
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOncogenic KRAS G12D mutation promotes dimerization through a second, phosphatidylserine–dependent interface: a model for KRAS oligomerization†
Ki-Young
Lee
a,
Masahiro
Enomoto
a,
Teklab
Gebregiworgis
a,
Geneviève M. C.
Gasmi-Seabrook
a,
Mitsuhiko
Ikura
 *ab and
Christopher B.
Marshall
*a
*ab and
Christopher B.
Marshall
*a
aPrincess Margaret Cancer Centre, University Health Network, Toronto, Ontario M5G 1L7, Canada. E-mail: mitsu.ikura@uhnresearch.ca; chris.marshall@uhnresearch.ca
bDepartment of Medical Biophysics, University of Toronto, Toronto, Ontario M5G 1L7, Canada
First published on 7th September 2021
Abstract
KRAS forms transient dimers and higher-order multimers (nanoclusters) on the plasma membrane, which drive MAPK signaling and cell proliferation. KRAS is a frequently mutated oncogene, and while it is well known that the most prevalent mutation, G12D, impairs GTP hydrolysis, thereby increasing KRAS activation, G12D has also been shown to enhance nanoclustering. Elucidating structures of dynamic KRAS assemblies on a membrane has been challenging, thus we have refined our NMR approach that uses nanodiscs to study KRAS associated with membranes. We incorporated paramagnetic relaxation enhancement (PRE) titrations and interface mutagenesis, which revealed that, in addition to the symmetric ‘α–α’ dimerization interface shared with wild-type KRAS, the G12D mutant also self-associates through an asymmetric ‘α–β’ interface. The ‘α–β’ association is dependent on the presence of phosphatidylserine lipids, consistent with previous reports that this lipid promotes KRAS self-assembly on the plasma membrane in cells. Experiments using engineered mutants to spoil each interface, together with PRE probes attached to the membrane or free in solvent, suggest that dimerization through the primary ‘α–α’ interface releases β interfaces from the membrane promoting formation of the secondary ‘α–β’ interaction, potentially initiating nanoclustering. In addition, the small molecule BI-2852 binds at a β–β interface, stabilizing a new dimer configuration that outcompetes native dimerization and blocks the effector-binding site. Our data indicate that KRAS self-association involves a delicately balanced conformational equilibrium between transient states, which is sensitive to disease-associated mutation and small molecule inhibitors. The methods developed here are applicable to biologically important transient interactions involving other membrane-associated proteins.
1 Introduction
RAS GTPases are peripheral membrane proteins that function as binary molecular switches, cycling between an inactive GDP-loaded state, and an active GTP-loaded state that stimulates multiple effectors to drive proliferative cell signaling pathways.1,2 KRAS is a RAS isoform of paramount clinical significance since it is mutated in approximately 30% of all human cancers, particularly pancreatic, colon, and lung cancers, and mutants are implicated in resistance to current cancer therapies. Notably, the G12D mutant has been recognized as the most common oncogenic mutant, occurring in a quarter of KRAS-driven cancers.3,4Recently accumulating data support a new paradigm for RAS signaling in which lateral segregation of wild-type or mutant KRAS into transient dimers or higher-order multimers (i.e., nanoclusters) at the plasma membrane is essential for recruiting and activating downstream RAF kinases, to stimulate mitogen-activated protein kinase (MAPK) signaling and cell proliferation.5–7 In particular, the predominant oligomeric state of KRAS at physiological expression levels was found to be a dimer,6 which is considered the basic unit capable of promoting functional dimerization of RAF. This assembly process has emerged as an additional layer of KRAS mutant-mediated oncogenesis, in conjunction with the long-known effect of mutations that impair GTP hydrolysis and lock KRAS in a constitutively active GTP-bound state. However, despite the biological and pathological importance of KRAS assembly, the mechanisms underlying the formation of different oligomeric states of KRAS remain unclear; it has been proposed that higher order multimers assemble from dimeric subunits.
