
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
‘Reverse biomimetic’ synthesis of L-arogenate and its stabilized analogues from L-tyrosine†
Louise
Eagling
 a,
Daniel J.
Leonard
a,
Maria
Schwarz
a,
Iñaki
Urruzuno
a,
Grace
Boden
a,
J. Steven
Wailes
b,
John W.
Ward
a and
Jonathan
Clayden
*a
a,
Daniel J.
Leonard
a,
Maria
Schwarz
a,
Iñaki
Urruzuno
a,
Grace
Boden
a,
J. Steven
Wailes
b,
John W.
Ward
a and
Jonathan
Clayden
*a
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol BS8 1TS, UK. E-mail: j.clayden@bristol.ac.uk
bJealott's Hill International Research Centre, Bracknell RG42 6EY, UK
First published on 30th July 2021
Abstract
L-Arogenate (also known as L-pretyrosine) is a primary metabolite on a branch of the shikimate biosynthetic pathway to aromatic amino acids. It plays a key role in the synthesis of plant secondary metabolites including alkaloids and the phenylpropanoids that are the key to carbon fixation. Yet understanding the control of arogenate metabolism has been hampered by its extreme instability and the lack of a versatile synthetic route to arogenate and its analogues. We now report a practical synthesis of L-arogenate in seven steps from O-benzyl L-tyrosine methyl ester in an overall yield of 20%. The synthetic route also delivers the fungal metabolite spiroarogenate, as well as a range of stable saturated and substituted analogues of arogenate. The key step in the synthesis is a carboxylative dearomatization by intramolecular electrophilic capture of tyrosine's phenolic ring using an N-chloroformylimidazolidinone moiety, generating a versatile, functionalizable spirodienone intermediate.
Introduction
The aromatic amino acids L-Tyr and L-Phe are biosynthesized through the shikimate pathway (Scheme 1), in which the non-aromatic precursor prephenate is reductively aminated and aromatized to yield tyrosine and phenylalanine.1–3 Variants of the pathway exist in which either aromatization or reductive amination occurs first. The former passes through phenylpyruvate or its 4-hydroxy derivative, while the latter (discovered by Stenmark in 1974 (ref. 3)) passes through L-arogenate, 1 (also known as pretyrosine) as the final non-aromatic intermediate.4 The shikimate pathway is present in plants (where it may process >30% of photosynthetically fixed carbon), bacteria, fungi, and some other organisms, but is absent from animals, which must obtain aromatic amino acids from their diet. The pathway is therefore an attractive target for development of new herbicides, antimicrobial agents, and live vaccines.5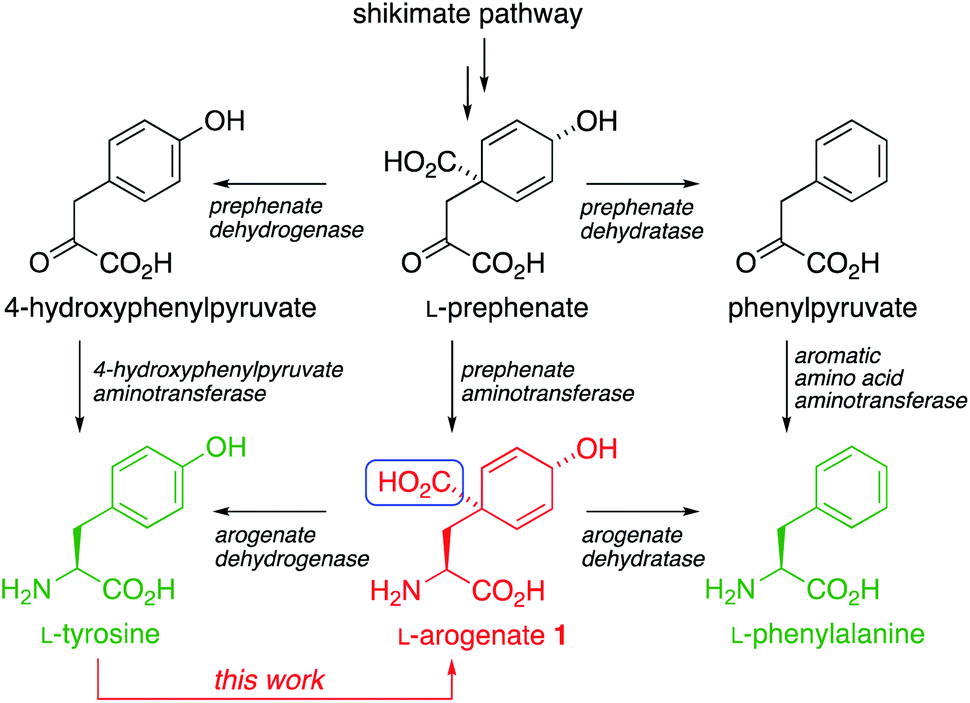 | ||
