
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcitation ratiometric chloride sensing in a standalone yellow fluorescent protein is powered by the interplay between proton transfer and conformational reorganization†
Cheng
Chen
 a,
Jasmine N.
Tutol
b,
Longteng
Tang
a,
Liangdong
Zhu
a,
Whitney S. Y.
Ong
b,
Sheel C.
Dodani
*b and
Chong
Fang
*a
a,
Jasmine N.
Tutol
b,
Longteng
Tang
a,
Liangdong
Zhu
a,
Whitney S. Y.
Ong
b,
Sheel C.
Dodani
*b and
Chong
Fang
*a
aDepartment of Chemistry, Oregon State University, 153 Gilbert Hall, Corvallis, OR 97331-4003, USA. E-mail: Chong.Fang@oregonstate.edu; Web: https://fanglab.oregonstate.edu/
bDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, 800 West Campbell Road, Richardson, TX 75080, USA. E-mail: Sheel.Dodani@utdallas.edu; Web: https://lab.utdallas.edu/dodani/
First published on 21st July 2021
Abstract
Natural and laboratory-guided evolution has created a rich diversity of fluorescent protein (FP)-based sensors for chloride (Cl−). To date, such sensors have been limited to the Aequorea victoria green fluorescent protein (avGFP) family, and fusions with other FPs have unlocked ratiometric imaging applications. Recently, we identified the yellow fluorescent protein from jellyfish Phialidium sp. (phiYFP) as a fluorescent turn-on, self-ratiometric Cl− sensor. To elucidate its working mechanism as a rare example of a single FP with this capability, we tracked the excited-state dynamics of phiYFP using femtosecond transient absorption (fs-TA) spectroscopy and target analysis. The photoexcited neutral chromophore undergoes bifurcated pathways with the twisting-motion-induced nonradiative decay and barrierless excited-state proton transfer. The latter pathway yields a weakly fluorescent anionic intermediate 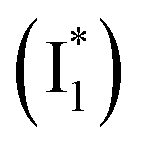 , followed by the formation of a red-shifted fluorescent state
, followed by the formation of a red-shifted fluorescent state 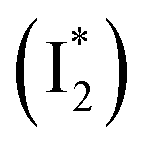 that enables the ratiometric response on the tens of picoseconds timescale. The redshift results from the optimized π–π stacking between chromophore Y66 and nearby Y203, an ultrafast molecular event. The anion binding leads to an increase of the chromophore pKa and ESPT population, and the hindrance of
that enables the ratiometric response on the tens of picoseconds timescale. The redshift results from the optimized π–π stacking between chromophore Y66 and nearby Y203, an ultrafast molecular event. The anion binding leads to an increase of the chromophore pKa and ESPT population, and the hindrance of 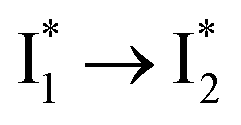 conversion. The interplay between these two effects determines the turn-on fluorescence response to halides such as Cl− but turn-off response to other anions such as nitrate as governed by different binding affinities. These deep mechanistic insights lay the foundation for guiding the targeted engineering of phiYFP and its derivatives for ratiometric imaging of cellular chloride with high selectivity.
conversion. The interplay between these two effects determines the turn-on fluorescence response to halides such as Cl− but turn-off response to other anions such as nitrate as governed by different binding affinities. These deep mechanistic insights lay the foundation for guiding the targeted engineering of phiYFP and its derivatives for ratiometric imaging of cellular chloride with high selectivity.
Introduction
Chloride (Cl−) is the most abundant physiological anion and serves important roles in various biological processes including regulation of the cell volume and intracellular pH, stabilization of the resting membrane potential, and neurotransmission.1–3 Dysregulation of Cl− gradients is associated with many human diseases such as cystic fibrosis, nephrolithiasis, and epilepsy,4–7 so the pathology and potential treatment demand an accurate detection of intracellular Cl− concentrations. Fluorescence imaging with fluorescent indicators has been widely used and made significant contributions to this important aim.8–10 Chemical fluorescent probes such as quinolinium-based dyes and bioconjugates thereof have been reported to exhibit high sensitivity to Cl− with excellent binding affinity, and are pH-independent.11–14 These properties make them suitable to monitor Cl− in the physiological concentration range of 3–80 mM.8 However, these small chemical indicators often exhibit shortcomings that include photobleaching due to UV excitation and gradual leakage of loaded dyes from the labeled cells.An alternative to these exogenous indicators is the endogenously expressed fluorescent proteins (FPs). This endeavor has been mostly limited to the Aequorea victoria green fluorescent protein (avGFP) family.15 The yellow variant avYFP16 and enhanced GFP (EGFP) mutant E2GFP17 are two major players, both of which incorporate the key mutation T203Y for halide ion binding. In avYFPs, Cl− binding quenches the fluorescence of the anionic chromophore by shifting the ground-state equilibrium to the neutral form due to the increased pKa.18,19 It makes avYFP and its mutants (e.g., avYFP-H148Q) turn-off fluorescence sensors. The residue Y203 and three other polar residues, Q69, R96, and Q183, constitute the Cl− binding pocket (see the crystal structure in Fig. 1b). Notably, this binding pocket is located at the opposite end of the chromophore Y66 protonation site and is close to the carbonyl group of the imidazolinone ring. The pKa increase of the avYFP chromophore was thus attributed to the suppression of the phenolate negative charge delocalization upon Cl− binding.19 Y203 plays an indispensable role in Cl− binding, supported by the drastically weakened binding affinity of the Y203T mutant.19 Meanwhile, the antiparallel π–π stacking between Y203 and chromophore Y66 leads to a spectral redshift from GFP. In contrast, the turn-off fluorescence in E2GFP was ascribed to a static quenching mechanism wherein Cl− binding leads to a nonfluorescent protein population.17
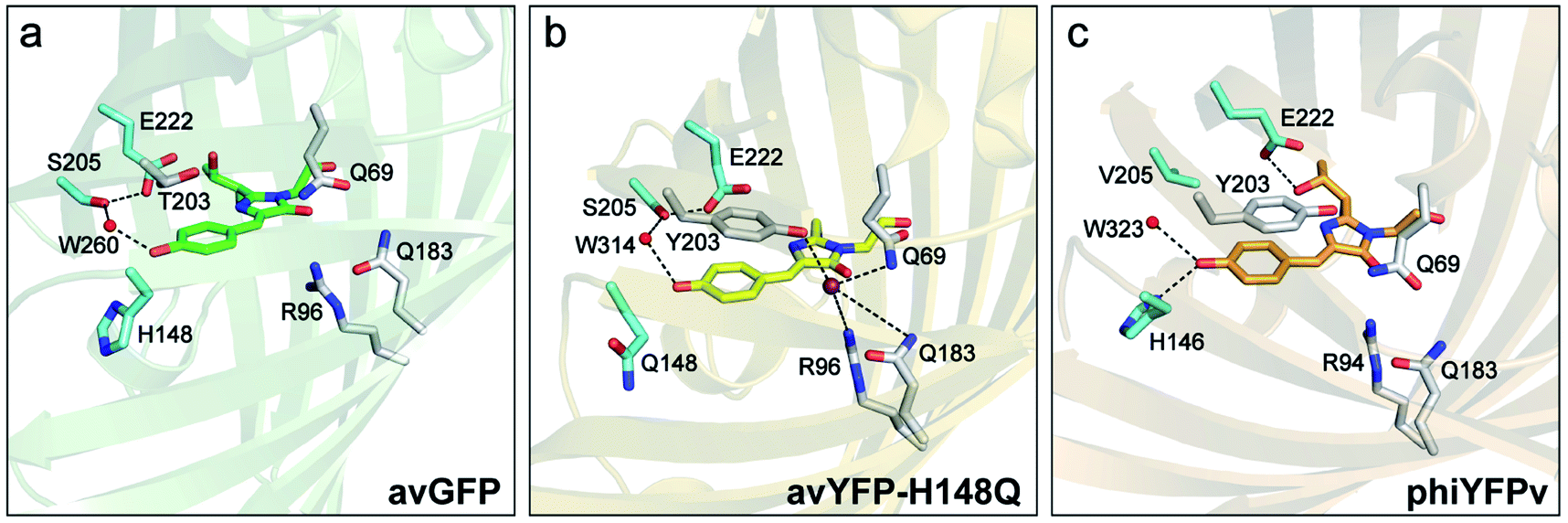 | ||
| Fig. 1 Crystal structures of (a) avGFP (PDB ID: 1EMB),60 (b) avYFP-H148Q bound with iodide (purple sphere) (PDB ID: 1F09),19 and (c) phiYFPv (PDB ID: 4HE4).47 The chromophore is shown as sticks in green for avGFP, yellow for avYFP-H148Q, and orange for phiYFPv, while water molecules (W) are shown as red spheres. The oxygen and nitrogen atoms are colored in red and blue, respectively. The highly ordered residues that could participate in the excited-state proton transfer pathway are highlighted in cyan. Similar residues constituting the anion-binding pocket (a tightly packed environment) in avYFP-H148Q and phiYFPv are shown in light gray in panels (b) and (c), reminiscent of the avGFP residues in panel (a). | ||
Indeed, protein engineering methods have been used to improve avYFP and E2GFP in intensity or ratiometric-based imaging applications.9 To achieve the latter, fusions with Cl−-insensitive or pH-independent FPs14 have enabled ratiometric sensing to account for variations in expression, irradiation intensity, photobleaching, or pH.20–26 For example, Clomeleon is a fusion of avYFP with cyan fluorescent protein (CFP) based on a Förster resonance energy transfer (FRET) mechanism, where the CFP and YFP units are the energy donor and acceptor, respectively.20 Other Cl− sensors include, but are not limited to, Cl-Sensor,22 SuperClomeleon,24 ClopHensor,23 and LSSmClopHensor.26
Recently, the yellow fluorescent protein from the jellyfish Phialidium sp. (phiYFP) was identified as an excitation ratiometric Cl− sensor, which has long been overlooked.27 phiYFP has a high sequence identity to avYFP-H148Q (52%) and has the exact same residues in the anion binding pocket: Q69, R94, Q183, and Y203, while Y203 π-stacks with the chromophore Y66 (see Fig. 1b and c). The resemblance causes quenched fluorescence of the anionic chromophore due to an increased pKa in the presence of Cl−. Interestingly, upon excitation of the neutral chromophore, phiYFP displays enhanced fluorescence of the anionic form with the addition of Cl− (Fig. S1a in the ESI†), indicative of excited-state proton transfer (ESPT).28,29 In contrast, phiYFP shows turn-off (e.g., NO3−) or insensitive (e.g., phosphate) fluorescence to other oxyanions.27 The underlying mechanism remains elusive since fluorescence is an ultrafast process,30 which is further complicated by two fluorescence bands from the anionic chromophore. Moreover, the wild-type phiYFP is impractical for cellular imaging due to the low operational pH and Cl− binding affinity (Kd ∼ 300–400 mM at pH = 5–6). To enable rational engineering of phiYFP for live-cell imaging applications, a mechanistic understanding of the excited-state dynamics31,32 of phiYFP has become crucial.
In this work, we implemented time-resolved femtosecond transient absorption (fs-TA) spectroscopy to reveal ultrafast ESPT dynamics and molecular origins of anionic species that underpin the excitation ratiometric response of phiYFP. Two representative cases (Cl− and NO3−) were investigated with 400 nm excitation. Using model-specific global analysis,33,34 we revealed key bifurcated pathways for the photoexcited neutral chromophore: ultrafast ESPT and formation of a dark state. The ESPT pathway yields two anionic species, 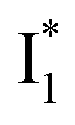 and
and 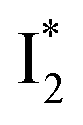 , the latter of which contributes to the ratiometric response. Notably,
, the latter of which contributes to the ratiometric response. Notably, 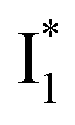 and
and 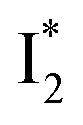 are both anionic states with the π–π stacking between the chromophore Y66 and nearby Y203 unoptimized and optimized, respectively. Anion binding inhibits the
are both anionic states with the π–π stacking between the chromophore Y66 and nearby Y203 unoptimized and optimized, respectively. Anion binding inhibits the 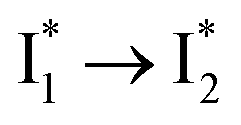 conversion. The interplay between the ESPT population increase and inhibition of
conversion. The interplay between the ESPT population increase and inhibition of 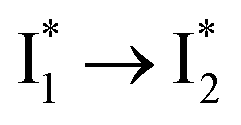 conversion leads to high selectivity to Cl−. Based on these fresh and deep mechanistic insights, we propose strategies to engineer phiYFP for cellular imaging by improving Cl− binding affinity and operational pH.
conversion leads to high selectivity to Cl−. Based on these fresh and deep mechanistic insights, we propose strategies to engineer phiYFP for cellular imaging by improving Cl− binding affinity and operational pH.
