
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Combining palladium and ammonium halide catalysts for Morita–Baylis–Hillman carbonates of methyl vinyl ketone: from 1,4-carbodipoles to ion pairs†
Yang
Yang
a,
Bo
Zhu
a,
Lei
Zhu
 b,
Ying
Jiang
a,
Chun-Ling
Guo
b,
Jing
Gu
b,
Qin
Ouyang
*b,
Wei
Du
*a and
Ying-Chun
Chen
*ab
b,
Ying
Jiang
a,
Chun-Ling
Guo
b,
Jing
Gu
b,
Qin
Ouyang
*b,
Wei
Du
*a and
Ying-Chun
Chen
*ab
aKey Laboratory of Drug-Targeting and Drug Delivery System of the Ministry of Education, West China School of Pharmacy, and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu 610041, China. E-mail: duweiyb@scu.edu.cn; ycchen@scu.edu.cn; Fax: +86 28 85502609
bCollege of Pharmacy, Third Military of Medical University, Chongqing 400038, China. E-mail: ouyangq@tmmu.edu.cn
First published on 22nd July 2021
Abstract
Here we report that Morita–Baylis–Hillman carbonates from diverse aldehydes and methyl vinyl ketones can be directly utilised as palladium-trimethylenemethane 1,4-carbodipole-type precursors, and both reactivity and enantioselectivity are finely regulated by adding a chiral ammonium halide as the ion-pair catalyst. The newly assembled intermediates, proposed to contain an electronically neutral π-allylpalladium halide complex and a reactive compact ion pair, efficiently undergo asymmetric [4 + 2] annulations with diverse activated alkenes or isatins, generally with high regio-, diastereo- and enantio-selectivity, and even switchable regiodivergent or diastereodivergent annulations can be well realised by tuning the substrate or catalyst assemblies. An array of control experiments, including UV/Vis absorption study and density functional theory calculations, are conducted to rationalise this new double activation mode combining a palladium complex and an ammonium halide as an ion-pair catalyst.
Introduction
With the development of transition metal catalysis, zwitterionic species possessing a π-allylmetal complex moiety, which could be generated from allylic alcohol derivatives in situ, have been extensively employed for the construction of enantioenriched cyclic frameworks.1 The corresponding hetero 1,n-dipoles can be readily generated from various allylic alcohol-derived carbonate, carbamate, or ester derivatives.2 In addition, all-carbon-based dipoles have also been well explored,3 and those having a palladium-trimethylenemethane (Pd-TMM) motif received special attention.4 While fruitful scaffolds have been constructed by using Pd-TMM 1,3-carbodipoles,5 1,4-carbodiploes from allylic carbonates, generally with two electron-withdrawing groups at the homoallylic position, could be efficiently utilised (Scheme 1a).6 However, despite the significant progress in this field, the substitution patterns of the TMM moiety are not expandable, and the 1,4-carbodipole precursors are generally limited to those possessing a gem-diactivated carbon centre to meet the required C–H acidity for deprotonation and stabilisation for anion intermediates. In addition, the stereocontrol for the related annulations usually relies on the selection of properly designed chiral P-centred ligands, generally with crowded architectures. Moreover, for more flexible 1,4-dipoles, a Pd(II)-coordination site (including carbonyl, ester, and pyridine) embedding in the substrates is necessary for achieving high levels of enantioselectivity, by forming six- or seven-membered Pd(II)-ligated intermediates (Scheme 1a). As a result, the development of an alternative catalytic system, rather than a sole Pd-based chiral complex, would be desirable for the reactions of some challenging substrates. | ||
| Scheme 1 Diverse catalytic strategies involved in various Pd-based dipole species. | ||
It has been documented that adding nBu4Cl (TBAC) could significantly enhance the [3 + 2] annulation reaction of Pd(II)-based 1,3-C,N-dipoles and activated alkenes, by possibly forming anionic Pd complexes (Scheme 1b, left).7 Ooi even designed a chiral BINOL-derived ammonium–phosphine hybrid ligand, which enabled fine stereocontrol in the construction of vicinal quaternary stereogenic centres in the annulation reaction of similar dipoles from oxazolidinones, via proposed doubly ion-pairing intermediates (Scheme 1b, right).8 Nevertheless, combining the double activation catalysis of ammonium halides with Pd-TMM carbodipole chemistry and deeply understanding the relevant activation mechanism still remain to be elucidated.
