
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti PCET in symmetrically substituted benzimidazoles†
Emmanuel
Odella
 a,
Maxim
Secor
b,
Mackenna
Elliott
a,
Thomas L.
Groy
a,
Thomas A.
Moore
a,
Sharon
Hammes-Schiffer
*b and
Ana L.
Moore
*a
a,
Maxim
Secor
b,
Mackenna
Elliott
a,
Thomas L.
Groy
a,
Thomas A.
Moore
a,
Sharon
Hammes-Schiffer
*b and
Ana L.
Moore
*a
aSchool of Molecular Sciences, Arizona State University, Tempe, Arizona 85287-1604, USA. E-mail: amoore@asu.edu
bDepartment of Chemistry, Yale University, New Haven, Connecticut 06520-8107, USA. E-mail: sharon.hammes-schiffer@yale.edu
First published on 23rd August 2021
Abstract
Proton-coupled electron transfer (PCET) reactions depend on the hydrogen-bond connectivity between sites of proton donors and acceptors. The 2-(2′-hydroxyphenyl) benzimidazole (BIP) based systems, which mimic the natural TyrZ-His190 pair of Photosystem II, have been useful for understanding the associated PCET process triggered by one-electron oxidation of the phenol. Substitution of the benzimidazole by an appropriate terminal proton acceptor (TPA) group allows for two-proton translocations. However, the prototropic properties of substituted benzimidazole rings and rotation around the bond linking the phenol and the benzimidazole can lead to isomers that interrupt the intramolecular hydrogen-bonded network and thereby prevent a second proton translocation. Herein, a strategic symmetrization of a benzimidazole based system with two identical TPAs yields an uninterrupted network of intramolecular hydrogen bonds regardless of the isomeric form. NMR data confirms the presence of a single isomeric form in the disubstituted system but not in the monosubstituted system in certain solvents. Infrared spectroelectrochemistry demonstrates a two-proton transfer process associated with the oxidation of the phenol occurring at a lower redox potential in the disubstituted system relative to its monosubstituted analogue. Computational studies support these findings and show that the disubstituted system stabilizes the oxidized two-proton transfer product through the formation of a bifurcated hydrogen bond. Considering the prototropic properties of the benzimidazole heterocycle in the context of multiple PCET will improve the next generation of novel, bioinspired constructs built by concatenated units of benzimidazoles, thus allowing proton translocations at nanoscale length.
Introduction
Nature employs a tyrosine Z-histidine (TyrZ-His190) pair as a redox relay that couples proton transfer to the redox process between the reaction-center P680 and the water oxidizing catalyst in photosystem II (PSII). The high efficiency in this redox process relies on the coupled movement of both protons and electrons, which imparts an energetic advantage over the single electron transfer process by lowering the activation barrier.1 This reaction, better known as proton-coupled electron transfer (PCET), has been experimentally and theoretically studied for years, inspiring many researchers to design a collection of artificial models capable of mimicking the crucial role of the TyrZ-His190 pair.1–3A benzimidazole attached covalently to a phenol derivative (BIP 1, Fig. 1A) is a versatile platform that closely emulates several aspects of the TyrZ-His190 pair and its unique PCET event (the benzimidazole models His190 and the phenol models TyrZ4–6). Variants of this model have been reported in which BIP is covalently linked to a high potential porphyrin.7–9 In our recent work, a low or barrierless pathway for PCET with the concerted process occurring in ∼100 fs has been identified using 2D electronic vibrational spectroscopy.10
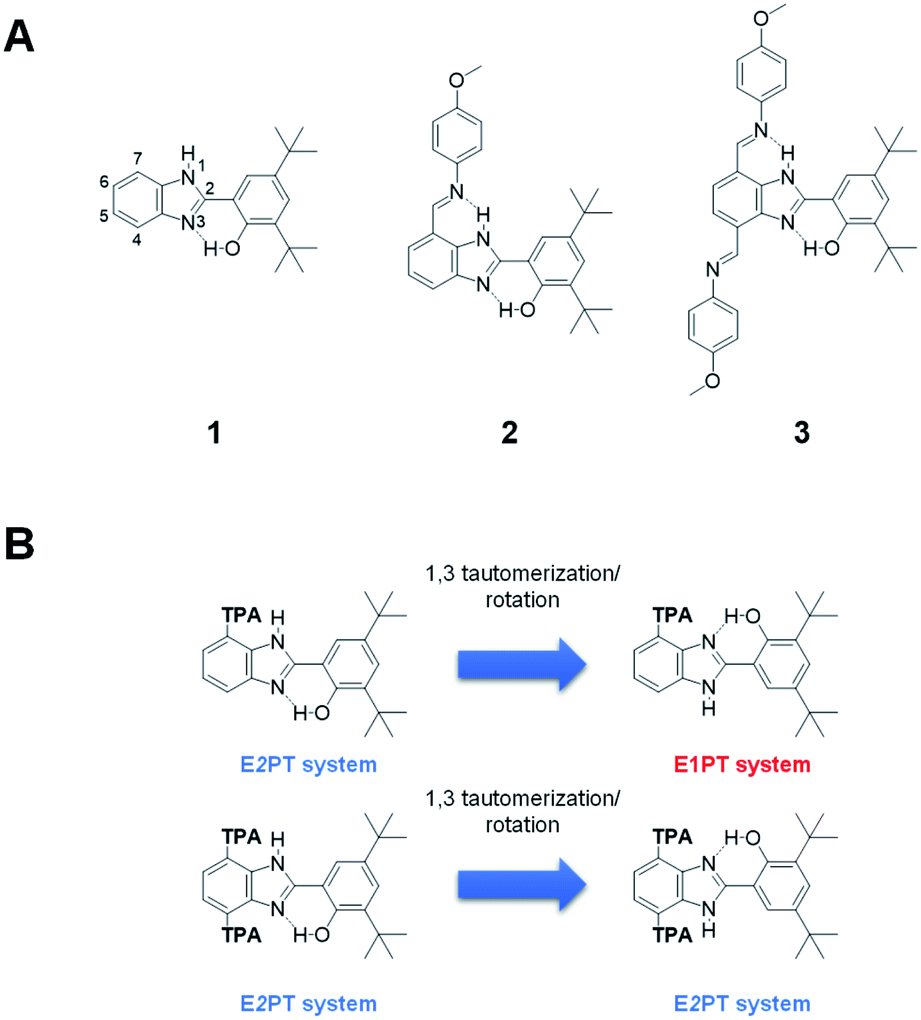 | ||
| Fig. 1 (A) Molecular structures of compounds 1–3. (B) 1,3-Annular tautomerization/rotation process in BIP monosubstituted at 7-position (top) with a terminal proton acceptor (TPA) and disubstituted at 4,7 positions (bottom) with an identical TPA. | ||
Upon electrooxidation of 1, a reversible one-electron coupled with one-proton transfer (E1PT) occurs.2,11 This proton displacement between the phenol and the proximal benzimidazole nitrogen takes place in a well-defined and strong intramolecular hydrogen bond connecting both groups. An expansion in the proton translocation distance was successfully achieved by proton wires constructed by covalently intercalating a series of benzimidazole units between the oxidation center (the phenol) and the terminal proton acceptor (TPA), in which electron transfer from the phenol is coupled to up to four proton translocations that span a total distance of ∼16 Å.12
The success of these biologically inspired proton wires relies on the uninterrupted sequence of intramolecular hydrogen bonds, which creates a network over which protons move reversibly between acidic and basic sites within the molecule via a Grotthuss-type mechanism. Furthermore, a clear example of the consequences associated with the disruption of a hydrogen-bonded network has been described in our recent work. The absence of a crucial hydrogen bond in the network causes a pronounced change in the overall reversibility and thermodynamics of the PCET process.13 The discontinuity of the hydrogen-bonded network, and its potential consequences on PCET, is also evidenced in a less explored feature—the intrinsic prototropic property (annular 1,3 tautomerism) of benzimidazole.14–16 BIP substituted at any of the remaining benzimidazole positions (4 to 7 positions, see numbering in 1, Fig. 1A) consists of a mixture of isomers, whose presence can be clearly detected by NMR spectroscopy under certain conditions.17–19 It is known that systems based on 2-(2′-hydroxyphenyl)benzimidazole (BIP without tert-butyl groups) may exist in three different enol conformers and a keto tautomeric structure.15,18,20,21 Among them, the closed cis-enol species, featuring an intramolecular hydrogen bond between the phenol and the lone pair of the benzimidazole nitrogen, is the most stable conformation in the ground state in non-polar or low polarity solvents. However, the dielectric constant and hydrogen bonding ability of the medium play an important role in the population of the closed cis-enol conformer relative to the other species in solution. Solvents acting as hydrogen bond donors or acceptors stabilize preferentially the keto, open cis-enol, and trans-enol forms over the intramolecularly hydrogen bonded closed cis-enol.15,18,20,21
Conformational isomers are energetically accessible to the bioinspired constructs that significantly hinder the PCET process. Specifically, rotamers associated with the bond between the phenol and benzimidazole moieties flip the system over and remove the possibility of multi-proton translocation. Indeed, the annular 1,3-tautomerization facilitates the rotation around this bond by stabilizing the system with the formation of the hydrogen bond between the phenol and the lone pair of the other nitrogen atom of the benzimidazole. Therefore, these structures are interconvertible via tautomerism followed by rotation along the bond connecting the phenol and benzimidazole moieties (Fig. 1B).