Results and discussion
Excited-state dynamics upon Cl− binding
We examined the excited-state dynamics of phiYFP in pH = 5.5 buffer where phiYFP shows the best ratiometric response to Cl− binding. In the presence of 400 mM Cl− (the apparent dissociation constant Kd is 384 mM),27 ground-state phiYFP is populated by neutral (A) and anionic (B) forms of similar concentrations (pKa = 5.7) with the peak absorption at 403 and 526 nm, respectively (Fig. 2a).28 Notably, the apo protein with no ions bound (pKa = 4.9) shows a negligible A peak at pH = 5.5 (see Fig. S3a and d in the ESI†).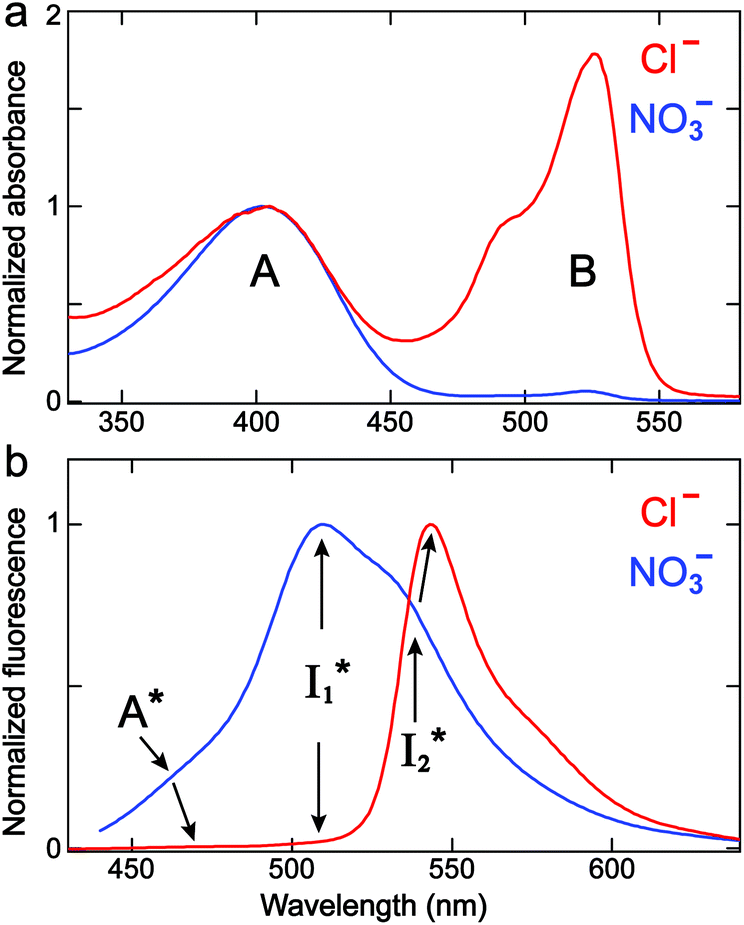 | ||
| Fig. 2 Normalized steady-state (a) absorption and (b) emission spectra (λex = 400 nm) of phiYFP in 50 mM MES buffer (pH = 5.5) with 400 mM NaCl (red) and NaNO3 (blue). The absorption spectra are normalized at the protonated chromophore A band maximum. The multiple emission species are labeled with peak positions indicated. | ||
Upon 400 nm excitation of the A form, phiYFP with 400 mM Cl− is dominated by an emission band at 542 nm (Fig. 2b), which is the same as that with 490 nm excitation of B (Fig. S2c in the ESI†) and hence attributed to the anionic form. We term this emitting species 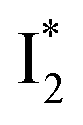 (“I” denotes the anionic intermediate and the asterisk denotes the excited state) due to the unrelaxed protein environment which is common for FPs with ESPT.28,31 Two other emission peaks are also identified with much weaker intensities (Table 1 and Fig. S2b in the ESI†), further validated by the emission spectra with 400 mM NO3− (Fig. 2b, S1c, d and S2d in the ESI†) or at lower pH values.27 The peak at 470 nm (better shown in Fig. S2b, ESI†) originates from A* emission, which is similar to avGFP and expected for the neutral chromophore with a Stokes shift of ∼70 nm.31,35 Meanwhile, since the anionic
(“I” denotes the anionic intermediate and the asterisk denotes the excited state) due to the unrelaxed protein environment which is common for FPs with ESPT.28,31 Two other emission peaks are also identified with much weaker intensities (Table 1 and Fig. S2b in the ESI†), further validated by the emission spectra with 400 mM NO3− (Fig. 2b, S1c, d and S2d in the ESI†) or at lower pH values.27 The peak at 470 nm (better shown in Fig. S2b, ESI†) originates from A* emission, which is similar to avGFP and expected for the neutral chromophore with a Stokes shift of ∼70 nm.31,35 Meanwhile, since the anionic 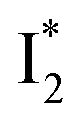 (B*-like) is already present, the emission peak at 505 nm likely arises from an
(B*-like) is already present, the emission peak at 505 nm likely arises from an 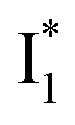 species due to its spectral departure from A*. Interestingly, with decreasing pH, the photoexcited phiYFP becomes trapped in
species due to its spectral departure from A*. Interestingly, with decreasing pH, the photoexcited phiYFP becomes trapped in 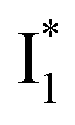 while
while 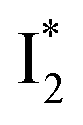 emission is diminished (spectra shown in our previous report).27 At pH = 4.5 (below its pKa) and upon 400 nm excitation, only the
emission is diminished (spectra shown in our previous report).27 At pH = 4.5 (below its pKa) and upon 400 nm excitation, only the 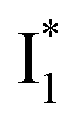 emission is observable with a shift in the peak maximum (Fig. S4 and Table S1 in the ESI†). This behavior stems from the pH-dependent fluorescence response of phiYFP, reminiscent of avYFPs and E2GFP that are also pH-dependent Cl− indicators.17,19
emission is observable with a shift in the peak maximum (Fig. S4 and Table S1 in the ESI†). This behavior stems from the pH-dependent fluorescence response of phiYFP, reminiscent of avYFPs and E2GFP that are also pH-dependent Cl− indicators.17,19
| FPs | Absorption (nm) | Emissiona (nm) | pKa | ||||
|---|---|---|---|---|---|---|---|
| A | B | A* |
|
|
B* | ||
a The A*, 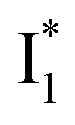 , and , and 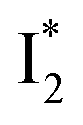 emission bands result from the excitation of A; the B* emission results from the excitation of B.
b Data are taken from the literature.19,56,57 avYFP and avYFP-H148Q values were measured in aqueous solution without any interfering anions (buffered with 150 mM gluconate).19
c The emission wavelengths at pH = 5.5 are extracted from the second-order derivative analysis35 (Fig. S2 in the ESI) with the anion concentration of 400 mM.
d The pKa of the apo phiYFP was measured27 to be 4.9 (see more details about the chromophore pKa determination using pH-dependent spectra and Fig. S3 in the ESI).
e n.a.: not available. emission bands result from the excitation of A; the B* emission results from the excitation of B.
b Data are taken from the literature.19,56,57 avYFP and avYFP-H148Q values were measured in aqueous solution without any interfering anions (buffered with 150 mM gluconate).19
c The emission wavelengths at pH = 5.5 are extracted from the second-order derivative analysis35 (Fig. S2 in the ESI) with the anion concentration of 400 mM.
d The pKa of the apo phiYFP was measured27 to be 4.9 (see more details about the chromophore pKa determination using pH-dependent spectra and Fig. S3 in the ESI).
e n.a.: not available.
|
|||||||
| avGFP-S65Tb | 394 | 489 | 459 | n.a. | 508 | 510 | 6.0 |
| avYFPb | 392 | 514 | Negligible | n.a. | n.a. | 528 | 5.4 |
| avYFP-H148Qb | 396 | 515 | n.a. | n.a. | n.a. | 529 | 6.7 |
| phiYFP (Cl−)c | 403 | 526 | 470 | 504 | 542 | 542 | 5.7d |
| phiYFP (NO3−)c | 402 | 524 | 463 | 506 | 534 | 534 | 6.6d |
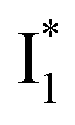
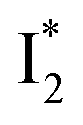
To track molecular events that lead to the biosensor fluorescence, we collected the fs-TA spectra of phiYFP with Cl− by 400 nm excitation of the neutral chromophore (pH = 5.5, Fig. 3a and b). The spectral evolution can be divided into four stages. Stage I involves the ultrafast formation (within the cross-correlation time of ∼80 fs) of 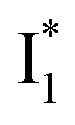 as evinced by the rise of the stimulated emission (SE) band at ∼510 nm. Meanwhile, the excited-state absorption (ESA) band at ∼450 nm increases and becomes broader, leading to a reversal of the intensity change from stage I to II at ∼500 nm (Fig. 3a, see Fig. S5a in the ESI† for detailed dynamics), which indicates the interference from another state or species (e.g., a dark state). Therefore, the apparent SE band decay at ∼500 nm in stage I is due to the spectral overlap of a broader absorption feature in this region (i.e., manifesting the transient ESA band rise dynamics). Stage II shows opposite dynamics of the ESA (∼450 nm, decay) and SE (500–530 nm, rise) bands (Fig. 3a), and their similar time constants imply the decay of the tentative dark state (Fig. S5 and Table S2 in the ESI†). Such a dark state evolving on the fs-to-ps timescale can be uncovered from TA global analysis and control experiments (see below).
as evinced by the rise of the stimulated emission (SE) band at ∼510 nm. Meanwhile, the excited-state absorption (ESA) band at ∼450 nm increases and becomes broader, leading to a reversal of the intensity change from stage I to II at ∼500 nm (Fig. 3a, see Fig. S5a in the ESI† for detailed dynamics), which indicates the interference from another state or species (e.g., a dark state). Therefore, the apparent SE band decay at ∼500 nm in stage I is due to the spectral overlap of a broader absorption feature in this region (i.e., manifesting the transient ESA band rise dynamics). Stage II shows opposite dynamics of the ESA (∼450 nm, decay) and SE (500–530 nm, rise) bands (Fig. 3a), and their similar time constants imply the decay of the tentative dark state (Fig. S5 and Table S2 in the ESI†). Such a dark state evolving on the fs-to-ps timescale can be uncovered from TA global analysis and control experiments (see below).