Morita–Baylis–Hillman (MBH) adducts, easily accessible by condensation between activated alkenes and carbonyl compounds, have been explored in asymmetric allylic alkylation reactions by forming π-allylpalladium intermediates.9 Typical MBH adducts from β-keto esters have been recently employed as Pd-TMM-type 1,4-carbodipole precursors in asymmetric [4 + 2] annulations via the deprotonation strategy;6e nevertheless, the common analogues derived from simple enones, such as methyl vinyl ketone (MVK), and with broader substitution patterns (Scheme 1c), are much more challenging for similar transformations, and no success has been achieved yet. It was proposed that the corresponding intermediates, either 1,4-carbodipoles I or Pd-ligated five-membered species II10 after the oxidative addition/deprotonation process, might encounter several disadvantages, such as low stability or poor reactivity; especially, the stereocontrol would be quite challenging because of the flexibility of the skeleton together with the long distance between the nucleophilic site and the chiral ligand. Therefore, we envisaged that the addition of ammonium halide salts, as ion-pair catalysts (IPCs),11 would result in the formation of ion-pairing intermediates III from unstable 1,4-carbodipole species I, which would be potentially helpful for the subsequent reaction: (1) the cationic π-allylpalladium moiety would be neutralised by the halide of the IPC, especially when Cl− or Br− is used as a strong coordination ligand;12 (2) the formation of a compact chiral ion-pair would not only stabilise the labile enolate but also guarantee better reactivity and enantioselectivity in the initial key addition process; (3) the assembly and application of an individual chiral IPC and chiral ligand would be easily tunable, thus providing better synergistic stereocontrol and potentially realising diastereodivergent synthesis in the annulations; (4) such a double activation catalytic strategy would be different from the previously reported cooperative catalytic mode combining palladium and phase-transfer catalysts (Scheme 1c).13
Results and discussion
Condition optimization
Based on the above considerations, we conducted the reaction of the MBH carbonate 1a condensed from MVK and benzaldehyde with 2-benzylidene-1H-indene-1,3(2H)-dione 2a under the catalysis of palladium. After extensive screening, it was found that the expected [4 + 2] spirocyclic product143a with an exo-methylene group could be efficiently obtained with exclusive regio- and diastereo-selectivity, by employing Pd(OAc)2 and a bisphosphine ligand, such as BINAP L1 or SKP L2,9c suggesting that 1,4-carbodipole I could be generated.6e Unfortunately, very low enantiocontrol was observed, probably due to the remote reaction site (Table 1, entries 1 and 2). Phosphoramidite (R)-L3 was tested as well, but failed to afford any product (entry 3). We also tried the bifunctional ligand L4 bearing an ammonium salt,8a without success either (entry 4). In contrast, when cinchonine-derived ammonium salt C1 was added into the previously inert palladium/achiral phosphoramidite L5 catalytic system, high reactivity, albeit with low enantiocontrol, was observed, suggesting the potential formation of a reactive ion-pairing intermediate (entry 5). Chiral salts had apparent effects on both reactivity and enantioselectivity (entries 6–10), and C4 with a bulky substituent gave a higher ee value (entry 8).15 It should be noted that no reaction occurred without the phosphoramidite ligand even in the presence of C4 (entry 11). In contrast, the combination of a bisphosphine ligand L1 or L2 and C4 proved to be unsuccessful (entries 12 and 13). These results indicated that adding an ammonium halide would apparently change the previously formed Pd(II) species of 1a.10 Subsequently, a few chiral phosphoramidites were explored with chiral IPC C4. As a mismatch was observed for ligand (R)-L3 (entry 14), the enantioselectivity was slightly improved with (S)-L3 (entry 15). Moreover, better enantiocontrol was achieved by changing the N-substituent of the phosphoramidite to a bulkier one (entry 16), and the optimal results were obtained with L7 (entry 17). Meanwhile, the significance of IPC was further confirmed, since no reaction occurred without C4 (entry 18). Pleasingly, the reactivity and stereoselectivity were almost retained with lower loadings of the dipole precursor, chiral ligand and IPC (entry 19).10|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | C | Time (h) | Yieldb (%) | eec (%) |
| a Unless otherwise noted, reactions were carried out with MBH carbonate 1a (0.075 mmol), acceptor 2a (0.05 mmol), Pd(OAc)2 (5 mol%), L (10 mol%) and C (10 mol%) in toluene (1.0 mL) under Ar. b Isolated yield. c Determined by HPLC analysis on a chiral stationary phase. d With 1a (0.06 mmol), L7 (7.5 mol%) and C1 (7.5 mol%). | |||||
| 1 | L1 | — | 24 | 86 | −3 |
| 2 | L2 | — | 24 | 77 | 6 |
| 3 | (R)-L3 | — | 24 | NR | — |
| 4 | L4 | — | 24 | NR | — |
| 5 | L5 | C1 | 39 | 95 | −2 |
| 6 | L5 | C2 | 39 | 27 | −9 |
| 7 | L5 | C3 | 39 | 74 | 39 |
| 8 | L5 | C4 | 18 | 70 | 81 |
| 9 | L5 | C5 | 39 | 87 | 50 |
| 10 | L5 | C6 | 39 | <10 | −17 |
| 11 | — | C4 | 24 | NR | — |
| 12 | L1 | C4 | 24 | NR | — |
| 13 | L2 | C4 | 24 | <5 | — |
| 14 | (R)-L3 | C4 | 6 | 46 | 70 |
| 15 | (S)-L3 | C4 | 6 | 63 | 86 |
| 16 | L6 | C4 | 22 | 63 | 91 |
| 17 | L7 | C4 | 36 | 86 | 93 |
| 18 | L7 | — | 36 | NR | — |
| 19d | L7 | C4 | 36 | 83 | 92 |

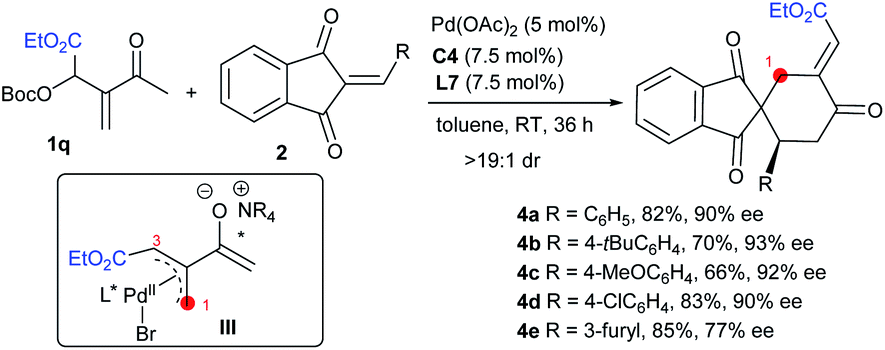
Substrate scope of regioselective [4 + 2] annulations
With the optimal conditions in hand, we next explored the substrate scope of the asymmetric [4 + 2] annulations of MBH carbonates 1 and 2-alkylidene indene-1,3-diones 2 under the double activation catalysis of Pd(OAc)2, ligand L7 and IPC C4. The results are summarised in Table 2. For MBH carbonates 1 derived from different aromatic aldehydes or cinnamaldehyde and MVK, good reactivity was observed even with 2.5 mol% loadings of a palladium catalyst in some cases, and the corresponding products 3a–3p were smoothly produced in high yields with excellent stereoselectivity (Table 2, entries 1–16). It should be noted that the halide substituents on the aryl ring could be well-tolerated. When acceptors 2 with a different aryl group were employed, similarly good results were generally obtained (entries 17–25). An alkyl-substituted substrate was also applicable albeit with moderate enantioselectivity (entry 26). In addition, product 3aa with an additional stereogenic centre was afforded with good diastereoselectivity from a perillaldehyde-derived enone (entry 27).16 In some cases, using Pd(OAc)2 showed low reactivity, and thus other palladium sources were applied for better results (entries 17, 26 and 27).|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) | eec (%) |
a Unless otherwise noted, reactions were carried out with MBH carbonate 1 (0.12 mmol), alkene 2 (0.10 mmol), Pd(OAc)2 (5 mol%), C4 (7.5 mol%) and L7 (7.5 mol%) in toluene (1.0 mL) at rt for 36 h under Ar.