An example of multiple PCET (MPCET) has been demonstrated by placing at the 7-position of the benzimidazole a substituted phenylimine as the TPA (2, Fig. 1A).22 However, in this system, only one of the isomers contains the necessary, uninterrupted series of intramolecular hydrogen bonds for facilitating a one-electron, two-proton transfer (E2PT) process. Furthermore, this tautomerization/rotation process creates potential interruptions in proton wires of arbitrary length by converting EnPT (n represents the number of protons, n > 1) systems to interrupted isomers so that only EnPT processes with n less than the number of protons are possible. One strategy to eliminate the effects of tautomerization/rotation is by alkylating the imidazole nitrogen. However, this removes the NH group, which is a participant in the second proton transfer process. Another approach is a symmetrization of the benzimidazole moiety by adding the same substitution at the 4 and 7 positions, as illustrated in Fig. 1B. In this case, the annular 1,3-tautomerization is not eliminated, but the resulting species from the proton exchange have equivalent structures with identical intramolecular hydrogen bonds. This removes the issues caused by tautomerization/rotation processes by making every species capable of forming the two intramolecular hydrogen bonds required for the E2PT process. The incorporation of a second, identical TPA on BIP (3, Fig. 1A) yielding a system invariant to this tautomerization/rotation is described here.
Results and discussion
Design and synthesis
The synthesis and structural characterization of molecules 1 and 2 have been previously reported.22,23 Molecule 3 was obtained following a seven-step synthetic strategy shown in Scheme 1. The synthesis begins with cyanation of the commercially available 4,7-dibromo-2,1,3-benzothiadiazole (A). Subsequently, sulfur extrusion by a reductive reaction of the dicyano derivative (B) employing the heterogeneous catalytic NaBH4/CoCl2·6H2O system gives 1,2-diamino-3,6-dicyanobenzene (C).24,25 The condensation reaction between the resulting aromatic diamino compound and 3,5-di-tert-butyl-2-hydroxybenzaldehyde in nitrobenzene results in the BIP-4,7 disubstituted platform (D). Alkaline hydrolysis of D produces the dicarboxylic acid derivative E. Reduction of E using LiAlH4 affords the diol (F), which is then oxidized to the dialdehyde (G) with activated MnO2. The final step includes the formation of the imine linkage using compound G and two equivalents of p-anisidine (4-methoxyaniline) following a known procedure.22,26 The complete synthetic procedure and structural characterization are provided in the ESI section.†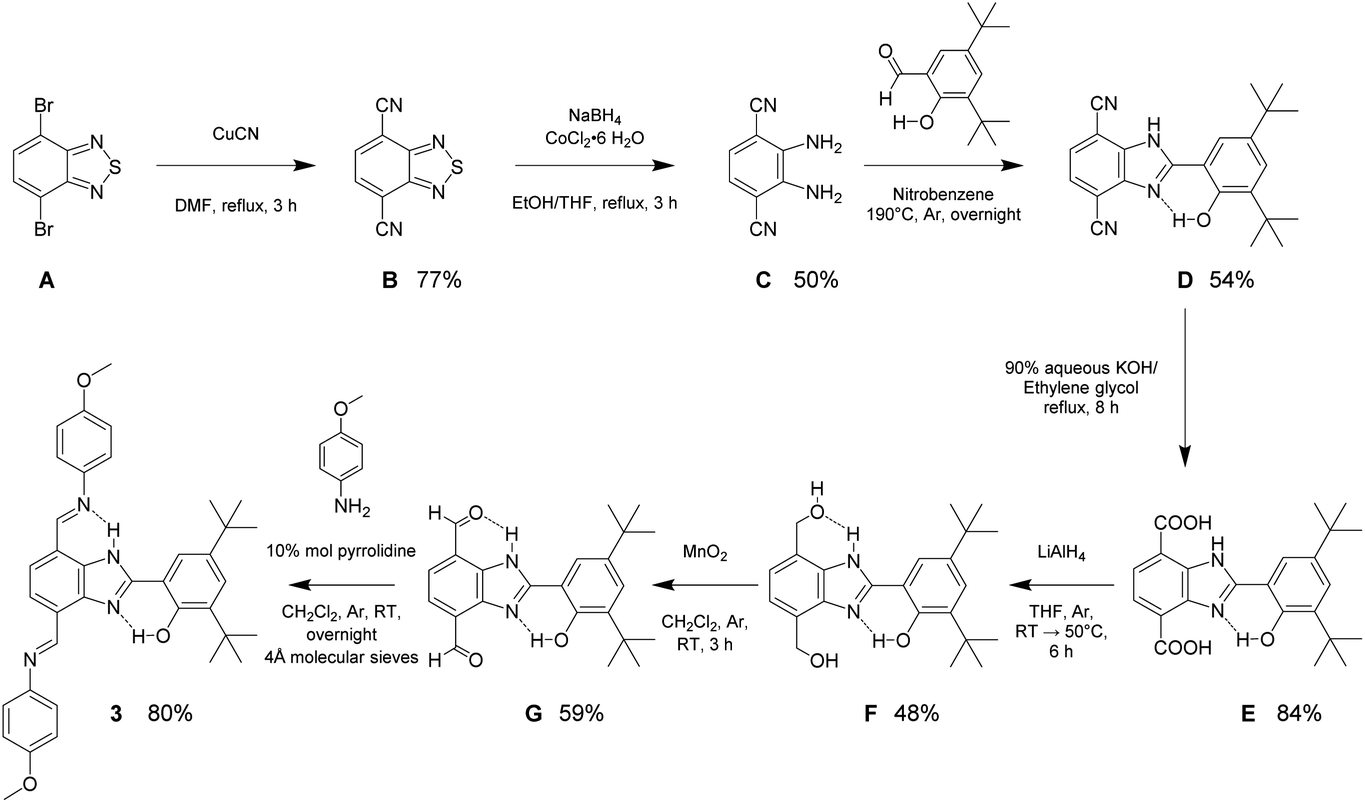 | ||
| Scheme 1 Synthetic strategy detailing reaction conditions, yields, and intermediate molecules for preparation of 3. | ||
Structural characterization
Crystallographic data is useful to establish how the hydrogen-bonded network is extended within the molecule. A nearly planar molecular framework across the phenol, the benzimidazole proton relay, and one of the TPA branches in 3 is observed, with dihedral angles between the phenol and benzimidazole moieties of 1.3°, and between the benzimidazole and the azomethine group of 3.6° (Fig. 2). The fairly short O⋯N (2.61 Å) and N⋯N (2.83 Å) distances are indicative of strong hydrogen bonds between the phenolic proton and the nitrogen lone pair of the benzimidazole, as well as the benzimidazole NH and the imine nitrogen of the TPA at the 7-postion. It is worth comparing the O⋯N and N⋯N distances found in the X-ray data of 3 with its monosubstituted analogue 2 (Fig. 2 and Table 1).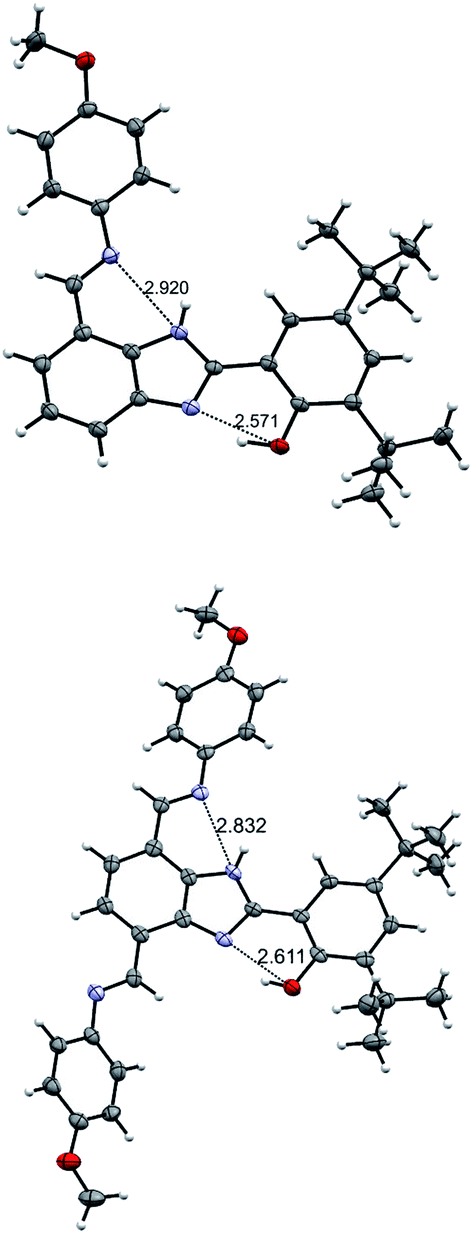 | ||
| Fig. 2 Crystal structures of 2 (top) and 3 (bottom). Carbon atoms are shown in gray, oxygen atoms in red, nitrogen atoms in light violet, and hydrogen atoms in white. Thermal ellipsoids are drawn at the 50% probability level. The crystal structure of 2 is adapted from ref. 22. | ||
| 2 | 3 | |
|---|---|---|