It is becoming increasingly clear that single-point mutations of Gly12 modulate the conformational dynamics of KRAS8–10 as well as the propensity for KRAS dimerization,11 suggesting potential mutation-specific modes of dimerization and possible differences between wild-type and mutant KRAS dimer structures. Various structures of RAS dimers have been predicted by different molecular dynamics (MD) models, which exhibit isoform-specific differences between H-, N-, and K-RAS dimers.12–16 Different MD methods have also produced distinct dimer models of the same isoforms. Further, the bound nucleotide is known to influence KRAS dimerization,11,14 and the structures of wild-type KRAS homodimers in the GTP versus GDP-bound states differ.17
We previously established a system for NMR studies using KRAS attached to a lipid bilayer nanodisc and a variety of paramagnetic relaxation enhancement (PRE) spin labels (in solution, or conjugated to either KRAS or lipid head groups) to determine the structures of transient dimers of wild-type KRAS on a membrane surface.17 Selective isotope labeling was used to overcome broadening and overlap of NMR cross-peaks from a >100 kDa KRAS-nanodisc complex. Nanodiscs provide homogenous and stable native-like membrane fragments of a size that enables NMR detection.18,19
Here, we have further developed these NMR methodologies, and applied them to KRAS G12D, specifically the KRAS4B splice variant (henceforth referred to as KRAS), which is more ubiquitously expressed and extensively characterized relative to KRAS4A. We previously showed that this oncogenic mutation alters the orientational dynamics of monomeric KRAS on the membrane,20 and others have demonstrated that it enhances the formation of nanoclusters in cells.5 In the dimeric state of KRAS–G12D, we observed PRE effects that are not compatible with the single dimer interface previously observed for wild-type KRAS. To deconvolute the overall PRE, we performed paramagnetic NMR titrations and structural model-guided mutagenesis of the interface (a schematic of these experimental approaches appears in Fig. S1†), which enabled us to identify two distinct modes of dimerization for KRAS–G12D. These include the specific, symmetric ‘α–α’ interface shared with wild-type KRAS, and a nonspecific, asymmetric ‘α–β’ interface that was detected only in the G12D mutant. The presence of two dimer interfaces could potentially promote the assembly of KRAS G12D dimers into nanoclusters.
2 Results and discussion
2.1. Two distinct modes of dimerization of the oncogenic KRAS–G12D mutant on the membrane
To explore the impact of the G12D mutation on the KRAS dimer structure, we covalently conjugated the C-terminal Cys185 (which would be farnesylated in cells) to a maleimide moiety on the head group of a lipid (PE-MCC). KRAS was conjugated to pre-assembled nanodiscs containing 5% PE-MCC as well as 20% unsaturated phosphatidylserine (PS) to mimic the plasma membrane composition that has been shown to promote KRAS dimerization,21 and is similar to that used in previous KRAS studies.17,20 The nanodisc comprises a lipid bilayer of diameter ∼76 Å encircled by two membrane scaffold proteins (MSP1D1), which can accommodate a maximum of two KRAS proteins (∼36 Å each).22,23 To observe transient intermolecular interactions between KRAS–G12D molecules in the activated, GTPγS (a non-hydrolyzable GTP analogue)-bound state, we employed PRE (Fig. S2†). Maleimide-conjugated KRAS–G12D (MC-KRAS–G12D) was isotopically 13C-labeled at single methyl groups of Ile, Leu, and Val (ILV, Cδ1, Cδ and Cγ, respectively), and the amides of Lys were 15N-labeled (Fig. S3†). Methyls were chosen for sensitive detection and Lys was chosen because it is abundant in the C-terminal membrane-anchoring hypervariable region (HVR). These MC-KRAS–G12D nanodiscs were mixed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with fully processed, farnesylated KRAS–G12D (FP-KRAS–G12D, also GTPγS-bound) engineered with single surface-exposed cysteine residues (Cys1, Cys39, Cys118, or Cys169) for attachment of a TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) spin label. Cys1 and Cys39 are in and near the β-sheet effector binding site (β interface), whereas Cys118 and Cys169 are on the opposite side at the periphery of the α4–α5 region (α interface).