b Isolated yields.
c Determined by HPLC analysis on a chiral stationary phase; generally dr > 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by 1H NMR analysis.
d For 72 h.
e With Pd(OAc)2 (2.5 mol%), C4 (3.75 mol%) and L7 (3.75 mol%) in toluene (1.0 mL).
f With Pd2(dba)3 (2.5 mol%).
g The absolute configuration of enantiopure 3s was determined by X-ray analysis. The other products were assigned by analogy.
h With PdCp(η3-allyl) (5 mol%).
i Dr = 15:1.
j Dr = 8:1. :1 by 1H NMR analysis.
d For 72 h.
e With Pd(OAc)2 (2.5 mol%), C4 (3.75 mol%) and L7 (3.75 mol%) in toluene (1.0 mL).
f With Pd2(dba)3 (2.5 mol%).
g The absolute configuration of enantiopure 3s was determined by X-ray analysis. The other products were assigned by analogy.
h With PdCp(η3-allyl) (5 mol%).
i Dr = 15:1.
j Dr = 8:1.
|
||||
| 1 | Ph | Ph | 3a, 83 | 92 |
| 2d | 3-CH3C6H4 | Ph | 3b, 76 | 93 |
| 3d | 4-CH3C6H4 | Ph | 3c, 78 | 93 |
| 4 | 4-PhC6H4 | Ph | 3d, 80 | 92 |
| 5 | 4-FC6H4 | Ph | 3e, 93 | 94 |
| 6e | 3,4-Cl2C6H3 | Ph | 3f, 70 | 93 |
| 7d | 2-BrC6H4 | Ph | 3g, 94 | 92 |
| 8 | 3-BrC6H4 | Ph | 3h, 90 | 92 |
| 9e | 4-BrC6H4 | Ph | 3i, 89 | 89 |
| 10 | 2-NO2C6H4 | Ph | 3j, 80 | 88 |
| 11 | 4-NO2C6H4 | Ph | 3k, 98 | 95 |
| 12 | 4-CNC6H4 | Ph | 3l, 83 | 95 |
| 13 | 2-Naphthyl | Ph | 3m, 84 | 92 |
| 14e | 2-Furyl | Ph | 3n, 81 | 90 |
| 15e | 3-Furyl | Ph | 3o, 88 | 91 |
| 16 | 2-Styryl | Ph | 3p, 75 | 92 |
| 17f | Ph | 2-CH3C6H4 | 3q, 96 | 94 |
| 18e | Ph | 4-CH3C6H4 | 3r, 90 | 93 |
| 19 | Ph | 4-tBuC6H4 | 3s, 90 | 93g |
| 20 | Ph | 3-MeOC6H4 | 3t, 82 | 96 |
| 21 | Ph | 2-ClC6H4 | 3u, 56 | 81 |
| 22 | Ph | 4-FC6H4 | 3v, 80 | 92 |
| 23 | Ph | 4-BrC6H4 | 3w, 77 | 95 |
| 24 | Ph | 3-NO2C6H4 | 3x, 61 | 92 |
| 25 | Ph | 3-Furyl | 3y, 93 | 85 |
| 26h | Ph | Cyclopropanyl | 3z, 96 | 61i |
| 27h | Ph |
|
3aa, 73 | —j |
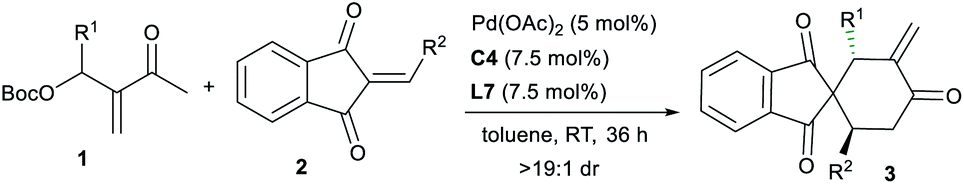
Remarkably, the MBH carbonate 1q derived from ethyl glyoxalate and MVK was compatible as well in the reactions with alkenes 2 under the standard conditions, and other types of [4 + 2] products 4a–4e with different regioselectivity were furnished, by switching the allylic alkylation step from C3 to C1 (Scheme 2, intermediate III), and probably the electronic effect of the substituents played a key role in the regioselectivity.10
 | ||
| Scheme 2 Regiodivergent [4 + 2] annulations between MBH carbonate 1q and alkenes 2. | ||
Substrate scope of asymmetric diastereodivergent [4 + 2] annulations
