palladium-catalyzed hydroaminocarbonylation reactions have been extensively developed over the past few decades. Recently, ligand-controlled regiodivergent palladium-catalyzed hydroaminocarbonylations, to access either linear or branched amides under relatively mild conditions, which involved palladium-hydride species, have been independently developed by the groups of Beller,7 Cole-Hamilton,8 Liu,9 Huang,10 and Alper11 (Fig. 1a).12 Despite these advances, asymmetric Pd-catalyzed hydroaminocarbonylations of alkenes with control of the regio- and enantio-selectivity are still less explored, and the substrate scope of amines is limited to arylamines. The only elegant work was reported by the Guan group, who presented a novel asymmetric Markovnikov hydroaminocarbonylation of alkenes with anilines enabled by Pd-monodentate phosphoramidite catalysis.13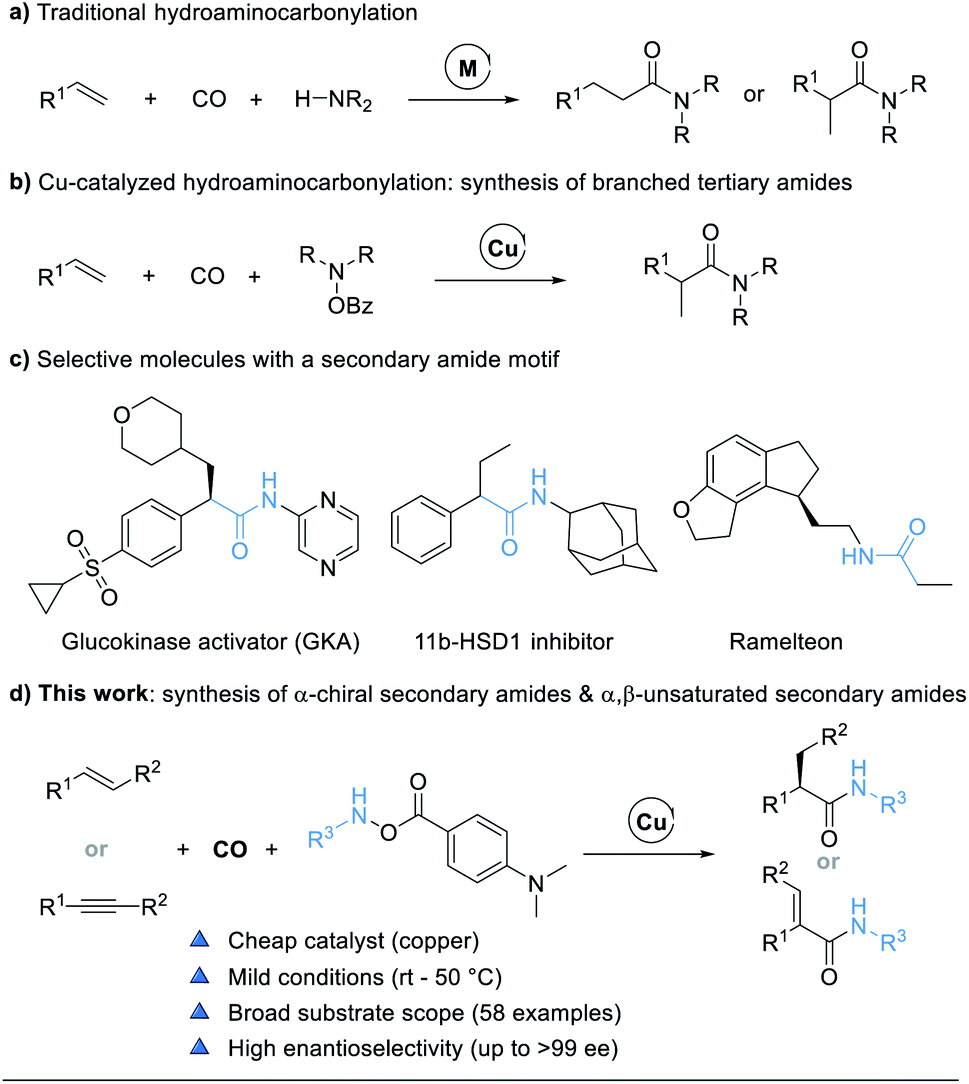 | ||
| Fig. 1 (a) Traditional hydroaminocarbonylation. (b) Cu-catalyzed hydroaminocarbonylation: synthesis of branched tertiary amides. (c) Selective molecules with a secondary amide motif. (d) Synthesis of α-chiral secondary amides and α,β-unsaturated secondary amides (this work). | ||
In general, the use of monodentate ligands favors Markovnikov selectivity in palladium-catalyzed hydroaminocarbonylations for the formation of branched amides.7c,9,10b,11,14 However, the challenge of monodentate ligand-assisted Pd-catalyzed enantioselective carbonylations is the critical competitive coordination between CO and chiral ligand-palladium species, resulting in no “chelate effect”, especially under high temperatures and pressures.15 To resolve this issue, an alternative approach is to change palladium into a metal with slightly weaker coordination ability to carbon monoxide and copper might be a choice. Recent developments in copper hydride chemistry have enabled a new approach to the hydroamination of alkenes based on the hydrocupration of the alkene and subsequent amination of the reactive alkyl copper intermediate with electrophilic amine reagents.16 We proposed to carry out the hydrocupration of alkenes with amine electrophiles under a CO atmosphere to achieve a selective hydroaminocarbonylation reaction. Indeed, we do realize that this Cu-catalyzed hydroaminocarbonylation reaction will prepare branched tertiary amides (Fig. 1b).17 Nevertheless, this approach to hydroaminocarbonylation has been limited to the synthesis of tertiary amides with hydroxyamines (R2NOBz, R = alkyl) as the dialkylamine transfer reagents.