| a Determined by X-ray diffraction measurements. b Determined by DFT calculations. c 1H NMR data in CDCl3. d Each computed redox potential assumes the maximum number of intramolecular proton transfers. The redox potentials corresponding to the intermediate proton transfer process (E1PT) are given in Table S13. e The potential for 2 in CH2Cl2 was used as the reference for all calculated potentials associated with an E2PT product, and therefore it agrees with the experimental value by construction. f The potential of the pseudoreference electrode was determined using the ferrocenium/ferrocene redox couple as an internal standard and adjusting to the saturated calomel electrode (SCE) scale (with E1/2 taken to be 0.46 V vs. SCE in CH2Cl2). g Measured at 100 mV s−1. | ||
| O⋯N distance (Å) | 2.57a, 2.57b | 2.61a, 2.58b |
| N⋯N distance (Å) | 2.92a, 2.84b | 2.83a, 2.83b |
| Phenol-benzimidazole torsional angle (°) | 3.4a, 2.4b | 1.3a, 3.3b |
| Benzimidazole-imine linkage torsional angle (°) | 1.9a, 0.1b | 3.6a, 0.6b |
| δ NH (ppm)c | 11.84 | 11.97 |
| δ OH (ppm)c | 13.30 | 13.14 |
| Calculated E1/2 (V vs. SCE)d | 0.95e | 0.85 |
| Experimental E1/2 (V vs. SCE)f | 0.95 | 0.87 |
| ΔEp (mV)g | 130 | 64 |
| i c/iag | 0.97 | 0.91 |
For the disubstituted molecule 3, the O⋯N distance is comparable to that obtained for 2 (2.61 Å vs. 2.57 Å). The N⋯N distance for 3 is also comparable to that of 2. Geometry optimizations were performed with density functional theory (DFT) using the B3LYP-D3(BJ) functional27–31 and a 6-31G** basis set32–35 (see details in the ESI†). The resulting optimized structures support the crystallographic data. The agreement in structure between experiment and computation is highlighted in selected bonds and angles (see Table 1).
The existence of strong intramolecular hydrogen bonds and the differences noticed between 2 and 3 are well-reflected in their 1H NMR spectra in dry non-polar, non-protic solvents such as CDCl3 and CD2Cl2. Resonances of benzimidazole NH (δNH) and phenolic (δOH) protons above 11.00 ppm and 13.00 ppm, respectively, are characteristic in BIP derivatives featuring strong hydrogen-bonded networks.22,36 Based on the chemical shifts in CDCl3, the hydrogen bond between the phenolic proton and the nitrogen lone pair of the benzimidazole in 3 is slightly weaker than that for 2, whereas the hydrogen bond between the benzimidazole NH and the imine N is slightly stronger in the case of the 4,7 disubstituted molecule (3). The strength of these intramolecular hydrogen bonds, qualitatively described by the δNH and δOH values, is consistent with the X-ray crystallographic distances found for both compounds (see Fig. 2 and Table 1). Interestingly, the X-ray data show that the p-OCH3 phenylimine substituent at the 4-position is rotated 180° relative to the same substituent at the 7-position (see Fig. 2, TPA hydrogen-bonded with the benzimidazole NH), and this conformation remains in both CDCl3 and CD2Cl2 solutions. Analysis of computationally obtained free energies reveal that this conformation represents ∼72% of the relevant, thermodynamically accessible isomers in the neutral state (vide infra). The proportion of isomers in solution originated by the annular 1,3-tautomerism and rotation of the bond linking the phenol and benzimidazole moieties is deeply affected by both the dielectric constant of the solvent and its capability to interact specifically by hydrogen bonding with the molecule, among other factors.15,18,20,21 In the case of 2, the isomer featuring the fully hydrogen-bonded network (see Fig. 1A) is the most stable conformation in CDCl3 solution. Only one set of 1H NMR resonances lines is accordingly detected.22 However, the switch of CDCl3 for acetone-d6 alters the equilibrium between isomers, and the presence of a minor population (isomeric ratio: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.20) stabilized by specific solvent–solute interactions is noticed (Fig. 3). The formation of intermolecular hydrogen bond interactions between the solvent (hydrogen bond acceptor) and either the free NH or OH (hydrogen bond donors) in the open cis-enol form in 2 could decrease the rate of inter-conversion between the two tautomeric species when acetone-d6 is used as solvent. The opposite is shown for 3, where only one species in solution is detected independently of the nature of the solvent, as a consequence of the additional substitution at the 4-position with an identical TPA group (Fig. 3).
