DOI:
10.1039/D1SC03958J
(Edge Article)
Chem. Sci., 2021,
12, 12419-12428
The power of trichlorosilylation: isolable trisilylated allyl anions, allyl radicals, and allenyl anions†
Received
20th July 2021
, Accepted 13th August 2021
First published on 13th August 2021
Abstract
Treatment of hexachloropropene (Cl2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Cl)–CCl3) with Si2Cl6 and [nBu4N]Cl (1
C(Cl)–CCl3) with Si2Cl6 and [nBu4N]Cl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1) in CH2Cl2 results in a quantitative conversion to the trisilylated, dichlorinated allyl anion salt [nBu4N][Cl2CC(SiCl3)–C(SiCl3)2] ([nBu4N][1]). Tetrachloroallene Cl2CCCCl2 was identified as the first intermediate of the reaction cascade. In the solid state, [1]− adopts approximate Cs symmetry with a dihedral angle between the planes running through the olefinic and carbanionic fragments of [1]− of CC–Si//Si–C–Si = 78.3(1)°. One-electron oxidation of [nBu4N][1] with SbCl5 furnishes the distillable blue radical 1˙. The neutral propene Cl2CC(SiCl3)–C(SiCl3)2H (2) was obtained by (i) protonation of [1]− with HOSO2CF3 (HOTf) or (ii) H-atom transfer to 1˙ from 1,4-cyclohexadiene. Quantitative transformation of all three SiCl3 substituents in 2 to Si(OMe)3 (2OMe) or SiMe3 (2Me) substituents was achieved by using MeOH/NMe2Et or MeMgBr in CH2Cl2 or THF, respectively. Upon addition of 2 equiv. of tBuLi, 2Me underwent deprotonation with subsequent LiCl elimination, 1,2-SiMe3 migration and Cl/Li exchange to afford the allenyl lithium compound Me3Si(Li)CCC(SiMe3)2 (Li[4]), which is an efficient building block for the introduction of Me, SiMe3, or SnMe3 (5) groups. The trisilylated, monochlorinated allene Cl3Si(Cl)CCC(SiCl3)2 (6), was obtained from [nBu4N][1] through Cl−-ion abstraction with AlCl3 and rearrangement in CH2Cl2 (1˙ forms as a minor side product, likely because the system AlCl3/CH2Cl2 can also act as a one-electron oxidant).
:4:1) in CH2Cl2 results in a quantitative conversion to the trisilylated, dichlorinated allyl anion salt [nBu4N][Cl2CC(SiCl3)–C(SiCl3)2] ([nBu4N][1]). Tetrachloroallene Cl2CCCCl2 was identified as the first intermediate of the reaction cascade. In the solid state, [1]− adopts approximate Cs symmetry with a dihedral angle between the planes running through the olefinic and carbanionic fragments of [1]− of CC–Si//Si–C–Si = 78.3(1)°. One-electron oxidation of [nBu4N][1] with SbCl5 furnishes the distillable blue radical 1˙. The neutral propene Cl2CC(SiCl3)–C(SiCl3)2H (2) was obtained by (i) protonation of [1]− with HOSO2CF3 (HOTf) or (ii) H-atom transfer to 1˙ from 1,4-cyclohexadiene. Quantitative transformation of all three SiCl3 substituents in 2 to Si(OMe)3 (2OMe) or SiMe3 (2Me) substituents was achieved by using MeOH/NMe2Et or MeMgBr in CH2Cl2 or THF, respectively. Upon addition of 2 equiv. of tBuLi, 2Me underwent deprotonation with subsequent LiCl elimination, 1,2-SiMe3 migration and Cl/Li exchange to afford the allenyl lithium compound Me3Si(Li)CCC(SiMe3)2 (Li[4]), which is an efficient building block for the introduction of Me, SiMe3, or SnMe3 (5) groups. The trisilylated, monochlorinated allene Cl3Si(Cl)CCC(SiCl3)2 (6), was obtained from [nBu4N][1] through Cl−-ion abstraction with AlCl3 and rearrangement in CH2Cl2 (1˙ forms as a minor side product, likely because the system AlCl3/CH2Cl2 can also act as a one-electron oxidant).
Introduction
Organosilanes are invaluable building blocks for advanced materials and multifaceted reagents for organic synthesis.1 Characteristic of the first application area is that the Si atoms remain as essential, function-determining parts in the molecular scaffolds (e.g., luminescent siloles,2 silsesquioxane cages,3 and silicone polymers4,5). The opposite is true for the second application area, because the silyl groups are no longer present in the final products after they served to transfer the organic fragment (e.g., Peterson olefination,6 Hiyama-type C–C-coupling,7 or Tamao oxidation8). Especially allylsilanes, which combine the reactivity of alkenes and metal-allyl reagents, have been termed “one of the most important building blocks in modern organic synthesis” (cf. the Hosomi–Sakurai allylation9).10 While a plethora of efficient routes to monosilylated allyl systems have been elaborated so far,11 higher silylated derivatives are still difficult to access, which is unfortunate since geminal disilyl compounds in particular can be involved in numerous synthetically useful transformations.12
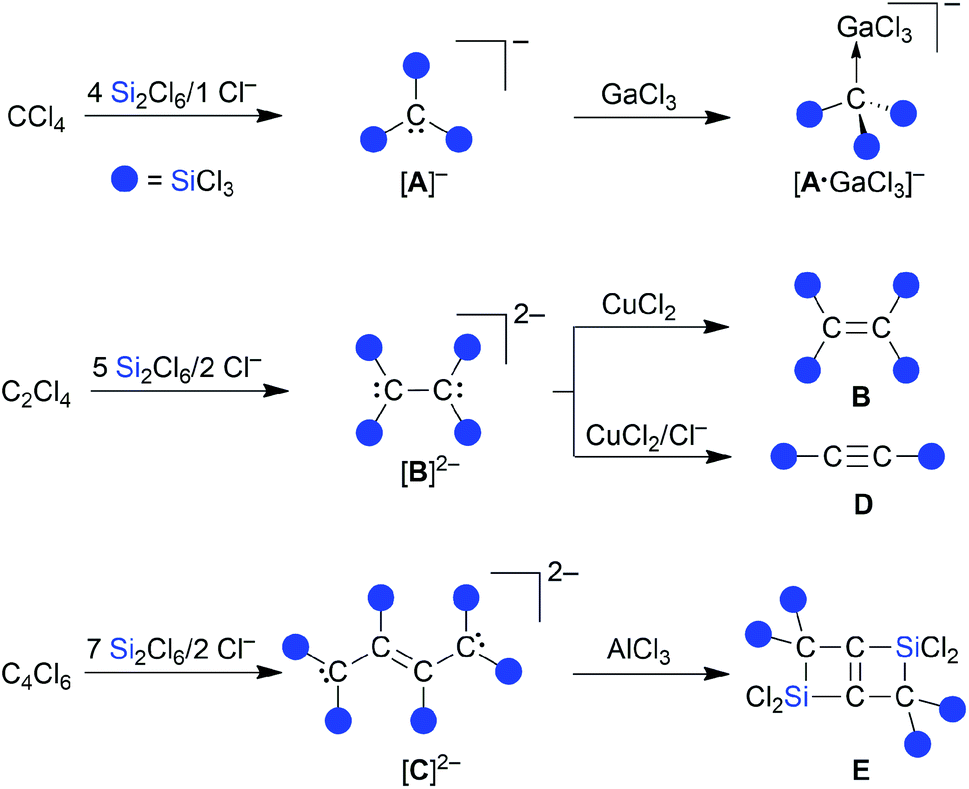
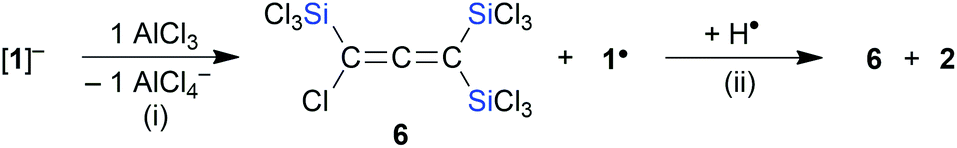
We have recently disclosed that the inexpensive chlorinated hydrocarbons CCl4, Cl2CCCl2, and Cl2CC(Cl)–C(Cl)CCl2 can be straightforwardly converted to the exhaustively trichlorosilylated anions [A]−–[C]2− upon treatment with Si2Cl6 in the presence of [nBu4N]Cl (Scheme 1).13–15 The actual active silylation reagent, the silanide [SiCl3]−,16–18 is generated by Cl−-induced heterolysis of the disilane. [SiCl3]− can either react as a nucleophile in a carbophilic or chlorophilic attack (abstraction of a chloronium ion to form SiCl4 and a carbanion), or behave as a Cl−-stabilized dichlorosilylene [SiCl2·Cl]−.13,16,18,19 The primary products, [A]−–[C]2−, have been further transformed into a variety of other multiply trichlorosilylated compounds with geminally and/or vicinally positioned SiCl3 substituents ([A·GaCl3]−–E; Scheme 1).13–15 An SiCl3 group differs from the more abundant SiMe3 group in a number of ways: (i) the Si–Cl bonds offer the possibility of extensive derivatizations, such as transformation to Si–H bonds, alkylation, or hydrolysis to (oligo)siloxanes. (ii) SiCl3 substituents are the better π acceptors and have a greater ability to stabilize adjacent carbanions (α effect).4 (iii) SiCl3 substituents are the stronger Lewis acids. Analogous to the above-mentioned release of [SiCl3]− from Si2Cl6/Cl−, also other main-group anions [RnE]− can readily be generated under mild conditions from precursor fragments RnE–SiCl3 upon addition of Lewis basic anions, such as F− or Cl− (E = e.g., C, Si, Ge, P, S).13,14,20–23
 |
| | Scheme 1 Reactions of CCl4, C2Cl4, or C4Cl6 with Si2Cl6/Cl− furnish the exhaustively trichlorosilylated carbanions [A]−, [B]2−, or [C]2−, which give access to a variety of further compounds, such as the adduct [A·GaCl3]−, the C2 compounds B and D, and the strained, edge-fused double silacyclobutene E. | |
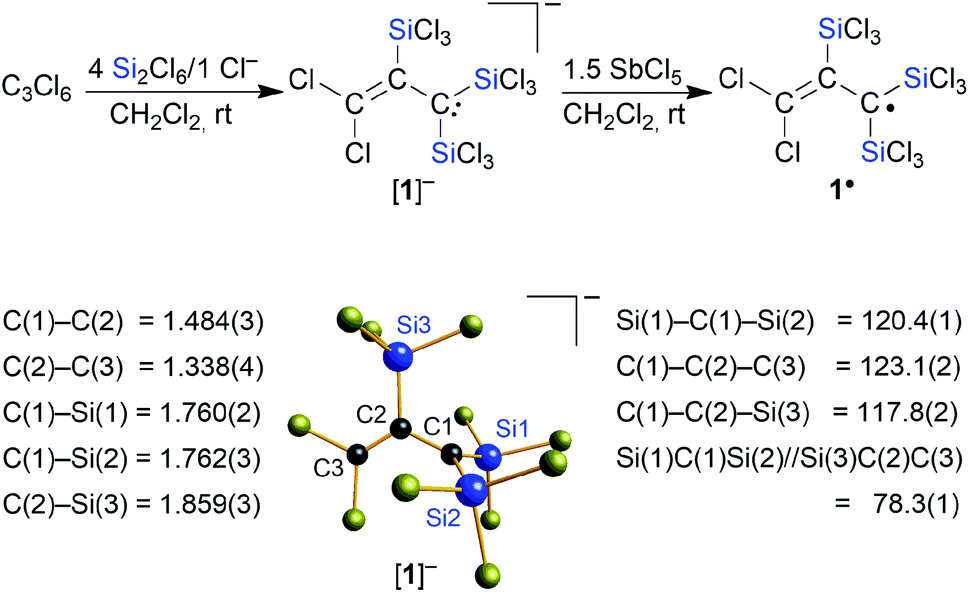
Herein, we fill the gap in the series of perchlorinated substrates and report on the reaction of the C3 substrate hexachloropropene (C3Cl6) with the Si2Cl6/Cl− system. In striking contrast to the previous cases (Scheme 1), there is now no quantitative Cl/SiCl3 exchange, but we rather obtain selectively the trisilylated, dichlorinated allyl anion [1]− (Scheme 2). One-electron oxidation of [1]− affords the distillable blue radical 1˙. Furthermore, we will show that [1]− is a valuable starting material for the synthesis of multiply silylated C3 products as diverse as propenes, cyclopropenes, and allenes.