Secondary amides (–NH–CO–), including chiral secondary amides, are omnipresent structural motifs in peptides, natural products, pharmaceuticals, and agrochemicals (Fig. 1c).18 It is therefore highly desired to develop an efficient and practical methodology that can achieve secondary amides with high regio- and enantio-selectivity by using cheap catalysts under mild conditions. Herein we report the development of a copper-catalyzed hydroaminocarbonylation process to directly synthesize α-chiral secondary amides from alkenes and modified amine transfer reagents. The method can also be extended to the synthesis of α,β-unsaturated secondary amides by using alkynes as the substrate (Fig. 1d).
Results and discussion
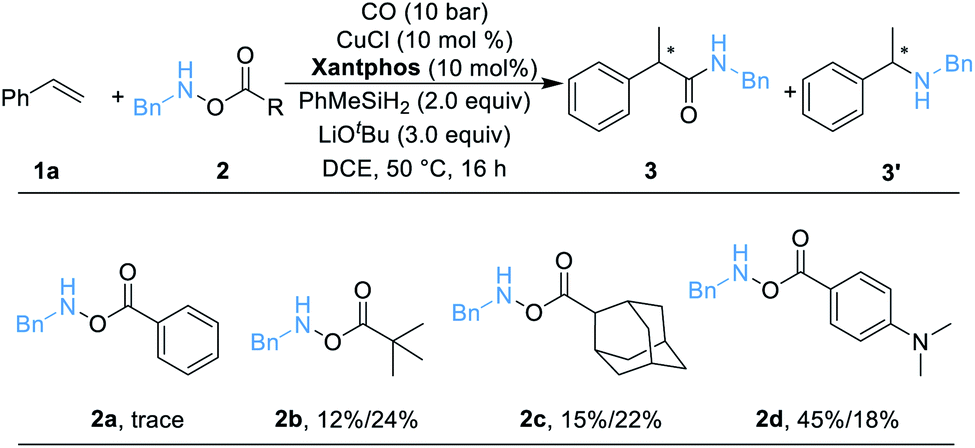
We began our work by studying the reaction between styrene 1a (1.2 equiv.) and O-benzoyl-N-benzylhydroxylamine 2a [BnN(H)OBz, 1.0 equiv.] utilizing our previously reported conditions (Table 1). Unfortunately, only a trace amount of the desired secondary amide 3 can be detected. We reasoned that this result might be caused by the poor stability of amine transfer reagent 2a, and the rapid nonproductive consumption of 2a by LCuH diminished the yield. Thus, we tested different amine transfer reagents for this hydroaminocarbonylation under the same conditions. The results are summarized in Scheme 1. We found that the use of 2d, an amine transfer reagent bearing a 4-(dimethylamino)benzoate group, delivered the desired secondary amide 3 in the highest yield, albeit the direct C–N coupling product 3′ was also generated in 18% yield.| a Standard conditions: 1a (0.12 mmol, 1.5 equiv.), 2 (0.1 mmol, 1.0 equiv.), [SiH] (0.2 mmol, 2.0 equiv.), CuCl (10 mol%), ligand (10 mol%), LiOtBu (0.3 mmol, 3.0 equiv.), CO (10 bar), DCE (0.5 mL), 50 °C, 16 h; yields are determined by GC analysis using hexadecane as an internal standard. |
|---|
|
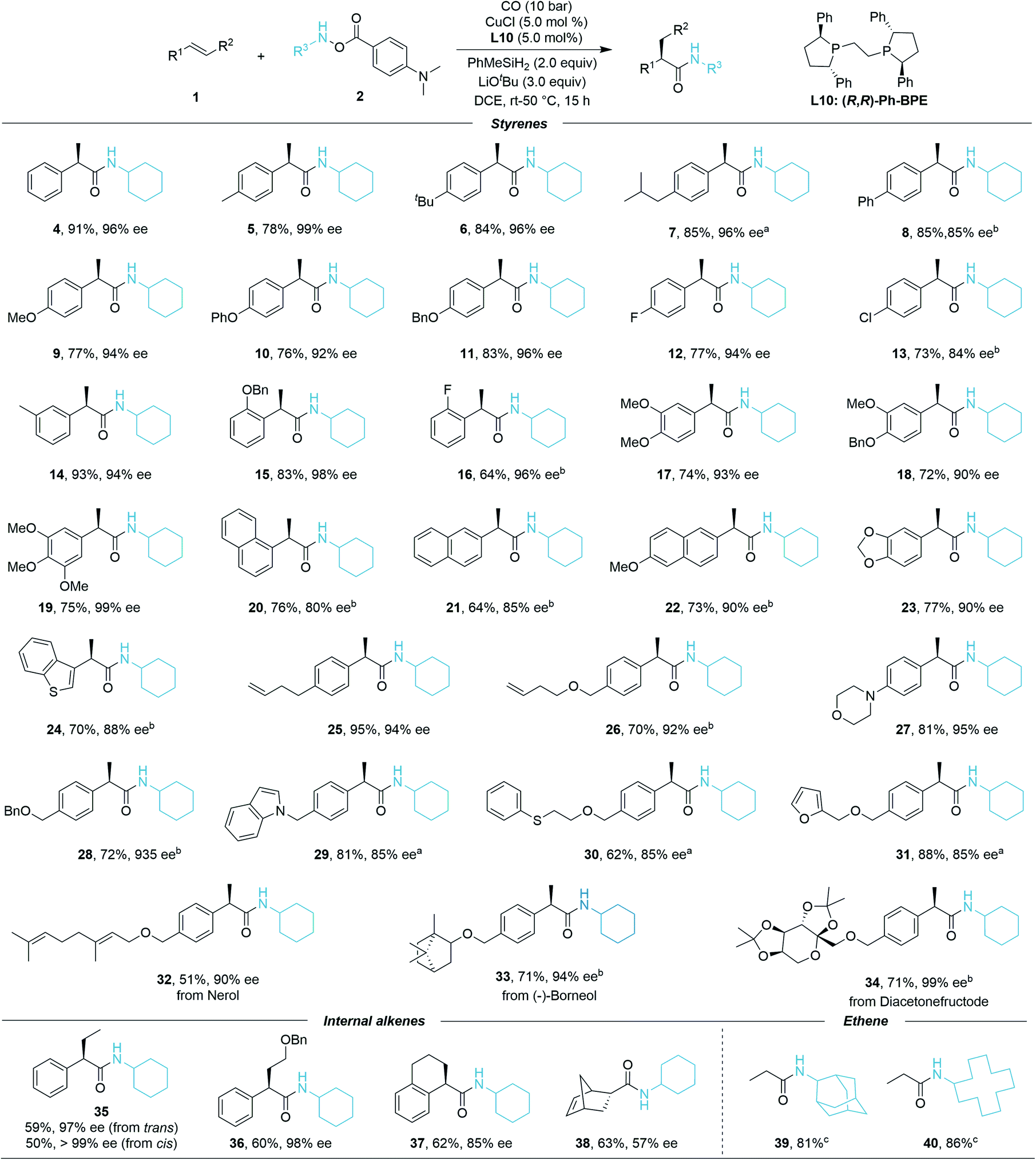 | ||