:1 with fully processed, farnesylated KRAS–G12D (FP-KRAS–G12D, also GTPγS-bound) engineered with single surface-exposed cysteine residues (Cys1, Cys39, Cys118, or Cys169) for attachment of a TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) spin label. Cys1 and Cys39 are in and near the β-sheet effector binding site (β interface), whereas Cys118 and Cys169 are on the opposite side at the periphery of the α4–α5 region (α interface).
The ratios of cross peak intensities before and after the spin label was quenched by reduction (Ipara/Idia) were translated into 1H transverse PRE rates (1H-Γ2) (ESI† and methods, eqn (1)). 1H-Γ2 values of the ILV methyl and Lys amide probes of MC-KRAS–G12D were plotted by residue (Fig. 1). The TEMPO spin labels at Cys118 or Cys169 in the α interface of FP-KRAS–G12D induced strong PRE (1H-Γ2 > 30 Hz) on probes in and near the α interface, as well as moderate PRE (10 Hz < 1H-Γ2 < 30 Hz) on probes in the β interface. Spin-labeled Cys169 induced moderate PRE on the polybasic HVR (K175–K179). Consistent with these observations, spin labels conjugated to Cys1 or Cys39, in and near the β interface, induced reciprocal moderate PRE on probes in and near the α interface. Representative spectra with peaks that exhibit differential PRE effects induced by these spin labels are shown in Fig. S4.† Mapping these paramagnetic interactions onto an arbitrary model of a KRAS dimer (comprising two subunits of a monomeric KRAS–G12D crystal structure) (Fig. 1F–H), indicates that no single dimer model can satisfy all the experimental data, but rather suggests that two distinct interfaces exist. Based on the location of the PRE hotspots on the surface, we designated a primary ‘α–α’ interface resembling that of the wild type,17 as well as a secondary ‘α–β’ (same as ‘β–α’) interface. With two dimer interfaces and asymmetric labeling, several possible molecular dimer configurations can be generated (Fig. 1E). The isotopically labeled MC-KRAS–G12D α interface (α*, where * designates NMR-observable protein) can interact with the spin-labeled FP-KRAS–G12D α or β interface to form α*–α or α*–β dimers. On the other hand, the isotopically labeled β interface (β*) can form a reciprocal β*–α dimer with the spin-labeled α interface. The probability of occurrence of α*–β and β*–α dimers would be expected to be equal. Note that no α–β PRE effects were observed in wild-type KRAS, which exhibited only an α–α dimer interface.17 Importantly, the PRE profile induced by FP-KRAS–G12D on MC-KRAS–G12D was fully consistent with the pattern observed in an experiment performed exclusively with fully processed KRAS (i.e., 13C ILVT-FP-KRAS–G12D plus Cys118-TEMPO-FP-KRAS–G12D in the presence of nanodiscs) (Fig. S5†). No intermolecular PRE was observed between FP-KRAS–G12D molecules in the absence of nanodiscs, demonstrating that like the wild type, KRAS–G12D dimerization is membrane dependent (Fig. S5A†). The MC-KRAS–G12D spectral changes upon dimerization with unlabeled FP-KRAS–G12D were nearly identical to those induced by the TEMPO-labeled FP-KRAS–G12D variants that had been quenched by reduction with vitamin C (i.e., PRE-inactive) (Fig. S6†), suggesting the TEMPO labels have negligible impact on dimerization.