| Scheme 1 The biosynthesis of L-Tyr and L-Phe, and (in red) our ‘reverse biomimetic’ synthesis of L-arogenate 1, introducing the highlighted carboxyl group by an intramolecular dearomatizing carboxylation. | ||
Two routes to L-arogenate 1 have been reported previously. In one,6 the key spirocyclic cyclohexadienol intermediate was formed from a non-biogenetic precursors by Diels–Alder cycloaddition. Conversion of the adduct into L-arogenate was confirmed by NMR, but the final product was not isolated. In the other,7 Birch reduction and Michael addition gave a dearomatised intermediate that was resolved and converted into a sample of arogenate. We now report the first asymmetric synthesis of an isolated sample of L-arogenate by a direct and practical route, using arogenate's biosynthetic product L-tyrosine as a starting material. We characterise this strategy as a ‘reverse biomimetic’ synthesis. The synthesis is rapid, efficient, scalable, and highly amenable to the synthesis of more stable arogenate analogues.
Our synthetic route employs the dearomatizing cyclization8 of tyrosine's phenolic ring as an atom-economic way to install the spirocyclohexadienol ring of the target. Dearomatising cyclisations – whether by nucleophilic, electrophilic, or radical methods – allow stereochemical complexity to be built from a simple planar precursor, and have been used in the syntheses of a number of natural products.9 Dearomatizing spirocyclisations of phenols have been widely exploited, yet remarkably few of these employ tyrosine, and all of them are oxidative. These examples produced spirolactones,10 spirolactams11 and a spirodienone carboxylic acid precursor to the natural product discorhabdin.12
Results and discussion
The biosynthetic decarboxylative aromatization that converts arogenate to tyrosine – a reaction we sought to run in reverse – involves the formal elimination of a one-carbon unit (boxed in blue in Scheme 1) at the formic acid oxidation level. In the course of an exploration of a Bischler–Napieralski type cyclization of some carbamoyl chloride derivatives of aromatic amino acids,13 which typically gives isoquinolinones without dearomatisation, we discovered reactivity unique to L-tyrosine. We found that tyrosine's imidazolidinone derivative 6 (Scheme 2) underwent a dearomatizing carbonylation that allowed this one-carbon unit could be reintroduced exactly as required for a possible synthesis of arogenate. | ||