| Scheme 1 Substrate scope of alkenes. Standard conditions: 1 (0.12 mmol, 1.2 equiv.), 2 (0.1 mmol, 1.0 equiv.), MePhSiH2 (0.2 mmol, 2.0 equiv.), CuCl (5.0 mol%), L10 (5.0 mol%), LiOtBu (0.3 mmol, 3.0 equiv.), CO (10 bar), DCE (0.5 mL), 50 °C, 15 h, isolated yields. ee values are determined by chiral-phase HPLC. a40 °C. brt (room temperature, 26 °C). cEthene gas (2 bar) was used instead of styrenes. | ||
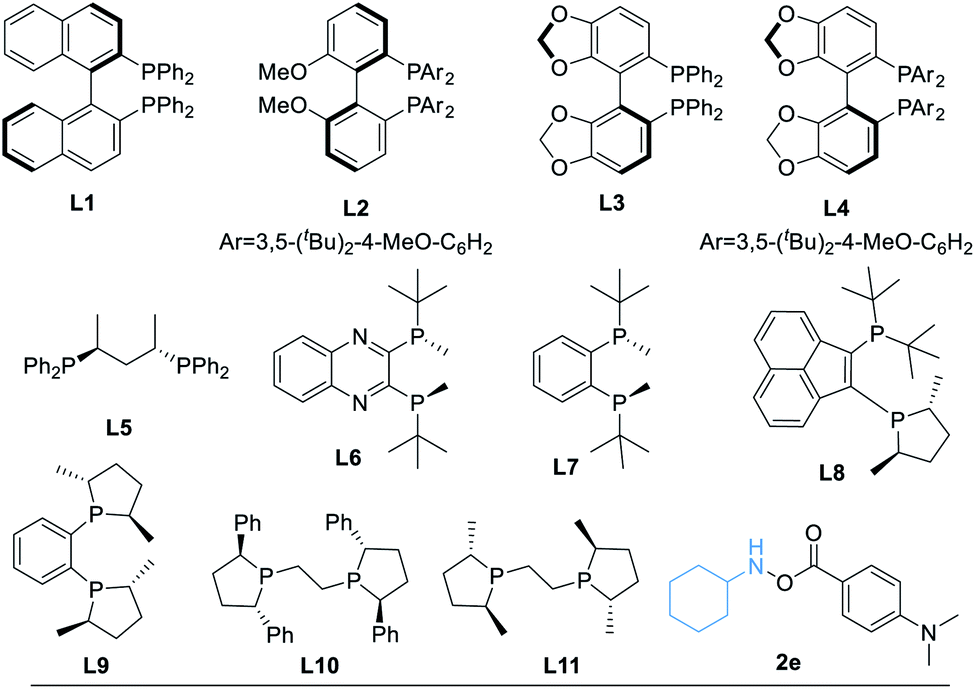
After determining the best amine transfer reagent 2d, we started to investigate the reaction by using chiral ligands for constructing α-chiral secondary amides. As shown in Table 2, chiral ligands L1–L3 only gave a trace amount of product 3. Bulky (S)-DTBM-Segphos (L4) and (S,S)-BDPP (L5) delivered the desired product in very low yields. The use of (S,S)-QuinoxP* L6 leads to 14% yield of the desired product 3 with moderate enantioselectivity (Table 2, entry 4, 73% ee). (S,S)-BenzP* (L7), L8, (R,R)-Me-DuPhos (L9), and (S,S)–Me-BPE (L11) were ineffective for this reaction. Fortunately, using (R,R)-Ph-BPE (L10) as the ligand provided 3 in 62% yield with excellent enantioselectivity (Table 2, entry 8, 99% ee). From these results, especially compared with the result between L9 and L11, we believe that the bite angle and the basicity of the ligand applied are crucial for the success of this transformation. Then different kinds of silane were tested, and low or no yield of product 3 was observed when Et3SiH, PMHS, or (EtO)2MeSiH was applied. Excellent ee of the carbonylated product can be obtained when Ph2SiH2 was used, but the yield of 3 decreased to 47%. Changing the amine transfer reagent 2d to 2e, the desired product 4 can be afforded in 89% yield with 95% ee (Table 2, entry 10). Notably, the yield of 4 would not be diminished by decreasing the catalyst loading to 5.0 mol%, and a trace amount of 4′ was detected (Table 2, entry 11, 91% yield, 96% ee). Surprisingly, lowering the temperature to room temperature (26 °C), the reaction could still proceed well, giving product 4 in 83% yield (Table 2, entry 12, 96% ee).