 |
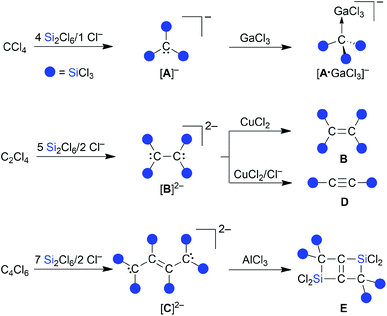
| | Scheme 2 Synthesis of [nBu4N][1] from C3Cl6 and Si2Cl6/[nBu4N]Cl; oxidation of [nBu4N][1] with SbCl5 generates the thermostable radical 1˙. Molecular structure of [Ph4P][1] in the solid state (the [Ph4P]+ cation is omitted for clarity); selected bond lengths [Å], bond angles [°], and dihedral angle [°]. | |
Results and discussion
Synthesis and characterization of the mixed Cl/SiCl3-substituted allyl anion [1]−
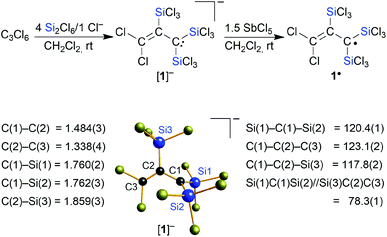
Similar to the cases of [A]−, [B]2−, and [C]2−, we initially aimed at the synthesis of a fivefold trichlorosilylated allyl anion [C3(SiCl3)5]−. To this end, we treated 1 equiv. of hexachloropropene (C3Cl6) with 6 equiv. of Si2Cl6 and 1 equiv. of [nBu4N]Cl in CH2Cl2 at room temperature; 5 equiv. of the disilane were supposed to provide the five SiCl3 substituents, while the 6th equiv. was meant to formally abstract a Cl+ cation and generate the negative charge. According to NMR spectroscopy, the reaction mixture contained no unconsumed C3Cl6, no CH moieties, and one major organosilicon product that gave rise to three 13C and two 29Si resonances. Two of the 13C resonances pointed toward the presence of an olefinic subunit (δ(13C) = 135.8, 137.3). The third 13C NMR signal possessed a chemical shift value of 50.3 ppm, which is close to the value of the signal of the carbanionic centers in [C]2− (50.2 ppm).15 However, the total numbers of NMR signals were not consistent with a [C3(SiCl3)5]− anion either of average C2v or Cs symmetry. We also noted smaller 29Si resonances characteristic of chloride diadducts of perchlorinated cyclohexasilanes (e.g. [Si6Cl12·2Cl]2−), typically formed from Si2Cl6/Cl− mixtures in the absence of an additional reactant.16,18,19,24 Taken together, these observations led to the working hypothesis that the obtained product was a partly trichlorosilylated/chlorinated allyl anion. To substantiate this conclusion, we repeated the reaction with 4 equiv. of Si2Cl6 and obtained the same organosilicon product in a quantitative fashion and this time without the cyclohexasilane contaminants (NMR-spectroscopic control; 90% yield). An aliquot of the isolated product in CH2Cl2 was subsequently quenched with excess MeOH and titrated with aqueous NaOH. The amount of HCl released was determined to be 8 equiv., in agreement with three SiCl3 substituents introduced (note that 1 equiv. of HCl is neutralized in situ by the carbanionic center). Instead of the aimed-for [nBu4N][C3(SiCl3)5], a salt of the form [nBu4N][C3(SiCl3)3Cl2] had obviously been prepared ([nBu4N][1]; Scheme 2). Neither an increase in the amount of Si2Cl6 added (up to 17 equiv.) nor an elevated reaction temperature (refluxing CH2Cl2) enforced a higher degree of silylation, but only resulted in more cyclohexasilane side products.
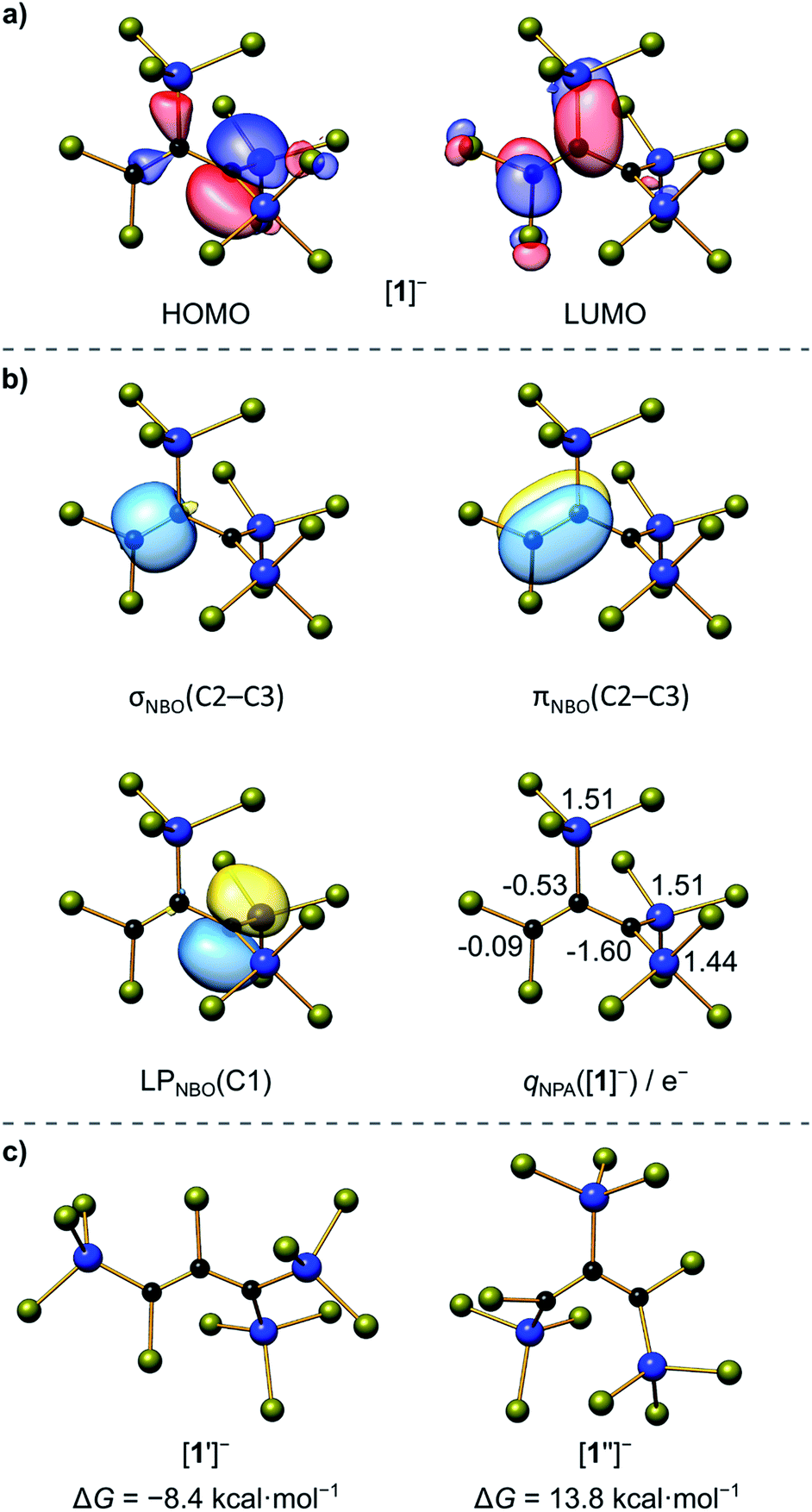
Finally, X-ray crystallography confirmed the number of three SiCl3 groups present in [1]− and unequivocally revealed their positions at the C3 chain: suitable single crystals of [Ph4P][1] were obtained from a 1:4:1 mixture of C3Cl6:Si2Cl6:[Ph4P]Cl in CH2Cl2 (Scheme 2). All three carbon atoms of [1]− have a planar configuration. C(1) and C(3) carry two SiCl3 substituents and two Cl atoms, respectively; the third SiCl3 substituent resides at C(2). The anion adopts an approximate Cs symmetry in the solid state with a dihedral angle Si(1)C(1)Si(2)//Si(3)C(2)C(3) of 78.3(1)°. The C(1)–C(2) bond (1.484(3) Å) is even longer than that of the formal C(sp2)–C(sp2) single bond in 1,3-butadiene (1.47 Å),25 while the C(2)C(3) bond length (1.338(4) Å) is typical of an isolated CC double bond (1.34 Å).25 Any charge delocalization along the C3 chain can obviously be neglected. Rather, the electron lone pair (LP) at C(1) is stabilized by the α effect of the two attached SiCl3 groups,4 which leads to a significant contraction of the C(1)–Si(1)/Si(2) bonds (1.760(2)/1.762(3) Å) compared to the C(2)–Si(3) bond (1.859(3) Å). The electronic structure of [1]− was further examined by quantum-chemical calculations at the ωB97X-D3(BJ)/ma-def2-QZVPP//ωB97X-D3(BJ)/ma-def2-TZVP(CPCM(CH2Cl2))26 level (cf. the frontier orbitals shown in Fig. 1a). The range-separated hybrid functional was applied to avoid self-interaction error-related problems for anionic systems and too weakly bound electron density. A natural bond orbital (NBO)27 analysis confirms a C(2)C(3) double bond and a LP localized at the C(1) atom (Fig. 1b). C(1) bears a highly negative charge of −1.60e− and C(2) is also significantly negatively charged (−0.53e−) while C(3) is almost neutral (−0.09e−). This trend is supported by a LP(C(1)) → σ*(C(2)–C(3)) charge-transfer stabilization estimate of E(2) = 10.3 kcal mol−1. Strong charge-transfer estimates are further observed from LP(C(1)) into the antibonding C(2)–Si(3) (E(2) = 7.6 kcal mol−1), Si(1)–Cl (ΣE(2) = 30.2 kcal mol−1), and Si(2)–Cl (ΣE(2) = 29.5 kcal mol−1) σ* orbitals. Accordingly, charge transfer from the LP to the adjacent silyl groups is a crucial factor for the stabilization of [1]−. In sum, repulsive but much smaller contributions are found for the interactions of the corresponding Si–C σ bonds with the CC π (anti-)bond (3.1 and 3.6 kcal mol−1). Nevertheless, the experimentally obtained isomer of [1]− is energetically disfavored relative to the isomer [1′]− bearing a Cl atom at C(2) and a trans-SiCl3 group at C(3) (with respect to C(1); Fig. 1c).