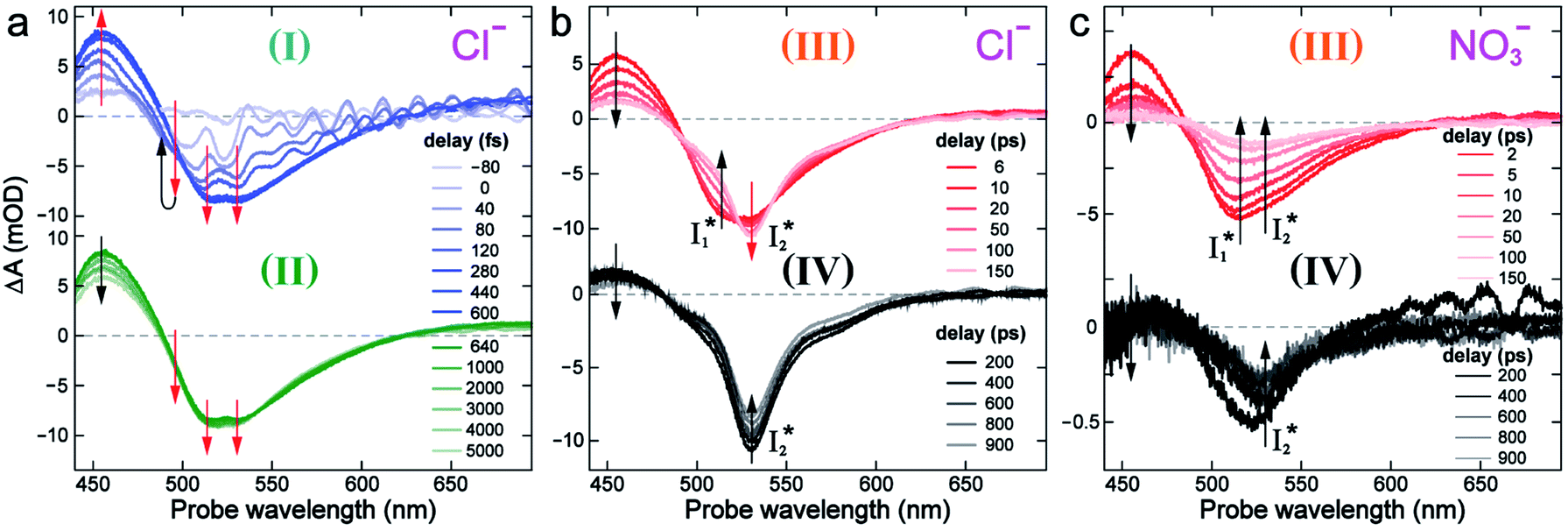 | ||
| Fig. 3 Time-resolved fs-TA spectra of phiYFP at pH = 5.5 with 400 mM (a and b) Cl−, and (c) NO3− following 400 nm photoexcitation. (I), (II), (III), and (IV) denote four distinct stages of spectral evolution. Stages (I) and (II) with 400 mM NO3− (Fig. S9 in the ESI†) are omitted due to the similarity to Cl−. Red and black arrows denote the transient electronic peak intensity magnitude rise and decay, respectively. mOD: milli-optical density. | ||
Stage III involves key processes for phiYFP to function as a turn-on fluorescence biosensor for Cl−. The 510 nm SE band 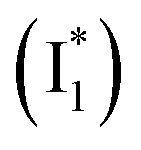 decay time constants (31 and 132 ps) largely match the rise time constants (36 and 406 ps) of the 530 nm SE band
decay time constants (31 and 132 ps) largely match the rise time constants (36 and 406 ps) of the 530 nm SE band 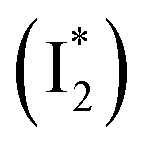 , indicating a two-state model (i.e.,
, indicating a two-state model (i.e., 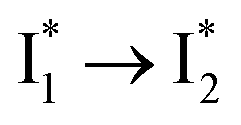 , see Fig. 4a). Mismatch of the longer component (132 ps versus 406 ps) may be caused by the spectral overlap of different molecular species or competing processes.32,36 This observation suggests that the turn-on fluorescence is enabled by
, see Fig. 4a). Mismatch of the longer component (132 ps versus 406 ps) may be caused by the spectral overlap of different molecular species or competing processes.32,36 This observation suggests that the turn-on fluorescence is enabled by 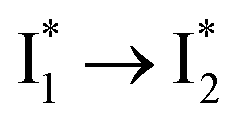 conversion in the presence of Cl− as
conversion in the presence of Cl− as 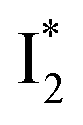 is the strongly fluorescent state. Stage IV mainly probes the radiative decay of
is the strongly fluorescent state. Stage IV mainly probes the radiative decay of 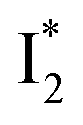 , reflected by the SE band decay at 530 nm (Fig. 3b). The nanosecond (ns) time constant agrees well with the high fluorescence quantum yield (FQY) of
, reflected by the SE band decay at 530 nm (Fig. 3b). The nanosecond (ns) time constant agrees well with the high fluorescence quantum yield (FQY) of 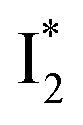 .27
.27
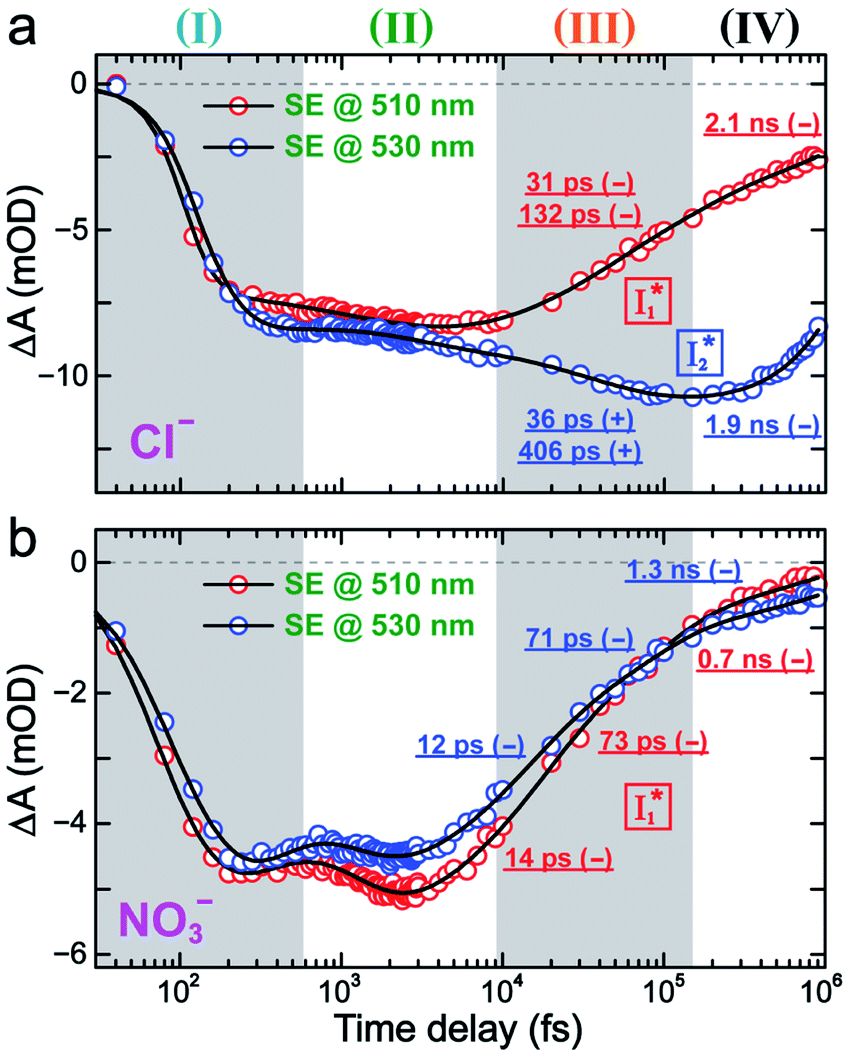 | ||
| Fig. 4 Kinetics of stimulated emission bands at 510 (red) and 530 nm (blue) in the fs-TA spectra of phiYFP at pH = 5.5 with 400 mM (a) Cl− and (b) NO3− after 400 nm photoexcitation. The time constants of stage (III) and (IV) are listed by the fits (solid black lines) with +/− signs denoting the peak intensity rise/decay, respectively. | ||
To deconvolute the spectral overlap that hinders direct visualization of the underlying reaction species and processes, global analysis is required to simultaneously treat both spectral and temporal dimensions and yield a topological landscape for the excited-state potential energy surface (PES).33 We performed the model-specific global analysis, also termed target analysis,34,37 to account for the observed transient dynamics of phiYFP. A branched kinetic model was used to examine the fs-TA dynamics wherein the photoexcited A* species bifurcates and evolves along two reaction coordinates: pathways P1 and P2 (Fig. 5a). P1 involves the nonradiative decay of A* likely through conformational twisting motions, whereas P2 involves an ESPT reaction that produces 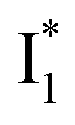 and
and 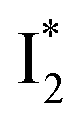 . Notably, the presence of P1 is corroborated by two observations. First, the 500 nm SE band (
. Notably, the presence of P1 is corroborated by two observations. First, the 500 nm SE band (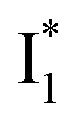 that converts from A*) shows complex dynamics within ∼5 ps (Fig. 3a) due to the overlapping electronic bands; otherwise its decay would be monotonic for a pure species/state in the unidirectional
that converts from A*) shows complex dynamics within ∼5 ps (Fig. 3a) due to the overlapping electronic bands; otherwise its decay would be monotonic for a pure species/state in the unidirectional 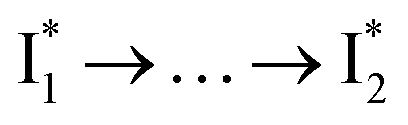 reaction (see Fig. S5, as well as Fig. S6† with global analysis of TA spectra in the ESI†). Second, P2 has a small weight that can be estimated from the FQY. The large difference in FQYs of
reaction (see Fig. S5, as well as Fig. S6† with global analysis of TA spectra in the ESI†). Second, P2 has a small weight that can be estimated from the FQY. The large difference in FQYs of 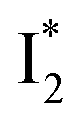 (B*-like) and B* upon excitation of A and B (i.e., 0.06 and 0.44, respectively)27 as well as similar intensity of
(B*-like) and B* upon excitation of A and B (i.e., 0.06 and 0.44, respectively)27 as well as similar intensity of 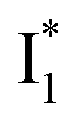 and
and 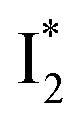 SE bands (an efficient
SE bands (an efficient 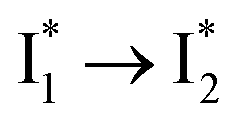 conversion in the presence of 400 mM Cl−) indicate that A* decays mainly via other nonradiative channels (e.g., P1) instead of ESPT (P2). The resultant species-associated difference spectra (SADS) from least-squares fits (see Fig. S7 and S8 in the ESI† for the fit residuals in our detection window) display expected spectral features for the examined kinetic model, which constitute an accurate representation of the fs-TA data.33
conversion in the presence of 400 mM Cl−) indicate that A* decays mainly via other nonradiative channels (e.g., P1) instead of ESPT (P2). The resultant species-associated difference spectra (SADS) from least-squares fits (see Fig. S7 and S8 in the ESI† for the fit residuals in our detection window) display expected spectral features for the examined kinetic model, which constitute an accurate representation of the fs-TA data.33
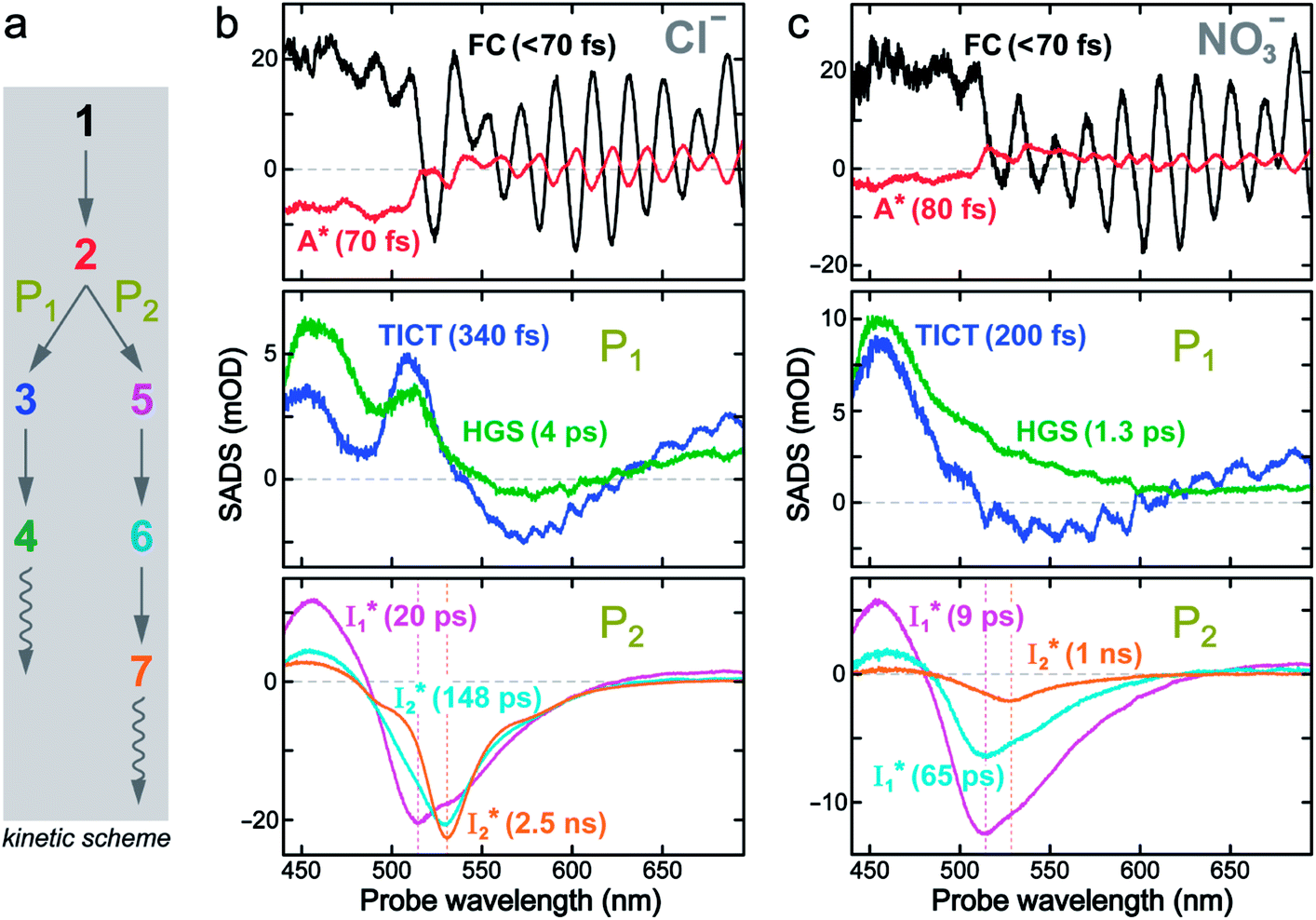 | ||
Fig. 5 Deconvolution of the excited-state dynamics of phiYFP by target analysis. (a) Kinetic model with reactant bifurcation. Straight and wavy arrows denote the state transition and self-decay process, respectively. Species-associated difference spectra (SADS) from the fs-TA data of phiYFP at pH = 5.5 with 400 mM (b) Cl− and (c) NO3−. Each SADS is color-coded as defined in (a). The associated lifetimes are labeled for the corresponding transient molecular species. Vertical dotted lines highlight the 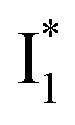 and and 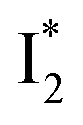 emissions. The zero OD lines are shown by horizontal gray dashed lines. FC: Franck–Condon, TICT: twisted intramolecular charge transfer, HGS: hot ground state. In top panels, the probe region above ∼520 nm is affected by coherent artefacts near time zero of photoexcitation; see Fig. S6† for the associated EADS and DADS analyses with an in-depth comparison. Note that for the P1 pathway, the rapidly formed state 3 (blue, the TICT state in middle panels) has distinct TA features from state 2 (red, the planar A* state in top panels) and a sub-ps decay time constant back to the electronic ground state (green, the HGS in middle panels) of the protonated chromophore. emissions. The zero OD lines are shown by horizontal gray dashed lines. FC: Franck–Condon, TICT: twisted intramolecular charge transfer, HGS: hot ground state. In top panels, the probe region above ∼520 nm is affected by coherent artefacts near time zero of photoexcitation; see Fig. S6† for the associated EADS and DADS analyses with an in-depth comparison. Note that for the P1 pathway, the rapidly formed state 3 (blue, the TICT state in middle panels) has distinct TA features from state 2 (red, the planar A* state in top panels) and a sub-ps decay time constant back to the electronic ground state (green, the HGS in middle panels) of the protonated chromophore. | ||
The black and red SADS are the Franck–Condon state (FC, <70 fs) and A* (70 fs) preceding the bifurcated reactions, respectively (Fig. 5a). P1 can be fit with two SADS attributed to a dark twisted intramolecular charge transfer state (TICT, blue trace) and a hot ground state (HGS, green trace, Fig. 5b) of the biosensor chromophore. This assignment is firmly supported by clear spectral resemblance to the GFP chromophore and its structural analogues that share the same flexible methine bridge. These chromophores in solution are nonfluorescent and characteristic of ultrafast ring-twisting-induced nonradiative decays.38–43 The aforementioned dark state is also consistent with various high-level calculations that predicted the twisted intermediate state(s) along the phenolic/phenolate (P-) ring and/or imidazolinone (I-) ring-twisting coordinates of the chromophore.39–42 In addition, for experimental corroboration, we obtained the fs-TA spectra of several GFP chromophore derivatives that we recently synthesized in solution (see Fig. S10 and S11 in the ESI†),44 which exhibit an early-time significantly red-shifted SE band from the fluorescence peak and a bluer ESA band around 450 nm (resembling Fig. 5b and c middle panels for the A*-like TICT state, twisting is involved).