:0.20) stabilized by specific solvent–solute interactions is noticed (Fig. 3). The formation of intermolecular hydrogen bond interactions between the solvent (hydrogen bond acceptor) and either the free NH or OH (hydrogen bond donors) in the open cis-enol form in 2 could decrease the rate of inter-conversion between the two tautomeric species when acetone-d6 is used as solvent. The opposite is shown for 3, where only one species in solution is detected independently of the nature of the solvent, as a consequence of the additional substitution at the 4-position with an identical TPA group (Fig. 3).
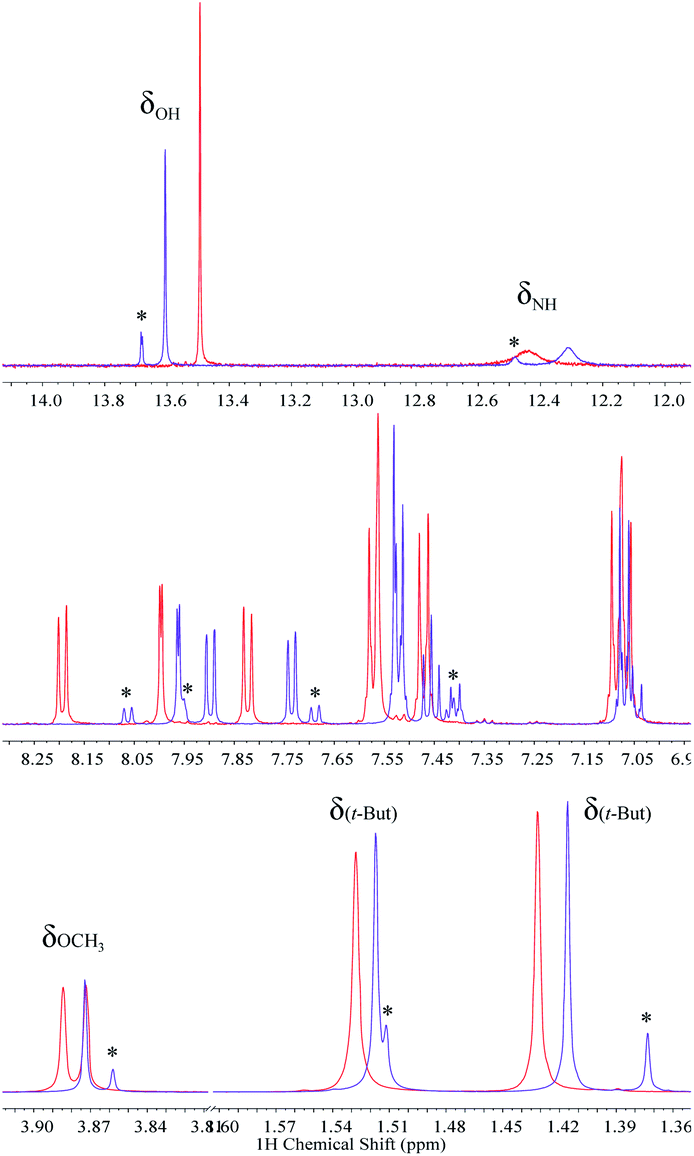 | ||
| Fig. 3 1H NMR spectra of 2 (purple) and 3 (red) in acetone-d6 displaying resonances in the downfield (top), aromatic (middle), and aliphatic (bottom) regions. The resonances of both OCH3 groups (δOCH3) in 3 appear at different ppm. Signals of the minor isomer of 2 are highlighted (*). | ||
1HNMR spectra of 3 in CDCl3, CD2Cl2 and acetone-d6 show that both TPA groups are magnetically nonequivalent (see full structural characterization in the ESI†). The phenol rotation and annular 1,3-tautomerism are slow on the NMR timescale because of the strong hydrogen-bonded network along the molecule. NOESY experiments performed in CDCl3 (Fig. 4) and CD2Cl2 (Fig. S15†) certainly allow the distinction of each TPA branch and are in agreement with the conformation adopted by the molecule in the crystallographic unit cell (see Fig. 2). The azomethine proton of the imine linkage exhibiting a hydrogen bond (Ha in Fig. 4B) interacts through space with the aromatic protons of both the benzimidazole (H6) and the p-OCH3 phenyl ring (Hc), whereas the azomethine proton of the TPA at the 4-position (Hb in Fig. 4B) only shows a cross peak with the aromatic proton of the p-OCH3 phenyl ring (Hc′).