It is interesting but challenging to switch the diastereoselectivity of the reactions by simply tuning the conditions, as chiral compound libraries with higher stereodiversity could be efficiently obtained from identical starting materials.17 Considering that two independent chiral sources were involved in the current catalytic system, we assumed that the diastereodivergent [4 + 2] annulations of MBH carbonates 1 might be realised. Although we failed to construct different diastereomers with 2-alkylidene indene-1,3-diones 2, 3-vinyl oxindoles 5 possessing an ester or a benzoyl group could successfully act as the counterparts in the switchable diastereodivergent [4 + 2] annulations. Under the catalysis of Pd(OAc)2, achiral ligand L8 and IPC C4, the major diastereomers 6a–6g, having three contiguous stereogenic centres, were generally separated directly in moderate yields with excellent enantioselectivity, as summarised in Scheme 3, though fair to moderate diastereocontrol was observed.10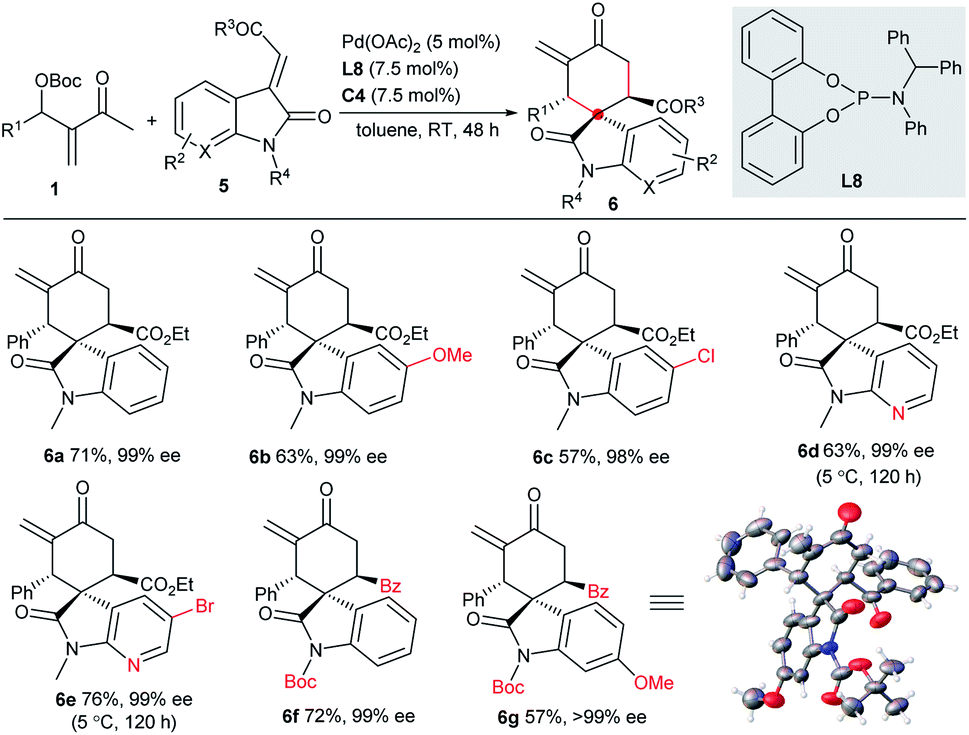 | ||
| Scheme 3 Asymmetric diastereodivergent [4 + 2] annulations between MBH carbonates 1 and 3-vinyl oxindoles 5. | ||
In contrast, the assembly of Pd(OAc)2, ligand (R)-L3 and chiral IPC C7 could switch the diastereoselectivity, and afford separable diastereomers 7a–7d in moderate yields and enantioselectivity (Scheme 4, conditions A). Although we could not achieve more satisfactory results by screening diverse catalyst combinations,10 it was found that the substitution pattern of the acceptors also had a dramatic effect on the outcome. Almost enantiopure diastereomers 9a–h were effectively isolated in moderate yields, by employing amide substrates 8 under the catalysis of Pd(OAc)2, ligand (S)-L3 and chiral IPC C4, also demonstrating the flexibility and tunability of the current double activation catalytic system (Scheme 4, conditions B).10
 | ||
| Scheme 4 Asymmetric diastereodivergent [4 + 2] annulation by tuning the catalytic system or substrate. | ||
Substrate scope of asymmetric oxa-[4 + 2] annulations
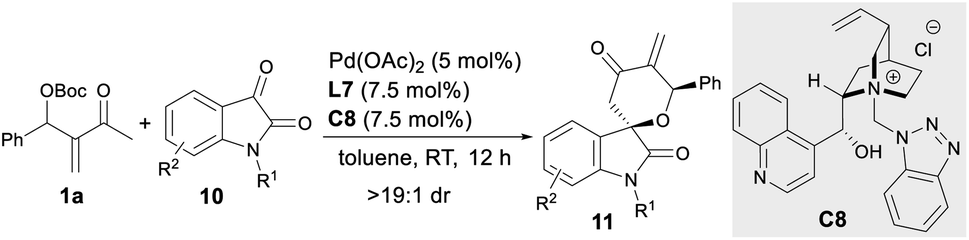
Besides activated alkenes, activated ketones, such as N-protected isatins 10,18 were applicable as two-atom partners in the asymmetric oxa-[4 + 2] annulations with MBH carbonate 1a. As summarised in Table 3, the assembly of Pd(OAc)2, ligand L7 and IPC C8 proved to be a reliable catalytic system, giving spiro[tetrahydropyran-3,3′-oxindole] 11a–11h in moderate to good yields with good to high enantioselectivity (Table 3, entries 1–8), even on a 1.0 mmol scale (entry 5).10|
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) | eec (%) |
|
a Unless otherwise noted, reactions were carried out with MBH carbonate 1 (0.12 mmol), isatin 10 (0.10 mmol), Pd(OAc)2 (5 mol%), C8 (7.5 mol%) and L7 (7.5 mol%) in toluene (1.0 mL) at rt for 12 h under Ar.