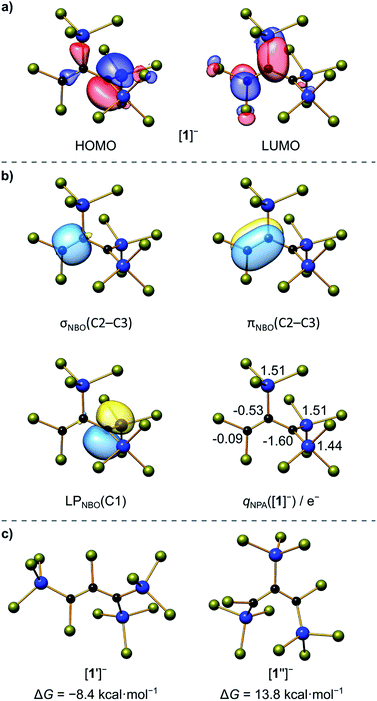 |
| | Fig. 1 (a) Selected Kohn–Sham molecular orbitals of [1]−; (b) NBOs involved in the bonding of the C3 chain (isosurface value = 0.05 e−1/2 Bohr−3/2) and natural charges at the Si and C atoms; (c) Gibbs free energies relative to [1]− (ΔG = 0 kcal mol−1) of isomers [1′]− and [1′′]− with different substitution patterns. Computed at the ωB97X-D3(BJ)/ma-def2-QZVPP//ωB97X-D3(BJ)/ma-def2-TZVP(CPCM(CH2Cl2)) level; see Scheme 2 for the atom numbering. | |
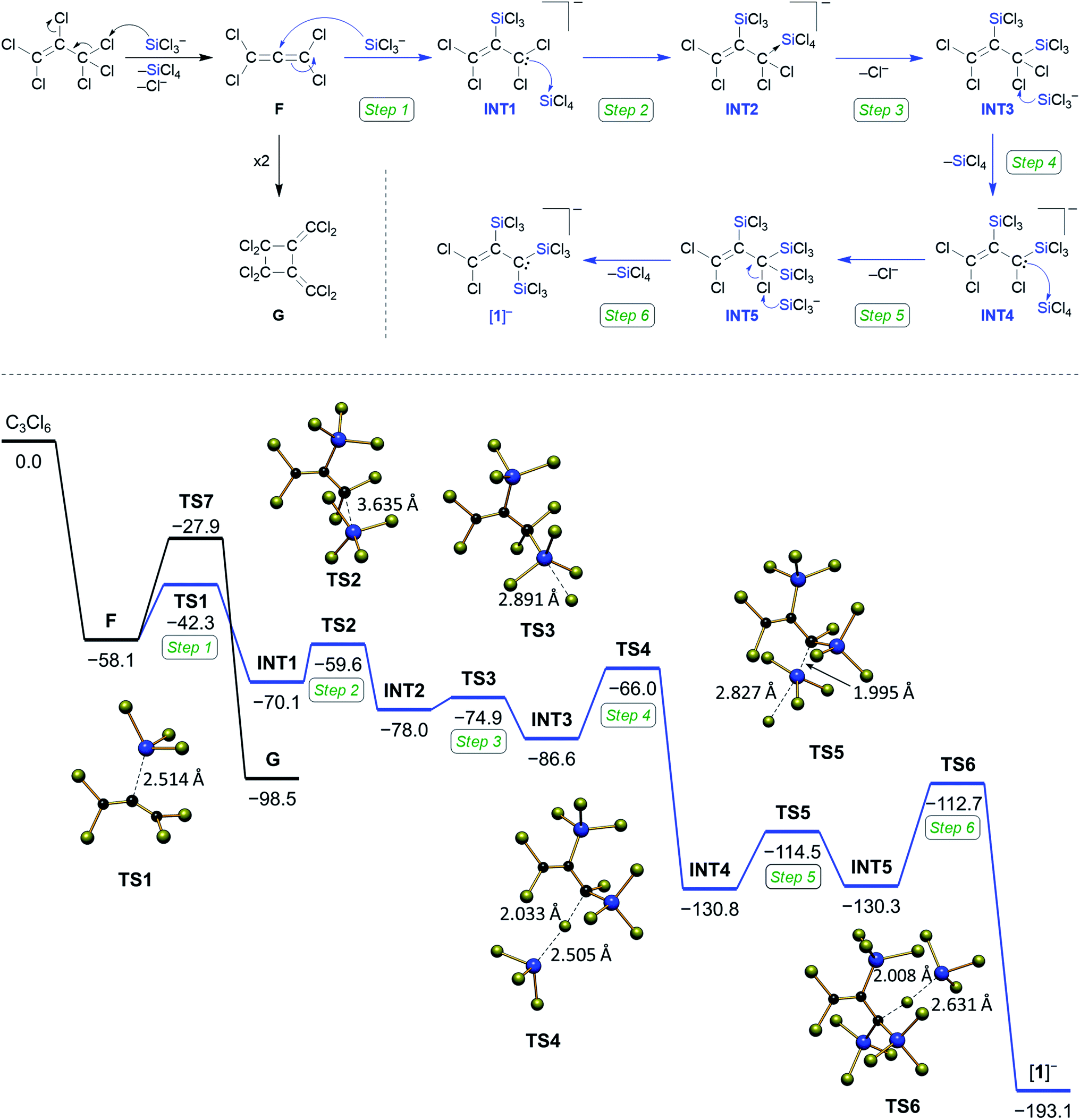
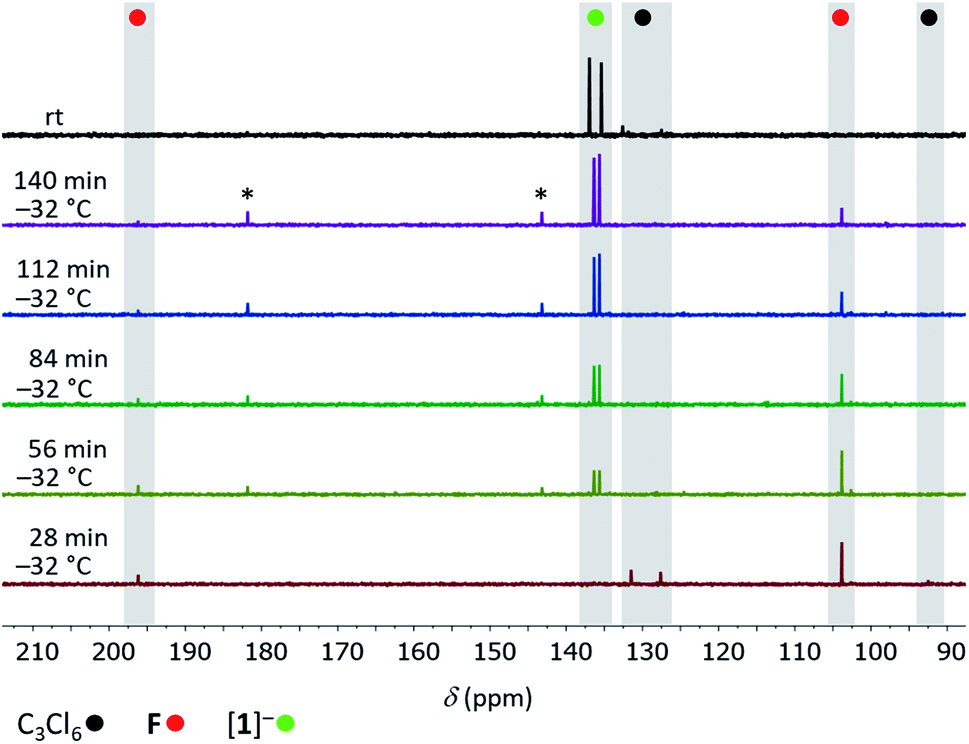
To gain some experimental insight into mechanistic details of the formation of [1]−, we performed two NMR-scale experiments. Experiment 1: a mixture C3Cl6:Si2Cl6:[nBu4N]Cl (1:1:0.07) in CD2Cl2 was prepared at room temperature. After 1.5 h, when most of the C3Cl6 and Si2Cl6 had been consumed, NMR-spectroscopic investigation of the reaction mixture revealed tetrachloroallene (F)28 and its dimer G29 as the sole organic products (Scheme 3). Experiment 2: a solid mixture C3Cl6:Si2Cl6:[nBu4N]Cl (1:4:1) in CD2Cl2 was prepared at liquid-nitrogen temperature and allowed to melt inside the pre-cooled NMR spectrometer (−32 °C). 13C{1H} NMR spectra were recorded at regular intervals of 28 min. During the first five intervals, the temperature of −32 °C was maintained; later, the sample was slowly brought to room temperature (see the ESI† for full details). After the first interval, F was detected besides residual starting material C3Cl6 (Fig. 2). After the second interval, the resonances of [1]− had emerged, those of F were still visible and those of C3Cl6 had vanished. As the reaction progressed, the amount of F in the mixture steadily decreased while that of [1]− increased, until finally (at room temperature) only [1]− was present. The dimer G was not observed at any point in time. Two small 13C resonances (δ = 182.2, 143.9), which appeared together with the signals of [1]− and disappeared again at the end of the conversion may be assignable to an unknown intermediate (Fig. 2). Experiment 1 suggests an initial dechlorination of C3Cl6via chloronium-ion abstraction by [SiCl3]− to form SiCl4 and F/G with concomitant release of Cl−.30 Two further conclusions can be drawn: (i) the activation barrier of the reaction C3Cl6 → F is even lower than that of the reaction 2 F → G,31 because F is replenished faster than it dimerizes to G. (ii) The overall barrier of the follow-up reaction F → [1]− is higher than that of C3Cl6 → F. Otherwise, the conversion C3Cl6 → F with only catalytic amounts of Cl− could not proceed quantitatively because the formation of [nBu4N][1] traps 1 equiv. of [nBu4N]Cl. The results of experiment 2 can be interpreted as follows: (i) also in the presence of 1 equiv. of Cl−, F remains the primary intermediate of the reaction between C3Cl6 and Si2Cl6 and is generated already at low temperatures. (ii) When the stoichiometrically required amounts of Si2Cl6/Cl− are present, the reaction F → [1]− runs to completion and the dimerization of F can no longer compete. (iii) Apart from F, no further intermediates are unequivocally identifiable by in situ NMR spectroscopy (but only the final product [1]−). Therefore, our mechanistic proposal for the formation of [1]− has to be based on previous experiences with related systems and quantum-chemical calculations at the ωB97X-D3(BJ)/ma-def2-QZVPP+COSMO-RS32,33//ma-def2-TZVP(CPCM(CH2Cl2)) level that fully support all the conclusions drawn (Scheme 3).34
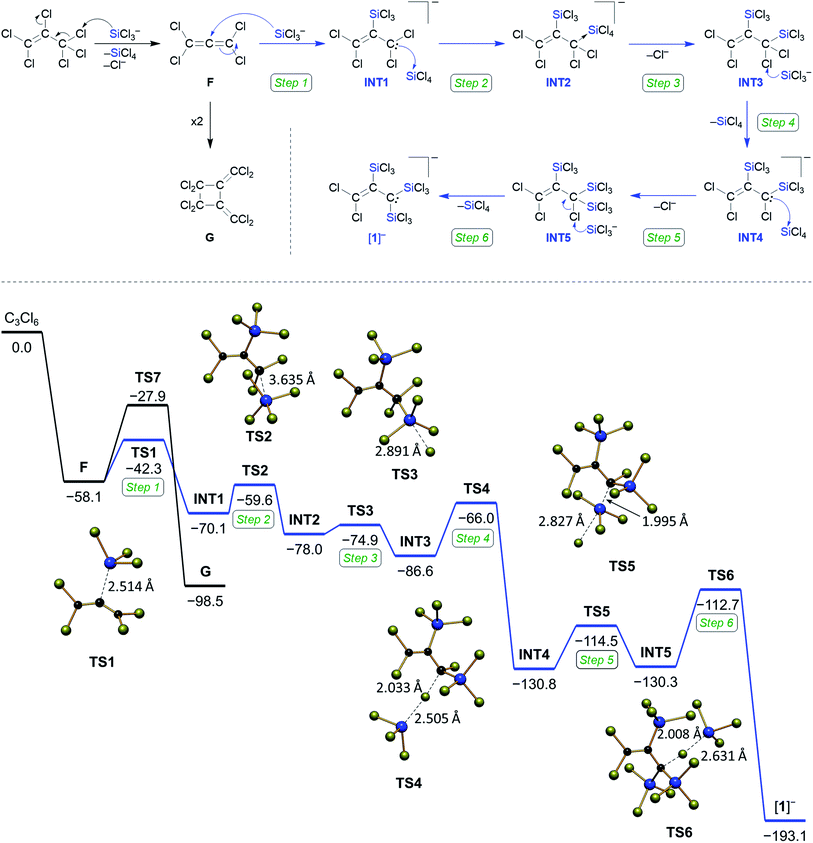 |
| | Scheme 3 Top: reaction of C3Cl6 with Si2Cl6 and Cl− furnishes perchloroallene F, which can either dimerize to G (black pathway) or react further to [1]− (blue pathway). Bottom: Gibbs free energy (kcal mol−1) diagram for the formation of [1]− computed at the ωB97X-D3(BJ)/ma-def2-QZVPP+COSMO-RS//ma-def2-TZVP(CPCM(CH2Cl2)) level of theory (standard conditions). | |
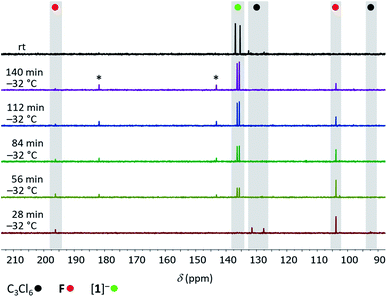 |
| | Fig. 2
13C{1H} NMR spectra recorded at −32 °C at 28 min intervals on the reaction mixture C3Cl6:Si2Cl6:[nBu4N]Cl (1:4:1; CD2Cl2) and at room temperature (top); (*) unknown intermediate. | |