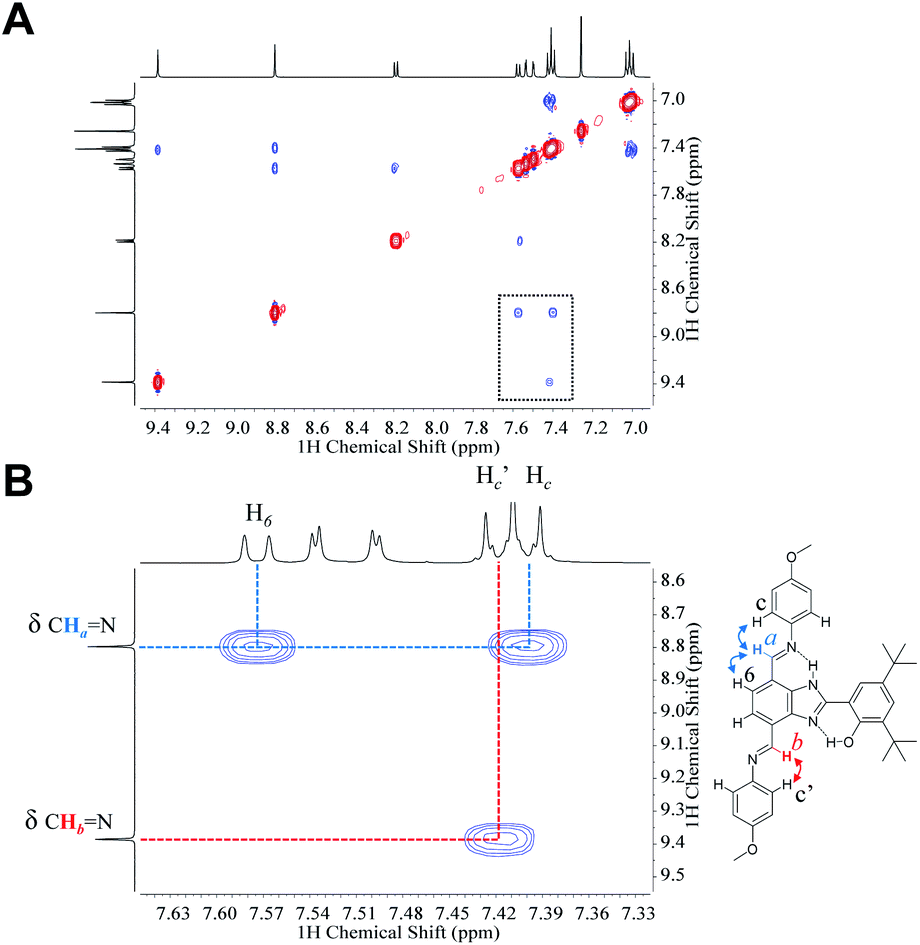 | ||
| Fig. 4 (A) NOESY of 3 in CDCl3 showing the downfield/aromatic regions of the spectrum. (B) Expansion of the selected area (dotted rectangle) in A. The arrows shown in the molecular structure describe the interactions through the space of the azomethine protons (Ha and Hb) of both TPA branches (color coded). | ||
Computational isomer analysis
Geometry optimizations and free energy calculations of 2 and 3 were performed at the DFT level of theory using the conductor-like polarizable continuum model (CPCM)37,38 to represent CH2Cl2 solvent. Both 2 and 3 have four rotamer species, each with various possible protonation states, as shown in Fig. 5A and B, respectively. All the possibilities shown in these figures were found to correspond to local minima on the potential energy surface. The relative free energies for all minima in both the oxidized and neutral states of 2 and 3 are reported in Tables S11 and S12,† respectively. The Boltzmann population for each state was calculated using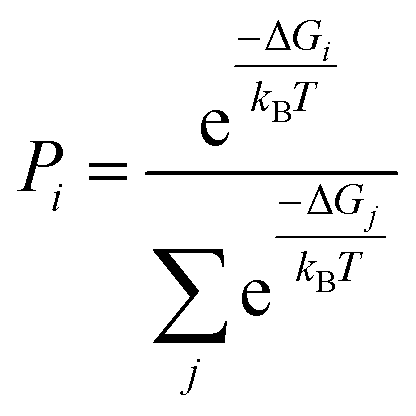 | (1) |
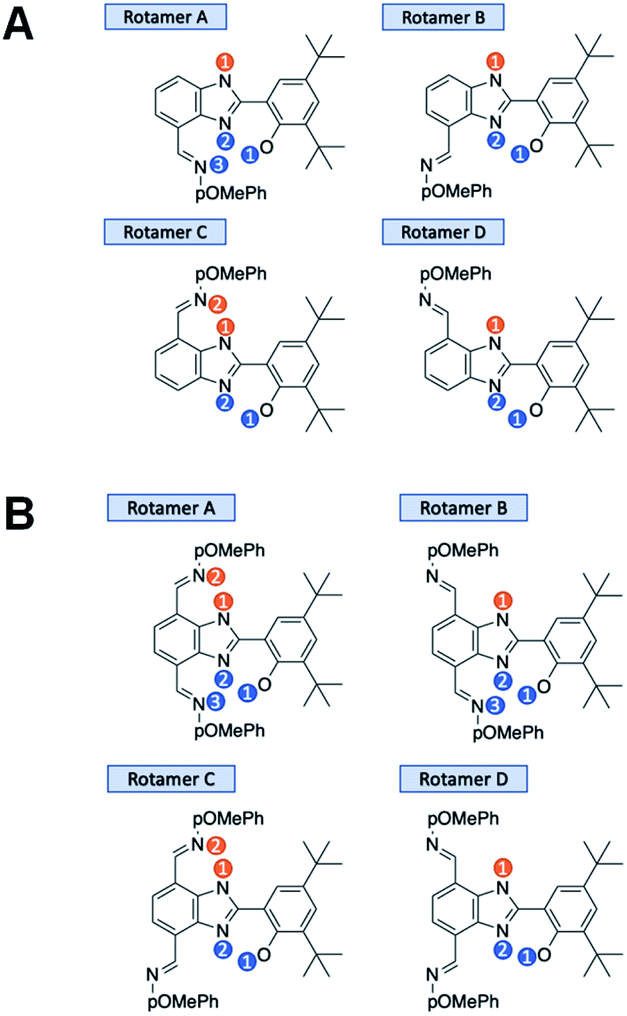 | ||
| Fig. 5 Rotamers, tautomers, and protonation states of 2 (A) and 3 (B). There are 11 and 15 possible arrangements of 2 and 3, respectively. The blue markers indicate possible positions of the proton initially on the phenol (before oxidation) within the hydrogen-bonded network. The orange markers indicate possible positions of the proton initially on benzimidazole (before oxidation) within the hydrogen-bonded network. | ||
In 2, the neutral system prior to oxidation is 98.6% rotamer C with both protons in position 1 and 1.2% rotamer B with both protons in position 1 (see Fig. 5A). In total, about 1.4% of 2 is initially in a rotamer state that cannot facilitate two-proton translocation. In 3, the neutral system prior to oxidation is 72.1% rotamer C with both protons in position 1 and 26.9% rotamer A with both protons in position 1 (see Fig. 5B). Thus, only 0.02% of 3 is initially in a rotamer state that cannot facilitate two-proton translocation. These calculations of the relative populations of the various states reveal an advantage of disubstituted, symmetrized bioinspired proton wires in terms of decreasing the populations of conformations that are not conducive to the E2PT process. Although compound 2 is mostly configured to facilitate E2PT (98.6%), thermally accessible rotamers could be a much greater obstacle in larger systems where more than two proton transfers require a consistent uninterrupted hydrogen-bonded network over multiple benzimidazole units. Longer proton wires typically have more possible rotamers lacking the ability to facilitate further proton transfer. As demonstrated here, the disubstituted system is populated 99.98% by rotamers with uninterrupted hydrogen-bonded networks allowing multiple proton transfers to occur.