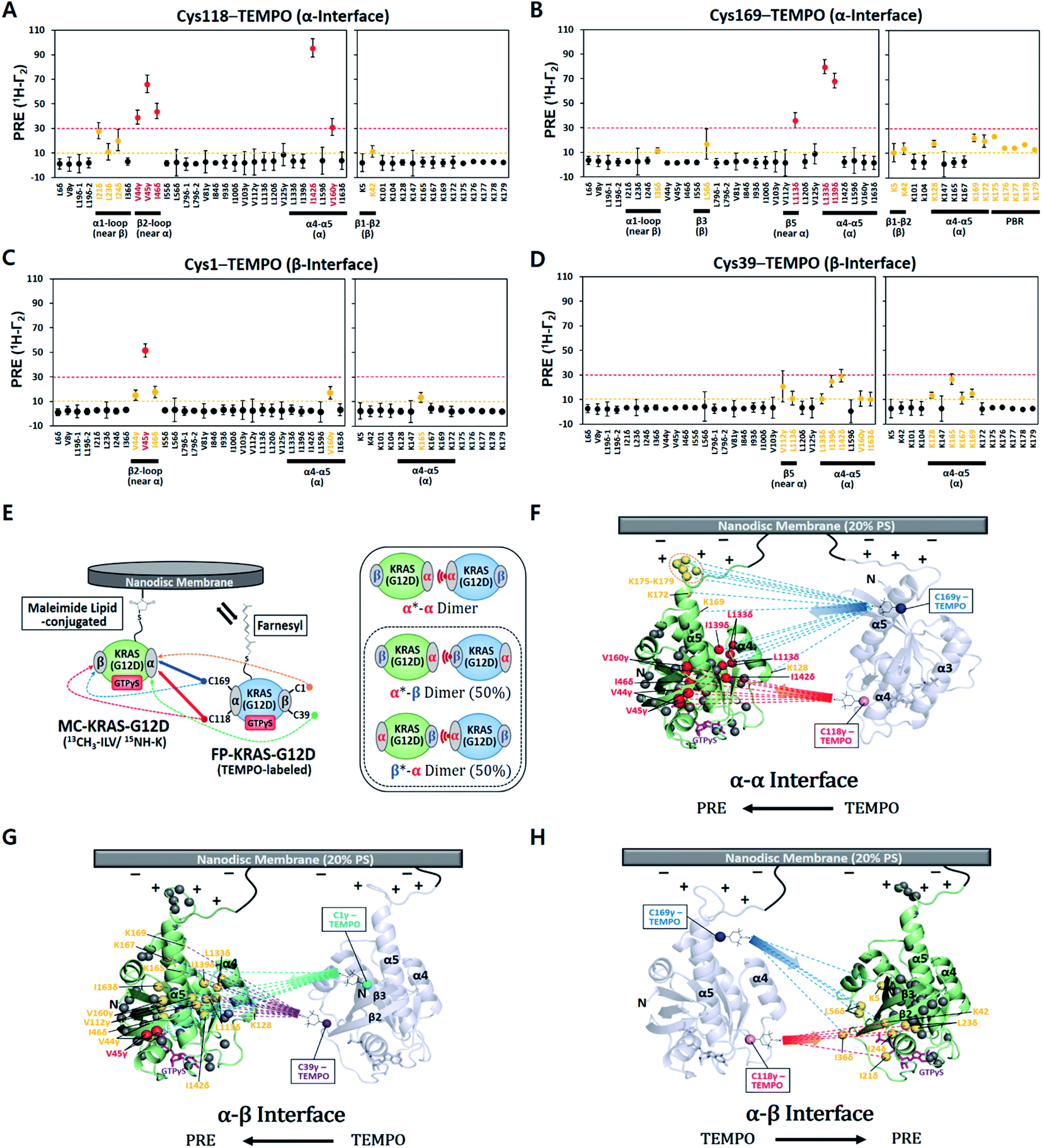 | ||
| Fig. 1 Intermolecular protein–protein PRE effects between KRAS–G12D molecules on the membrane. (A–D) 1H-Γ2 PRE rates for ILV 13C-methyls and Lys 15N-amides of MC-KRAS–G12D in the presence of FP-KRAS–G12D tagged with TEMPO at Cys118 (A), Cys169 (B), Cys1 (C), or Cys39 (D). Probes are coloured according to the PRE exhibited; 1H-Γ2 values > 10 s−1 are moderate (yellow) and >30 s−1 are strong (red). PBR; polybasic region (K175–K179). (E) Schematic of PRE effects from spin labels attached to four sites on FP-KRAS–G12D on isotopically labelled MC-KRAS–G12D (left panel). Solid and dotted lines represent strong and moderate PRE, respectively. The right panel shows the possible molecular configurations of MC/FP KRAS–G12D dimers; green: isotopically labelled MC-KRAS–G12D; blue: spin labelled FP-KRAS–G12D. The isotopically labeled interfaces exhibiting PRE effects are indicated with an asterisk in each label. α*–β and β*–α dimers are equally likely to form. (F–H) Mapping KRAS–G12D protein:protein PRE effects. PRE-affected probes at the α*–α (F), α*–β (G), or α–β* (H) dimer interfaces mapped onto the crystal structure of GTPγS-bound KRAS–G12D (PDB ID: 4DSO) in an arbitrary dimerization model on a membrane containing 20% phosphatidylserine (PS) lipid. ILV 13C-methyl and Lys 15N-amide probes that exhibit moderate and strong PRE effects are colored as in panels A–D. Dotted lines represent the PRE effects that arise from TEMPO conjugated to Cys118 (red), Cys169 (blue), Cys1 (cyan), or Cys39 (purple) in the opposing protomer (arbitrarily positioned). | ||
To compare site-specific binding affinities for KRAS dimerization through the α–α and the α–β interfaces, 13C ILV-labeled MC-KRAS–G12D was titrated with FP-KRAS–G12D bearing a TEMPO spin label at Cys118 on the α interface. Due to the complexity of the system, we made the assumption that the membrane association of free FP-KRAS–G12D molecules would be followed by its rapid dimerization with MC-KRAS–G12D as a result of the enhanced KRAS concentration on the highly restricted bilayer surface. The presence of two KRAS molecules on the ∼76 Å diameter nanodisc membrane corresponds to an extremely high two-dimensional concentration of ∼44000 molecules per μm2. Thus, these two sequential steps were approximated by a single step by which FP-KRAS–G12D is in equilibrium between solution and the membrane-bound dimeric state.