b Isolated yields.
c Determined by HPLC analysis on a chiral stationary phase; dr > 19:1 by 1H NMR analysis.
d Data in parentheses were obtained on a 1.0 mmol scale. The absolute configuration of 11e was determined by X-ray analysis after conversion to enantiopure 14 (Scheme 5). The other products were assigned by analogy.
|
||||
| 1 | Me | H | 11a, 62 | 87 |
| 2 | Me | 5-Me | 11b, 72 | 90 |
| 3 | Me | 5-Cl | 11c, 85 | 84 |
| 4 | Me | 6-Br | 11d, 62 | 84 |
| 5 | Me | 7-Me | 11e, 81 (72) | 92d (91) |
| 6 | Me | 7-Cl | 11f, 67 | 80 |
| 7 | Bn | H | 11g, 78 | 80 |
| 8 | Bn | 5-Cl | 11h, 65 | 80 |
Transformations of the [4 + 2] annulation products
The enone moiety in the products enables further transformations to access the products with higher molecular complexity. As illustrated in Scheme 5, the in situ generated 1,3-dipole from N-phenylbenzohydrazonoyl chloride underwent highly diastereoselective [3 + 2] cycloaddition with 3a,19 affording the spirocyclic framework 12 in an excellent yield. In addition, the heterocycle 13 could be efficiently constructed with a retained ee value from 3avia a [4 + 2] annulation with malononitrile. In addition, an interesting dimerisation of 11evia an oxa-Diels–Alder reaction gave polycyclic product 14 with excellent diastereoselectivity and enantioselectivity.20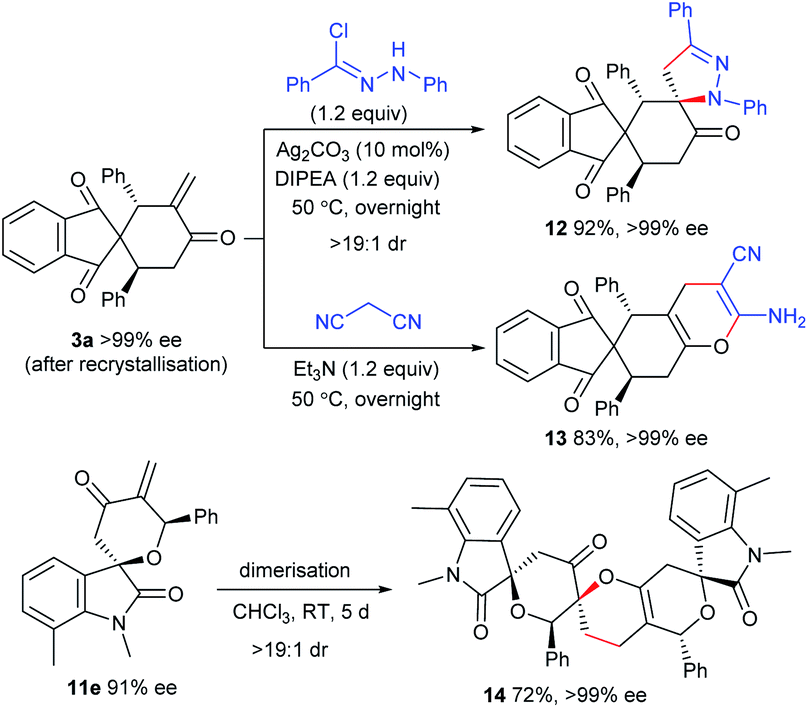 | ||
| Scheme 5 Synthetic transformations of the products. | ||
Mechanism studies
To gain some insight into the catalytic mechanism, several control experiments were further conducted. As summarised in Scheme 6a, the combination of Pd(OAc)2 and achiral phosphoramidite L8 failed to promoted the reaction of 1a and 2a in the absence of an IPC. In contrast, adding either TBAC or TBAB significantly enhanced the desired annulation reaction, whereas the iodide salt (TBAI) was less efficient, probably due to the coordinating effects of different halides.12,21 While we were not able to obtain some valuable information by 1H NMR or mass spectrometry analysis, we carried out UV-Vis absorption experiments to monitor the complexation process.22 As outlined in Scheme 6b, a slightly different absorption spectrum was observed after adding MBH carbonate 1a into the mixture of Pd(OAc)2 and L8. In contrast, apparent absorption changes were detected by further adding diverse ammonium halides. In particular, much stronger absorption in the 320–380 nm area was observed, indicating that some new complex species would be generated from the previously formed dipole I or ligated intermediate II (Scheme 1c) and an ammonium halide.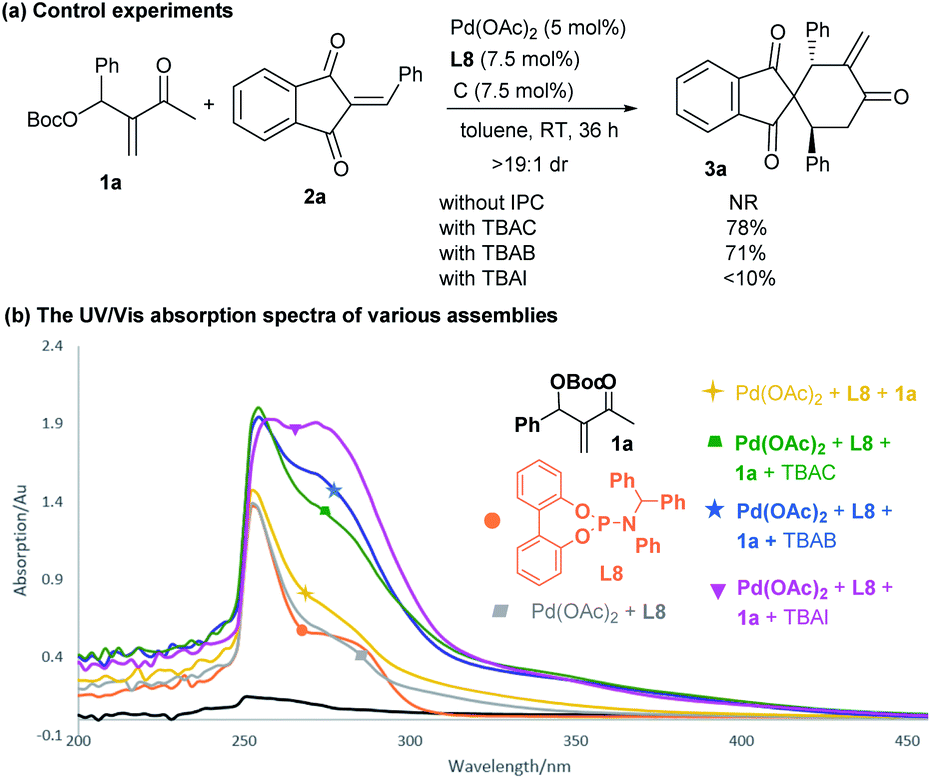 | ||
| Scheme 6 Control experiments with diverse IPCs and UV-Vis absorption analysis. | ||
In order to further rationalise the role of ammonium halides, the mechanism of the catalytic reaction was studied by density functional theory (DFT) calculations. To simplify the process, P(OPh)3 was selected as the ligand, since it gave similar results to those of phosphoramidite L8,10 and simple Me4NBr (TMAB) was employed as the PTC partner. As outlined in Scheme 7, after the oxidative insertion by Pd(0) and deprotonation, the five-membered ligated intermediate DL-INT1-B was found to be the more stable intermediate.10 The reactive 1,4-carbodipole DL-INT1-C had a much higher energy (18.6 kcal mol−1), and the subsequent addition to acceptor 2aviaDL-TS1 had an energy barrier of 25.0 kcal mol−1, which was found to be inert at room temperature (pathway A). In contrast, a ligand exchange of DL-INT1-B would easily occur after adding TMAB, giving intermediate INT1-B1-TMA1 (2.3 kcal mol−1) (pathway B).23 Moreover, after isomerisation to active ion-pair INT1-D1-TMA1 (12.9 kcal mol−1) having an electronically neutral Pd(II) complex, the subsequent Michael addition to 2aviaTS1 had an energy barrier of 20.4 kcal mol−1, which was significantly lower than that of DL-TS1. The resultant INT2 would be easily converted to the ion-pairing intermediate INT3, which would undergo allylic substitution viaTS2 with an energy barrier of 8.8 kcal mol−1, finally affording the intermediate INT4. We also compared the possible formation of a regioisomer viaTS2′ from ion-pair INT3′, which had a higher energy barrier of 10.6 kcal mol−1, also in good accordance with a predicted ratio (21:1). An obvious π–π interaction between the aromatic rings of 1a and 2a was observed in TS2 (Scheme 7), which might be one of the reasons for the reduction of the energy and the formation of regioisomer 3a.10
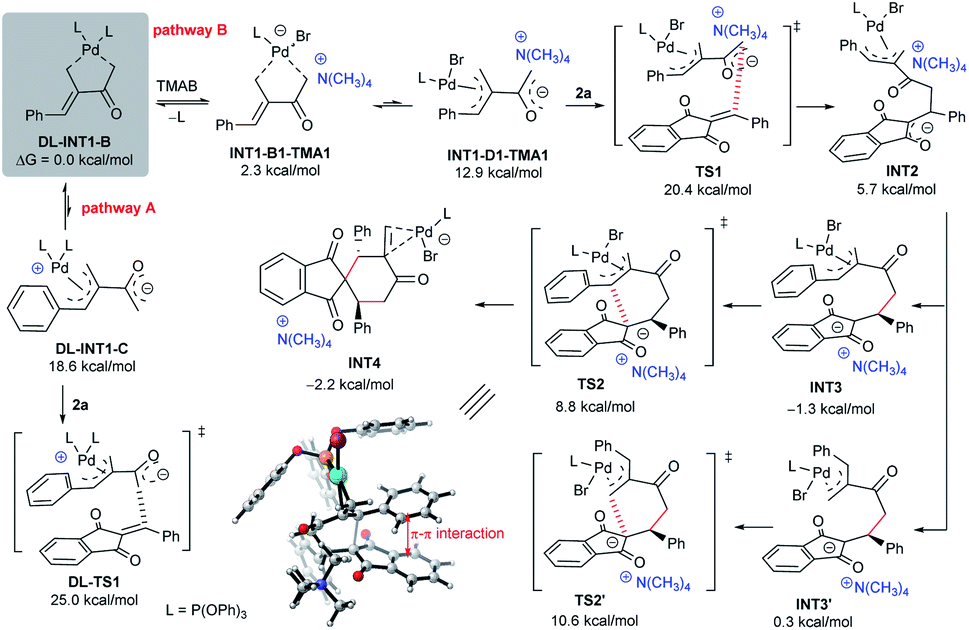 | ||
| Scheme 7 DFT calculations on the proposed catalytic mechanism, at the B3LYP-D3/6-311++G(d,p)//B3LYP-D3/6-31(d) SDD for the Pd (toluene) level. | ||
Conclusions
We have developed a new palladium/ammonium halide double activation catalytic system, which could facilitate the conversion of possible π-allylpalladium complex-based 1,4-carbodipoles from Morita–Baylis–Hillman (MBH) carbonates of vinyl methyl ketone to more reactive compact ion-pair species. Both palladium complex and ammonium halide components were easily tunable, which would be helpful for enhancing reactivity and stereocontrol on both nucleophilic and electrophilic sites of the newly formed intermediates in a cooperative manner. The catalytic efficacy of such a transition metal and organic ion-pair system has been well demonstrated in asymmetric [4 + 2] annulations of substantial MBH carbonates and 2-alkylidene indene-1,3-diones, affording densely functionalised spirocyclic compounds16 in moderate to good yields with excellent stereoselectivity, and even regioselective annulations could be switched by tuning the substitutions of the MBH carbonates. Furthermore, switchable diastereodivergent [4 + 2] annulations between MBH carbonates and 3-olefinic oxindoles have also been realised by modifying the flexible assembly of the palladium complex and ion-pairing agent. The catalytic system has been further utilised in asymmetric oxa-[4 + 2] annulations between MBH carbonates and isatins, affording an array of spiro[tetrahydropyran-3,3′-oxindole] architectures. Moreover, UV-Vis absorption experiments and DFT calculations have been conducted to elucidate the catalytic mechanism, especially for the possible role of ammonium halides. We believe that the flexible double activation system combining a palladium complex and an ion-pair catalyst would find more application in diverse asymmetric reactions.Data availability