| Scheme 2 Synthesis of L-arogenate 1 by dearomatizing cyclization of the N-chloroformylimidazolidinone derivative 6 of L-Tyr. Reagents and conditions: (a) MeNH2, 24 h; (b) t-BuCHO, MgSO4, 16 h; (c) COCl2, pyridine, THF, rt, 3h; (d) H2, Pd/C, THF, rt, 4 h; (e) Et3N, MeCN, 150 °C (mw), 10 min; (f) NaBH4, CeCl3·7H2O, MeOH, −78 °C, 30 min; (g) Ba(OH)2, H2O, 30 °C, 18 h; (h) Ba(OH)2, BaCO3, H2O, 80 °C, 5 h; (i) Reported conditions: NaOH, Na2CO3, EtOH, 70 °C, 20 h.6 | ||
The trans diastereoisomer of carbamoyl chloride 5 was synthesised selectively from O-benzyl L-tyrosine methyl ester 2 by formation of the methyl amide 3, condensation with pivalaldehyde, and phosgene-induced cyclization of the imine 4 under kinetic control.14,15 Heating benzyl ether 5 induces cyclization to an isoquinolinone ring,13 but quite different reactivity is observed with the free phenol 6 (X-ray crystal structure, confirming relative stereochemistry, shown in Fig. 1a) that was formed cleanly by hydrogenolysis of 5 in THF. These conditions preserved the (surprisingly stable15,16) carbamoyl chloride group.17
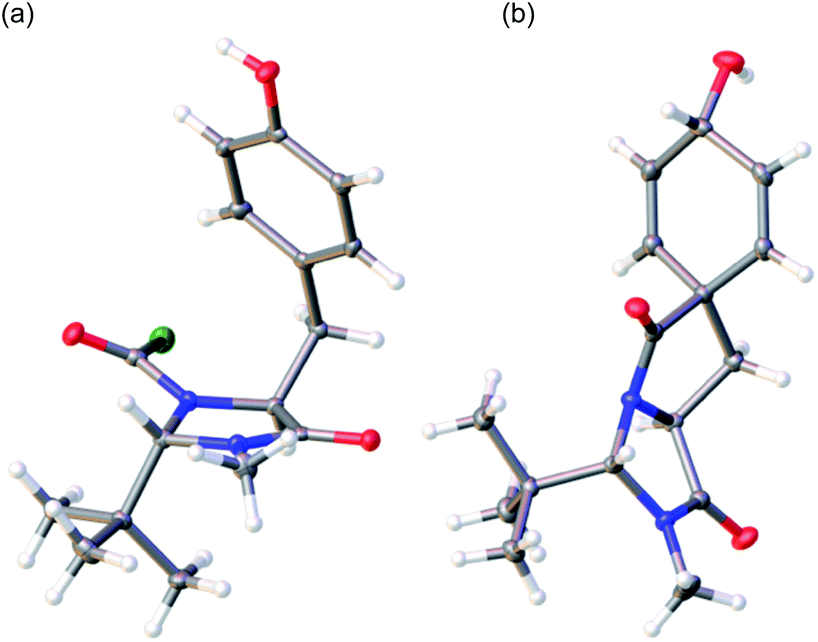 | ||
| Fig. 1 (a) X-ray crystal structure of 6; (b) X-ray crystal structure of minor diastereoisomer 8b.21 | ||
We found that heating 6 to 60 °C for 72 h in the presence of Et3N and KI induced dearomatizing spirocyclisation to the dienone 7, a compound with clear structural homology to arogenate 1, in 82% yield. Optimization of this cyclization (see ESI†) showed that microwave irradiation with Et3N in acetonitrile for 10 min at 150 °C gave 7 cleanly in quantitative yield from 6 without the need for chromatographic purification. Spirocycle 7 was fully bench stable over periods of months, but exposure to acid or base (including attempts to hydrolyse its lactam rings) led to decomposition products arising principally from rearomatization.
This spirocyclization contrasts with related electrophilic intramolecular acylations of non-phenolic compounds, which yield isoquinolinones13 without dearomatisation. There are no other reported acylative dearomatisations of phenols.18
To generate the dienol structure characteristic of arogenate, the spirocyclic dienone 7 was reduced cleanly under Luche conditions19 at −78 °C to a separable 3.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomeric alcohols 8a and 8b, from which 8a was isolated in 72% yield. The minor diastereoisomer 8b was crystalline, and an X-ray crystal structure (Fig. 1b) allowed us to assign the relative configurations of 8a and 8b. In the major product 8a, formed by approach of the reducing agent anti to the more hindered carbonyl and tert-butyl group, the hydroxyl group is orientated syn to the carbonyl group, as is also the case in arogenate and prephenate.