|
|
||||
|---|---|---|---|---|
| Entry | 2 | Ligand | Yield (%) | ee (%) |
| a Standard conditions: 1a (0.12 mmol, 1.2 equiv.), 2d (0.1 mmol, 1.0 equiv.), [SiH] (0.2 mmol, 2.0 equiv.), CuCl (10 mol%), ligand (10 mol%), LiOtBu (0.3 mmol, 3.0 equiv.), CO (10 bar), DCE (0.5 mL), 50 °C, 15 h; yields are determined by GC analysis using hexadecane as an internal standard. Ee values are determined by chiral-phase HPLC. n.d. = not determined. b Using 2e as a substrate, isolated yield. c CuCl (5 mol%), ligand (5 mol%). d rt (room temperature, 26 °C). | ||||
| 1 | 2d | L1–L3 | Trace (3/3′) | — |
| 2 | 2d | L4 | 8/6 (3/3′) | n.d |
| 3 | 2d | L5 | 10/7 (3/3′) | n.d |
| 4 | 2d | L6 | 14/2 (3/3′) | 73 (3) |
| 5 | 2d | L7 | Trace (3/3′) | — |
| 6 | 2d | L8 | Trace (3/3′) | — |
| 7 | 2d | L9 | Trace (3/3′) | — |
| 8 | 2d | L10 | 62/9 (3/3′) | 99 (3) |
| 9 | 2d | L11 | Trace (3/3′) | — |
| 10b | 2e | L10 | 89/Trace (4/4′) | 95 (4) |
| 11bc | 2e | L10 | 91/Trace (4/4′) | 96 (4) |
| 12b,c,d | 2e | L10 | 83/Trace (4/4′) | 96 (4) |
|
||||

After identification of the optimal conditions, we next investigated the generality of this asymmetric Cu-catalyzed hydroaminocarbonylation reaction. The reaction efficiency was first evaluated under the optimal reaction conditions with the scope of alkenes. As shown in Scheme 1, styrenes bearing different electron-donating or electron-withdrawing groups at the para position were successfully converted into the α-chiral secondary amides 4–13 in high yields and excellent enantiomeric ratios (73–91% yield, 85–99% ee). Meta-F, meta-Me, and ortho-OBn-substituted styrenes gave the corresponding products 14–16 in 64–93% yields and 94–98% ee, thus indicating that the reaction is insensitive to the steric configuration of the styrenes. Di- and tri-substituted styrenes also proceeded smoothly to generate the desired products 17–20 with high enantioselectivities (72–75% yield, 90–99% ee). To our delight, 1-vinylnaphthalene, 2-vinylnaphthalene, 2-methoxy-6-vinylnaphthalene, 5-vinylbenzo[d][1,3]dioxole, and 5-vinylbenzo[b]thiophene converted efficiently in the reaction even at room temperature (20–24, 64–77% yield, 80–90% ee). Gratifyingly, excellent enantiomeric ratios were observed even when using functionalized styrenes as the coupling partners (25–31, 62–95% yield, 85–95% ee). Moreover, the late-stage functionalization of bioactive molecules has been investigated. Nerol, (−)-borneol, and diacetonefructose derived styrenes all reacted well, generating the corresponding secondary amides in good yields and high er values (32–34, 51–71% yield, 90–99% ee). Furthermore, internal styrenes were also proved to be compatible in the catalytic system; trans-β-methylstyrene afforded product 35 in 59% yield with 97% ee, cis-β-methylstyrene gave 35 in 50% yield with >99% ee, product 36 can be delivered in good yield and excellent enantioselectivity (60% yield, 97% ee). When 1,2-dihydronaphthalene was employed, the desired product 37 was isolated in 62% yield with 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 er. The bicyclic strained alkene could also provide 38 in 63% yield but with moderate enantioselectivity (57% ee). Meanwhile, ethene gas can be applied as well and high yields of the corresponding products (39, 40) were achieved under standard conditions, which further demonstrated the generality of the reaction. However, no desired product could be detected when the other aliphatic alkenes were tested, but the decomposition of the hydroxylamine starting material.
:7 er. The bicyclic strained alkene could also provide 38 in 63% yield but with moderate enantioselectivity (57% ee). Meanwhile, ethene gas can be applied as well and high yields of the corresponding products (39, 40) were achieved under standard conditions, which further demonstrated the generality of the reaction. However, no desired product could be detected when the other aliphatic alkenes were tested, but the decomposition of the hydroxylamine starting material.
We then turned our attention to the compatibility of hydroxylamine electrophiles and were pleased to find a broad substrate scope. The reactivities of various mono alkyl-substituted amine transfer reagents were investigated for the synthesis of α-chiral secondary amides (Scheme 2). We observed that cyclic amine sources were tolerated with five-, seven-, and even twelve-membered rings, affording the desired products 41, 43, and 44 with high yields and excellent enantioselectivities (70–92% yield, 88–93% ee). The absolute configuration of 44 was confirmed by X-ray crystallographic analysis,19 and the configuration of the other compounds described in this work was assigned in analogy to 44. The (S)-configuration product 42 can be obtained in 83% yield (−90% ee) by using (S,S)-Ph-BPE. In the current system, amine transfer reagents were well tolerated regardless of the bulky adamantyl group (45), heterocyclic group (46), and indanyl group (47). An amine transfer reagent with a tertiary alkyl group substituent was a competent substrate, delivering product 48 in good yield and enantioselectivity (76% yield, 91% ee). Additionally, desired products 49–51 can also be isolated in 74–84% yields with indicated diastereomeric ratio (dr) values. In these cases, there are ee values, but we were not able to measure the values. Additionally, several other examples of substrates were prepared and tested as well, but very low or no desired product could be detected. This might be due to the decreased stability of the corresponding amine substrates. The attempt to prepare a substrate related with an aniline derivate failed.