Electrochemical studies
Cyclic voltammetry (CV) was used to gain insight into the thermodynamics of the PCET process. All potentials are given relative to the ferrocenium/ferrocene (Fc+/Fc) redox couple and are adjusted to the saturated calomel electrode (SCE) scale (Table 1). Redox potentials were calculated using DFT and methods previously benchmarked (see ESI†).39 The CV of 3 is shown in Fig. 6 and compared to that of 1, 2 and G, a precursor of 3 (see Scheme 1).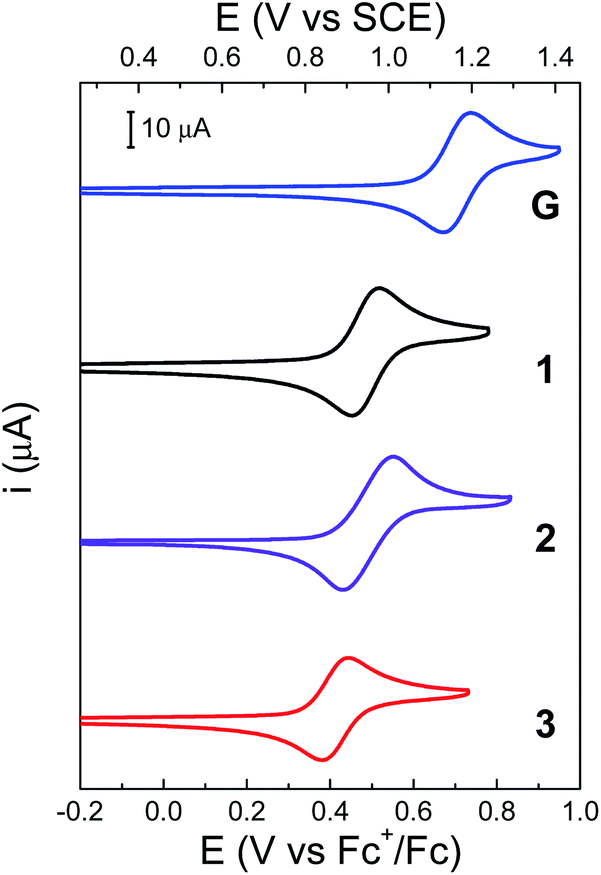 | ||
| Fig. 6 CVs of G (blue), unsubstituted BIP 1 (black), 2 (purple) and 3 (red). Concentration: 1 mM of the indicated BIPs, 0.1 M TBAPF6 supporting electrolyte in dry CH2Cl2. WE: glassy carbon. Pseudo RE: Ag wire (ferrocene as internal reference). CE: Pt wire. Scan rate, 100 mV s−1. | ||
Previous studies have shown that 2 exhibits a quasi-reversible one-electron oxidation process with a peak-to-peak separation (ΔEp) of 130 mV in CH2Cl2, while the chemical reversibility is evidenced by the high cathodic to anodic peak current ratio (ic/ia = 0.97, at a scan rate of 100 mV s−1).22 For 2, the experimental midpoint potential (E1/2, taken as the average of the anodic and cathodic peaks) of the phenoxyl radical/phenol redox couple is 0.95 V vs. SCE (see Table 1). For this molecule, the oxidation of the phenol is coupled with the intramolecular transfer of both protons by an E2PT process, leading to the protonation of the TPA and formation of the phenoxyl radical as the oxidized product.22
The electrochemical performance of 3 is clearly affected by the presence of an additional TPA. The ΔEp is reduced by a factor of two, which suggests faster electrode kinetics compared with 2. The chemical reversibility of 3 is also indicated by the ic/ia ratio approaching unity (ic/ia = 0.91, at 100 mV s−1, see Table 1). Furthermore, an appreciable cathodic shift is observed (ΔE1/2 = 80 mV, see Table 1) relative to 2, indicating that the phenol becomes easier to oxidize when the BIP has identical TPAs capable of accepting a second proton when the phenoxyl radical is formed. The computational analysis of rotamers and protonation states of 3 suggests that, at equilibrium after E2PT, the system is ∼97% in rotamer A with both protons in position 2 (see Fig. 7B). Thus, the oxidized product of 3 is stabilized by an extra intramolecular hydrogen bond relative to 2 (see Fig. 7), providing an explanation for the difference observed in the redox potentials.
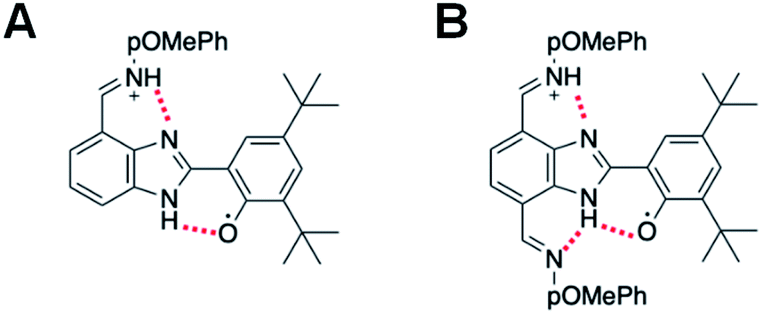 | ||
| Fig. 7 (A) Oxidized E2PT product of 2 lacking a stabilizing, bifurcated hydrogen bond. (B) Oxidized E2PT product of 3 containing a stabilizing, bifurcated hydrogen bond. | ||
A bifurcated hydrogen bond would be formed in compound 3 after an E2PT process. The new NH bond formed during proton transfer from the phenoxyl radical to the benzimidazole nitrogen in 3 now has two hydrogen bond acceptors, i.e., the oxygen of the phenol group from which it came, and the unprotonated nitrogen of the TPA at the 4-position. Based on the DFT geometry optimizations, the N⋯O and N⋯N distances associated with these hydrogen bonds are 2.65 Å and 2.86 Å, respectively, for the E1PT product and 2.67 Å and 2.86 Å, respectively, for the E2PT product. These hydrogen bonds stabilize both the E1PT and E2PT products of compound 3.
Bifurcated hydrogen bonds have been studied experimentally and theoretically40 and have been shown to stabilize PCET in substituted phenol systems designed to mimic the tyrosine radical redox system of PSII.41 The bifurcated hydrogen bond in compound 3 demonstrates similar character. The DFT calculations indicate that the E0PT, E1PT, and E2PT processes (i.e., oxidation with no proton transfer, one proton transfer, and two proton transfers, see Table S13†) are all more thermodynamically favorable for 3 than for 2. The calculations also indicate that the E2PT process is thermodynamically favorable for both 2 and 3 (i.e., the E2PT product is more stable than the E0PT and E1PT products for both compounds). These computational results are consistent with the experimental observation that both compounds undergo E2PT and that 2 (single TPA) has a higher redox potential than 3 (double TPA). In the case of G, the two electron-withdrawing aldehyde groups at the 4 and 7 positions cause an anodic shift of the E1/2 compared to 1 (Fig. 6, ΔE1/2 = 210 mV). In this case, the lower pKa of the protonated aldehyde group prevents the formation of a local minimum associated with the E2PT product (see Table S13†).
Analysis of the computational results provides further insight into the impact of the bifurcated hydrogen bond and the second TPA substituent in 3. Comparison of the free energies of the E2PT product of 3 for rotamer A, which has a bifurcated hydrogen bond, and rotamer C, which does not, indicates that the bifurcated hydrogen bond provides a stabilization of 3.25 kcal mol−1 (Table S12†). Moreover, the E2PT redox potential of 3 is 0.17 V lower for rotamer A than rotamer C, mainly due to the stabilization provided by the bifurcated hydrogen bond in the oxidized species. Comparison of the E2PT redox potentials of rotamer C for 2 and 3 indicates that the addition of the second TPA increases the redox potential by 0.03 V. Thus, the effect of the bifurcated hydrogen bond is much larger than the effect of the substituent on the E2PT redox potential.