:1 mixture of diastereoisomeric alcohols 8a and 8b, from which 8a was isolated in 72% yield. The minor diastereoisomer 8b was crystalline, and an X-ray crystal structure (Fig. 1b) allowed us to assign the relative configurations of 8a and 8b. In the major product 8a, formed by approach of the reducing agent anti to the more hindered carbonyl and tert-butyl group, the hydroxyl group is orientated syn to the carbonyl group, as is also the case in arogenate and prephenate.
Methods were sought for the conversion of 8a to arogenate 1 by hydrolysis of the imidazolidinone and lactam rings. Heating with acid led to rearomatization to products that included dihydroisoquinolone 10 and phenylalanine. Likewise, most bases promoted rearomatization to mixtures of phenylalanine- and tyrosine-derived products. Barium hydroxide gave the most encouraging results: treatment with 12 equiv. aqueous Ba(OH)2 for 18 h at 30 °C opened the imidazolidinone ring to yield a sample containing >50% by 1H NMR of the partially hydrolysed lactam 9, which is the natural product spiroarogenate.20
The purification of 9 and its separation from inorganic salts proved problematic. The ion-exchange chromatography reported6,7 for the purification of spiroarogenate and arogenate was ineffective, as a consequence of the dilution required by the barium salts. In acidic solution, 9 underwent rearomatizing fragmentation (with a half-life measured in hours at pH 5: see ESI†) to phenylalanine, preventing the use of standard reversed-phase chromatography. HPLC using hydrophilic interaction liquid chromatography (HILIC) columns was successful at basic pH, but separations were poor. Eventually, successful results were obtained using a porous graphitic carbon stationary phase in the form of a Hypercarb™ column, which successfully removed more retained aromatic impurities giving a 27% isolated yield of air-stable spiroarogenate 9. The 1H NMR spectrum of 9 ([α]23D = −40) matched that of spiroarogenate isolated from Neurospora crassa (Fig. 2).20
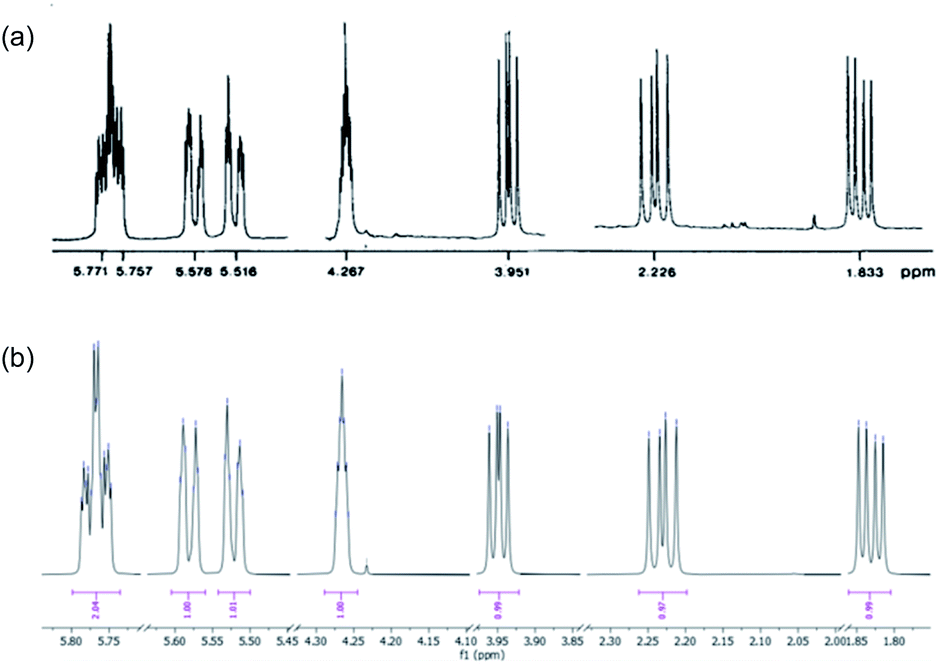 | ||
| Fig. 2 1H NMR spectrum of spiroarogenate 9 (a) isolated from Neurospora crassa (400 MHz in D2O);19 (b) synthesized by the method of Scheme 2 (400 MHz in D2O). | ||
Danishefsky et al. reported the hydrolysis of 9 to 1 using NaOH and Na2CO3,6 and we were able to form traces of arogenate from 9 by this method. However, purification of 9 using a gradient of 0–80% acetonitrile in 100 mM ammonium bicarbonate buffer revealed that the conditions that formed spiroarogenate 9 from 8a also formed a trace of arogenate 1, along with another by-product, the imidazolidinone 11. Raising the temperature of the hydrolysis of 8a with Ba(OH)2 to 70 °C for 5 hours increased the yield of arogenate to 55% (by 1H NMR). The free acid of arogenate 1 was even more unstable than spiroarogenate 9, decomposing instantly by rearomatization to phenylalanine. Use of a Na2CO3/NaHCO3 buffer (10 mM at pH 9.2) in place of the ammonium bicarbonate buffer solved this problem, with material produced by aqueous hydrolysis of 8a at 80 °C with Ba(OH)2 (3 equiv.) and BaCO3 (1 equiv.)22 giving a sample of the stable disodium salt of arogenate, 1-Na in 41% yield, calculated by quantitative NMR.