Notably, caution needs to be taken here that the blue SADS (assigned to TICT in Fig. 5b and c middle panels) may not be the true species spectrum of the TICT state but instead representation of a state/species possibly with overlapped TA features of the A* (e.g., with an SE band) and TICT (e.g., with an ESA band) states possessing the same time constant as a sequential model is used for this pathway. This point is partially aided by calculations that confirmed the dark-state nature of a twisted intermediate state of GFP chromophores (see above), and hence the observed red-shifted SE feature (P1 in Fig. 5, also see Fig. S10 and S11 in the ESI†) could arise from the overlap of A* SE and A*/TICT ESA bands. However, the significantly red-shifted SE peak with respect to the A* fluorescence peak in phiYFP (i.e., ∼460–470 nm → ∼550–570 nm, the latter SE peak is even redder than the deprotonated 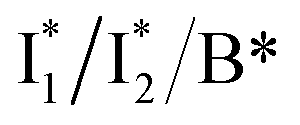 fluorescence peak), and likewise in GFP chromophore analogues (Fig. S11 in the ESI†) with a stagnant SE band position concomitant with a rising adjacent ESA band can hardly be attributed to an A* subpopulation with a planar conformation. In other words, when the initially excited planar A* state decays in the absence of ESPT (P2 pathway here) or steric restrictions, it does so on a sub-ps timescale (e.g., 340 fs in Fig. 5b and 200 fs in Fig. 5c) by motions along a barrierless twisting coordinate, which likely involves the P-ring twist that is not directly attached to the protein backbone (i.e., I-ring side) and could contribute to the observed TA features along the uncovered P1 pathway as an S1 potential energy minimum has been proposed in the literature.39–43 Nevertheless, more efforts are required to elucidate the exact chromophore (A*) twisting mechanisms in solution and the protein matrix after photoexcitation. The ultrafast formation (likely within the cross-correlation time of 70–80 fs, see Methods in the ESI†) and decay (340 fs for Cl− and 200 fs for NO3−, see Fig. 5b and c middle panels) of the TICT state are also hinted and supported by the fs-TA dynamics with global analysis featuring the TICT and HGS of three related GFP chromophore analogues (see Fig. S11 in the ESI†). In essence, this ultrafast twisting pathway (P1) directly competes with the similarly fast ESPT pathway (P2; <70 fs from the rise of the 510 nm
fluorescence peak), and likewise in GFP chromophore analogues (Fig. S11 in the ESI†) with a stagnant SE band position concomitant with a rising adjacent ESA band can hardly be attributed to an A* subpopulation with a planar conformation. In other words, when the initially excited planar A* state decays in the absence of ESPT (P2 pathway here) or steric restrictions, it does so on a sub-ps timescale (e.g., 340 fs in Fig. 5b and 200 fs in Fig. 5c) by motions along a barrierless twisting coordinate, which likely involves the P-ring twist that is not directly attached to the protein backbone (i.e., I-ring side) and could contribute to the observed TA features along the uncovered P1 pathway as an S1 potential energy minimum has been proposed in the literature.39–43 Nevertheless, more efforts are required to elucidate the exact chromophore (A*) twisting mechanisms in solution and the protein matrix after photoexcitation. The ultrafast formation (likely within the cross-correlation time of 70–80 fs, see Methods in the ESI†) and decay (340 fs for Cl− and 200 fs for NO3−, see Fig. 5b and c middle panels) of the TICT state are also hinted and supported by the fs-TA dynamics with global analysis featuring the TICT and HGS of three related GFP chromophore analogues (see Fig. S11 in the ESI†). In essence, this ultrafast twisting pathway (P1) directly competes with the similarly fast ESPT pathway (P2; <70 fs from the rise of the 510 nm 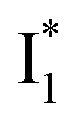 SE band, see Fig. 4 and Table S2 in the ESI†), echoing Meech et al. and our recent studies on the halogenated GFP chromophores (essentially nonfluorescent superphotoacids in nature) which demonstrate active competition between these two ultrafast pathways.35,45
SE band, see Fig. 4 and Table S2 in the ESI†), echoing Meech et al. and our recent studies on the halogenated GFP chromophores (essentially nonfluorescent superphotoacids in nature) which demonstrate active competition between these two ultrafast pathways.35,45
We also remark that the precise assignment of this TICT state with certain ring twisting motions (I- and/or P-ring) remains an active research subject particularly for various calculations and simulations of GFP-like chromophores in solution and a protein environment where the interplay between the chromophore and its surroundings needs to be accurately modeled and validated by experimental findings. In essence, this rapidly formed state along the P1 pathway can be considered A*-like (highlighting its protonated chromophore nature in S1 but with some characteristic TA signatures that differ from the planar A*, see Fig. 5 top and middle panels) and mainly involves the ultrafast ring-twisting motions with weak environmental frictional forces,38,43 in the absence of the ESPT reaction (i.e., P2 pathway herein, Fig. 5a). The main focus of this work is thus to identify the “hidden” P1-branching pathway for this protein chromophore in active competition with the ESPT reaction and gain fundamental insights into low FQY of phiYFP upon excitation of the protonated A form27 so we could interpret the complex TA spectral patterns and develop the rational design principles for chloride sensing from the bottom up (see below).
On the other hand, the best fit of P2 results in three SADS that dominantly track the 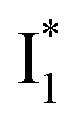 (magenta) to
(magenta) to 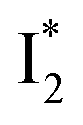 (orange) transition. Notably, the uncovered intermediate state (dark cyan) exhibits a similar SE band maximum to
(orange) transition. Notably, the uncovered intermediate state (dark cyan) exhibits a similar SE band maximum to 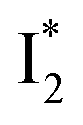 but with a broader bandwidth (Fig. 5b, bottom panel), and hence we surmise that it represents a vibrationally unrelaxed state within the PES well of
but with a broader bandwidth (Fig. 5b, bottom panel), and hence we surmise that it represents a vibrationally unrelaxed state within the PES well of 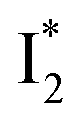 .30,32 Therefore, the major time constant for the
.30,32 Therefore, the major time constant for the 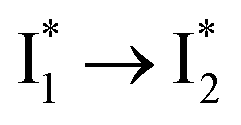 conversion is 20 ps inside phiYFP with 400 mM Cl−.
conversion is 20 ps inside phiYFP with 400 mM Cl−.
Anion selectivity: chloride versus nitrate
Similar to avYFPs, the deprotonated form B* fluorescence of phiYFP is sensitive to many anions due to the increased pKa (thus more protonated A form) and quenched B* fluorescence upon the excitation of the deprotonated B form.19,27,46 In this regard, phiYFP can be utilized as an intensiometric Cl− sensor. However, in terms of an excitation-ratiometric response, phiYFP exhibits notably higher sensitivities to halides (see Fig. S1, ESI†) than many oxyanions such as NO3−. Poor selectivity to NO3− lies in the significantly inhibited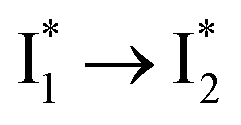 conversion (see the fs-TA results below), which leads to the apparent turn-off fluorescence of
conversion (see the fs-TA results below), which leads to the apparent turn-off fluorescence of 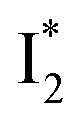 (Fig. S1c in the ESI†).
(Fig. S1c in the ESI†).
In the presence of 400 mM NO3−, the ground-state phiYFP chromophore at pH = 5.5 is dominated by the protonated form with a higher chromophore pKa than that in the case of 400 mM Cl− (Fig. 2a). This is in accord with the higher binding affinity of NO3− (Kd = 19 mM at pH = 5.5)27 that destabilizes the anionic chromophore due to electrostatic repulsion between the delocalized negative charge and the bound anion. In the electronic state after excitation of the A form, the NO3−-bound phiYFP exhibits similar early-time dynamics to the Cl−-bound phiYFP in stages I and II (see Fig. S9 in the ESI† for TA spectra). In stage III, the absence of a clear rise of the 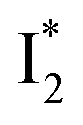 SE band at ∼530 nm indicates that NO3− significantly inhibits the
SE band at ∼530 nm indicates that NO3− significantly inhibits the 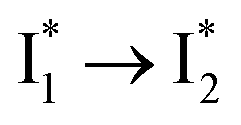 conversion and traps most ESPT product populations in the
conversion and traps most ESPT product populations in the 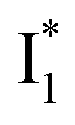 state (Fig. 3c). Only a small population of
state (Fig. 3c). Only a small population of 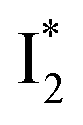 (less than 10% of
(less than 10% of 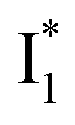 , derived from the SE band intensities assuming similar oscillator strengths for the SE transitions) is observed at late time in stage IV. As a result, the SE band intensities at 510 nm
, derived from the SE band intensities assuming similar oscillator strengths for the SE transitions) is observed at late time in stage IV. As a result, the SE band intensities at 510 nm 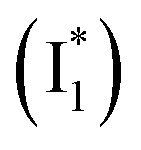 and 530 nm
and 530 nm 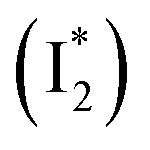 exhibit almost identical dynamics to the last ns component at a small weight reflecting the
exhibit almost identical dynamics to the last ns component at a small weight reflecting the 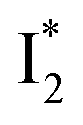 dynamics (0.7 ns decay at 510 nm, faster than 1.3 ns at 530 nm, Fig. 4b).
dynamics (0.7 ns decay at 510 nm, faster than 1.3 ns at 530 nm, Fig. 4b).
Target analysis of the NO3−-bound phiYFP reveals a similar topology of the excited-state PES as the Cl−-bound phiYFP. In the presence of NO3−, phiYFP bifurcates into a TICT-featured nonradiative pathway and ESPT, the latter pathway differing from the Cl−-bound phiYFP in the 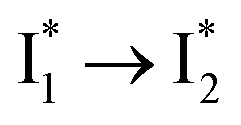 conversion. In Fig. 5c, the magenta and dark cyan SADS show similar SE bands at ∼510 nm (the peak intensity decreases on the ps timescale), tracking the
conversion. In Fig. 5c, the magenta and dark cyan SADS show similar SE bands at ∼510 nm (the peak intensity decreases on the ps timescale), tracking the 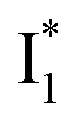 species. The existence of two
species. The existence of two 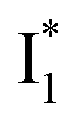 SADS suggests that
SADS suggests that 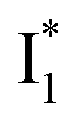 might decay along two reaction coordinates. Notably,
might decay along two reaction coordinates. Notably, 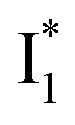 is a much less fluorescent state than
is a much less fluorescent state than 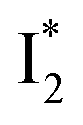 and phiYFP is mostly trapped in the
and phiYFP is mostly trapped in the 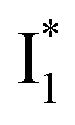 state when NO3− is present (Fig. 3c). Therefore, the magenta SADS with a lifetime of 9 ps likely reflects the major nonradiative decay channel that is similar to that of A*. This pathway effectively competes with the
state when NO3− is present (Fig. 3c). Therefore, the magenta SADS with a lifetime of 9 ps likely reflects the major nonradiative decay channel that is similar to that of A*. This pathway effectively competes with the 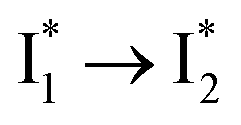 reaction as represented by the dark cyan to orange SADS transition with a lifetime of 65 ps. The slower rate of this latter pathway leads to a limited accumulation of
reaction as represented by the dark cyan to orange SADS transition with a lifetime of 65 ps. The slower rate of this latter pathway leads to a limited accumulation of 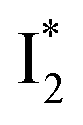 . This is in contrast to Cl− binding for which the
. This is in contrast to Cl− binding for which the 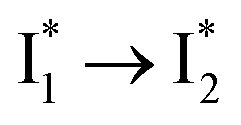 conversion with a time constant of 20 ps is more efficient (Fig. 5b, bottom panel). Therefore, the lower reaction barrier of the
conversion with a time constant of 20 ps is more efficient (Fig. 5b, bottom panel). Therefore, the lower reaction barrier of the 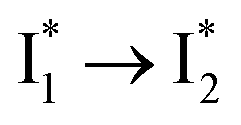 conversion underlies the turn-on fluorescence response for Cl−. The working mechanism of phiYFP as a Cl− sensor is summarized in Fig. 6, wherein the anion selectivity results from the
conversion underlies the turn-on fluorescence response for Cl−. The working mechanism of phiYFP as a Cl− sensor is summarized in Fig. 6, wherein the anion selectivity results from the 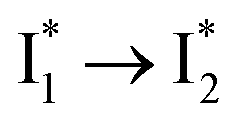 barrier that is dictated by the efficient reorganization of π–π stacking between Y203 and chromophore Y66 (see below).
barrier that is dictated by the efficient reorganization of π–π stacking between Y203 and chromophore Y66 (see below).