We measured 1H-Γ2 PRE rates of probes in both the α and β interfaces of MC-KRAS–G12D at each point of the titration (Fig. 2 and S7†). PRE titration curves for probes on the α interface (i.e., α*–α) fit well with a one-site specific binding model with an average apparent KD of 141 μM, thus its dimerization affinity is ∼4-fold higher than wild-type KRAS.17 Notably, β2-loop residues (44–46), located at the boundary between the α and β interfaces, exhibit a titration pattern similar to the α interface, thus this region is designated part of the α interface in this study. Considering that Gly12 is away from the α interface, the G12D mutation may induce long-range changes in the conformation or dynamics of the α interface that promote α–α dimerization. In contrast to the α interface, titration curves for the β interface do not fit a specific binding model, rather they are better fitted by a nonspecific binding model in which 1H-Γ2 rates correlate linearly with the concentration of FP-KRAS–G12D (total concentration minus the fraction calculated to adopt the α–α dimeric state on the basis of the KD value for the α–α interaction), with an average binding constant (i.e., slope) of 0.34 Hz μM−1. Note that the α–α and α–β dimers would be mutually exclusive with the 2:1 stoichiometry of KRAS per nanodisc surface22,23 used in this study. Potential cooperativity of both interactions to form higher-order multimers remains to be studied, but requires larger nanodiscs, and detection of NMR signals from such a supramolecular complex is technically challenging. The nanodisc-based PRE titration developed here is a unique tool to detect residues involved in weak, nonspecific but biologically important protein–protein interactions on the membrane. The nonspecific α–β interaction between KRAS protomers is implicated in formation of transient signaling-competent nanoclusters (see the Section 2.7).