The extreme instability of arogenate towards aromatization prompted us to explore modifications to the route that might provide more stable analogues with potential future utility as probes or modifiers of the function of arogenate-metabolising enzymes. Two alternative approaches, both starting from the spirodienone 7, are shown in Scheme 3. Hydrogenation of 7 gave the cyclohexanone 12, which was reduced with remarkably high diastereoselectivity (>95:5) to alcohol 13a in 93% yield using K-selectride, or less selectively to a separable 47:53 mixture of 13a and its crystalline alcohol epimer 13b with NaBH4. The X-ray crystal structure of 13b (Fig. 3a) confirmed the relative stereochemistry of this pair of diastereoisomers. Heating 13a with 3 eq. NaOH in H2O at 90 °C gave tetrahydrospiroarogenate 14 in a yield of 80%. Modification of the hydrolysis conditions to 12 equiv. Ba(OH)2 and 1 eq. BaCO3 in H2O at 80 °C led directly to tetrahydroarogenate 15, which was isolated after reversed-phase column chromatography as its TFA salt in 75% yield. Importantly, no elimination or other decomposition products were observed from these saturated analogues, giving them potential value in future biological studies.23
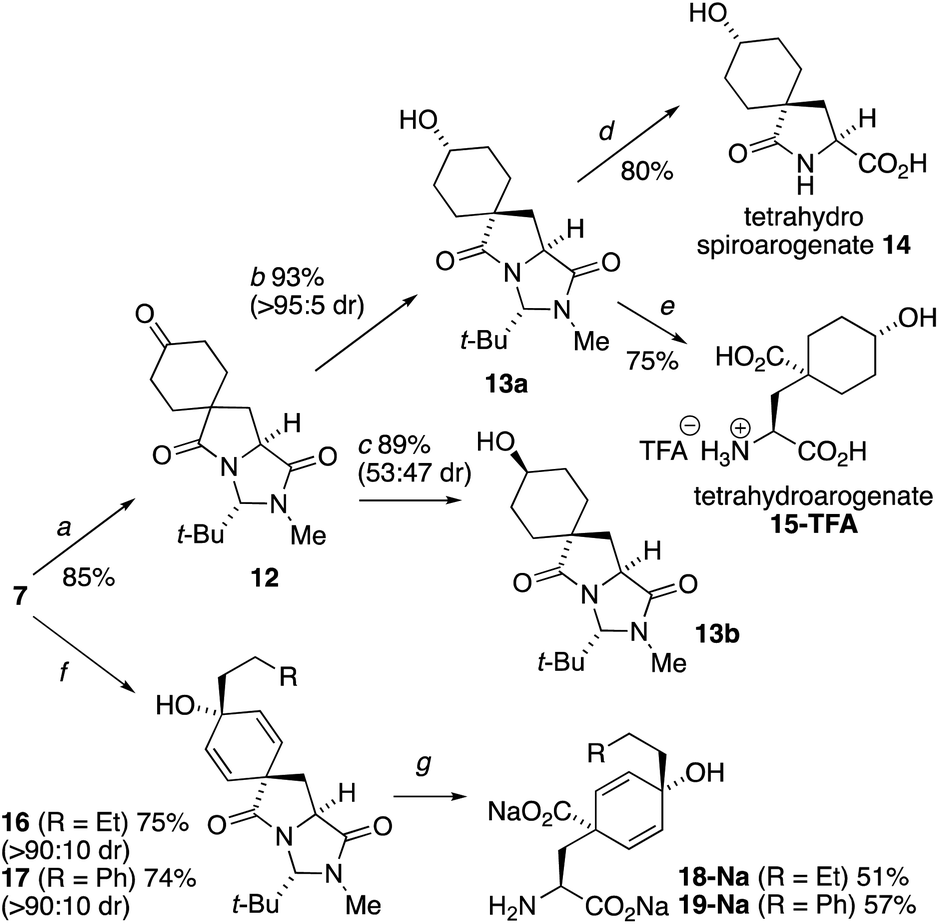 | ||
| Scheme 3 Arogenate analogues stabilized towards aromatization. Reagents and conditions: (a) H2, Pd/C, THF, rt, 16 h; (b) K-selectride (1.6 equiv.), THF, −78 °C, 1 h; (c) NaBH4 (3 equiv.), MeOH, 0 °C – rt, 18 h; (d) NaOH, H2O, 90 °C, 16 h; (e) Ba(OH)2, BaCO3, H2O, 80 °C, 20 h; (f) RCH2CH2MgBr, THF, −78 °C – rt, 2 h; (g) NaOH, H2O, EtOH, 70 °C, 20 h. | ||
 | ||
| Fig. 3 (a) X-ray crystal structure of 13b; (b) X-ray crystal structure of 24d. | ||