Step 1: we assume a nucleophilic attack of [SiCl3]− on the central carbon atom of F, which produces the allyl anion INT1. Step 2: INT1 and SiCl4 form a pentacoordinated adduct INT2. Step 3: INT2 readily releases a Cl− ion yielding the disilylated prop-1-ene INT3. Step 4: chlorophilic attack of [SiCl3]− on a Cl–C(sp3) bond of INT3 generates the allyl anion INT4 (a similar chloronium-ion abstraction initiates also the reactions C3Cl6 → F and CCl4 → [A]− (Scheme 1)13,14,20). Step 5: due to the α effect of the SiCl3 substituent, the generated electron lone pair will preferentially reside on the silylated C-terminus, which straightforwardly explains why the third SiCl3 group is attached at this position to give the neutral prop-1-ene INT5, even though this step is slightly endergonic. Step 6: a chlorophilic attack of [SiCl3]− on the remaining Cl–C(sp3) bond of INT5 releases the product [1]− and provides a huge thermodynamic driving force. The reaction likely stops at this stage for kinetic reasons, because [1]− is already a sterically crowded compound: quantum-chemical calculations on the putative pentasilylated allyl anion [C3(SiCl3)5]− show a highly strained structure, even though the reaction [1]− + 2 [SiCl3]− → [C3(SiCl3)5]− + 2 Cl− is predicted to be exergonic by ΔG = −82 kcal mol−1. The situation in hexasilylated [C]2− is different, since its two carbanionic termini adopt orthogonal conformations with respect to the central CC double bond, avoiding unfavorable vicinal SiCl3⋯SiCl3 interactions that should be a major issue in the case of [C3(SiCl3)5]−.
Targeted synthesis of the thermostable allyl radical 1˙
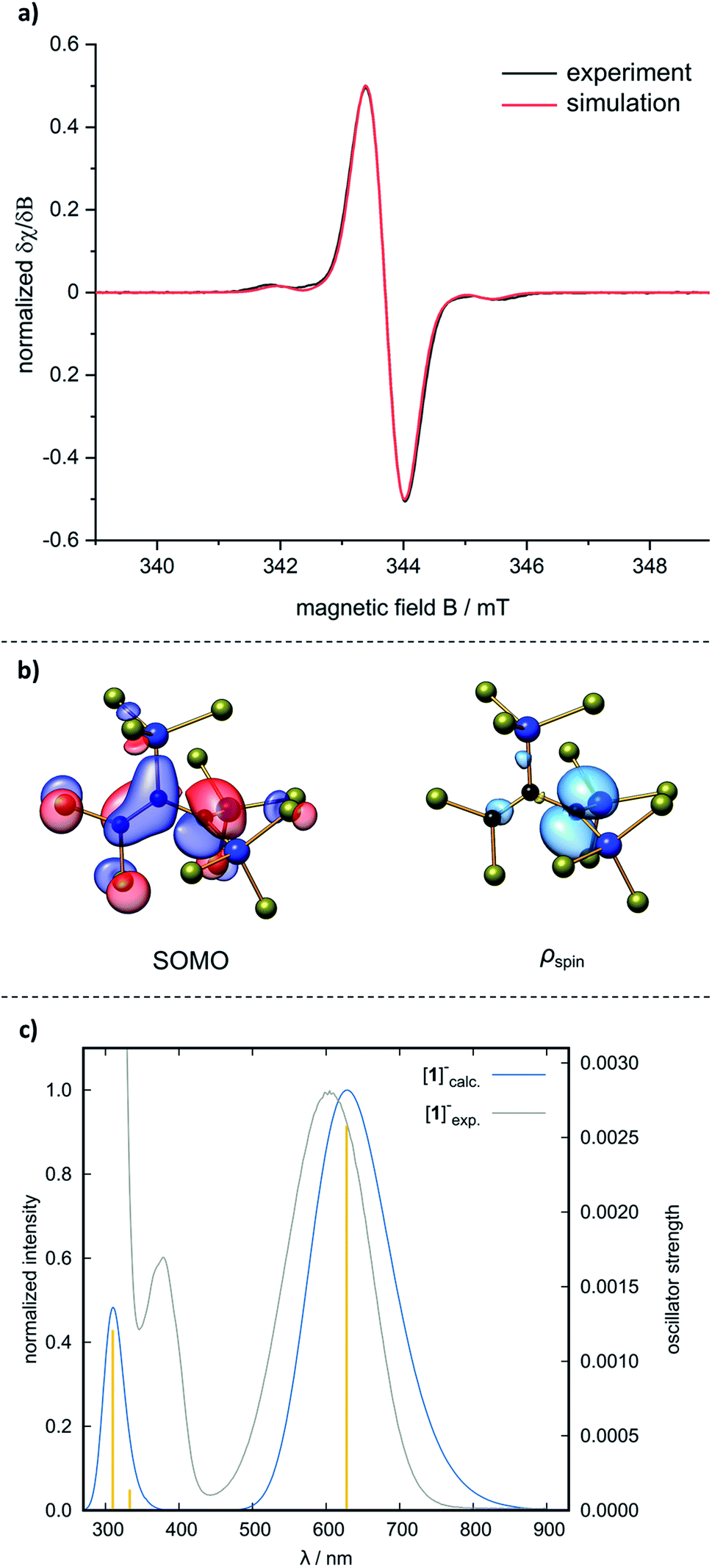
Cyclic voltammetry on [nBu4N][1] showed a (quasi)reversible redox wave with a half-wave potential of E1/2 = 0.06 V (vs. FcH/FcH+; CH2Cl2, supporting electrolyte: [nBu4N][B(C6F5)4]), indicating that the radical 1˙ may be synthetically accessible (we found no indication of further oxidation of 1˙ to the corresponding allyl cation [1]+). After an extensive screening of numerous oxidants, SbCl5 was identified as the reagent of choice (for more detailed information on the oxidizing agents used and the respective reactions, see the ESI†):35 when SbCl5 was added at room temperature to a brown solution of [nBu4N][1] in CH2Cl2, the color immediately changed to green. After 5 min of stirring, n-hexane was added to precipitate all insoluble salts, the CH2Cl2/n-hexane mixture was evaporated, and the blue 1˙ was distilled off (10−3 mbar, 90 °C; 78% yield, Scheme 2). The exceptional (thermal) stability of distillable 1˙ under inert conditions stands out among most other organic radicals.36 The identity of 1˙ was proven by a quenching experiment with the H-atom donor 1,4-cyclohexadiene, which gave the corresponding prop-1-ene 2 (cf.Scheme 6) and C6H6 in a 2:1 ratio. An EPR spectrum, recorded at room temperature on a solution of 1˙ in CH2Cl2, is in good agreement with a simulated spectrum obtained using the hyperfine coupling constants computed at the ωB97X-D3(BJ)/ma-def2-TZVP(CPCM(CH2Cl2)) level of theory (Fig. 3a). The singly occupied molecular orbital (SOMO) of 1˙ is plotted in Fig. 3b; a corresponding spin-density plot shows that the odd electron is mainly localized in the pz orbital of C(1). The blue color of 1˙ arises from an absorption band at λmax = 605 nm (ε = 11023 M−1 cm−1) in CH2Cl2, which is reasonably reproduced by quantum-chemical calculations applying the Tamm–Dancoff approximation of time-dependent DFT (TDA-DFT)37 and molecule-dependent optimal tuning38 (Fig. 3c; cf. [nBu4N][B˙]: λmax = 650 nm (ref. 13)). UV/vis spectroscopy can thus be applied as a useful diagnostic tool to assess the stability of 1˙ toward air and moisture. A cuvette was charged with a dilute solution of 1˙ in CH2Cl2 under inert conditions. A UV/vis spectrum was recorded, the cap was opened to the ambient atmosphere and closed again to avoid evaporation of the solvent. Measurements were repeated in regular intervals of 30 min and showed a linear decrease in absorbance. After 4 h, the sample still had an intense blue color; after 14 h, it was colorless.