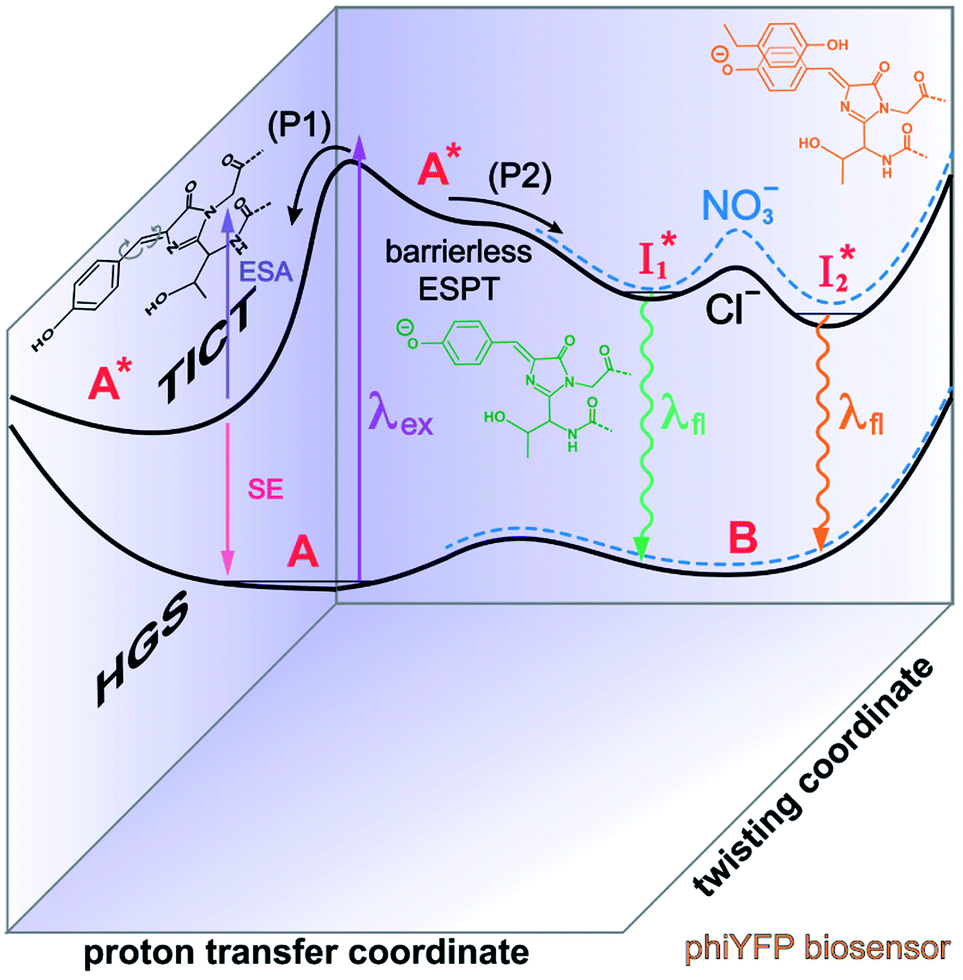 | ||
Fig. 6 Schematic potential energy surfaces of phiYFP as a chloride sensor. The excited-state dynamics are governed by two reaction coordinates: a twisting coordinate (P1) and a proton transfer coordinate (P2) as substantiated by target analysis in Fig. 5. The magenta and violet upward arrows denote the photoexcitation and excited-state absorption (ESA) of the neutral chromophore (mainly from the TICT state), respectively. The red downward arrow could represent weak stimulated emission (SE) of the TICT state (protonated chromophore, A*-like). The green and orange wavy arrows denote radiative emission of the 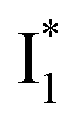 and and 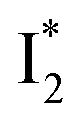 states, respectively. In the inset, the representative chromophore structures/conformations of states, respectively. In the inset, the representative chromophore structures/conformations of 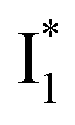 , , 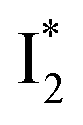 , and TICT states are depicted in green, orange, and black, respectively, by their potential energy wells. Both twisting angles of the methine bridge are depicted to represent a likely pathway for nonradiative relaxation of the initially bifurcated A* species that enters a dark state (left side). The associated ESA and red-shifted SE bands are characteristic of GFP-like chromophores in the S1 state, which differ from the transient electronic features when ESPT occurs. , and TICT states are depicted in green, orange, and black, respectively, by their potential energy wells. Both twisting angles of the methine bridge are depicted to represent a likely pathway for nonradiative relaxation of the initially bifurcated A* species that enters a dark state (left side). The associated ESA and red-shifted SE bands are characteristic of GFP-like chromophores in the S1 state, which differ from the transient electronic features when ESPT occurs. | ||
Origins of the emitting species 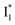 and
and 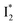
The fs-TA results show that the conversion from 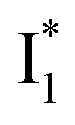
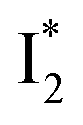
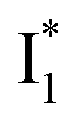 to
to 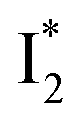 (both with deprotonated chromophore species), preceded by ESPT, is key to the excitation ratiometric response of phiYFP to Cl−. The identical emission wavelength of
(both with deprotonated chromophore species), preceded by ESPT, is key to the excitation ratiometric response of phiYFP to Cl−. The identical emission wavelength of 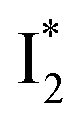 and B* (Table 1) infers that
and B* (Table 1) infers that 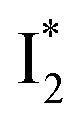 has the highest structural resemblance to B*. The red-shifted wavelength (∼540 nm) versus avGFP (∼510 nm) is caused by π–π stacking interactions between the chromophore Y66 and adjacent Y203 (Fig. 1c), which is also characteristic of avYFP (Fig. 1b). We note that the chromophore is deprotonated (buffer pH is higher than the chromophore pKa) in the crystal structure (Fig. 1c).47 To interpret
has the highest structural resemblance to B*. The red-shifted wavelength (∼540 nm) versus avGFP (∼510 nm) is caused by π–π stacking interactions between the chromophore Y66 and adjacent Y203 (Fig. 1c), which is also characteristic of avYFP (Fig. 1b). We note that the chromophore is deprotonated (buffer pH is higher than the chromophore pKa) in the crystal structure (Fig. 1c).47 To interpret 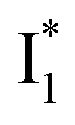 without a direct observation of the ground-state I1, we obtained the correlated spectral and computational results of phiYFP that shed light on the structural difference between
without a direct observation of the ground-state I1, we obtained the correlated spectral and computational results of phiYFP that shed light on the structural difference between 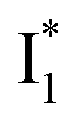 and
and 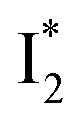 , and we can attribute
, and we can attribute 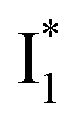 to the anionic chromophore with insignificant π–π stacking interactions with the proximal Y203 as follows.
to the anionic chromophore with insignificant π–π stacking interactions with the proximal Y203 as follows.
First, the absorption or emission of A with anion binding exhibits a decent redshift from GFP (by ∼500 cm−1 or less, Table 1), in contrast to 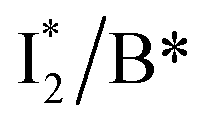 where π–π stacking plays a major role in red-shifting the peak by ∼1000 cm−1 or more.48 This implies that a neutral chromophore might experience weak π–π stacking interactions, consistent with the unchanged A peak wavelength from the apo (with 400 mM gluconate) to the 400 mM Cl−-bound phiYFP.27 Density functional theory (DFT) calculations for the neutral chromophore of phiYFP in the absence of anions show that the π–π stacking between two phenolic rings could intrinsically red-shift the S0–S1 gap to an appreciable extent (Fig. S12 in the ESI†), and the π–π stacking configurations could be characteristic of certain anion binding events near the chromophore (e.g., see Fig. 1b and c). Interestingly for comparison, a red-shifted absorption was observed for the avYFP-H148Q neutral chromophore (416 nm) in the absence of any interacting anions.18 The addition of Cl− then blue-shifts the absorption by 10–20 nm, indicating that the π–π stacking interaction of the neutral chromophore in avYFP-H148Q is likely disrupted or weakened by Cl− binding. This finding is corroborated by the crystal structures of apo and iodide-bound avYFP-H148Q.19 Compared to the apo structure, I− binding causes the Y203 hydroxyl to move toward I− due to the strong H-bond and thus distorts the bond parallelity between the phenolic rings of chromophore Y66 and Y203 (Fig. S13a, side view, ESI†), in accord with the observed electronic absorption peak blueshift.
where π–π stacking plays a major role in red-shifting the peak by ∼1000 cm−1 or more.48 This implies that a neutral chromophore might experience weak π–π stacking interactions, consistent with the unchanged A peak wavelength from the apo (with 400 mM gluconate) to the 400 mM Cl−-bound phiYFP.27 Density functional theory (DFT) calculations for the neutral chromophore of phiYFP in the absence of anions show that the π–π stacking between two phenolic rings could intrinsically red-shift the S0–S1 gap to an appreciable extent (Fig. S12 in the ESI†), and the π–π stacking configurations could be characteristic of certain anion binding events near the chromophore (e.g., see Fig. 1b and c). Interestingly for comparison, a red-shifted absorption was observed for the avYFP-H148Q neutral chromophore (416 nm) in the absence of any interacting anions.18 The addition of Cl− then blue-shifts the absorption by 10–20 nm, indicating that the π–π stacking interaction of the neutral chromophore in avYFP-H148Q is likely disrupted or weakened by Cl− binding. This finding is corroborated by the crystal structures of apo and iodide-bound avYFP-H148Q.19 Compared to the apo structure, I− binding causes the Y203 hydroxyl to move toward I− due to the strong H-bond and thus distorts the bond parallelity between the phenolic rings of chromophore Y66 and Y203 (Fig. S13a, side view, ESI†), in accord with the observed electronic absorption peak blueshift.
In analogy, the absorption peak at ∼400 nm with no blueshift upon halide binding in phiYFP27 indicates that the neutral chromophore is likely in an unoptimized π–π stacked conformation even in the apo phiYFP. Further evidence could be provided by X-ray crystallography performed at a lower pH than the chromophore pKa value. In our fs-TA experiments, due to the ultrafast ESPT reaction that does not allow significant environment reorganization, the nascent photoproduct 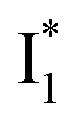 and nearby residues would remain in a similar conformation to A*.29,32,36 The fact that
and nearby residues would remain in a similar conformation to A*.29,32,36 The fact that 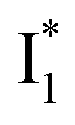 emits at a similar wavelength (∼500–510 nm) to the anionic GFP chromophore (Table 1) also implies the absence or significant lack of π–π stacking for
emits at a similar wavelength (∼500–510 nm) to the anionic GFP chromophore (Table 1) also implies the absence or significant lack of π–π stacking for 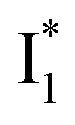 versus
versus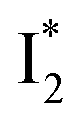 . Further support arises from our DFT calculations for the phiYFP anionic chromophore. As the phenolic ring of Y203 is rotated away from the optimal antiparallel position with chromophore Y66, the calculated S0–S1 energy gap is shifted to bluer wavelengths (Fig. S14 in the ESI†). Though the calculations do not involve the anions explicitly, we focus on the intrinsic effect and general implications (without specific consideration of a unique protein matrix or employing the high-level full-scale QM/MM calculations from the electronic ground to excited states) of π–π stacking configurations that sample the chromophore phase space directly modulated by the anion binding events.
. Further support arises from our DFT calculations for the phiYFP anionic chromophore. As the phenolic ring of Y203 is rotated away from the optimal antiparallel position with chromophore Y66, the calculated S0–S1 energy gap is shifted to bluer wavelengths (Fig. S14 in the ESI†). Though the calculations do not involve the anions explicitly, we focus on the intrinsic effect and general implications (without specific consideration of a unique protein matrix or employing the high-level full-scale QM/MM calculations from the electronic ground to excited states) of π–π stacking configurations that sample the chromophore phase space directly modulated by the anion binding events.