Infrared spectroelectrochemistry studies
Fig. 8 displays the experimental infrared spectroelectrochemistry (IRSEC) spectra of 3 recorded under oxidative conditions generating increasing amounts of the phenoxyl radical from the neutral phenol species. DFT normal-mode analysis of 3 has been employed for the assignment of selected vibrational modes (Fig. S17 and Table S14†). Substantial changes in the absorbance and band positions associated with the oxidation are indicated with arrows. It is interesting to point out the existence of well-defined isosbestic points across the spectra, suggesting that the neutral species are being progressively converted into the oxidized species. The band at 1614 cm−1 in the neutral species is assigned to the stretching C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of the imine linkage (Fig. 8A), with a similar frequency of that observed for the monosubstituted 2.22 The decrease in the intensity of this band upon electrooxidation indicates that the neutral species of 3 is being converted into its oxidized form. Some of the new bands shown in the spectra are characteristic of vibrational modes affected by protonation after PCET. In particular, the new band at 1651 cm−1 corresponds to the large component of the protonated imine group stretching, which is a distinctive IR marker displayed by other BIP derivatives bearing a substituted imine group as the TPA.12,22,42 This is evidence that an E2PT product is being formed upon phenol oxidation. Computational spectra (Fig. S17†) corroborate protonation of the TPA in the oxidized species. Specifically, the peak at 1651 cm−1 appears in the simulated IRSEC of the E2PT product but does not appear in the simulated IRSEC of the E1PT product (Fig. S17B and A,† respectively), confirming that the experimental IRSEC corresponds to the E2PT product. Computed IRSEC spectra also show a band at 1593 cm−1 assigned to a breathing mode of the TPA, which includes a bending motion of the protonated azomethine. This band corresponds to the band at 1591 cm−1 in the experimental spectra. The band growing at 1560 cm−1 in the computed spectra, associated with the in-plane bending of both transferred protons, could explain the appearance of the band at 1558 cm−1 in the experimental spectra.
N of the imine linkage (Fig. 8A), with a similar frequency of that observed for the monosubstituted 2.22 The decrease in the intensity of this band upon electrooxidation indicates that the neutral species of 3 is being converted into its oxidized form. Some of the new bands shown in the spectra are characteristic of vibrational modes affected by protonation after PCET. In particular, the new band at 1651 cm−1 corresponds to the large component of the protonated imine group stretching, which is a distinctive IR marker displayed by other BIP derivatives bearing a substituted imine group as the TPA.12,22,42 This is evidence that an E2PT product is being formed upon phenol oxidation. Computational spectra (Fig. S17†) corroborate protonation of the TPA in the oxidized species. Specifically, the peak at 1651 cm−1 appears in the simulated IRSEC of the E2PT product but does not appear in the simulated IRSEC of the E1PT product (Fig. S17B and A,† respectively), confirming that the experimental IRSEC corresponds to the E2PT product. Computed IRSEC spectra also show a band at 1593 cm−1 assigned to a breathing mode of the TPA, which includes a bending motion of the protonated azomethine. This band corresponds to the band at 1591 cm−1 in the experimental spectra. The band growing at 1560 cm−1 in the computed spectra, associated with the in-plane bending of both transferred protons, could explain the appearance of the band at 1558 cm−1 in the experimental spectra.
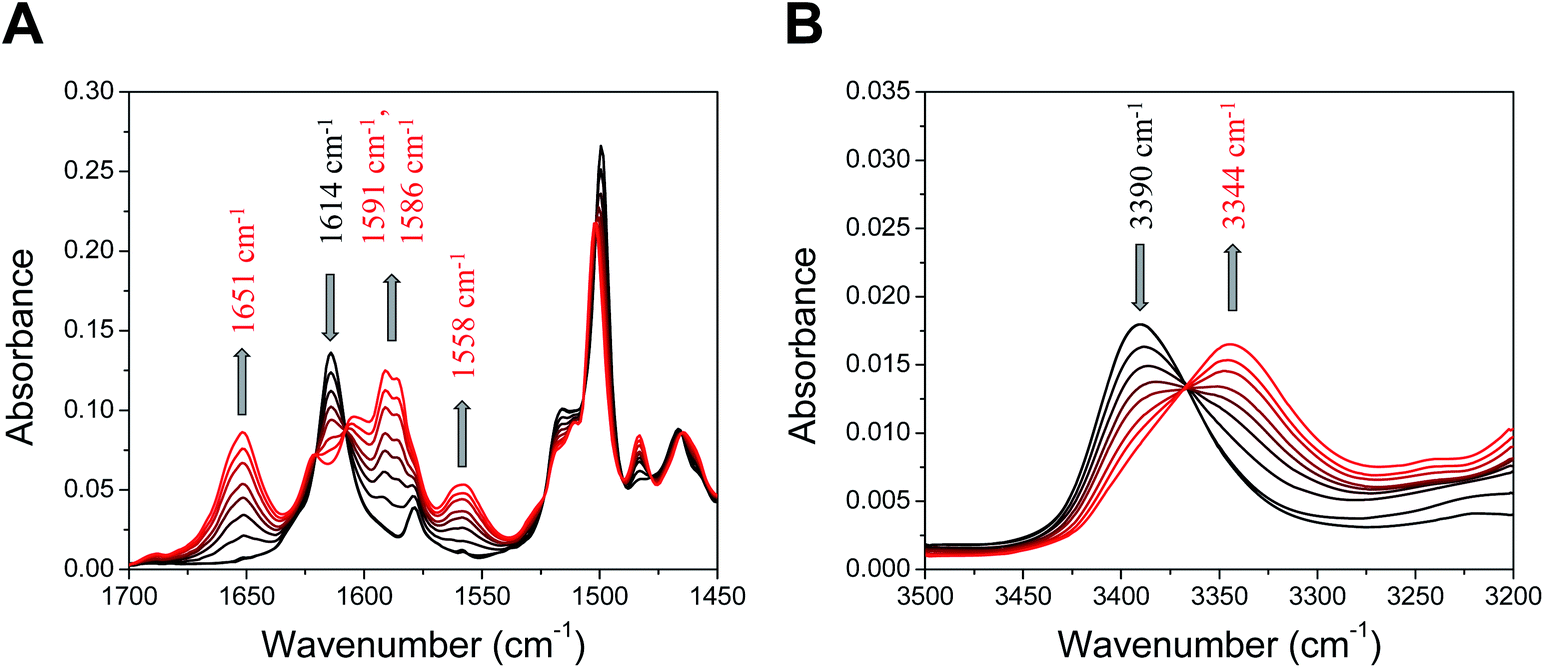 | ||
| Fig. 8 IRSEC spectra of 3 recorded under electro-oxidative conditions in (A) the middle (1700–1450 cm−1) and (B) the high (3500–3200 cm−1) frequency regions. Solvent: dry CH2Cl2, 0.1 M TBAPF6. Characteristic bands displaying changes under polarization are indicated with upward and downward grey arrows. | ||
Evidence of proton translocation to form the E2PT product is also observed in the high frequency region, where the NH stretching mode (νNH) is detected (Fig. 8B).22,23 In the neutral species, the νNH for 3 appears at 3390 cm−1, whereas the frequency of this mode is situated at 3394 cm−1 for 2;22 the slight shift of the νNH is in agreement with the downfield shift of the δNH observed in the 1H NMR (see Table 1). Upon phenol oxidation, the band at 3390 cm−1 decreases and a new band at 3344 cm−1 arises, which is assigned to the νNH of the proximal benzimidazole NH. In the case of the monosubstituted analogue 2, the νNH in the oxidized species is centered at higher frequency (3360 cm−1),22 suggesting that the decrease in the νNH observed for oxidized 3 is most likely due to the bifurcated hydrogen bond.