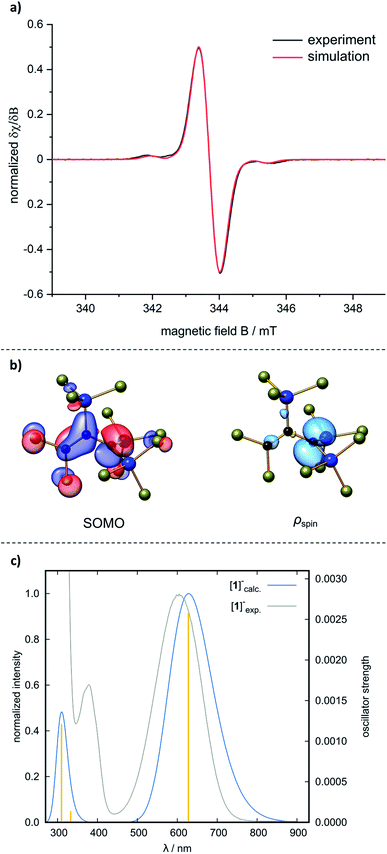 |
| | Fig. 3 (a) Experimental EPR spectrum of 1˙ (black; 0.48 mM in CH2Cl2, room temperature). Simulated EPR spectrum (red), based on the experimentally obtained g value of 2.0036 and computed hyperfine coupling constants (hfc) calculated at the ωB97X-D3(BJ)/ma-def2-TZVP(CPCM(CH2Cl2)) level of theory: 1 × a(13C) = −8.8 G, 1 × a(13C) = 18.0 G, 1 × a(13C) = 27.8 G, 2 × a(29Si) = 14.1 G, 1× a(29Si) = −29.3 G, 2 × a(35Cl/37Cl) = 0.4 G, 3 × a(35Cl/37Cl) = 0.7 G, 6 × a(35Cl/37Cl) = 1.0 G; linewidth = 1.0 G. All exptl. hfc have been scaled by the respective gyromagnetic ratios and natural abundances using the program easyspin.39 (b) Singly occupied Kohn–Sham molecular orbital (SOMO) and Mulliken spin-density plot of 1˙. Isosurface values = 0.05 e−1/2 Bohr−3/2 (MOs) and 0.005 e Bohr−1/2 (ρspin). (c) Normalized computed (blue), experimental (grey) UV/vis spectrum of 1˙ and calculated transitions (yellow) at the TDA-DFT ωB97X-D3(BJ)/ma-def2-TZVP(CPCM(CH2Cl2)) level, applying molecule-dependent optimal tuning. | |
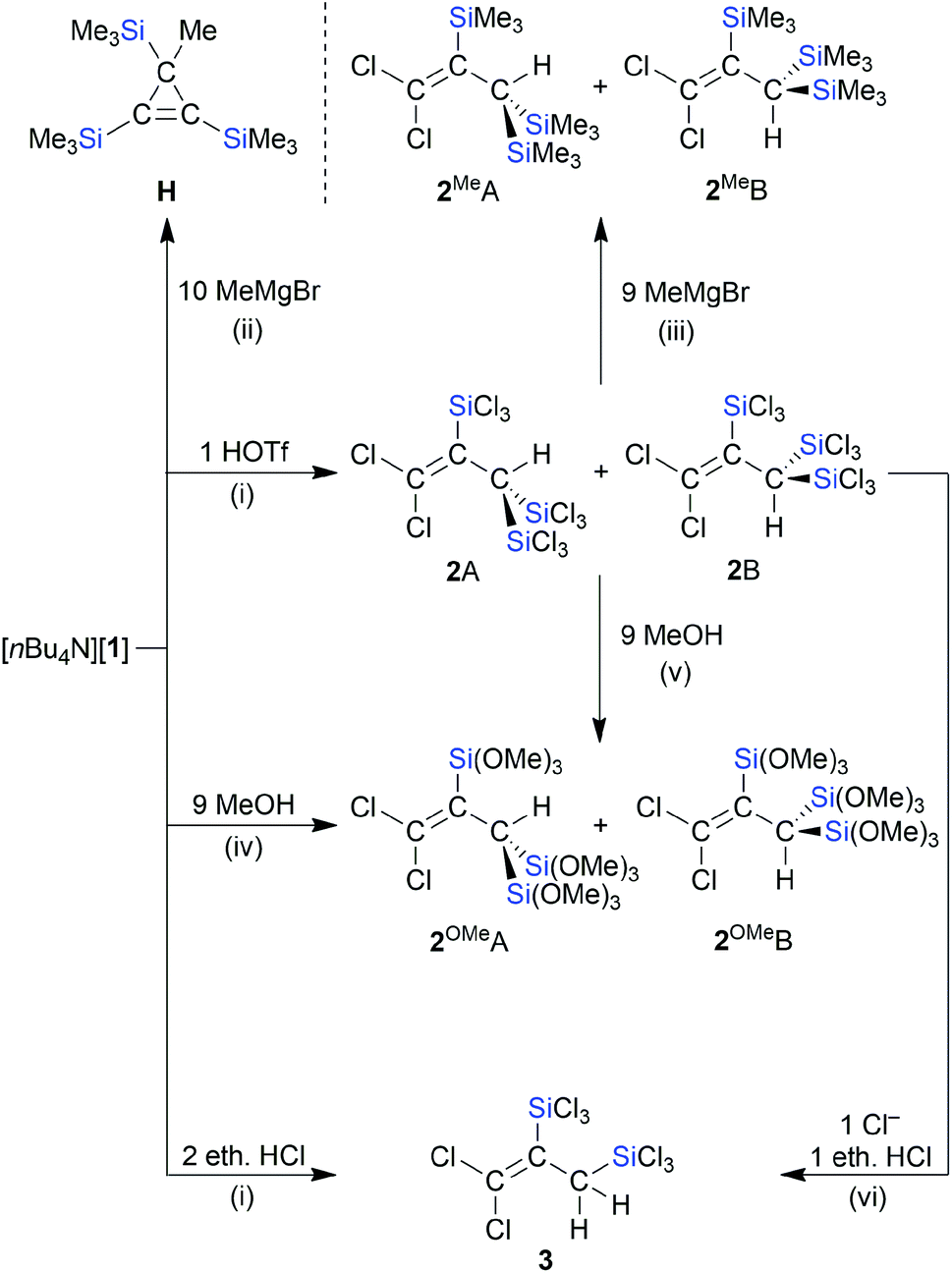
Synthesis of the mixed Cl/SiR3-substituted propenes 2, 2OMe, and 2Me (R = Cl, OMe, Me)
We next converted [nBu4N][1] to the neutral propene through protonation with HOSO2CF3 (HOTf) in CD2Cl2 (Scheme 4). In the 1H NMR spectrum, the product mixture gave rise to two singlets at 3.75 and 4.09 ppm with relative intensities of 2:1 (the 1H resonances of the CH units in doubly protonated [C]2− appear at 3.49 ppm (ref. 15)). The 13C{1H} and 29Si NMR spectra were also characterized by a major and minor set of signals, both compatible with the molecular structure of the target compound 2. An explanation of the observed NMR features lies in the formation of rotamers, 2A and 2B, due to restricted rotation about the sterically encumbered C–C single bond (cf. the comparable situation in (Cl3Si)2(H)C–C(H)(SiCl3)2).13,40 This interpretation gained support from GC-MS measurements, which gave the same molecular masses for 2A/2B and were in line with a chemical formula of Cl2CC(SiCl3)–C(H)(SiCl3)2 (m/z = 511.60, [M˙]+). Quantum-chemical calculations (ωB97X-D3(BJ)/ma-def2-QZVPP+COSMO-RS//ma-def2-TZVP(CPCM(CH2Cl2))) indeed predict a high barrier of rotation about the C(1)–C(2) bond of 2 (ΔG‡ = 33.5 kcal mol−1; the corresponding computed rotational barrier of [1]− is ΔG‡ = 30.1 kcal mol−1 and the rotation activation enthalpy of the parent allyl anion was calculated to ΔH‡ = 19 kcal mol−1 according to ref. 41). Since a mere rotamer mixture should yield solely [1]− as the deprotonation product, we treated 2A/2B with various strong bases. A selective transformation was achieved by using 1 equiv. of [B]2− as the proton-abstracting reagent, which indeed regenerated [1]− quantitatively (see the ESI† for more details).42 In contrast to the reaction with HOTf, the reaction of [1]− with 2 equiv. ethereal HCl in CD2Cl2 gives a product that shows only one set of signals in the NMR spectra (use of 1 equiv. ethereal HCl generates a mixture of the same product and [1]−). These signals can be assigned to compound 3, which carries only two SiCl3 groups and is formed through C-protonation accompanied by protodesilylation (Scheme 4). The latter reduces the steric overload and, as a consequence, the rotation about the C–C single bond is no longer restricted. As previously observed in related cases, the choice of the acid obviously has a major influence on the reaction outcome (non-nucleophilic counteranion OTf−vs. nucleophilic counteranion Cl−).13,15
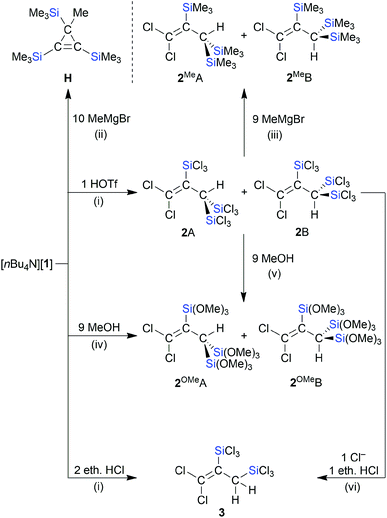 |
| | Scheme 4 Conversions of [nBu4N][1] to the propenes 2A/2B, 2OMeA/2OMeB, or 3 by treatment with HOSO2CF3 (HOTf), MeOH, or ethereal HCl, respectively; the reactions of [nBu4N][1] or 2A/2B with MeMgBr provide the respective products H or 2MeA/2MeB (deviating from the balanced reaction equations, HOTf and MeMgBr were used in excess amounts). (i) CH2Cl2, room temperature; (ii) THF, 60 °C in a sealed NMR tube, 21 h; (iii) THF, reflux temperature, 1 d; (iv) 8 equiv. NMe2Et, CH2Cl2, room temperature; (v) 9 equiv. NMe2Et, CH2Cl2, 0 °C to room temperature; (vi) 1 equiv. [nBu4N]Cl, CD2Cl2, 60 °C, 3 d. | |
A controlled methanolysis of 2A/2B again provided two isomers, 2OMeA/2OMeB (1:1), and added useful NMR handles (Scheme 4):43 (i) the OMe and CH integrals confirmed the presence of one unique Si(OMe)3 group, two chemically equivalent Si(OMe)3 groups, and one CH proton in 2OMeA/2OMeB. (ii) Selective NOESY NMR experiments enabled an assignment of the individual signal sets to rotamer 2OMeA or 2OMeB. With a similar motivation as in the case of the methanolysis reaction, 2A/2B were also treated with excess MeMgBr in THF. After workup, we isolated 2MeA/2MeB in a ratio of 2:1 (Scheme 4; computed rotational barrier about the C–C single bond: ΔG‡ = 34.0 kcal mol−1). The fact that 2MeA is the dominant rotamer was confirmed by a NOESY-NMR experiment.