Second, the dimness of 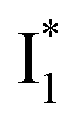 suggests a weak π–π stacking interaction. The high FQY of B* (0.44 in the presence of 400 mM Cl−)27 suggests that π–π stacking suppresses nonradiative decay pathways. This mechanism is nicely supported by site-directed mutagenesis of phiYFP: the mutation of Y203 by residues such as valine, threonine, and serine results in reduced fluorescence.47,49 We note that phiYFPv was crystallized from aqueous buffer containing 20 mM Tris–HCl at pH = 8 with 200 mM NaCl,47 while wild-type phiYFP was dissolved in 20 mM Tris–HCl buffer at pH = 8 with 100 mM NaCl in an earlier study,49 so the associated spectral data of phiYFP and its mutants taken in such buffer solutions could involve Cl− binding and its effect on the chromophore environment. Meanwhile, these mutations blue-shift the absorption and emission with respect to the wild-type phiYFP,49,50 supporting the assignment of
suggests a weak π–π stacking interaction. The high FQY of B* (0.44 in the presence of 400 mM Cl−)27 suggests that π–π stacking suppresses nonradiative decay pathways. This mechanism is nicely supported by site-directed mutagenesis of phiYFP: the mutation of Y203 by residues such as valine, threonine, and serine results in reduced fluorescence.47,49 We note that phiYFPv was crystallized from aqueous buffer containing 20 mM Tris–HCl at pH = 8 with 200 mM NaCl,47 while wild-type phiYFP was dissolved in 20 mM Tris–HCl buffer at pH = 8 with 100 mM NaCl in an earlier study,49 so the associated spectral data of phiYFP and its mutants taken in such buffer solutions could involve Cl− binding and its effect on the chromophore environment. Meanwhile, these mutations blue-shift the absorption and emission with respect to the wild-type phiYFP,49,50 supporting the assignment of 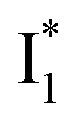 and
and 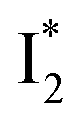 whose electronic transition wavelengths are sensitive to π–π stacking interactions. Our steady-state (FQY) and fs-TA (lifetime) results demonstrate that both A* and
whose electronic transition wavelengths are sensitive to π–π stacking interactions. Our steady-state (FQY) and fs-TA (lifetime) results demonstrate that both A* and 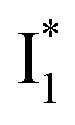 are much less fluorescent than
are much less fluorescent than 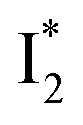 (Fig. 2 and 3). Weak fluorescence of A* is correlated with bifurcated pathways where nonradiative twisting motions and ultrafast ESPT constitute the dominant decay dynamics (Fig. 5 and 6). Similarly, weak fluorescence of
(Fig. 2 and 3). Weak fluorescence of A* is correlated with bifurcated pathways where nonradiative twisting motions and ultrafast ESPT constitute the dominant decay dynamics (Fig. 5 and 6). Similarly, weak fluorescence of 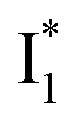 could be a consequence of two competing nonradiative channels: one involves the TICT-mediated decay characterized by the magenta → dark cyan SADS on the tens of ps timescale (see Fig. 5 bottom panels, showing low/high weight with Cl−/NO3− binding, respectively); the other is the
could be a consequence of two competing nonradiative channels: one involves the TICT-mediated decay characterized by the magenta → dark cyan SADS on the tens of ps timescale (see Fig. 5 bottom panels, showing low/high weight with Cl−/NO3− binding, respectively); the other is the 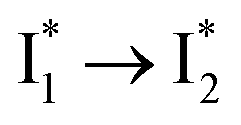 conversion (more prominent with Cl− than NO3−), highlighted by a notable SE peak redshift on the 20 ps timescale in the presence of 400 mM Cl− (Fig. 5b bottom panel). The former pathway is rationalized by the
conversion (more prominent with Cl− than NO3−), highlighted by a notable SE peak redshift on the 20 ps timescale in the presence of 400 mM Cl− (Fig. 5b bottom panel). The former pathway is rationalized by the 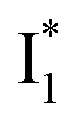 conformation: it is produced by an ultrafast (<100 fs) ESPT reaction that yields a largely intact nuclear arrangement as A* (particularly with weak π–π stacking).32 The corresponding local environment presumably favors the chromophore ring-twisting-induced decay pathways, which become more kinetically dominant when the competing pathway, i.e., optimization of the π–π stacking, occurs on a slower timescale (especially in the NO3−-bound state, see Fig. 5c).
conformation: it is produced by an ultrafast (<100 fs) ESPT reaction that yields a largely intact nuclear arrangement as A* (particularly with weak π–π stacking).32 The corresponding local environment presumably favors the chromophore ring-twisting-induced decay pathways, which become more kinetically dominant when the competing pathway, i.e., optimization of the π–π stacking, occurs on a slower timescale (especially in the NO3−-bound state, see Fig. 5c).
Correlations between anion binding and ratiometric fluorescence response
The excellent ratiometric fluorescence response differentiates phiYFP from the previously reported Cl− sensors such as avYFP-H148Q, which by itself can only work as an intensiometric indicator and lacks selectivity between halides and other anions such as NO3−. In our study, the excited-state dynamics of phiYFP have been elucidated with a key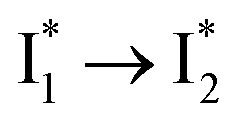 conversion governing the excitation ratiometric response that is highly sensitive to Cl− binding. Notably, NO3− leads to turn-off fluorescence of
conversion governing the excitation ratiometric response that is highly sensitive to Cl− binding. Notably, NO3− leads to turn-off fluorescence of 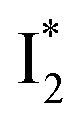 that essentially causes poor ratiometric responses. A closer inspection of the fluorescence spectra shows that the
that essentially causes poor ratiometric responses. A closer inspection of the fluorescence spectra shows that the 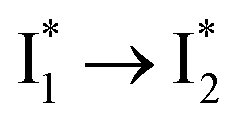 conversion is slightly hindered by Cl− binding. Such a hindrance becomes more significant for Br−, I−, and NO3− and is in the order of Cl− < Br− < I− < NO3− (Fig. S1 in the ESI†). This result is in line with their binding affinities,27 suggesting that anion binding commonly impedes the
conversion is slightly hindered by Cl− binding. Such a hindrance becomes more significant for Br−, I−, and NO3− and is in the order of Cl− < Br− < I− < NO3− (Fig. S1 in the ESI†). This result is in line with their binding affinities,27 suggesting that anion binding commonly impedes the 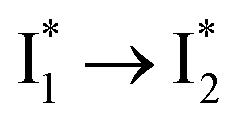 conversion, i.e., optimization of π–π stacking. It is rationalizable in that the anion binding would distort the antiparallel conformation between the chromophore Y66 phenolate and Y203 phenolic rings (see Fig. 1c for phiYFPv, and Fig. S13 in the ESI† for detailed comparisons between the phiYFPv and avYFP-H148Q crystal structures) due to the electrostatic interaction between the anion and Y203 phenolic hydroxyl, thus exerting an energy barrier for the optimization of the π–π stacking interactions to reach the
conversion, i.e., optimization of π–π stacking. It is rationalizable in that the anion binding would distort the antiparallel conformation between the chromophore Y66 phenolate and Y203 phenolic rings (see Fig. 1c for phiYFPv, and Fig. S13 in the ESI† for detailed comparisons between the phiYFPv and avYFP-H148Q crystal structures) due to the electrostatic interaction between the anion and Y203 phenolic hydroxyl, thus exerting an energy barrier for the optimization of the π–π stacking interactions to reach the 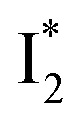 state.
state.
Meanwhile, the addition of Cl− results in a simultaneous increase of 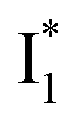 and
and 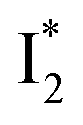 fluorescence bands (Fig. S1a in the ESI†), indicative of an increased
fluorescence bands (Fig. S1a in the ESI†), indicative of an increased 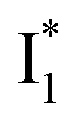 population as a result of ESPT, following excitation of the A form at 400 nm. This is because more neutral populations are excited due to the increased pKa, which also occurs for other anions in the order by potencies: Cl− < Br− < I− ∼ NO3− (see Fig. S1 in the ESI†). In aggregate, these results show that the different ratiometric responses of phiYFP to Cl− and NO3− originate from an intrinsic competition between the pKa increase (thus more A* and then
population as a result of ESPT, following excitation of the A form at 400 nm. This is because more neutral populations are excited due to the increased pKa, which also occurs for other anions in the order by potencies: Cl− < Br− < I− ∼ NO3− (see Fig. S1 in the ESI†). In aggregate, these results show that the different ratiometric responses of phiYFP to Cl− and NO3− originate from an intrinsic competition between the pKa increase (thus more A* and then 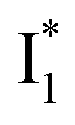 generation) and the inhibition of
generation) and the inhibition of 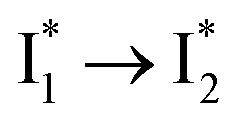 conversion. The dominance of the former factor leads to the turn-on response of phiYFP upon Cl− binding. This finding also explains why the best ratiometric performance can be achieved at pH values around the chromophore pKa where population interchange between the neutral and anionic forms is the most significant (with the largest slope in the titration curve when pH = pKa).
conversion. The dominance of the former factor leads to the turn-on response of phiYFP upon Cl− binding. This finding also explains why the best ratiometric performance can be achieved at pH values around the chromophore pKa where population interchange between the neutral and anionic forms is the most significant (with the largest slope in the titration curve when pH = pKa).
We propose that anion binding affinity is the dominant factor for both chromophore pKa and 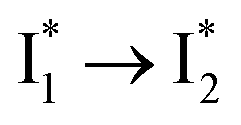 conversion in phiYFP. First, the pKa increase is reflective of the free energy change in the ground state. Anions with higher binding affinities suppress the charge delocalization of the anionic chromophore to a larger extent and thus increase pKa more (Table 1). Second, anion binding hinders the
conversion in phiYFP. First, the pKa increase is reflective of the free energy change in the ground state. Anions with higher binding affinities suppress the charge delocalization of the anionic chromophore to a larger extent and thus increase pKa more (Table 1). Second, anion binding hinders the 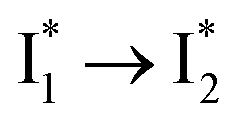 conversion because of greater destabilization of
conversion because of greater destabilization of 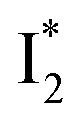 than
than 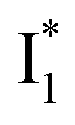 and hence a decreased free energy change. This is corroborated by our spectral observation that
and hence a decreased free energy change. This is corroborated by our spectral observation that 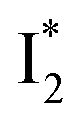 with Cl− binding emits at a redder wavelength (542 nm) than with NO3− binding (534 nm), whereas
with Cl− binding emits at a redder wavelength (542 nm) than with NO3− binding (534 nm), whereas 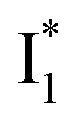 emits at similar wavelengths (∼505 nm) for both cases (Fig. S2 in the ESI,† and Table 1). Observations at a lower pH of 4.5 yielded similar trends (Fig. S4 and Table S1, ESI†). The destabilization of
emits at similar wavelengths (∼505 nm) for both cases (Fig. S2 in the ESI,† and Table 1). Observations at a lower pH of 4.5 yielded similar trends (Fig. S4 and Table S1, ESI†). The destabilization of 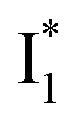 by anion binding (see Fig. S4e in the ESI†) is due to the repulsive interaction between the anion and delocalized negative charge of the chromophore. The extra destabilization effect on
by anion binding (see Fig. S4e in the ESI†) is due to the repulsive interaction between the anion and delocalized negative charge of the chromophore. The extra destabilization effect on 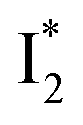 (similar to B* in Fig. S4f, ESI†), besides electrostatic repulsion, may result from the weakening of π–π stacking due to anion binding that changes the relative conformation of Y203 and the nearby chromophore (e.g., Fig. S13a in the ESI†).51–53 Regarding the anion binding affinity, previous studies on avYFP-H148Q suggested it to be correlated with, but not limited to, the ion dehydration energy: Cl− > Br− > NO3− > I−.18,54,55 The protein interaction usually increases with decreasing dehydration energy. Other factors such as the size, electrostatic configuration, and symmetry of anions (e.g., trigonal nitrate versus spherical halide)18 may also contribute to the radiative emission properties of the chromophore embedded in the protein matrix that hosts the bound ions.
(similar to B* in Fig. S4f, ESI†), besides electrostatic repulsion, may result from the weakening of π–π stacking due to anion binding that changes the relative conformation of Y203 and the nearby chromophore (e.g., Fig. S13a in the ESI†).51–53 Regarding the anion binding affinity, previous studies on avYFP-H148Q suggested it to be correlated with, but not limited to, the ion dehydration energy: Cl− > Br− > NO3− > I−.18,54,55 The protein interaction usually increases with decreasing dehydration energy. Other factors such as the size, electrostatic configuration, and symmetry of anions (e.g., trigonal nitrate versus spherical halide)18 may also contribute to the radiative emission properties of the chromophore embedded in the protein matrix that hosts the bound ions.
Difference between phiYFP and avYFPs
Despite the high similarity of phiYFP and avYFPs, the latter proteins cannot be effectively used in an excitation ratiometric mode without fusion with another FP. This is probably a result of low ESPT efficiency that leads to weak fluorescence of the anionic chromophore. Weak fluorescence also occurs for the neutral form due to out-of-plane torsions as suggested.56 Recently, avYFP-H148Q was examined upon the neutral form excitation (at 400 nm) and showed turn-off fluorescence in response to Cl− binding without two anionic species such as in phiYFP (i.e.,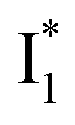 and
and 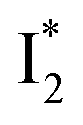 ).27 The contrasting excited-state dynamics highlight the difference in the local environment of neutral chromophores of the two FPs.
).27 The contrasting excited-state dynamics highlight the difference in the local environment of neutral chromophores of the two FPs.
As unraveled from the above spectral analysis, the neutral chromophore of phiYFP undergoes ultrafast ESPT (faster than our ∼80 fs instrument response time, see Experimental methods in the ESI†) in competition with a twisting-motion-induced nonradiative decay pathway. The Cl− binding raises the pKa and consequently leads to an increased ESPT product 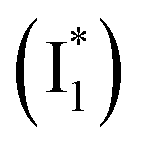 due to photoexcitation of more A species. In contrast, avYFP-H148Q exhibits a hindered ESPT reaction as reflected by the decreasing anionic form fluorescence, even though Cl− binding raises the pKa.27 This finding indicates that, for some reason, Cl− binding could reduce the ESPT efficiency, which requires further time-resolved spectral characterization of avYFP. In contrast, the observed ultrafast ESPT rate of phiYFP is insensitive to anion binding (e.g., Cl− and NO3−) and reminiscent of the avGFP mutant S65T/H148D, wherein ultrafast ESPT occurs and a short H-bond was identified between the chromophore hydroxyl and D148 in the crystal structure (see Fig. 1a for avGFP).57–59 Unfortunately, the crystal structure of phiYFP at low pH (having dominant neutral chromophores) remains unavailable.
due to photoexcitation of more A species. In contrast, avYFP-H148Q exhibits a hindered ESPT reaction as reflected by the decreasing anionic form fluorescence, even though Cl− binding raises the pKa.27 This finding indicates that, for some reason, Cl− binding could reduce the ESPT efficiency, which requires further time-resolved spectral characterization of avYFP. In contrast, the observed ultrafast ESPT rate of phiYFP is insensitive to anion binding (e.g., Cl− and NO3−) and reminiscent of the avGFP mutant S65T/H148D, wherein ultrafast ESPT occurs and a short H-bond was identified between the chromophore hydroxyl and D148 in the crystal structure (see Fig. 1a for avGFP).57–59 Unfortunately, the crystal structure of phiYFP at low pH (having dominant neutral chromophores) remains unavailable.