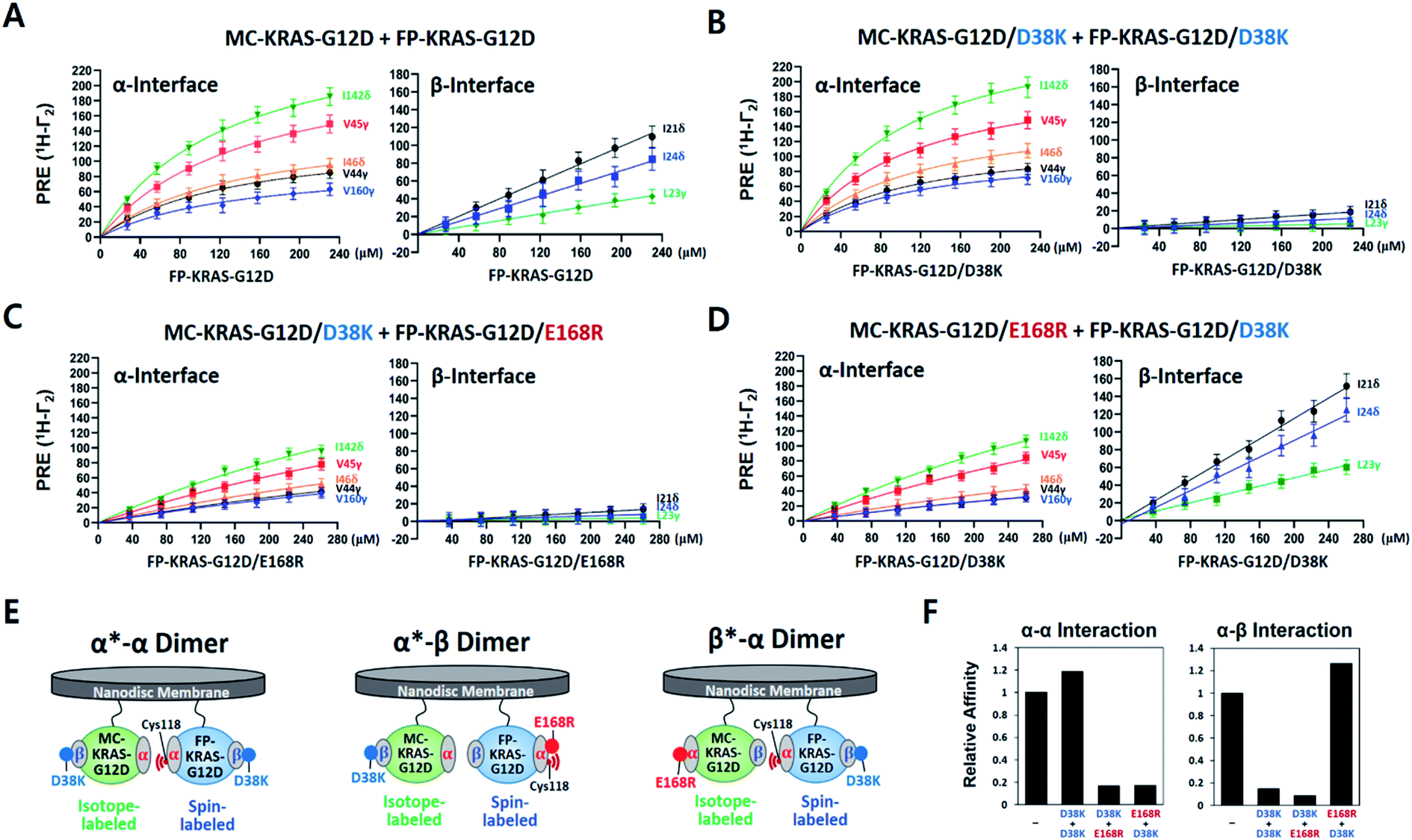 | ||
| Fig. 2 KRAS–G12D α–α dimers associate in a specific manner while α–β association is non-specific. (A) PRE rates of ILV 13C-methyl probes in the α and β interfaces of MC-KRAS–G12D as a function of the concentration of FP-KRAS–G12D bearing a TEMPO spin label at Cys118 in the α interface. (B–D) PRE rates obtained with three combinations of interface mutants of KRAS–G12D, as indicated. (E) Schematic of the molecular configurations of the KRAS–G12D dimers favored by the interface mutants in panels B–D (α*–α, α*–β, or β*–α): green; isotopically labelled MC-KRAS–G12D: blue; spin labelled FP-KRAS–G12D. The isotopically labeled interfaces exhibiting PRE effects are indicated with an asterisk in each label. (F) Relative affinities for α–α and α–β interactions of KRAS–G12D and the engineered mutants indicated derived from data in panels A–D. Relative affinities (1/KD) are normalized to the interaction between MC-KRAS–G12D and FP-KRAS–G12D in panel A. | ||
To determine whether wild-type KRAS can form an α–β dimer in a more saturating condition, we performed PRE experiments with Cys118-TEMPO-FP-KRAS–GTPγS at two-fold excess over 13C-labeled MC-KRAS–GTPγS, but still no PRE effects were observed on the β interface (Fig. S8†). In addition, a TEMPO spin label on Cys1 in the wild-type β interface did not induce any PRE on the α interface, confirming that the G12D mutation promotes α–β interaction and demonstrating that this binding is not simply a result of co-localization of protomers on a nanodisc. Wild-type and mutant KRAS exhibited similar PRE patterns on their α interfaces, suggesting both KRAS constructs share the α–α dimer interface, and the lower overall PRE in the wild type is consistent with its lower affinity for α–α dimerization.17
2.2. HADDOCK cluster analysis of KRAS–G12D dimers and validation by interface mutagenesis
PRE-derived restraints were used to determine dimer structures in the HADDOCK 2.2 program. Since visual inspection of the PRE effects on KRAS illustrated that a single dimer structure would not satisfy all the PRE-derived distance restraints, they were converted into a set of ‘ambiguous’ restraints in HADDOCK calculations. We used a total of 40 restraints (Table S1†) to generate structural models and analyzed them using the standard cluster analysis tool in HADDOCK. Not surprisingly, the cluster analysis revealed two major distinct clusters of structures (Fig. S9†), each of which satisfies a subset of