All experimental procedures, characterization, computational data, copies of NMR and HRMS spectra for all new compounds and HPLC chromatograms related to this study, can be found in the ESI.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the NSFC (21971166, 21931006, and 21921002) and National Key Research and Development Program of China (2018YFA0507900).Notes and references
- (a) N. De and E. J. Yoo, ACS Catal., 2018, 8, 48–58 CrossRef CAS; (b) B. D. W. Allen, C. P. Lakeland and J. P. A. Harrity, Chem.–Eur. J., 2017, 23, 13830–13857 CrossRef CAS PubMed; (c) T.-R. Li, Y.-N. Wang, W.-J. Xiao and L.-Q. Lu, Tetrahedron Lett., 2018, 59, 1521–1530 CrossRef CAS.
- (a) F. Jiang, F.-R. Yuan, L.-W. Jin, G.-J. Mei and F. Shi, ACS Catal., 2018, 8, 10234–10240 CrossRef CAS; (b) S. Singha, E. Serrano, S. Mondal, C. G. Daniliuc and F. Glorius, Nat. Catal., 2020, 3, 48–54 CrossRef CAS; (c) T.-R. Li, F. Tan, L.-Q. Lu, Y. Wei, Y.-N. Wang, Y.-Y. Liu, Q.-Q. Yang, J.-R. Chen, D.-Q. Shi and W.-J. Xiao, Nat. Commun., 2014, 5, 5500–5509 CrossRef CAS PubMed; (d) L. A. Leth, F. Glaus, M. Meazza, L. Fu, M. K. Thøgersen, E. A. Bitsch and K. A. Jørgensen, Angew. Chem., Int. Ed., 2016, 55, 15272–15276 CrossRef CAS PubMed; (e) Q. Cheng, H.-J. Zhang, W.-J. Yue and S.-L. You, Chem, 2017, 3, 428–436 CrossRef CAS; (f) S. Singha, T. Patra, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2018, 140, 3551–3554 CrossRef CAS PubMed; (g) Z.-C. Chen, Z. Chen, Z.-H. Yang, L. Guo, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2019, 58, 15021–15025 CrossRef CAS PubMed; (h) Y. Wei, S. Liu, M.-M. Li, Y. Li, Y. Lan, L.-Q. Lu and W.-J. Xiao, J. Am. Chem. Soc., 2019, 141, 133–137 CrossRef CAS PubMed.
- (a) B. M. Trost and Z. Zuo, Angew. Chem., Int. Ed., 2021, 60, 5806–5810 CrossRef CAS PubMed; (b) B. M. Trost, P. J. Morris and S. J. Sprague, J. Am. Chem. Soc., 2012, 134, 17823–17831 CrossRef CAS PubMed; (c) Q. Cheng, J.-H. Xie, Y.-C. Weng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed.
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1986, 25, 1–20 CrossRef; (b) B. M. Trost and G. Mata, Acc. Chem. Res., 2020, 53, 1293–1305 CrossRef CAS PubMed; (c) D. M. T. Chan, Recent Advances in Palladium-Catalyzed Cycloadditions Involving Trimethylenemethane and its Analogs, in Cycloaddition Reactions in Organic Synthesis ed. S. Kobayashi and K. A. Jørgensen, Wiley-VCH, Weinheim, 2002, pp. 57–83 Search PubMed.
- (a) B. M. Trost, Y. Wang and C. Hung, Nat. Chem., 2020, 12, 294–301 CrossRef CAS PubMed; (b) B. M. Trost and Y.-L. A. Wang, Angew. Chem., Int. Ed., 2018, 57, 11025–11029 CrossRef CAS PubMed; (c) B. M. Trost and G. Mata, Angew. Chem., Int. Ed., 2018, 57, 12333–12337 CrossRef CAS PubMed; (d) Y.-Z. Liu, Z. Wang, Z. Huang, X. Zheng, W.-L. Yang and W.-P. Deng, Angew. Chem., Int. Ed., 2020, 59, 1238–1242 CrossRef CAS PubMed.
- (a) R. Shintani, S. Park and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 14866–14867 CrossRef CAS PubMed; (b) R. Shintani, M. Murakami and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 12356–12357 CrossRef CAS PubMed; (c) L.-C. Yang, Y.-N. Wang, R. Liu, Y. Luo, N. X. Qian, B. Yang, Z.-Q. Rong, Y. Lan, Z. Shao and Y. Zhao, Nat. Chem., 2020, 12, 860–868 CrossRef CAS PubMed; (d) B. M. Trost, Z. Jiao, Y. Liu, C. Min and C.-I. Hung, J. Am. Chem. Soc., 2020, 142, 18628–18636 CrossRef CAS PubMed; (e) B. M. Trost and Z. Jiao, J. Am. Chem. Soc., 2020, 142, 21645–21650 CrossRef CAS PubMed.