Conversion of [1]− and 2Me to the silylated cyclopropene H and the silylated allenes Li[4]–6
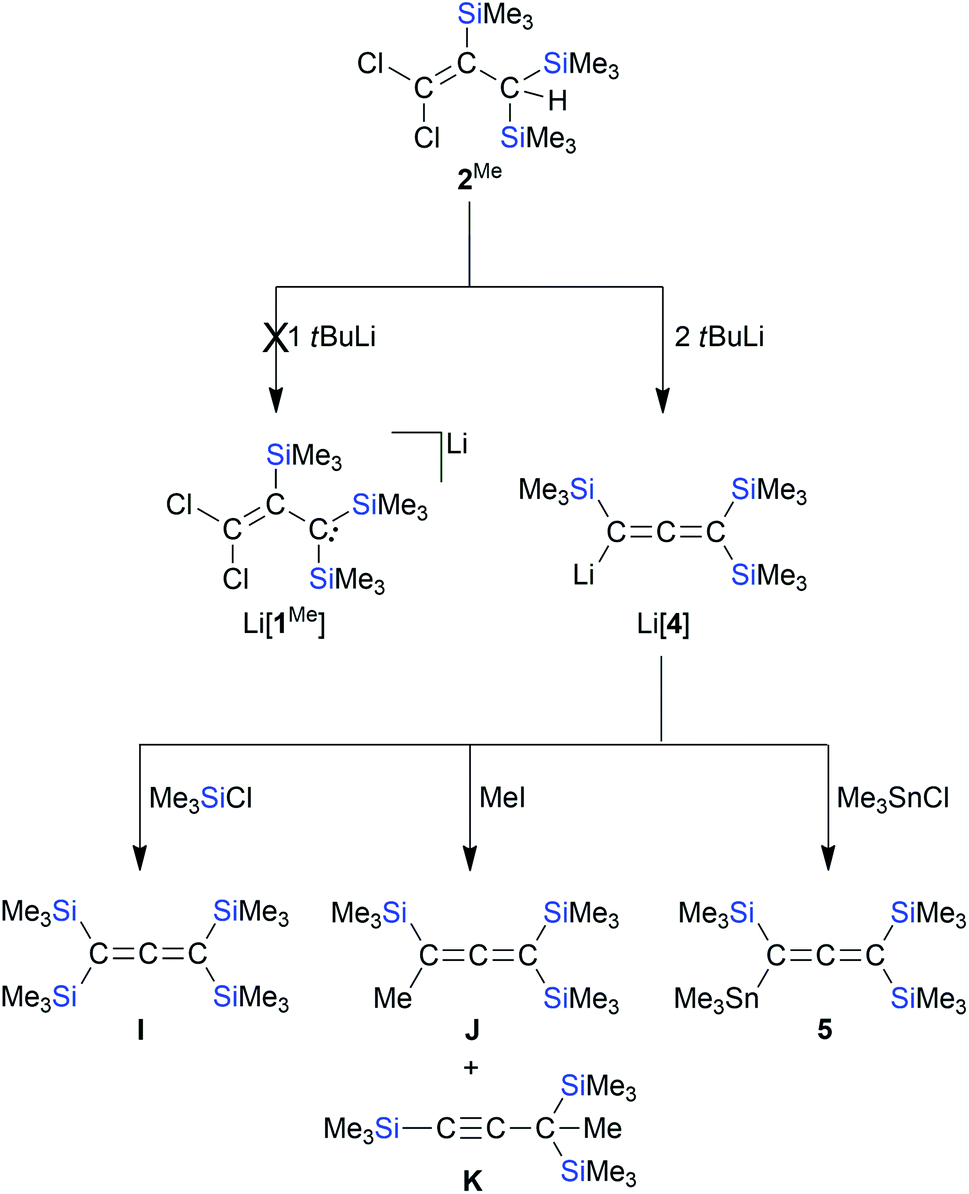
According to in situ NMR spectroscopy, the Grignard reagent does not deprotonate the propene cores of 2MeA/2MeB: a putative tris(trimethylsilyl) derivative [1Me]− should have a less stabilized carbanionic center than [1]−, because the α effect of a SiMe3 fragment is smaller than that of an SiCl3 unit, which decreases the acidity of 2Me relative to 2.4,13 The anticipated higher reactivity of [1Me]− makes this anion another worthwhile synthesis target. In a first attempt, 2A/2B were replaced by [nBu4N][1] in the Grignard reaction, which, however, did not furnish [1Me]− but rather the cyclopropene H44 (Scheme 4): As expected, all three SiCl3 groups were converted to SiMe3 groups. Against a priori expectations, the nucleophilicity of the carbanion was increased to the point that it underwent an intramolecular Cl− substitution to close the three-membered ring. Moreover, migration of one silyl group occurred45 and the second C-bonded Cl atom was replaced by a Me substituent. Silylated cyclopropenes are valuable building blocks for organic synthesis.46 Derivative H was previously obtained via five steps in ∼10% yield.44,47 In comparison, our two-step synthesis (80% yield) represents a significant improvement. Coming back to [1Me]−, we next employed the extremely basic tBuLi to accomplish the deprotonation of 2Me. The most selective reaction was reached with 2 equiv. of tBuLi, which gave the lithiated allene Li[4]48 (quantitative conversion; Scheme 5).49 In terms of the reaction mechanism, we propose that an initial proton abstraction generates [1Me]−, which, similar to the case of the reaction [nBu4N][1] + MeMgBr → H (Scheme 4), cyclizes to an H-type cyclopropene carrying a CCl instead of the CMe fragment. With the bulky tBuLi, the chlorocyclopropene does not undergo Cl/tBu but rather Cl/Li exchange. Ample precedence exists for the rearrangement of (lithiated) cyclopropenes to (lithiated) allenes.28,50 Li[4] straightforwardly reacts with Me3SiCl, MeI, or Me3SnCl to furnish the derivatives I,48J/K,48 and 5, respectively (Scheme 5). Also allenes, especially the silylated ones, are attracting great attention as building blocks in organic synthesis and in the field of materials science.51 The time- and cost-efficient new protocol reported here is therefore a potentially valuable addition to the existing toolbox of allene syntheses.52 Along these lines, it would be desirable to include [1]− as starting material for the synthesis of perchlorinated Li[4]-type compounds.50 Given that [nBu4N][1] per se has no tendency to eliminate Cl− ions even at elevated temperatures, we added AlCl3 in CH2Cl2 to support Cl− abstraction (cf.Scheme 1).15 After stirring at room temperature overnight, the reaction mixture had adopted a deep green color. The [nBu4N][AlCl4] formed was precipitated by addition of n-hexane, all volatiles were removed from the filtrate under reduced pressure, and the green oily residue was subjected to distillation. Only one fraction was obtained (10−3 mbar, 70 °C), which possessed a bright blue color; some yellow-brown material remained in the distillation sump. The blue distillate collected showed no signal in the 1H NMR spectrum, three 13C resonances and two 29Si NMR signals. One of the C nuclei was strongly deshielded (δ(13C) = 216.7, CD2Cl2), as is characteristic for central allene-C atoms. A typical allene stretching band was observed at 1928.5 cm−1 in the IR spectrum. A GC-MS measurement confirmed the formation of the trifold SiCl3-substituted chloroallene 6 (m/z = 473.60, [M˙]+; Scheme 6). The apparent blue color of 6 would be unique among comparable allene species (cf. the colorless compounds I, J, and 5) and, on the other hand, is reminiscent of the radical 1˙. Indeed, an EPR spectrum of the blue fraction in CH2Cl2 affirmed the presence of this NMR-silent radical in addition to 6. In order to gently quench 1˙, the solution was treated with 1,4-cyclohexadiene, whereupon the blue color disappeared over the course of 2 h. NMR spectroscopy on the colorless solution still showed the resonances of 6, but also the signals of 2 and C6H6 (Scheme 6). After removal of all volatile components, the mixture of 6 and 2 was dissolved in Et2O and treated with excess MeMgBr to produce the corresponding derivatives (Me3Si)(Cl)CCC(SiMe3)2 and 2Me, respectively. From the proton integral values in the 1H NMR spectrum of this blend, it was then estimated that the fraction of 2Me was about 20% – which, in turn, should also be true for the fraction of 1˙ in the blue mixture with 6. We finally succeeded in the isolation of 6 (24% yield) by performing the 1,4-cyclohexadiene quench on a preparative scale: while 6/1˙ cannot be separated by fractional distillation, this is conveniently possible for 6/2. How was 1˙ formed? Bock et al. have already advertised the system AlCl3/CH2Cl2 as a strong oxidizing agent and proposed the chloromethylium salt [CH2Cl][AlCl4] as the actual electron acceptor.53 So far, we never observed such reactivity (e.g., in the synthesis of E; Scheme 1),14,15 but in the present case, it may well play a role.
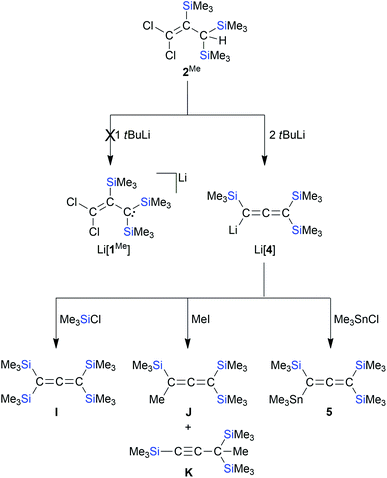 |
| | Scheme 5 The reaction of 2Me with tBuLi does not lead to Li[1Me] but to Li[4]. Li[4] reacts with Me3SiCl, MeI, or Me3SnCl to form the derivatives I, J/K, or 5, respectively (THF, room temperature). | |
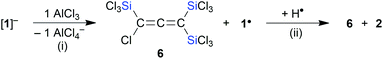 |
| | Scheme 6 Synthesis of 6 in a mixture with side product 1˙ through reaction of [1]− with AlCl3; the identity of 1˙ was confirmed by an H-atom abstraction reaction, which gave 2 and C6H6. (i) CH2Cl2, room temperature; (ii) excess 1,4-cyclohexadiene, CD2Cl2, room temperature; C6H6 was observed as byproduct. | |
Conclusions
We found that hexachloropropene (Cl2CC(Cl)–CCl3) reacts with the Si2Cl6/Cl− system (1:4:1) to give the trisilylated allyl anion [Cl2CC(SiCl3)–C(SiCl3)2]− ([1]−). The remarkable coexistence of a C-centered nucleophile with three electrophilic Si atoms in the same molecule is a consequence of the pronounced α effect of the SiCl3 groups, which electronically stabilize the electron lone pair. The Cl-to-SiCl3 exchange underlying the formation of [1]− involves [SiCl3]− ions as key intermediates and stops at the stage of the triply silylated product presumably due to steric constraints. As a result, [1]− presents four types of synthetically useful functional groups (i.e., Cl, SiCl3, CC, [R3C:]−) that can be employed in further derivatizations. Indeed, starting from readily available [1]−, a variety of still highly functionalized C3 compounds are accessible, such as the thermostable blue radical 1˙, the propenes Cl2CC(SiR′3)–C(SiR′3)2H (R′ = Cl, Me, OMe; 2, 2Me, 2OMe), or the allene Cl3Si(Cl)CCC(SiCl3)2 (6). The silylation protocol and follow-up reactions presented here thus represent a time- and cost-efficient addition to the currently available toolbox for the preparation of organosilicon building blocks.