restraints. They are the symmetric α–α dimer (69%) and the secondary asymmetric α–β dimer (28%). The G12D α–α dimer is essentially identical to the wild-type KRAS dimer structure previously reported using the same approach.17 However, the co-existence of a secondary α–β dimer was unexpected and raised further questions.First, we sought to design interface-specific mutants to disrupt either the α–α or α–β interface, thereby favouring assembly of the alternate dimer. In the HADDOCK models, D38 in the β interface is exclusively involved in the α–β interaction, while E168 in the α interface is exclusive to the α–α interaction. To promote formation of each dimer configuration, we introduced the interface-specific mutations D38K or E168R into both MC- and FP-KRAS–G12D. These mutant constructs were mixed to prepare samples including (i) MC-KRAS–G12D/D38K and FP-KRAS–G12D/D38K, (ii) MC-KRAS–G12D/D38K and FP-KRAS–G12D/E168R, and (iii) MC-KRAS–G12D/E168R and FP-KRAS–G12D/D38K, which are predicted to favour the α*–α, α*–β, and β*–α dimers, respectively. PRE titrations were performed with each pair of mutants using isotopically labeled MC-KRAS and FP-KRAS with Cys118-TEMPO in the α interface. Compared to intact MC- and FP-KRAS–G12D, these mutations selectively reduced PRE effects on the specific interfaces predicted (Fig. 2 and S7†). In sample (i), the D38K mutation reduced the PRE on the β interface, and reduced the average binding constant 7 fold (0.05 Hz μM−1), without affecting the PRE on the α interface (average KD 119 μM). In sample (ii), the combination of the D38K and E168R mutations reduced the PRE on both the α and β interfaces, with an average KD of 841 μM and an average binding constant of 0.03 Hz μM−1. When the same two mutations were introduced into a pair of proteins with the reverse labeling scheme (sample iii), the PRE on the α interface decreased (KD 818 μM) while PRE for the β interface increased (0.43 Hz μM−1). Collectively, in samples (i) and (ii), the D38K mutation in the β interface of MC-KRAS–G12D reduced the PRE induced by α–β dimerization, while in samples (ii) and (iii), the PRE induced by α–α dimerization was reduced by E168R mutations in the α interface. These observations demonstrate that residues E168 and D38 are critical for the α–α and α–β interactions, respectively. Single-point mutations at either the α–α or α–β dimer interface are sufficient to reduce dimerization through the corresponding interface while preserving the alternate dimer species.