An alternative approach to stabilized arogenate analogues entailed the addition of an additional alkyl substituent at the 4-position of the dienol ring of 1. A phenethyl moiety was selected with the possibility that analogues of this type may also mimic NADP in the binding pocket of arogenate dehydrogenase (see ESI†).2416 and 17 were made (dr > 9:1) by 1,2-addition of Grignard reagents to the carbonyl group of the dienone 7.25 The major diastereomers 16a and 17a were subjected to hydrolysis conditions. Heating 16a and 17a with 6 eq. NaOH in EtOH:H2O 2:1 at 70 °C gave the alkylated arogenate derivatives, 18 and 19, in 51% and 57% yield.2619 had increased stability in basic conditions but attempted isolation of the free acid forms of these products led to instantaneous rearomatization to their respective phenylalanine derivatives.
Other opportunities to synthesize ring-substituted arogenate derivatives entailed the spirocyclization of starting materials carrying substituted phenolic rings (Scheme 4).27 Starting from the protected form 20 of 3-iodo-L-tyrosine, a Suzuki coupling using methylboronic acid installed the 3-methyl substituent of 21. Conversion of this compound into the chloroformylimidazolidinone 22 followed straightforwardly under the conditions used in Scheme 2. Spirocyclization of 22 was unaffected by the substitution on the ring, and heating with Et3N for 10 min at 150 °C in the microwave gave a 1:1 mixture of the two diastereoisomers of 23. Reduction of these gave four diastereoisomers 24a–d which were separated using standard column chromatography and were characterized through a combination of NOE and X-ray crystallography (the X-ray crystal structure of 24d is shown in Fig. 3b). Hydrolysis using Ba(OH)2 at 40 °C yielded samples of the methylated spiroarogenate and arogenate analogues 25 and 26. The free acid form of 26 showed significantly greater stability towards rearomatization than arogenate itself.