Conclusions
The effect on PCET due to prototropic tautomerism of benzimidazole-phenol based systems has been investigated. Substitution of the benzimidazole core with two identical TPAs removes the existence of isomeric species possessing interrupted hydrogen-bonded networks that prevent the system from two intramolecular proton transfers corresponding to an E2PT process. Incorporation of the second TPA in the bioinspired construct not only ensures an uninterrupted hydrogen-bonded network but also causes a cathodic shift of the phenoxyl radical/phenol redox couple relative to its monosubstituted analogue. The formation of a bifurcated hydrogen bond, only possible in the disubstituted molecule, contributes to the relative stabilization of the E2PT product and explains the decrease in redox potential. The current findings, which combine theoretical and experimental approaches, are crucial for future designs of proton wires having multiple translocating protons triggered by a single electron transfer in a Grotthuss-type process over an uninterrupted hydrogen-bonded network including many benzimidazole units.Data availability
All the details of the experimental and theoretical part are in the ESI.† The crystal structure data have been deposited in the CCDC.Author contributions
EO and ME performed the synthesis and characterization of the compounds. MS performed computational work. TLG performed X-ray diffraction characterization. All authors analysed and interpreted the obtained results. EO and MS prepared figures for publication. EO and MS wrote the manuscript with input from TAM, SH-S and ALM. TAM, SH-S and ALM supervised and coordinated the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-FG02-03ER15393. The theoretical portion of this research was supported as part of the Center for Molecular Electro-catalysis, an Energy Frontier Research Center, funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences.Notes and references
- R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS.
- S. J. Mora, E. Odella, G. F. Moore, D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2018, 51, 445–453 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS.
- L. Benisvy, R. Bittl, E. Bothe, C. D. Garner, J. McMaster, S. Ross, C. Teutloff and F. Neese, Angew. Chem., Int. Ed., 2005, 44, 5314–5317 CrossRef CAS PubMed.
- M. Orio, O. Jarjayes, B. Baptiste, C. Philouze, C. Duboc, J. Mathias, L. Benisvy and F. Thomas, Chem.–Eur. J., 2012, 18, 5416–5429 CrossRef CAS.
- G. F. Moore, M. Hambourger, G. Kodis, W. Michl, D. Gust, T. A. Moore and A. L. Moore, J. Phys. Chem. B, 2010, 114, 14450–14457 CrossRef CAS.
- S. J. Mora, D. A. Heredia, E. Odella, U. Vrudhula, D. Gust, T. A. Moore and A. L. Moore, J. Porphyrins Phthalocyanines, 2019, 23, 1336–1345 CrossRef CAS.
- W. D. Guerra, E. Odella, M. N. Urrutia, P. A. Liddell, T. A. Moore and A. L. Moore, J. Porphyrins Phthalocyanines, 2021, 25, 674–682 CrossRef CAS.
- G. F. Moore, M. Hambourger, M. Gervaldo, O. G. Poluektov, T. Rajh, D. Gust, T. A. Moore and A. L. Moore, J. Am. Chem. Soc., 2008, 130, 10466–10467 CrossRef CAS PubMed.
- Y. Yoneda, S. J. Mora, J. Shee, B. L. Wadsworth, E. A. Arsenault, D. Hait, G. Kodis, D. Gust, G. F. Moore, A. L. Moore, M. Head-Gordon, T. A. Moore and G. R. Fleming, J. Am. Chem. Soc., 2021, 143, 3104–3112 CrossRef CAS.
- E. Odella, T. A. Moore and A. L. Moore, Photosynth. Res. DOI:10.1007/s11120-020-00815-x.
- E. Odella, B. L. Wadsworth, S. J. Mora, J. J. Goings, M. T. Huynh, D. Gust, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, J. Am. Chem. Soc., 2019, 141, 14057–14061 CrossRef CAS.
- W. D. Guerra, E. Odella, M. Secor, J. J. Goings, M. N. Urrutia, B. L. Wadsworth, M. Gervaldo, L. E. Sereno, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, J. Am. Chem. Soc., 2020, 142, 21842–21851 CrossRef CAS.
- F. Su, Z. Sun, W. Su and X. Liang, J. Mol. Struct., 2018, 1173, 690–696 CrossRef CAS.
- H. K. Sinha and S. K. Dogra, Chem. Phys., 1986, 102, 337–347 CrossRef CAS.
- A. R. Katritzky and J. M. Lagowski, in Prototropic Tautomerism of Heteroaromatic Compounds: IV. Five-Membered Rings with Two or More Hetero Atoms, ed. A. R. Katritzky, Academic Press, 1963, vol. 2, pp. 27–81 Search PubMed.
- V. Sridharan, S. Saravanan, S. Muthusubramanian and S. Sivasubramanian, Magn. Reson. Chem., 2005, 43, 551–556 CrossRef CAS.
- M. Forés, M. Duran, M. Solà, M. Orozco and F. J. Luque, J. Phys. Chem. A, 1999, 103, 4525–4532 CrossRef.
- C. I. Nieto, P. Cabildo, M. Á. García, R. M. Claramunt, I. Alkorta and J. Elguero, Beilstein J. Org. Chem., 2014, 10, 1620–1629 CrossRef.
- K. Das, N. Sarkar, A. K. Ghosh, D. Majumdar, D. N. Nath and K. Bhattacharyya, J. Phys. Chem., 1994, 98, 9126–9132 CrossRef CAS.
- K. Das, N. Sarkar, D. Majumdar and K. Bhattacharyya, Chem. Phys. Lett., 1992, 198, 443–448 CrossRef CAS.
- E. Odella, S. J. Mora, B. L. Wadsworth, M. T. Huynh, J. J. Goings, P. A. Liddell, T. L. Groy, M. Gervaldo, L. E. Sereno, D. Gust, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, J. Am. Chem. Soc., 2018, 140, 15450–15460 CrossRef CAS.
- M. T. Huynh, S. J. Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A.-L. Teillout, C. W. Machan, C. P. Kubiak, D. Gust, T. A. Moore, S. Hammes-Schiffer and A. L. Moore, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- B. A. D. Neto, A. S. Lopes, M. Wüst, V. E. U. Costa, G. Ebeling and J. Dupont, Tetrahedron Lett., 2005, 46, 6843–6846 CrossRef.
- J. M. Aslan, D. J. Boston and F. M. MacDonnell, Chem.–Eur. J., 2015, 21, 17314–17323 CrossRef CAS.
- S. Morales, F. G. Guijarro, J. L. García Ruano and M. B. Cid, J. Am. Chem. Soc., 2014, 136, 1082–1089 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. S. Ashvar, C. F. Chabalowski and M. J. Frisch, Faraday Discuss., 1994, 99, 103–119 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- T. F. Markle, I. J. Rhile, A. G. DiPasquale and J. M. Mayer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8185–8190 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- B. H. Solis and S. Hammes-Schiffer, Inorg. Chem., 2014, 53, 6427–6443 CrossRef CAS.
- I. Rozas, I. Alkorta and J. Elguero, J. Phys. Chem. A, 1998, 102, 9925–9932 CrossRef CAS.
- F. Thomas, O. Jarjayes, H. Jamet, S. Hamman, E. Saint-Aman, C. Duboc and J.-L. Pierre, Angew. Chem., Int. Ed., 2004, 43, 594–597 CrossRef CAS.
- E. Odella, S. J. Mora, B. L. Wadsworth, J. J. Goings, M. A. Gervaldo, L. E. Sereno, T. L. Groy, D. Gust, T. A. Moore, G. F. Moore, S. Hammes-Schiffer and A. L. Moore, Chem. Sci., 2020, 11, 3820–3828 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthesis and structural characterization, nuclear magnetic resonance data, crystal structure and X-ray data, and computational methods. CCDC 2094827. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03782j |
| This journal is © The Royal Society of Chemistry 2021 |