Data availability
Experimental and computational data associated with this article have been provided in the ESI.†
Author contributions
I. G. synthesized and characterized the compounds. M. Bu. performed the quantum-chemical calculations. B. E. performed the EPR measurement and related simulation. M. Bo. performed the X-ray crystal structure analysis. H.-W. L., S. G. and M. W. supervised the project. The manuscript was written by M. W. and I. G. and edited by all the co-authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to the Evonik Operations GmbH, Rheinfelden (Germany), for the generous donation of Si2Cl6. I. G. wishes to thank the Evonik Foundation for a PhD grant. This work was partially funded by the Bundesministerium für Wirtschaft und Energie through the WIPANO grant number 03THW10K19. The German Research Foundation (DFG) is gratefully acknowledged for financial support through a Gottfried Wilhelm Leibniz prize to S. G.
Notes and references
-
T. Hiyama and M. Oestreich, Organosilicon Chemistry, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed.
-
(a) D. Yan, J. Mohsseni-Ala, N. Auner, M. Bolte and J. W. Bats, Chem.–Eur. J., 2007, 13, 7204–7214 CrossRef CAS;
(b) D. Yan, M. Bolte and N. Auner, J. Organomet. Chem., 2008, 693, 908–916 CrossRef CAS;
(c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
-
(a) R. H. Baney, M. Itoh, A. Sakakibara and T. Suzuki, Chem. Rev., 1995, 95, 1409–1430 CrossRef CAS;
(b) D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431–1442 CrossRef CAS;
(c) R. Murugavel, A. Voigt, M. G. Walawalkar and H. W. Roesky, Chem. Rev., 1996, 96, 2205–2236 CrossRef CAS;
(d) H. C. L. Abbenhuis, Chem.–Eur. J., 2000, 6, 25–32 CrossRef CAS PubMed;
(e) K. J. Shea and D. A. Loy, Chem. Mater., 2001, 13, 3306–3319 CrossRef CAS;
(f) D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS;
(g) F. Wang, X. Lu and C. He, J. Mater. Chem., 2011, 21, 2775–2782 RSC;
(h)
T. Baumgartner and F. Jäkle, Main Group Strategies towards Functional Hybrid Materials, John Wiley & Sons, Ltd., Chichester, 1st edn, 2018 Search PubMed;
(i) F. Dong, L. Lu and C.-S. Ha, Macromol. Chem. Phys., 2019, 220, 1800324 CrossRef;
(j) F. Seidi, M. Jouyandeh, A. Taghizadeh, M. Taghizadeh, S. Habibzadeh, Y. Jin, H. Xiao, P. Zarrintaj and M. R. Saeb, Surf. Innovations, 2021, 9, 3–16 CrossRef.
-
M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, John Wiley & Sons, New York, 2000, vol. 1 Search PubMed.
-
(a) U. Eduok, O. Faye and J. Szpunar, Prog. Org. Coat., 2017, 111, 124–163 CrossRef CAS;
(b) F. Liravi and E. Toyserkani, Addit. Manuf., 2018, 24, 232–242 CAS;
(c) P. Mazurek, S. Vudayagiri and A. L. Skov, Chem. Soc. Rev., 2019, 48, 1448–1464 RSC;
(d) T. J. Wallin, L.-E. Simonsen, W. Pan, K. Wang, E. Giannelis, R. F. Shepherd and Y. Mengüç, Nat. Commun., 2020, 11, 4000 CrossRef CAS PubMed.
-
(a) D. J. Peterson, J. Org. Chem., 1968, 33, 780–784 CrossRef CAS;
(b) L. F. van Staden, D. Gravestock and D. J. Ager, Chem. Soc. Rev., 2002, 31, 195–200 RSC.
-
(a) Y. Hatanaka and T. Hiyama, Synlett, 1991, 1991, 845–853 CrossRef;
(b) S. E. Denmark and R. F. Sweis, Chem. Pharm. Bull., 2002, 50, 1531–1541 CrossRef CAS PubMed;
(c) T. Hiyama, J. Organomet. Chem., 2002, 653, 58–61 CrossRef CAS;
(d) T. Hiyama and E. Shirakawa, Top. Curr. Chem., 2002, 219, 61–85 CrossRef CAS;
(e) S. E. Denmark and M. H. Ober, Aldrichimica Acta, 2003, 36, 75–85 CAS.
-
(a) K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694–1696 CrossRef CAS;
(b) K. Tamao, T. Kakui, M. Akita, T. Iwahara, R. Kanatani, J. Yoshida and M. Kumada, Tetrahedron, 1983, 39, 983–990 CrossRef CAS;
(c) K. Tamao and N. Ishida, J. Organomet. Chem., 1984, 269, C37–C39 CrossRef CAS;
(d) K. Tamao, M. Kumada and K. Maeda, Tetrahedron Lett., 1984, 25, 321–324 CrossRef CAS.
-
(a) A. Hosomi and H. Sakurai, Tetrahedron Lett., 1976, 17, 1295–1298 CrossRef;
(b) A. Hosomi and K. Miura, Bull. Chem. Soc. Jpn., 2004, 77, 835–851 CrossRef CAS;
(c) J. H. Lee, Tetrahedron, 2020, 76, 131351 CrossRef CAS.
- N. Selander, J. R. Paasch and K. J. Szabó, J. Am. Chem. Soc., 2011, 133, 409–411 CrossRef CAS.
-
(a) H. Watanabe, M. Saito, N. Sutou and Y. Nagai, J. Chem. Soc., Chem. Commun., 1981, 617–618 RSC;
(b) T. K. Sarkar, Synthesis, 1990, 1990, 969–983 CrossRef;
(c) T. K. Sarkar, Synthesis, 1990, 1990, 1101–1111 CrossRef;
(d) W. E. Crowe, D. R. Goldberg and Z. J. Zhang, Tetrahedron Lett., 1996, 37, 2117–2120 CrossRef CAS;
(e) L. Chabaud, P. James and Y. Landais, Eur. J. Org. Chem., 2004, 3173–3199 CrossRef CAS;
(f) A. Barbero and F. J. Pulido, Acc. Chem. Res., 2004, 37, 817–825 CrossRef CAS;
(g) M. Oestreich and G. Auer, Adv. Synth. Catal., 2005, 347, 637–640 CrossRef CAS;
(h) D. J. Vyas and M. Oestreich, Chem. Commun., 2010, 46, 568–570 RSC;
(i) D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2010, 49, 8513–8515 CrossRef CAS;
(j) L. B. Delvos, D. J. Vyas and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 4650–4653 CrossRef CAS;
(k) W. Xue and M. Oestreich, Synthesis, 2019, 51, 233–239 CrossRef CAS;
(l) W.-L. Yu, Y.-C. Luo, L. Yan, D. Liu, Z.-Y. Wang and P.-F. Xu, Angew. Chem., Int. Ed., 2019, 58, 10941–10945 CrossRef CAS;
(m) J.-H. Zeng, J.-J. Chen, L. Chen and Z.-P. Zhan, Org. Chem. Front., 2020, 7, 1132–1136 RSC.
-
(a) L. Gao, X. Lin, J. Lei, Z. Song and Z. Lin, Org. Lett., 2012, 14, 158–161 CrossRef CAS PubMed;
(b) L. Gao, Y. Zhang and Z. Song, Synlett, 2013, 24, 139–144 CAS;
(c) S. Xu, R. Chen, Z. Fu, Y. Gao and J. Wang, J. Org. Chem., 2018, 83, 6186–6192 CrossRef CAS.
- I. Georg, J. Teichmann, M. Bursch, J. Tillmann, B. Endeward, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, J. Am. Chem. Soc., 2018, 140, 9696–9708 CrossRef CAS.
- J. Teichmann, C. Kunkel, I. Georg, M. Moxter, T. Santowski, M. Bolte, H.-W. Lerner, S. Bade and M. Wagner, Chem.–Eur. J., 2019, 25, 2740–2744 CrossRef CAS.
- I. Georg, M. Bursch, J. B. Stückrath, E. Alig, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, Angew. Chem., Int. Ed., 2020, 59, 16181–16187 CrossRef CAS.
- J. Tillmann, J. H. Wender, U. Bahr, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2015, 54, 5429–5433 CrossRef CAS PubMed.
- J. Teichmann, M. Bursch, B. Köstler, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, Inorg. Chem., 2017, 56, 8683–8688 CrossRef CAS.
- For a review article on the chemistry developed so far with the Si2Cl6/Cl− system, see: J. Teichmann and M. Wagner, Chem. Commun., 2018, 54, 1397–1412 RSC.
- J. Tillmann, M. Moxter, M. Bolte, H.-W. Lerner and M. Wagner, Inorg. Chem., 2015, 54, 9611–9618 CrossRef CAS PubMed.
- U. Böhme, M. Gerwig, F. Gründler, E. Brendler and E. Kroke, Eur. J. Inorg. Chem., 2016, 2016, 5028–5035 CrossRef.
- M. Olaru, M. F. Hesse, E. Rychagova, S. Ketkov, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2017, 56, 16490–16494 CrossRef CAS.
- M. B. Geeson and C. C. Cummins, Science, 2018, 359, 1383–1385 CrossRef CAS PubMed.
- M. B. Geeson, P. Ríos, W. J. Transue and C. C. Cummins, J. Am. Chem. Soc., 2019, 141, 6375–6384 CrossRef CAS PubMed.
- J. Tillmann, L. Meyer, J. I. Schweizer, M. Bolte, H.-W. Lerner, M. Wagner and M. C. Holthausen, Chem.–Eur. J., 2014, 20, 9234–9239 CrossRef CAS.
-
M. A. Fox and J. K. Whitesell, Organic Chemistry, Jones and Bartlett Publishers, Sudbury, Mass., 3rd edn, 2004 Search PubMed.