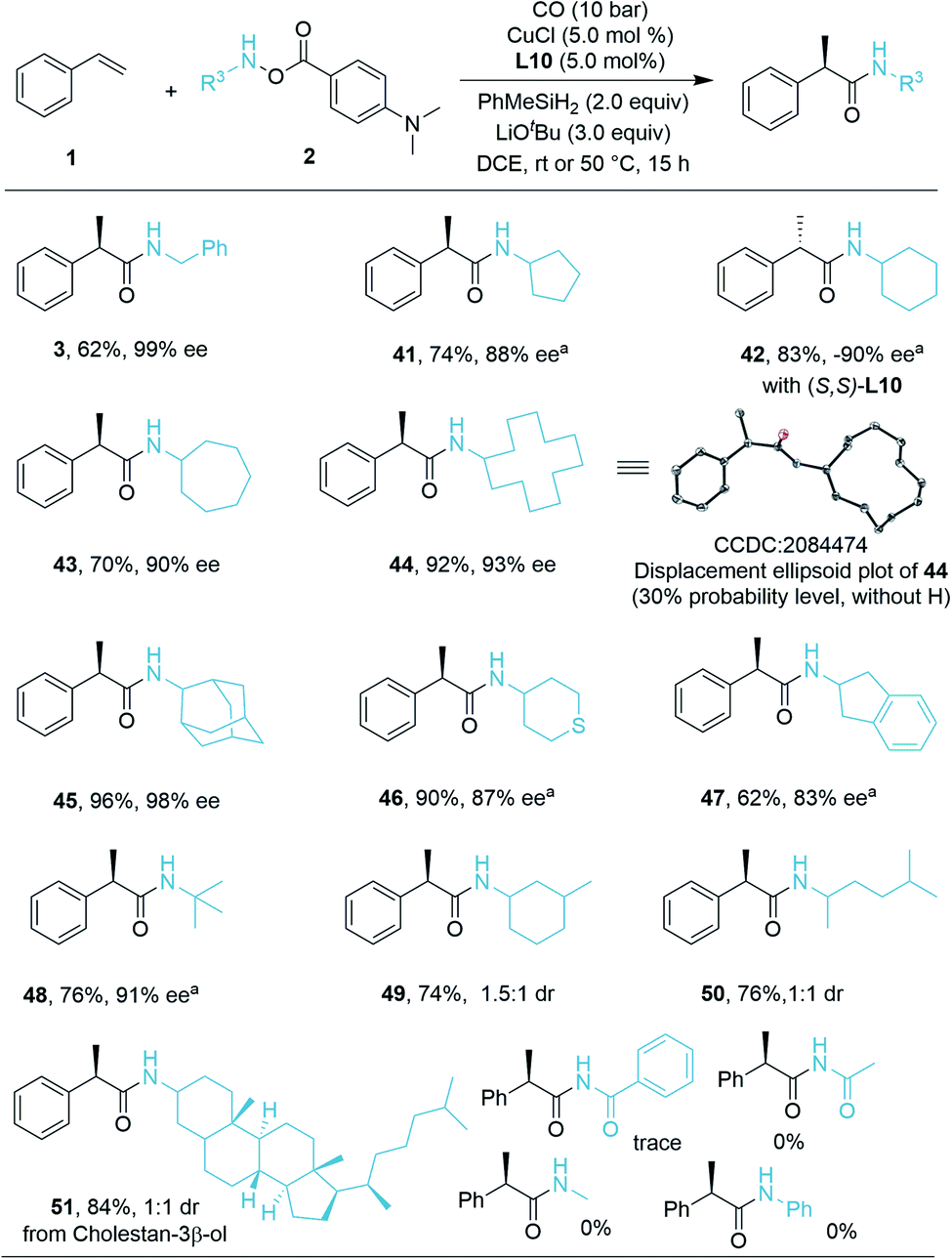 | ||
| Scheme 2 Substrate scope of hydroxylamines. Reaction conditions: 1 (0.12 mmol, 1.2 equiv.), 2 (0.1 mmol, 1.0 equiv.), MePhSiH2 (0.2 mmol, 2.0 equiv.), CuCl (5.0 mol%), L10 (5.0 mol%), LiOtBu (0.3 mmol, 3.0 equiv.), CO (10 bar), DCE (0.5 mL), 50 °C, 15 h, isolated yields; ee values are determined by chiral-phase HPLC, diastereoselectivities determined by 1H NMR analysis. art (room temperature, 26 °C). | ||
As a demonstration of the robustness and practicality of this method, the hydroaminocarbonylation process could be easily conducted on the 1.0 mmol scale with styrene and hydroxylamine electrophiles 2h (Table 3, eqn (1)). Using reduced catalyst loading, 2.5 mol% CuCl and (R,R)-Ph-BPE-phos, 44 could be synthesized in 87% isolated yield with high stereoselectivity (92% ee). Furthermore, the (R)-52 product (52 is an 11b-HSD1 inhibitor)18b can also be obtained easily in 69% yield and 96% ee by using trans-β-methylstyrene and 2i under standard conditions (Table 3, eqn (2)).