To mitigate this issue, we dissect the high-pH crystal structure (with an anionic chromophore, Fig. 1c)47 to gain more insights into ESPT. The H-bonding chain, chromophore → water → S205 → E222, has been widely accepted to enable ESPT in wild-type GFP and its many mutants.29,60 However, it is unlikely to be the ESPT route in phiYFP due to truncation of this H-bonding chain by V205 (Fig. 1c), which is incompatible with the ultrafast ESPT pathway starting from the phenolic hydroxyl group.28,61,62 Therefore, it is highly plausible that H146 with a short distance to the chromophore hydroxyl (2.8 Å for N⋯O at pH = 8 with a deprotonated chromophore) acts as the proton acceptor by forming a short H-bond with the chromophore hydroxyl in its neutral form, thus supporting an ultrafast ESPT as observed (Fig. 3 and 4). Meanwhile, we cannot exclude the possibility that ESPT could occur through an adjacent water molecule and then V205 or T144 (H-bonded to the water molecule with distances of 3.1 and 2.8 Å, respectively, at pH = 8), connecting to the exterior of protein. The low-pH crystal structure of phiYFP is expected to further elucidate such a barrierless ESPT reaction from a neutral chromophore. Moreover, the ESPT occurrence upon H148Q mutation (confirmed by the observation of emission from the anionic chromophore by excitation of the neutral chromophore)27,31 implies that the chromophore → water → S205 → E222 H-bonding chain constitutes the main ESPT route for avYFP-H148Q, which likely occurs on the ∼10 ps timescale28,29,63 that is much longer than that in phiYFP (see Fig. 5 and 6).
Perspectives on protein engineering for live-cell imaging
The wild-type phiYFP exhibits the reddest emission maximum of all FPs with an unmodified p-HBDI chromophore (GFP chromophore)27,49 and shows high selectivity to halides in the excitation ratiometric measurements, which makes it a promising standalone Cl− indicator. On the FP color tuning side, phiYFP emission is reminiscent of Boxer's recent work on the anionic GFP chromophore that adopts a diabatic two-state model to explain the color-tuning mechanism.64 Any effort to destabilize the negative charge on the phenolate leads to a spectral redshift. Increasing the electron density around or at the phenolate ring such as incorporating π–π stacking (e.g., an adjacent Y203 here) or electron-donating groups would red-shift the anionic chromophore emission.44,65 On the ion-sensing front, the low binding affinity (Kd ∼ 300–400 mM at pH = 5–6) makes the current phiYFP unsuitable for intracellular detection of Cl− concentrations without further engineering. In order to improve the binding affinity, one direct approach is to strengthen the electrostatic interaction between the anion and protein residues that constitute the binding pocket. Recent mutagenesis of phiYFP at Q69 showed that the replacement with less polar residues such as leucine reduces the binding affinity and consequently weakens ratiometric responses.27 Conversely, we envision the mutation of Q69 by a more polar residue such as lysine, which has similar sterics to glutamine and does not induce other significant conformational changes in the pocket near the chromophore imidazolinone ring (Fig. 1c), may help to improve the halide binding affinity.66It is also noteworthy that the anion binding is closely correlated with the protonation state of the chromophore, which accounts for the pH-dependent Cl− binding affinity of phiYFP as well as avYFPs and E2GFP.19,20 This is typical for many host/guest systems in which the binding free energy is changed by the electrostatics of the binding partners due to the specific protonation state.67,68 The quantitative description of Cl−/protein (the embedded chromophore with different charge states) binding can be found in the literature.19,69 Analogous to avYFPs, the protonated phiYFP chromophore has a much higher binding affinity to Cl− than the deprotonated chromophore. The apparent binding affinity lies somewhere between the two extremes and is higher at low pH.19,27 Furthermore, the best ratiometric performance of phiYFP (by the ratio of 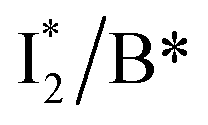 fluorescence with A and B excitations) is at pH values around the chromophore pKa due to a pronounced A/B population interchange. This is because the ratiometric response is governed by an interplay between the
fluorescence with A and B excitations) is at pH values around the chromophore pKa due to a pronounced A/B population interchange. This is because the ratiometric response is governed by an interplay between the 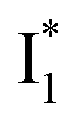 population increase due to the greater pKa and the
population increase due to the greater pKa and the 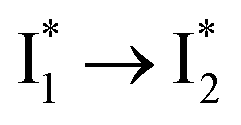 inhibition. It also agrees with the observation that the phiYFP ratiometric response is diminished at pH values below 5 (e.g., Fig. S4 in the ESI†) owing to the small increase of
inhibition. It also agrees with the observation that the phiYFP ratiometric response is diminished at pH values below 5 (e.g., Fig. S4 in the ESI†) owing to the small increase of 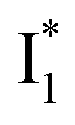 population and a more drastic
population and a more drastic 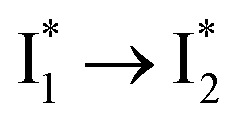 inhibition caused by stronger binding of anions.27 Therefore, upshifting the chromophore pKa (ideally with a higher slope of pKa increase from apo to anion-bound proteins, see Table 1 for the trend) is beneficial for the binding affinity and operational pH for the physiological Cl− detection.
inhibition caused by stronger binding of anions.27 Therefore, upshifting the chromophore pKa (ideally with a higher slope of pKa increase from apo to anion-bound proteins, see Table 1 for the trend) is beneficial for the binding affinity and operational pH for the physiological Cl− detection.
To raise the chromophore pKa and hence the operational pH, modifications of electrostatics in the chromophore Y66 moiety would be more effective than that in the T65 moiety.70,71 The comparison of phiYFP with avYFPs and other GFP variants with ESPT capability indicates the importance of residue 148 (146 for phiYFP) and 205 in both ground- and excited-state proton transfer.57,58,61 Since V205 in phiYFP interrupts the common H-bonding chain (with S205) necessary for ESPT, while the hydrophobic V205 was shown to stabilize the conformational state of Y203 (Fig. 1c) and maintain high brightness,47 the proximal H146 could be an effective target for future mutagenesis. For instance, H146Q may simultaneously increase the chromophore pKa and the Cl− binding affinity, reminiscent of avYFP-H148Q;19,27 however, any significant disruption of the aforementioned ESPT pathway (due to the replacement of H146 by a much weaker proton acceptor Q146) may abolish the ratiometric response of the protein. Alternatively, since the GFP variant S65T/H148D establishes a short H-bond between the chromophore hydroxyl and D148 and leads to ultrafast ESPT with an increased pKa (∼8),58 this mechanism is potentially applicable for phiYFP wherein the barrierless ESPT (Fig. 5 and 6) implies a short H-bond to promptly generate 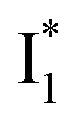 species.72 We can anticipate that the slightly less steric aspartate (i.e., H146D mutation) might retain an ultrafast ESPT reaction while increasing the same TYG (T65–Y66–G67) chromophore pKa by establishing a similar H-bonding interaction in phiYFP to that in avGFP-S65T/H148D. In addition, monomerization73,74 and increase of the FQY9,32,70 with multiple correlated mutations could further enhance the phiYFP usability in live-cell imaging applications.9,14
species.72 We can anticipate that the slightly less steric aspartate (i.e., H146D mutation) might retain an ultrafast ESPT reaction while increasing the same TYG (T65–Y66–G67) chromophore pKa by establishing a similar H-bonding interaction in phiYFP to that in avGFP-S65T/H148D. In addition, monomerization73,74 and increase of the FQY9,32,70 with multiple correlated mutations could further enhance the phiYFP usability in live-cell imaging applications.9,14
Conclusions
By implementing fs-TA spectroscopy and target analysis, aided by a series of key control samples and quantum calculations, we investigated the excited-state dynamics of the wild-type yellow fluorescent protein from jellyfish Phialidium sp., phiYFP. It is to date the only reported standalone FP to act as an excitation ratiometric biosensor for Cl−. It demonstrates high selectivity to halides over many other anions. The probe-dependent electronic dynamics in combination with target analysis of fs-TA spectra delineate the previously hidden branched reaction pathways of the photoexcited phiYFP, which effectively powers the appealing turn-on fluorescence with Cl− binding. Upon photoexcitation, the neutral chromophore promptly bifurcates and evolves along two reaction coordinates: the twisting motion-induced nonradiative decay (major)38–43,75 and ultrafast barrierless ESPT (minor). The ESPT pathway yields a nascent anionic product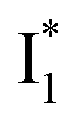 that converts to a red-shifted
that converts to a red-shifted 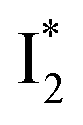 state on the tens of ps timescale. The
state on the tens of ps timescale. The 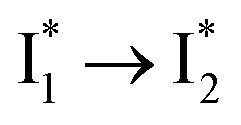 conversion is intrinsically inhibited by anion binding, the extent of which depends on the binding affinity. The higher binding affinity of NO3− than Cl− thus yields a more reduced
conversion is intrinsically inhibited by anion binding, the extent of which depends on the binding affinity. The higher binding affinity of NO3− than Cl− thus yields a more reduced 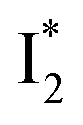 population. Meanwhile, anion binding raises the chromophore pKa and leads to a larger ESPT population (hence more deprotonated
population. Meanwhile, anion binding raises the chromophore pKa and leads to a larger ESPT population (hence more deprotonated 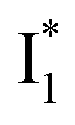 generation after excitation of the neutral A species).
generation after excitation of the neutral A species).
In essence, the turn-on/turn-off fluorescence of 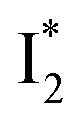 , which determines the anion selectivity in the ratiometric response, is a result of active competition between the ESPT population increase and inhibition of
, which determines the anion selectivity in the ratiometric response, is a result of active competition between the ESPT population increase and inhibition of 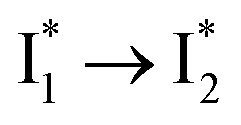 conversion. When the former factor dominates, phiYFP exhibits a turn-on fluorescence response and acts as an excellent excitation ratiometric biosensor for anions such as Cl−. In addition, the correlated spectral and crystal structure results uncover the origins of
conversion. When the former factor dominates, phiYFP exhibits a turn-on fluorescence response and acts as an excellent excitation ratiometric biosensor for anions such as Cl−. In addition, the correlated spectral and crystal structure results uncover the origins of 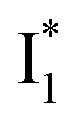 and
and 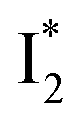 :
: 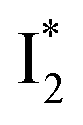 is the anionic chromophore with an optimized π–π stacking interaction between the chromophore Y66 phenolate and Y203 phenol, whereas
is the anionic chromophore with an optimized π–π stacking interaction between the chromophore Y66 phenolate and Y203 phenol, whereas 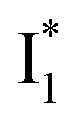 is the anionic chromophore with a lack of π–π stacking. Based on these fundamental insights, we proposed rational design strategies that focus on active sites to improve the anion binding affinity and raise the operational pH for cellular imaging advances. Therefore, our work allows comprehensive understanding of the working mechanism of phiYFP as a turn-on fluorescence, excitation ratiometric biosensor for Cl−. Further rational engineering of phiYFP, versatile and effective as a standalone fluorescent protein without fusion with other proteins relying on FRET, is expected to make timely and significant contributions in a broad range of fields from photophysics, protein engineering/de novo design, to biosensing applications.
is the anionic chromophore with a lack of π–π stacking. Based on these fundamental insights, we proposed rational design strategies that focus on active sites to improve the anion binding affinity and raise the operational pH for cellular imaging advances. Therefore, our work allows comprehensive understanding of the working mechanism of phiYFP as a turn-on fluorescence, excitation ratiometric biosensor for Cl−. Further rational engineering of phiYFP, versatile and effective as a standalone fluorescent protein without fusion with other proteins relying on FRET, is expected to make timely and significant contributions in a broad range of fields from photophysics, protein engineering/de novo design, to biosensing applications.
Data availability
The datasets with analysis and discussions supporting this article have been incorporated and presented as part of the comprehensive ESI.†Author contributions
Conceptualization, C. F. and S. C. D.; methodology, C. C., J. N. T., L. T., L. Z. and W. S. Y. O.; software, C. C. and L. T.; formal analysis, C. C., J. N. T., L. T. and W. S. Y. O.; investigation, C. C., J. N. T., L. T., L. Z. and W. S. Y. O.; visualization, C. C., J. N. T., L. T. and C. F.; writing—original draft, C. C., J. N. T. and L. T.; writing—review and editing, C. F. and S. C. D.; supervision, C. F. and S. C. D.; funding acquisition, C. F. and S. C. D. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the U.S. NSF grants CHE-2003550 and MCB-1817949 (to C. F.), the University of Texas at Dallas startup funds, the Welch Foundation grant AT-1918-20170325, and the NIH grant R35GM128923 (to S. C. D.). The Wei Family Private Foundation Scholarship (2014–2020) and Department of Chemistry Graduate Fellowship (Summer 2020) at Oregon State University (to C. C.) are also gratefully acknowledged.Notes and references
- T. J. Jentsch, V. Stein, F. Weinreich and A. A. Zdebik, Physiol. Rev., 2002, 82, 503–568 CrossRef CAS PubMed.