 | ||
| Scheme 4 Synthesis of 3-methylarogenate 26 from 3-iodotyrosine. Reagents and conditions: (a) MeB(OH)2, Cs2CO3, SPhosPdG2, dioxane, 90 °C, 16 h; (b) MeNH2, 24 h; (c) t-BuCHO, MgSO4, 16 h; (d) COCl2, pyridine, THF, RT, 3 h; (e) H2, Pd/C, THF, RT, 4 h; (f) Et3N, MeCN, 150 °C (mw), 10 min; (g) NaBH4, CeCl3·7H2O, MeOH, −78 °C, 30 min; (h) Ba(OH)2, H2O, 40 °C, 16 h. | ||
Conclusions
In summary, we report a synthesis of the primary metabolite arogenate 1 in seven steps and an overall yield of 20%, an order of magnitude improvement on the maximum yield previously published.7 We also report the synthesis of the natural product spiroarogenate 9. Both syntheses proceed through a synthetically versatile dienone intermediate 7, amenable to regiodiverse functionalization. Arogenate and spiroarogenate display significant instability towards rearomatization under acidic or basic conditions, but by modifying the synthetic route to introduce saturation or alkyl substituents we were able to make analogues that are significantly more stable. Additionally, the ability to convert Tyr back to arogenate efficiently opens up the possibility of making isotopically labelled samples of arogenate for use in biosynthetic studies. Exploration of the potential utility of these analogues and isotopomers as tools for the investigation and modification of plant metabolism are under way.Author contributions
LE, DJL, JWW, and JC conceived the project. JC directed the work. JSW provided scientific and practical input and advice. LE, DJL, MS, IU, GB and JWW carried out the experiments. LE, DJL, JWW and JC wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the EPSRC through the Bristol Chemical Synthesis Centre for Doctoral Training (EP/L015366/1), the Bristol Centre for Doctoral Training in Technology-Enhanced Chemical Synthesis (EP/S024107/1), research grant EP/L018527, and an Impact Acceleration Award; by Syngenta through a CASE award (to L. E)., by the ERC through Advanced Grant ROCOCO and Proof of Concept Grant QUATERMAIN, and by the Basque Government through a fellowship (to I. U.). We thank Prof Maxwell Crossley for informative discussions. We acknowledge the preliminary investigations carried out by Greg McHugh and we dedicate this paper to his memory.Notes and references
- J. R. Coggins, Herbic. Plant Metab., 1989, 38, 97–112 CAS.
- H. Maeda and N. Dudareva, Annu. Rev. Plant Biol., 2012, 63, 73–105 CrossRef CAS PubMed.
- (a) S. L. Stenmark, D. L. Pierson, R. A. Jensen and G. I. Glover, Nature, 1974, 247(5439), 290–292 CrossRef CAS PubMed; (b) L. O. Zamir, R. A. Jensen, B. H. Arison, A. W. Douglas, G. Albers-Schönberg and J. R. Bowen, J. Am. Chem. Soc., 1980, 102, 4499–4504 CrossRef CAS.
- A. M. Fazel, J. R. Bowen and R. A. Jensen, Proc. Natl. Acad. Sci. U. S. A., 1980, 77(3), 1270–1273 CrossRef CAS PubMed.
- (a) A. D. Baylis, Pest Manage. Sci., 2000, 56(4), 299–308 CrossRef CAS; (b) P. Rippert and M. Matringe, Eur. J. Biochem., 2002, 269(19), 4753–4761 CrossRef CAS PubMed.
- S. Danishefsky, J. Morris and L. A. Clizbe, J. Am. Chem. Soc., 1981, 103(6), 1602–1604 CrossRef CAS.
- M. J. Crossley and R. C. J. Reid, J. Chem. Soc., Chem. Commun., 1994, 2237 RSC.
- (a) J. Clayden and M. N. Kenworthy, Synthesis, 2004, 1721–1736 CrossRef CAS; (b) H. Brice, J. Clayden and S. D. Hamilton, Beilstein J. Org. Chem., 2010, 6, 22 Search PubMed; (c) J. Clayden, C. J. Menet and D. J. Mansfield, Org. Lett., 2000, 2(26), 4229–4232 CrossRef CAS PubMed; (d) J. Clayden and M. N. Kenworthy, Org. Lett., 2002, 4(5), 787–790 CrossRef CAS PubMed.