-
(a) J. Da Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC;
(b) Y.-S. Lin, G.-D. Li, S.-P. Mao and J.-D. Chai, J. Chem. Theory Comput., 2013, 9, 263–272 CrossRef CAS PubMed;
(c) J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed;
(d) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef;
(e) S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS;
(f) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS;
(g)
ORCA: An ab initio, density functional and semiempirical program package, V. 4.2.1, MPI für Kohlenforschung: Mülheim a. d. Ruhr, Germany, 2019 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS.
- W. E. Billups and R. E. Bachman, Tetrahedron Lett., 1992, 33, 1825–1826 CrossRef CAS.
- C. Korhummel and M. Hanack, Chem. Ber., 1989, 122, 2187–2192 CrossRef CAS.
- A related reaction, in which [CCl3]− replaces [SiCl3]− as chloronium-abstracting reagent in the conversion of C3Cl6 to F has been published: F. Boberg and H. Khalaf, Justus Liebigs Ann. Chem., 1970, 741, 153–156. The facile dimerization of F to G in the absence of other reaction partners is also well documented:
(a) A. Roedig, G. Märkl and B. Heinrich, Angew. Chem., Int. Ed. Engl., 1963, 2, 47 CrossRef;
(b) A. Roedig and B. Heinrich, Chem. Ber., 1967, 100, 3716–3724 CrossRef CAS.
- The calculated activation barrier of the reaction 2 F → G (ΔG = 30.2 kcal mol−1; cf.Scheme 3) is considerably higher than the experimentally determined barrier (ΔH‡ = 10.6 ± 3.1 kcal mol−1): N. Detzer and A. Roedig, Tetrahedron, 1971, 27, 5697–5703 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- F. Eckert and A. Klamt, AIChE J., 2002, 48, 369–385 CrossRef CAS.
- The reaction of perchlorocyclopropene, cyclo-C3Cl4, with Si2Cl6/Cl− (1:4:1) in CD2Cl2 is unselective and leads to a complex mixture of products, which contain not even traces of [1]− (13C{1H} NMR-spectroscopic control). Therefore, cyclo-C3Cl4 can be excluded as an intermediate in the reaction C3Cl6 → [1]−.
- A related example is the blue radical [B˙]−, which is accessible via comproportionation of [B]2− and B.13 Attempts at the targeted synthesis of 1˙ by H-atom abstraction with azobis(isobutyronitrile) (AIBN) failed.
- For selected other remarkably inert organic radicals, see:
(a) U. Groß, S. Rüdiger and A. Dimitrov, J. Fluorine Chem., 1996, 76, 139–144 CrossRef;
(b) K. Goto, T. Kubo, K. Yamamoto, K. Nakasuji, K. Sato, D. Shiomi, T. Takui, M. Kubota, T. Kobayashi, K. Yakusi and J. Ouyang, J. Am. Chem. Soc., 1999, 121, 1619–1620 CrossRef CAS;
(c) R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC;
(d) A. Ueda, H. Wasa, S. Nishida, Y. Kanzaki, K. Sato, D. Shiomi, T. Takui and Y. Morita, Chem.–Eur. J., 2012, 18, 16272–16276 CrossRef CAS;
(e) J. P. Peterson, M. R. Geraskina, R. Zhang and A. H. Winter, J. Org. Chem., 2017, 82, 6497–6501 CrossRef CAS;
(f) K. Kato, K. Furukawa, T. Mori and A. Osuka, Chem.–Eur. J., 2018, 24, 572–575 CrossRef CAS PubMed;
(g) K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS;
(h) B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem. Sci., 2020, 11, 1192–1204 RSC;
(i) M. Harada, M. Tanioka, A. Muranaka, T. Aoyama, S. Kamino and M. Uchiyama, Chem. Commun., 2020, 56, 9565–9568 RSC.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291 CrossRef CAS.
-
(a) T. Stein, L. Kronik and R. Baer, J. Chem. Phys., 2009, 131, 244119 CrossRef;
(b) M. Rubešová, E. Muchová and P. Slavíček, J. Chem. Theory Comput., 2017, 13, 4972–4983 CrossRef.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
- G. Fritz, S. Lauble, R. Befurt, K. Peters, E.-M. Peters and F. H. G. von Schnering, Z. Anorg. Allg. Chem., 1993, 619, 1494–1511 CrossRef CAS.
- R. González-Luque, I. Nebot-Gil, M. Merchán and F. Tomás, Theor. Chim. Acta, 1986, 69, 101–106 CrossRef.
- The use of 0.5 equiv. of [nBu4N]2[B] yielded a 1:1:1 mixture of 2, [nBu4N][1], and [nBu4N][(Cl3Si)2(H)C–C(SiCl3)2] ([nBu4N][BH]). Because of the two immediately adjacent negative charges, [B]2− is apparently a stronger base than [1]− (proton transfer from 2 is nevertheless slow); the Brønsted basicity of [BH]− is no longer high enough to deprotonate 2.
- A comparably selective methanolysis reaction leads from [nBu4N][1] to 2OMeA/2OMeB and is the basis for the titration experiment discussed.
- G. Maier, J. Neudert, O. Wolf, D. Pappusch, A. Sekiguchi, M. Tanaka and T. Matsuo, J. Am. Chem. Soc., 2002, 124, 13819–13826 CrossRef CAS PubMed.
-
(a) A. J. Ashe, J. Am. Chem. Soc., 1970, 92, 1233–1235 CrossRef CAS;
(b) C. W. Spangler, Chem. Rev., 1976, 76, 187–217 CrossRef CAS;
(c)
M. Kira and T. Iwamoto, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Ltd., Chichester, 2003, vol. 3, pp. 853–948 Search PubMed.
-
(a) H. A. Buchholz, G. K. S. Prakash, D. Deffieux and G. A. Olah, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10003–10005 CrossRef CAS PubMed;
(b) S. Chuprakov, D. A. Malyshev, A. Trofimov and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 14868–14869 CrossRef CAS;
(c) E. A. F. Fordyce, Y. Wang, T. Luebbers and H. W. Lam, Chem. Commun., 2008, 1124–1126 RSC;
(d) T. J. Thomas, B. A. Merritt, B. E. Lemma, A. M. McKoy, T. Nguyen, A. K. Swenson, J. L. Mills and M. G. Coleman, Org. Biomol. Chem., 2016, 14, 1742–1747 RSC;
(e) R. Vicente, Synthesis, 2016, 48, 2343–2360 CrossRef CAS.
- G. Maier, D. Volz and J. Neudert, Synthesis, 1992, 1992, 561–564 CrossRef.
- R. West and P. C. Jones, J. Am. Chem. Soc., 1969, 91, 6156–6161 CrossRef CAS.
- The use of 1 equiv. of tBuLi afforded the fourfold SiMe3-substituted allene I, leaving some of the starting material 2Me unconsumed. We assume that Li[4] is formed as an intermediate that takes up one SiMe3 group from residual 2Me to give I.
-
(a) R. Kostikov and A. de Meijere, J. Chem. Soc., Chem. Commun., 1984, 1528–1529 RSC;
(b) P. Binger, P. Müller, R. Wenz and R. Mynott, Angew. Chem., Int. Ed. Engl., 1990, 29, 1037–1038 CrossRef;
(c) R. Walsh, S. Untiedt and A. de Meijere, Chem. Ber., 1994, 127, 237–245 CrossRef CAS;
(d) A. de Meijere, D. Faber, U. Heinecke, R. Walsh, T. Müller and Y. Apeloig, Eur. J. Org. Chem., 2001, 6, 663–680 CrossRef;
(e) I. Zrinski, N. Novak-Coumbassa and M. Eckert-Maksic, Organometallics, 2004, 23, 2806–2809 CrossRef CAS;
(f) J. Li, C. Sun, S. Demerzhan and D. Lee, J. Am. Chem. Soc., 2011, 133, 12964–12967 CrossRef CAS;
(g) Y. Liu, D. Zhang and S. Bi, Organometallics, 2012, 31, 4769–4778 CrossRef CAS;
(h) A. C. Voukides, K. J. Cahill and R. P. Johnson, J. Org. Chem., 2013, 78, 11815–11823 CrossRef CAS PubMed.
-
(a) R. Zimmer, C. U. Dinesh, E. Nandanan and F. A. Khan, Chem. Rev., 2000, 100, 3067–3125 CrossRef CAS;
(b) M. A. Shengming, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef;
(c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783–816 RSC;
(d) T. M. Gregg, R. F. Algera, J. R. Frost, F. Hassan and R. J. Stewart, Tetrahedron Lett., 2010, 51, 6429–6432 CrossRef CAS;
(e) F. Lõpez and J. L. Mascareñas, Chem.–Eur. J., 2011, 17, 418–428 CrossRef;
(f) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS;
(g) P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef CAS;
(h) T. Lechel, F. Pfrengle, H.-U. Reissig and R. Zimmer, ChemCatChem, 2013, 5, 2100–2130 CrossRef CAS;
(i) C. S. Adams, C. D. Weatherly, E. G. Burke and J. M. Schomaker, Chem. Soc. Rev., 2014, 43, 3136–3163 RSC;
(j) M. D. Jovanovic, M. R. Petkovic and V. M. Savic, Synthesis, 2021, 53, 1035–1045 CrossRef CAS.
-
(a) J. A. Marshall and C. M. Grant, J. Org. Chem., 1999, 64, 8214–8219 CrossRef CAS;
(b) L. K. Sydnes, Chem. Rev., 2003, 103, 1133–1150 CrossRef CAS;
(c) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 795–818 CrossRef;
(d) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384–5418 RSC;
(e) T. Hashimoto, K. Sakata, F. Tamakuni, M. J. Dutton and K. Maruoka, Nat. Chem., 2013, 5, 240–244 CrossRef CAS;
(f) S. Wu, X. Huang, W. Wu, P. Li, C. Fu and S. Ma, Nat. Commun., 2015, 6, 7946 CrossRef;
(g) W.-D. Chu, Y. Zhang and J. Wang, Catal. Sci. Technol., 2017, 7, 4570–4579 RSC.
-
(a) H. Bock, G. Brähler, U. Henkel, R. Schlecker and D. Seebach, Chem. Ber., 1980, 113, 289–301 CrossRef CAS;
(b) H. Bock, G. Brähler, D. Dauplaise and J. Meinwald, Chem. Ber., 1981, 114, 2622–2631 CrossRef CAS;
(c) H. Bock and W. Kaim, Acc. Chem. Res., 1982, 15, 9–17 CrossRef CAS;
(d) H. Bock, U. Stein and P. Rittmeyer, Angew. Chem., Int. Ed. Engl., 1982, 21, 533–534 CrossRef;
(e) H. Bock and U. Lechner-Knoblauch, J. Organomet. Chem., 1985, 294, 295–304 CrossRef CAS;
(f) H. Bock, Angew. Chem., Int. Ed. Engl., 1989, 28, 1627–1650 CrossRef;
(g) H. Bock and B. Solouki, Chem. Rev., 1995, 95, 1161–1190 CrossRef CAS;
(h) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2078581. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03958j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Burkhard
Endeward
b,
Burkhard
Endeward