| a Isolated yield, ee values are determined by chiral-phase HPLC. |
|---|
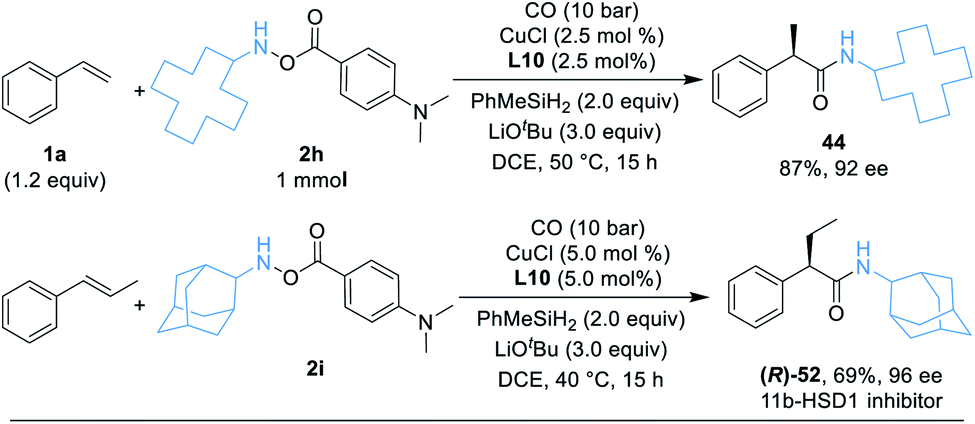
|
The reaction mechanism is proposed according to previous literature (Scheme 3).16,20 Initial formation of (L)CuOtBu from Cu(I), ligand, and LiOtBu, followed by reaction with [Si]H, generates an active copper hydride species, (L)Cu–H. Subsequently, (L)Cu–H insertion into an alkene provides alkylcopper intermediates A. After oxidative addition with electrophile hydroxylamine, the intermediates B is formed, which undergoes CO insertion to give intermediates C. Then, reductive elimination occurs to deliver the desired product, together with an (L)Cu–X complex, D. Finally, ligand exchange with LiOtBu regenerates the (L)CuOtBu species for the next catalytic cycle.
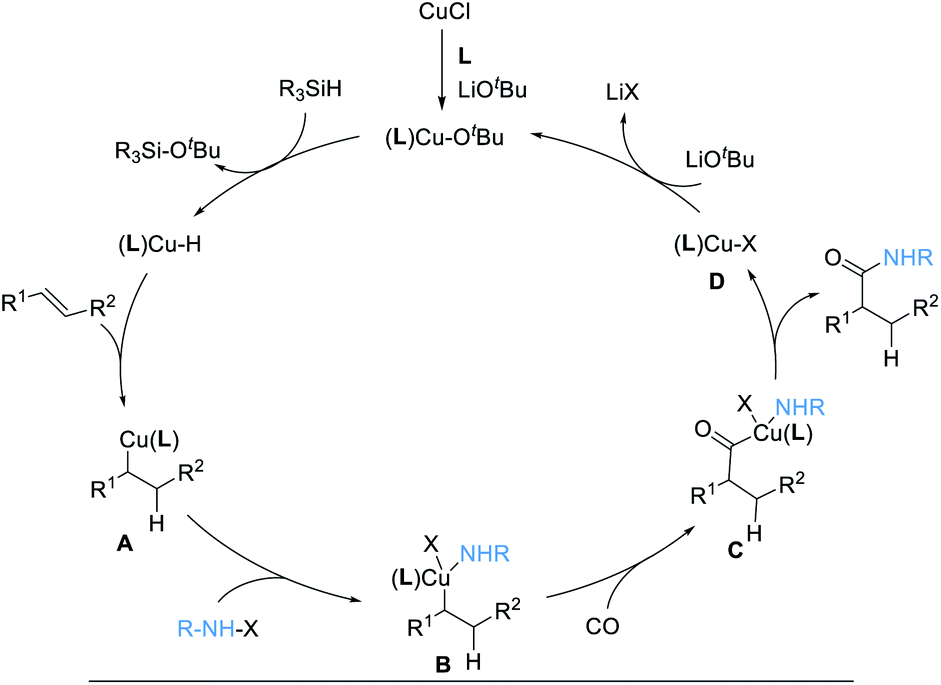 | ||
| Scheme 3 Proposed catalytic cycle. | ||
Alkynes are synthetically versatile and useful starting materials, because they can be prepared by a range of strategies.21 In addition, the potential reactivity of the two π-bonds increases their flexibility in multistep reaction sequences. Naturally, we then applied alkynes as the substrate in a similar catalytic system. A series of desired α,β-unsaturated secondary amides can be successfully delivered in moderate yields (53–59) as we anticipated and, as shown in Scheme 4, the products were produced as single geometric isomers.
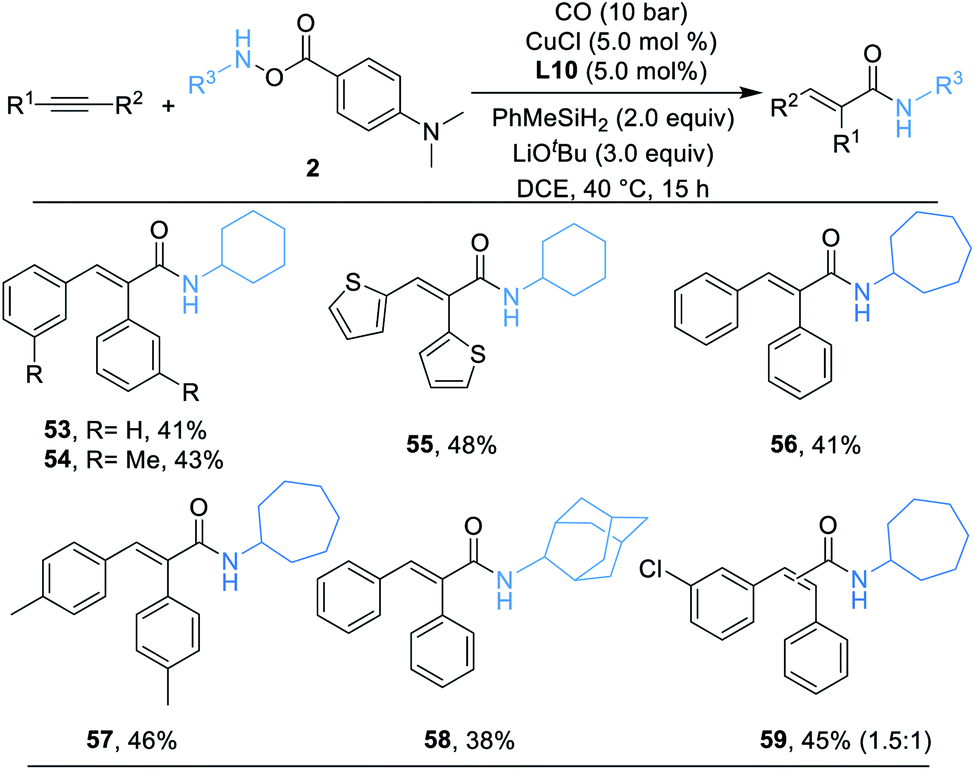 | ||
| Scheme 4 Substrate scope of alkynes. Reaction conditions: alkynes (0.1 mmol, 1.0 equiv.), 2 (0.12 mmol, 1.2 equiv.), MePhSiH2 (0.2 mmol, 2.0 equiv.), CuCl (5.0 mol%), L10 (5.0 mol%), LiOtBu (0.3 mmol, 3.0 equiv.), CO (10 bar), DCE (0.5 mL), 40 °C, 15 h, isolated yields. | ||
Conclusions
In conclusion, we have developed a copper-catalyzed regioselective and enantioselective intermolecular hydroaminocarbonylation of alkenes with electrophilic hydroxylamines. An electrophilic amine transfer reagent possessing a 4-(dimethylamino)benzoate group was the key to this process. The use of these reagents enabled the development of a general method to directly convert styrenes and even challenging β-substituted styrenes to α-chiral secondary amides. The method displays broad functional group tolerance and proceeds under very mild conditions, producing the desired α-chiral secondary amides in high yields with excellent enantioselectivities (up to >99% ee). We believe that this Cu-catalyzed enantioselective synthesis of α-chiral secondary amides will provide a new idea in the study of asymmetric hydroaminocarbonylation. Furthermore, alkynes were also compatible substrates in this catalytic system, giving the corresponding α,β-unsaturated secondary amide products.Author contributions