- (a) S. P. Roche and J. A. Porco, Angew. Chem., Int. Ed., 2011, 50(18), 4068–4093 CrossRef CAS PubMed; (b) L. Pouységu, D. Deffieux and S. Quideau, Tetrahedron, 2010, 66(13), 5908–5917 CrossRef; (c) G. Lemière, S. Sedehizadeh, J. Toueg, N. Fleary-Roberts and J. Clayden, Chem. Commun., 2011, 47(13), 3745–3747 RSC; (d) J. Clayden, C. J. Menet and K. Tchabanenko, Tetrahedron, 2002, 58(23), 4727–4733 CrossRef CAS; (e) J. Clayden, M. N. Kenworthy and M. Helliwell, Org. Lett., 2003, 5(6), 831–834 CrossRef CAS PubMed; (f) A. Ahmed, R. A. Bragg, J. Clayden and K. Tchabanenko, Tetrahedron Lett., 2001, 42(20), 3407–3410 CrossRef CAS; (g) J. Clayden, F. E. Knowles and I. R. Baldwin, J. Am. Chem. Soc., 2005, 127, 2412–2413 CrossRef CAS PubMed.
- S. Ficht, M. Mülbaier and A. Giannis, Tetrahedron, 2001, 57(23), 4863–4866 CrossRef CAS.
- N. A. Braun, M. A. Ciufolini, K. Peters and E.-M. Peter, Tetrahedron Lett., 1998, 39(26), 4667–4670 CrossRef CAS.
- H. Tohma, Y. Harayama, M. Hashizume, M. Iwata, M. Egi and Y. Kita, Angew. Chem., Int. Ed., 2002, 41(2), 348 CrossRef CAS PubMed.
- M. M. Amer, A. C. Carrasco, D. J. Leonard, J. W. Ward and J. Clayden, Org. Lett., 2018, 20(24), 7977–7981 CrossRef CAS PubMed.
- R. Naef and D. Seebach, Helv. Chim. Acta, 1985, 68, 135–143 CrossRef CAS.
- M. M. Amer, H. Abas, D. J. Leonard, J. W. Ward and J. Clayden, J. Org. Chem., 2019, 84(11), 7199–7206 CrossRef CAS PubMed.
- H. Abas, M. M. Amer, O. Olaizola and J. Clayden, Org. Lett., 2019, 21(6), 1908–1911 CrossRef CAS PubMed.
- Under these conditions no Rosenmund reduction to the formamide was observed..
- Very few examples of dearomatizing reactions of phenols with non-oxidising electrophiles are known: for an example, see A. Venkatram, T. Colley, J. DeRuiter and F. Smith, J. Heterocycl. Chem., 2005, 42, 297–301 CrossRef CAS.
- A. L. Gemal and J.-L. Luche, J. Am. Chem. Soc., 1981, 103(18), 5454–5459 CrossRef CAS.
- L. O. Zamir, R. Tiberio, E. Jung and R. A. Jensen, J. Biol. Chem., 1983, 258, 6486–6491 CrossRef CAS.
- X-ray crystal structures have been deposited with the CCDC: Deposition numbers 6: 2070018, 8b: 2070019, 13b: 2070023, 24d: 2070024..
- Previous work (ref. 6) had noted that addition of carbonate salts accelerated the hydrolysis of 9..
- We observed slow lactonization of 15 on standing in solution..
- P. Legrand, R. Dumas, M. Seux, P. Rippert, R. Ravelli, J.-L. Ferrer and M. Matringe, Structure, 2006, 14, 767–776 CrossRef CAS PubMed.
- The stereochemistry of the major diastereoisomers of 16 and 17 was assigned by analogy to the major diastereoisomer 8a.
- About 20% of the equivalent spiroarogenate analogue was also isolated from these hydrolyses, but no aromatized material was evident in the crude reaction product..
- Further exploration of possible routes to arogenate analogues is outlined in the ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2070018, 2070019, 2070023 and 2070024. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03554a |
| This journal is © The Royal Society of Chemistry 2021 |