- (a) J. G. Knight, P. A. Stoker, K. Tchabanenko, S. J. Harwood and K. W. M. Lawrie, Tetrahedron, 2008, 64, 3744–3750 CrossRef CAS; (b) M. A. Lowe, M. Ostovar, S. Ferrini, C. C. Chen, P. G. Lawrence, F. Fontana, A. A. Calabrese and V. K. Aggarwal, Angew. Chem., Int. Ed., 2011, 50, 6370–6374 CrossRef CAS PubMed.
- (a) K. Ohmatsu, N. Imagawa and T. Ooi, Nat. Chem., 2014, 6, 47–51 CrossRef CAS PubMed; (b) K. Ohmatsu, S. Kawai, N. Imagawa and T. Ooi, ACS Catal., 2014, 4, 4304–4306 CrossRef CAS.
- (a) B. M. Trost, H.-C. Tsui and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 3534–3535 CrossRef CAS; (b) X. Wang, F. Meng, Y. Wang, Z. Han, Y.-J. Chen, L. Liu, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 9276–9282 CrossRef CAS PubMed; (c) X. Wang, P. Guo, Z. Han, X. Wang, Z. Wang and K. Ding, J. Am. Chem. Soc., 2014, 136, 405–411 CrossRef CAS PubMed; (d) X. Wang, Z. Han, Z. Wang and K. Ding, Acc. Chem. Res., 2021, 54, 668–684 CrossRef CAS PubMed; (e) X. Wang, X. Wang, Z. Han, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2017, 56, 1116–1119 CrossRef CAS PubMed; (f) J. Liu, Z. Han, X. Wang, F. Meng, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2017, 56, 5050–5054 CrossRef CAS PubMed.
- For more details, see the ESI.†.
- (a) D. Qian and J. Sun, Chem.–Eur. J., 2019, 25, 3740–3751 CrossRef CAS PubMed; (b) D. Uraguchi and T. Ooi, J. Synth. Org. Chem., Jpn., 2018, 76, 1144–1153 CrossRef CAS; (c) L. Zong and C.-H. Tan, Acc. Chem. Res., 2017, 50, 842–856 CrossRef CAS PubMed.
- R. Jagadeesan, G. Velmurugana and P. Venuvanalingam, RSC Adv., 2015, 5, 80661–80667 RSC.
- (a) G.-S. Chen, Y.-J. Deng, L.-Z. Gong, A.-Q. Mi, X. Cui, Y.-Z. Jiang, M. C. K. Choi and A. S. C. Chan, Tetrahedron: Asymmetry, 2001, 12, 1567–1571 CrossRef CAS; (b) M. Nakoji, T. Kanayama, T. Okino and Y. Takemoto, Org. Lett., 2001, 3, 3329–3331 CrossRef CAS PubMed; (c) G.-Y. Ran, X.-X. Yang, J.-F. Yue, W. Du and Y.-C. Chen, Angew. Chem., Int. Ed., 2019, 58, 9210–9214 CrossRef CAS PubMed.
- D. Pizzirani, M. Roberti, S. Grimaudo, A. Di Cristina, R. M. Pipitone, M. Tolomeo and M. Recanatini, J. Med. Chem., 2009, 52, 6936–6940 CrossRef CAS PubMed.
- Y. Yang, Y. Jiang, W. Du and Y.-C. Chen, Chem.–Eur. J., 2020, 26, 1754–1758 CrossRef CAS PubMed.
- Significant cytotoxic activity against the human gastric cancer cell line MGC803 was observed for some spirocyclic products 3. See the ESI† for more details.
- (a) S. Krautwald and E. M. Carreira, J. Am. Chem. Soc., 2017, 139, 5627–5639 CrossRef CAS PubMed; (b) M. Bihani and J. C. G. Zhao, Adv. Synth. Catal., 2017, 359, 534–575 CrossRef CAS.
- R. Shintani, S. Hayashi, M. Murakami, M. Takeda and T. Hayashi, Org. Lett., 2009, 11, 3754–3756 CrossRef CAS PubMed.
- Q.-Q. Yang, X. Yin, X.-L. He, W. Du and Y.-C. Chen, ACS Catal., 2019, 9, 1258–1263 CrossRef CAS.
- (a) D. Liao, H. Li and X. Lei, Org. Lett., 2012, 14, 18–21 CrossRef CAS PubMed; (b) K. Fagnou and M. Lautens, Angew. Chem., Int. Ed., 2002, 41, 26–47 CrossRef CAS.
- A. Jutand, Appl. Organomet. Chem., 2004, 18, 574–582 CrossRef CAS.
- X. Huang, B. Wang, Y. Wang, G. Jiang, J. Feng and H. Zhao, Nature, 2020, 584, 69–74 CrossRef CAS PubMed.
- The ammonium salt may be located at different positions, and thus computation work is too large to calculate each pose for different conformations with an ammonium salt. To evaluate the coordination models with TMAB, PB-INT1-B1 and PB-INT1-D1 were selected as the representative conformations for η4- and η3-complexation, respectively.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data for new compounds, NMR, HRMS spectra and HPLC chromatograms, and CIF files of enantiopure products CCDC 2073270 (3s), 2073271 (6g), 2073272 (9c), and 2073273 (14). CCDC 2073270–2073273. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03517g |
| This journal is © The Royal Society of Chemistry 2021 |