XFW directed this project and revised the manuscript. YY performed all the experiments and prepared the manuscript. FZ assisted in products purification and SI preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Chinese Scholarship Council (CSC) for financial support. We thank the analytical team of LIKAT for their very kind support. We also thank Dr Anke Spannenberg for the X-ray crystal analysis.Notes and references
- (a) W. B. Motherwell, Appl. Organomet. Chem., 2000, 14, 170 CrossRef CAS; (b) D. B. G. Williams, M. L. Shaw, M. J. Green and C. W. Holzapfel, Angew. Chem., Int. Ed., 2008, 47, 560–563 CrossRef CAS PubMed; (c) A. Brennführer, H. Neumann and M. Beller, ChemCatChem, 2009, 1, 28–41 CrossRef; (d) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (e) M. Amézquita-Valencia and H. Alper, J. Org. Chem., 2016, 81, 3860–3867 CrossRef PubMed; (f) K. Dong, X. Fang, S. Gülak, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed; (g) H. Matsubara, T. Kawamoto, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2018, 51, 2023–2035 CrossRef CAS PubMed; (h) J.-B. Peng, F.-P. Wu and X.-F. Wu, Chem. Rev., 2019, 119, 2090–2127 CrossRef CAS PubMed; (i) X. Wang, B. Wang, X. Yin, W. Yu, Y. Liao, J. Ye, M. Wang, L. Hu and J. Liao, Angew. Chem., Int. Ed., 2019, 58, 12264–12270 CrossRef CAS PubMed; (j) J. Yang, J. Liu, H. Neumann, R. Franke, R. Jackstell and M. Beller, Science, 2019, 366, 1514 CrossRef CAS PubMed.
- (a) X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1–35 CrossRef CAS PubMed; (b) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed; (c) J. Liu, C. Schneider, J. Yang, Z. Wei, H. Jiao, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2021, 60, 371–379 CrossRef CAS PubMed.
- A. Striegler and J. Weber, J. Prakt. Chem., 1965, 29, 281–295 CrossRef CAS.
- (a) P. Pino and P. Paleari, Gazz. Chim. Ital., 1951, 81, 64 Search PubMed; (b) P. Pino and R. Magri, Chim. Ind., 1952, 34, 511 CAS; (c) S. I. Lee, S. U. Son and Y. K. Chung, Chem. Commun., 2002, 1310–1311 RSC.
- W. Reppe and H. Main, Chem. Abstr., 1953, 47, 5428 Search PubMed.
- Y. Tsuji, T. Ohsumi, T. Kondo and Y. Watanabe, J. Organomet. Chem., 1986, 309, 333–344 CrossRef CAS.
- (a) X. Fang, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14089–14093 CrossRef CAS PubMed; (b) H. Li, K. Dong, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2015, 54, 10239–10243 CrossRef CAS PubMed; (c) J. Liu, H. Li, A. Spannenberg, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 13544–13548 CrossRef CAS PubMed.
- C. Jiménez-Rodriguez, A. A. Núñez-Magro, T. Seidensticker, G. R. Eastham, M. R. L. Furst and D. J. Cole-Hamilton, Catal. Sci. Technol., 2014, 4, 2332–2339 RSC.
- H. Liu, N. Yan and P. J. Dyson, Chem. Commun., 2014, 50, 7848–7851 RSC.
- (a) G. Zhang, B. Gao and H. Huang, Angew. Chem., Int. Ed., 2015, 54, 7657–7661 CrossRef CAS PubMed; (b) Y. Hu, Z. Shen and H. Huang, ACS Catal., 2016, 6, 6785–6789 CrossRef CAS; (c) J. Zhu, B. Gao and H. Huang, Org. Biomol. Chem., 2017, 15, 2910–2913 RSC; (d) B. Gao, G. Zhang, X. Zhou and H. Huang, Chem. Sci., 2018, 9, 380–386 RSC; (e) J. Li, S. Wang, S. Zou and H. Huang, Commun. Chem., 2019, 2, 14 CrossRef.