- M. Suzuki, T. Morita and T. Iwamoto, Cell. Mol. Life Sci., 2005, 63, 12 CrossRef PubMed.
- H. Pasantes-Morales, R. A. Lezama, G. Ramos-Mandujano and K. L. Tuz, Am. J. Med., 2006, 119, S4–S11 CrossRef CAS PubMed.
- B. Kerem, J. M. Rommens, J. A. Buchanan, D. Markiewicz, T. K. Cox, A. Chakravarti, M. Buchwald and L. C. Tsui, Science, 1989, 245, 1073–1080 CrossRef CAS PubMed.
- S. E. Lloyd, S. H. S. Pearce, S. E. Fisher, K. Steinmeyer, B. Schwappach, S. J. Scheinman, B. Harding, A. Bolino, M. Devoto, P. Goodyer, S. P. A. Rigden, O. Wrong, T. J. Jentsch, I. W. Craig and R. V. Thakker, Nature, 1996, 379, 445–449 CrossRef CAS PubMed.
- H. Lerche, Y. G. Weber, K. Jurkat-Rott and F. Lehmann-Horn, Curr. Pharm. Des., 2005, 11, 2737–2752 CrossRef CAS PubMed.
- A. S. Verkman and L. J. V. Galietta, Nat. Rev. Drug Discovery, 2009, 8, 153–171 CrossRef CAS PubMed.
- P. Bregestovski, T. Waseem and M. Mukhtarov, Front. Mol. Neurosci., 2009, 2, 15 Search PubMed.
- D. Arosio and G. M. Ratto, Front. Cell. Neurosci., 2014, 8, 258 Search PubMed.
- T. D. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2015, 44, 4547–4595 RSC.
- A. S. Verkman, Am. J. Physiol.: Cell Physiol., 1990, 259, C375–C388 CrossRef CAS PubMed.
- S. Jayaraman, J. Biwersi and A. S. Verkman, Am. J. Physiol.: Cell Physiol., 1999, 276, C747–C757 CrossRef CAS PubMed.
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS.
- M. Zajac, K. Chakraborty, S. Saha, V. Mahadevan, D. T. Infield, A. Accardi, Z. Qiu and Y. Krishnan, J. Cell Sci., 2020, 133, jcs240390 CrossRef CAS PubMed.
- R. Y. Tsien, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- R. M. Wachter, M.-A. Elsliger, K. Kallio, G. T. Hanson and S. J. Remington, Structure, 1998, 6, 1267–1277 CrossRef CAS PubMed.
- D. Arosio, G. Garau, F. Ricci, L. Marchetti, R. Bizzarri, R. Nifosì and F. Beltram, Biophys. J., 2007, 93, 232–244 CrossRef CAS PubMed.
- S. Jayaraman, P. Haggie, R. M. Wachter, S. J. Remington and A. S. Verkman, J. Biol. Chem., 2000, 275, 6047–6050 CrossRef CAS PubMed.
- R. M. Wachter, D. Yarbrough, K. Kallio and S. J. Remington, J. Mol. Biol., 2000, 301, 157–171 CrossRef CAS PubMed.
- T. Kuner and G. J. Augustine, Neuron, 2000, 27, 447–459 CrossRef CAS PubMed.
- E. A. Souslova and D. M. Chudakov, Microsc. Res. Tech., 2006, 69, 207–209 CrossRef CAS PubMed.
- O. Markova, M. Mukhtarov, E. Real, Y. Jacob and P. Bregestovski, J. Neurosci. Methods, 2008, 170, 67–76 CrossRef CAS PubMed.
- D. Arosio, F. Ricci, L. Marchetti, R. Gualdani, L. Albertazzi and F. Beltram, Nat. Methods, 2010, 7, 516–518 CrossRef CAS PubMed.
- J. S. Grimley, L. Li, W. Wang, L. Wen, L. S. Beese, H. W. Hellinga and G. J. Augustine, J. Neurosci., 2013, 33, 16297–16309 CrossRef CAS PubMed.
- S. Saha, V. Prakash, S. Halder, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 645–651 CrossRef CAS PubMed.
- J. M. Paredes, A. I. Idilli, L. Mariotti, G. Losi, L. R. Arslanbaeva, S. S. Sato, P. Artoni, J. Szczurkowska, L. Cancedda, G. M. Ratto, G. Carmignoto and D. Arosio, ACS Chem. Biol., 2016, 11, 1652–1660 CrossRef CAS PubMed.
- J. N. Tutol, W. Peng and S. C. Dodani, Biochemistry, 2019, 58, 31–35 CrossRef CAS PubMed.
- M. Chattoraj, B. A. King, G. U. Bublitz and S. G. Boxer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8362–8367 CrossRef CAS PubMed.
- C. Fang, R. R. Frontiera, R. Tran and R. A. Mathies, Nature, 2009, 462, 200–204 CrossRef CAS PubMed.
- C. Fang, L. Tang and C. Chen, J. Chem. Phys., 2019, 151, 200901 CrossRef PubMed.
- B. G. Oscar, W. Liu, Y. Zhao, L. Tang, Y. Wang, R. E. Campbell and C. Fang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10191–10196 CrossRef CAS PubMed.
- C. Fang and L. Tang, Annu. Rev. Phys. Chem., 2020, 71, 239–265 CrossRef CAS PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- C. Chen, L. Zhu, S. A. Boulanger, N. S. Baleeva, I. N. Myasnyanko, M. S. Baranov and C. Fang, J. Chem. Phys., 2020, 152, 021101 CrossRef CAS PubMed.
- S. R. Meech, Chem. Soc. Rev., 2009, 38, 2922–2934 RSC.
- C. Chen, W. Liu, M. S. Baranov, N. S. Baleeva, I. V. Yampolsky, L. Zhu, Y. Wang, A. Shamir, K. M. Solntsev and C. Fang, J. Phys. Chem. Lett., 2017, 8, 5921–5928 CrossRef CAS PubMed.
- D. Mandal, T. Tahara and S. R. Meech, J. Phys. Chem. B, 2004, 108, 1102–1108 CrossRef CAS.
- M. E. Martin, F. Negri and M. Olivucci, J. Am. Chem. Soc., 2004, 126, 5452–5464 CrossRef CAS PubMed.
- P. Altoe, F. Bernardi, M. Garavelli, G. Orlandi and F. Negri, J. Am. Chem. Soc., 2005, 127, 3952–3963 CrossRef CAS PubMed.
- I. V. Polyakov, B. L. Grigorenko, E. M. Epifanovsky, A. I. Krylov and A. V. Nemukhin, J. Chem. Theory Comput., 2010, 6, 2377–2387 CrossRef CAS PubMed.
- A. Svendsen, H. V. Kiefer, H. B. Pedersen, A. V. Bochenkova and L. H. Andersen, J. Am. Chem. Soc., 2017, 139, 8766–8771 CrossRef CAS PubMed.
- M. A. Taylor, L. Zhu, N. D. Rozanov, K. T. Stout, C. Chen and C. Fang, Phys. Chem. Chem. Phys., 2019, 21, 9728–9739 RSC.
- C. Chen and C. Fang, Chem.–Asian J., 2020, 15, 1514–1523 CrossRef CAS PubMed.
- S. P. Laptenok, J. Conyard, P. C. B. Page, Y. Chan, M. You, S. R. Jaffrey and S. R. Meech, Chem. Sci., 2016, 7, 5747–5752 RSC.
- R. M. Wachter and S. J. Remington, Curr. Biol., 1999, 9, R628–R629 CrossRef CAS PubMed.
- N. V. Pletneva, V. Z. Pletnev, E. Souslova, D. M. Chudakov, S. Lukyanov, V. I. Martynov, S. Arhipova, I. Artemyev, A. Wlodawer, Z. Dauter and S. Pletnev, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1005–1012 CrossRef CAS PubMed.
- R. Nifosì, B. Mennucci and C. Filippi, Phys. Chem. Chem. Phys., 2019, 21, 18988–18998 RSC.
- A. A. Pakhomov and V. I. Martynov, Biochem. Biophys. Res. Commun., 2011, 407, 230–235 CrossRef CAS PubMed.
- A. A. Pakhomov, S. A. Tretyakova and V. I. Martynov, Dokl. Biochem. Biophys., 2012, 445, 207–209 CrossRef CAS PubMed.
- S. A. Arnstein and C. D. Sherrill, Phys. Chem. Chem. Phys., 2008, 10, 2646–2655 RSC.
- J.-I. Seo, I. Kim and Y. S. Lee, Chem. Phys. Lett., 2009, 474, 101–106 CrossRef CAS.
- C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC.
- D. M. Chipman, J. Chem. Phys., 2003, 118, 9937–9942 CrossRef CAS.
- D. M. Camaioni, M. Dupuis and J. Bentley, J. Phys. Chem. A, 2003, 107, 5778–5788 CrossRef CAS.
- M.-A. Elsliger, R. M. Wachter, G. T. Hanson, K. Kallio and S. J. Remington, Biochemistry, 1999, 38, 5296–5301 CrossRef CAS PubMed.
- X. Shu, K. Kallio, X. Shi, P. Abbyad, P. Kanchanawong, W. Childs, S. G. Boxer and S. J. Remington, Biochemistry, 2007, 46, 12005–12013 CrossRef CAS PubMed.
- X. Shi, P. Abbyad, X. Shu, K. Kallio, P. Kanchanawong, W. Childs, S. J. Remington and S. G. Boxer, Biochemistry, 2007, 46, 12014–12025 CrossRef CAS PubMed.
- M. Kondo, I. A. Heisler, D. Stoner-Ma, P. J. Tonge and S. R. Meech, J. Am. Chem. Soc., 2010, 132, 1452–1453 CrossRef CAS PubMed.
- K. Brejc, T. K. Sixma, P. A. Kitts, S. R. Kain, R. Y. Tsien, M. Ormö and S. J. Remington, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2306–2311 CrossRef CAS PubMed.
- L. Tang, Y. Wang, L. Zhu, K. Kallio, S. J. Remington and C. Fang, Phys. Chem. Chem. Phys., 2018, 20, 12517–12526 RSC.
- L. Tang, L. Zhu, M. A. Taylor, Y. Wang, S. J. Remington and C. Fang, Molecules, 2018, 23, 2226 CrossRef PubMed.
- J. T. M. Kennis, D. S. Larsen, I. H. M. van Stokkum, M. Vengris, J. J. van Thor and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17988–17993 CrossRef CAS PubMed.
- C.-Y. Lin, M. G. Romei, L. M. Oltrogge, I. I. Mathews and S. G. Boxer, J. Am. Chem. Soc., 2019, 141, 15250–15265 CrossRef CAS PubMed.
- C. Chen, M. S. Baranov, L. Zhu, N. S. Baleeva, A. Y. Smirnov, S. Zaitseva, I. V. Yampolsky, K. M. Solntsev and C. Fang, Chem. Commun., 2019, 55, 2537–2540 RSC.
- S. Zhong, D. Navaratnam and J. Santos-Sacchi, PLoS One, 2014, 9, e99095 CrossRef PubMed.
- M. M. J. Smulders, S. Zarra and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 7039–7046 CrossRef CAS PubMed.
- T. J. Paul, J. Z. Vilseck, R. L. Hayes and C. L. Brooks III, J. Phys. Chem. B, 2020, 124, 6520–6528 CrossRef CAS PubMed.
- C. R. Cantor and P. R. Schimmel, Biophysical Chemistry: Part III: The Behavior of Biological Macromolecules, Macmillan, 1980 Search PubMed.
- C. Chen, L. Zhu, M. S. Baranov, L. Tang, N. S. Baleeva, A. Y. Smirnov, I. V. Yampolsky, K. M. Solntsev and C. Fang, J. Phys. Chem. B, 2019, 123, 3804–3821 CrossRef CAS PubMed.
- C.-Y. Lin and S. G. Boxer, J. Am. Chem. Soc., 2020, 142, 11032–11041 CrossRef CAS PubMed.
- L. M. Oltrogge and S. G. Boxer, ACS Cent. Sci., 2015, 1, 148–156 CrossRef CAS PubMed.
- H.-w. Ai, M. A. Baird, Y. Shen, M. W. Davidson and R. E. Campbell, Nat. Protoc., 2014, 9, 910–928 CrossRef CAS PubMed.
- T. M. Wannier, S. K. Gillespie, N. Hutchins, R. S. McIsaac, S.-Y. Wu, Y. Shen, R. E. Campbell, K. S. Brown and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E11294–E11301 CrossRef CAS PubMed.
- S. A. Boulanger, C. Chen, L. Tang, L. Zhu, N. S. Baleeva, I. N. Myasnyanko, M. S. Baranov and C. Fang, Phys. Chem. Chem. Phys., 2021, 23(27), 14636–14648 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational methods, supplementary Fig. S1–S14 and Tables S1 and S2 with additional discussions on steady-state and time-resolved electronic spectroscopic results, second-derivative analysis and global/target analysis, calculated electronic transition gaps, and comparative crystal structures. See DOI: 10.1039/d1sc00847a |
| This journal is © The Royal Society of Chemistry 2021 |