- T. Xu, F. Sha and H. Alper, J. Am. Chem. Soc., 2016, 138, 6629–6635 CrossRef CAS PubMed.
- For selected examples of Rh-catalyzed hydroamination, see: (a) A. Behr, D. Levikov and E. Nürenberg, Catal. Sci. Technol., 2015, 5, 2783–2787 RSC; (b) K. Dong, X. Fang, R. Jackstell, G. Laurenczy, Y. Li and M. Beller, J. Am. Chem. Soc., 2015, 137, 6053–6058 CrossRef CAS PubMed.
- Y.-H. Yao, H.-Y. Yang, M. Chen, F. Wu, X.-X. Xu and Z.-H. Guan, J. Am. Chem. Soc., 2021, 143, 85–91 CrossRef CAS PubMed.
- H.-Y. Yang, Y.-H. Yao, M. Chen, Z.-H. Ren and Z.-H. Guan, J. Am. Chem. Soc., 2021, 143, 7298–7305 CrossRef CAS PubMed.
- (a) T. Suzuki, Y. Uozumi and M. Shibasaki, J. Chem. Soc., Chem. Commun., 1991, 1593–1595 RSC; (b) Y. Yan, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2006, 128, 16058–16061 CrossRef CAS PubMed; (c) C. Godard, B. K. Muñoz, A. Ruiz and C. Claver, Dalton Trans., 2008, 853–860 RSC; (d) B. Yang, Y. Qiu, T. Jiang, W. D. Wulff, X. Yin, C. Zhu and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2017, 56, 4535–4539 CrossRef CAS PubMed; (e) M. Chen, X. Wang, P. Yang, X. Kou, Z.-H. Ren and Z.-H. Guan, Angew. Chem., Int. Ed., 2020, 59, 12199–12205 CrossRef CAS PubMed; (f) D. M. Hood, R. A. Johnson, A. E. Carpenter, J. M. Younker, D. J. Vinyard and G. G. Stanley, Science, 2020, 367, 542–548 CrossRef CAS PubMed.
- (a) C. Deutsch, N. Krause and B. H. Lipshutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed; (b) Y. Miki, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 10830–10834 CrossRef CAS PubMed; (c) S.-L. Shi and S. L. Buchwald, Nat. Chem., 2015, 7, 38–44 CrossRef CAS PubMed; (d) D. Niu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 9716–9721 CrossRef CAS PubMed; (e) R. Y. Liu and S. L. Buchwald, Acc. Chem. Res., 2020, 53, 1229–1243 CrossRef CAS PubMed; (f) L.-J. Cheng and N. P. Mankad, J. Am. Chem. Soc., 2017, 139, 10200–10203 CrossRef CAS PubMed; (g) L.-J. Cheng, S. M. Islam and N. P. Mankad, J. Am. Chem. Soc., 2018, 140, 1159–1164 CrossRef CAS PubMed.
- Y. Yuan, F.-P. Wu, C. Schünemann, J. Holz, P. C. J. Kamer and X.-F. Wu, Angew. Chem., Int. Ed., 2020, 59, 22441–22445 CrossRef CAS PubMed.
- (a) A. Greenberg, C. M. Breneman, J. F. Liebman, The Amide Linkage: Structural Significance In Chemistry, Biochemistry, And Materials Science, John Wiley & Sons, 2002 Search PubMed; (b) N. A. Magnus, T. M. Braden, J. Y. Buser, A. C. DeBaillie, P. C. Heath, C. P. Ley, J. R. Remacle, D. L. Varie and T. M. Wilson, Org. Process Res. Dev., 2012, 16, 830–835 CrossRef CAS; (c) O. Uchikawa, K. Fukatsu, R. Tokunoh, M. Kawada, K. Matsumoto, Y. Imai, S. Hinuma, K. Kato, H. Nishikawa, K. Hirai, M. Miyamoto and S. Ohkawa, J. Med. Chem., 2002, 45, 4222–4239 CrossRef CAS PubMed; (d) Janssen Pharmaceutica N.V.- WO2004/56744, International Patent, 2004, A1 Search PubMed; (e) S. Duquesne, D. Destoumieux-Garzón, J. Peduzzi and S. Rebuffat, Nat. Prod. Rep., 2007, 24, 708–734 RSC; (f) D. J. Greenblatt, J. S. Harmatz and A. Karim, J. Clin. Pharmacol., 2007, 47, 485–496 CrossRef CAS PubMed; (g) E. A. Ilardi and A. Zakarian, Chem.–Asian J., 2011, 6, 2260–2263 CrossRef CAS PubMed; (h) D. Fiorito, Y. Liu, C. Besnard and C. Mazet, J. Am. Chem. Soc., 2020, 142, 623–632 CrossRef CAS PubMed.
- Deposition Number 2084474 (for 44) contains the supplementary crystallographic data for this paper. The absolute configuration was assigned as R which is consistent with the determined Flack parameter [0.01(12)].
- (a) T. Tsuda, T. Hashimoto and T. Saegusa, J. Am. Chem. Soc., 1972, 94, 658–659 CrossRef; (b) T. H. Lemmen, G. V. Goeden, J. C. Huffman, R. L. Geerts and K. G. Caulton, Inorg. Chem., 1990, 29, 3680–3685 CrossRef CAS.
- (a) B. M. Trost and A. H. Weiss, Adv. Synth. Catal., 2009, 351, 963–983 CrossRef CAS PubMed; (b) D. Habrant, V. Rauhala and A. M. P. Koskinen, Chem. Soc. Rev., 2010, 39, 2007–2017 RSC; (c) R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: General information, general procedures, analytical data, and NMR spectra. CCDC 2084474. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04210f |
| This journal is © The Royal Society of Chemistry 2021 |