2.3. Membrane orientations of α–α and α–β dimers of KRAS–G12D
To examine the orientation of MC-KRAS–G12D with respect to the membrane, we employed complementary ‘solvent’ and ‘membrane’ PRE experiments using Gd–DTPA–BMA in the bulk solvent and Gd3+-chelated head groups of PE-DTPA lipids in nanodiscs, respectively, as previously described.17 In both experiments, the 1H-Γ2 PRE rates were measured for ILV 13C-methyls of MC-KRAS–G12D on nanodiscs (where it is almost exclusively monomeric), and in the presence of an equal amount of unlabeled FP-KRAS–G12D, which favours the dimeric state of KRAS. In the monomeric state, relatively high membrane-PRE was observed in the α interface (Fig. S10†), consistent with our previous report that the KRAS–G12D monomer favours an orientation in which the α4–α5 surface (α-interface) contacts the membrane while the effector binding site (β-interface) is exposed,20 whereas wild-type KRAS exhibits the reverse preference.20 Van et al. recently reported membrane-PRE experiments using uniformly 15N-labeled wild-type FP-KRAS, in which an orientational preference for the α and β interfaces was not clearly observed.24 This may be related to (i) the intrinsic lower sensitivity of 15N-amide protons relative to 13C-methyl probes and/or (ii) less favorable association with the smaller lipid surface of the nanodisc employed. MSP1D1-ΔH5 has a diameter of ∼63 Å whereas that of MSP1D1 used in the current study is ∼76 Å. Note that the maximal length of the KRAS surface would be ∼70 Å, if the intrinsically disordered HVR (residues 172–185) assumes a fully elongated conformation. Membrane association of free FP-KRAS is much more dynamic than our lipid-conjugated MC-KRAS, thus the population of membrane-associated FP-KRAS states would be lower and may be below the level of detection.Relative PRE changes upon dimerization were assessed by calculating the 1H-Γ2 difference divided by the monomeric state 1H-Γ2 (i.e., Δ1H-Γ2,di-mono/1H-Γ2,mono) (Fig. 3 and S11–S14†). These experiments were also performed with the three combinations of the D38K and E168R mutations described above. It should be noted that even in the presence of FP-KRAS–G12D, PRE observations derive from a mixture of monomeric and dimeric states in equilibrium.
According to the magnitude of the Δ1H-Γ2,di-mono/1H-Γ2,mono ratios, MC-KRAS–G12D probes were classified into four groups: (i) large PRE increases (ratio > 0.6), (ii) moderate PRE increases (0.3 to 0.6), (iii) moderate PRE reductions (−0.6 to −0.3), and (iv) large PRE reductions (<−0.6). Our interpretation of these dimerization-induced ratios follows: (a) PRE changes with negative values for both the solvent and membrane PRE represent a dimerization site, which thus has reduced accessibility to both the solvent and the membrane, (b) PRE changes with positive values for the solvent PRE and negative values for membrane PRE represent increased solvent accessibility of a non-interacting site in the dimer away from the membrane, whereas (c) PRE changes with negative solvent and positive membrane PRE values represent sites of the dimer that face the membrane, which shields them from solvent.
Upon addition of FP-KRAS–G12D, both the α and β interfaces of MC-KRAS–G12D exhibited (a)-type PRE changes (Fig. S12–S14†), consistent with the TEMPO-tagged protein-induced PRE patterns, which showed that dimerization is mediated by the α–α and α–β interfaces. The interface mutants further supported this model. The presence of the D38K mutation on both protomers (i.e., condition (i), which disrupted β*–α and α*–β dimers) induced (a)-type PRE changes on the α interface, (b)-type for the β interface, and (c)-type for the α3-loop region. This demonstrates that, like wild-type KRAS-GTPγS,17 α–α dimerization induces an orientation in which the β interface is exposed and the α3-loop region is proximal to the membrane. The combination of E168R with D38K* (condition (ii), which inhibited both α*–α and β*–α dimerization) induced (a)-type PRE changes on the α interface and (b)-type on the β interface and α3-loop. When D38K was added to E168R* (condition (iii), in which both α*–α and α*–β dimerization were impaired), the (a)-type PRE change pattern was induced on the β interface while the α interface and α3-loop exhibited (b)-type changes. These observations demonstrate that α–β dimerization induces an orientation in which the non-dimerizing α and β sites are exposed to solvent away from the membrane. Dimerization-induced changes in the membrane and solvent PRE for each dimeric state are mapped onto membrane-bound KRAS–G12D α–α and α–β dimers in Fig. 3.
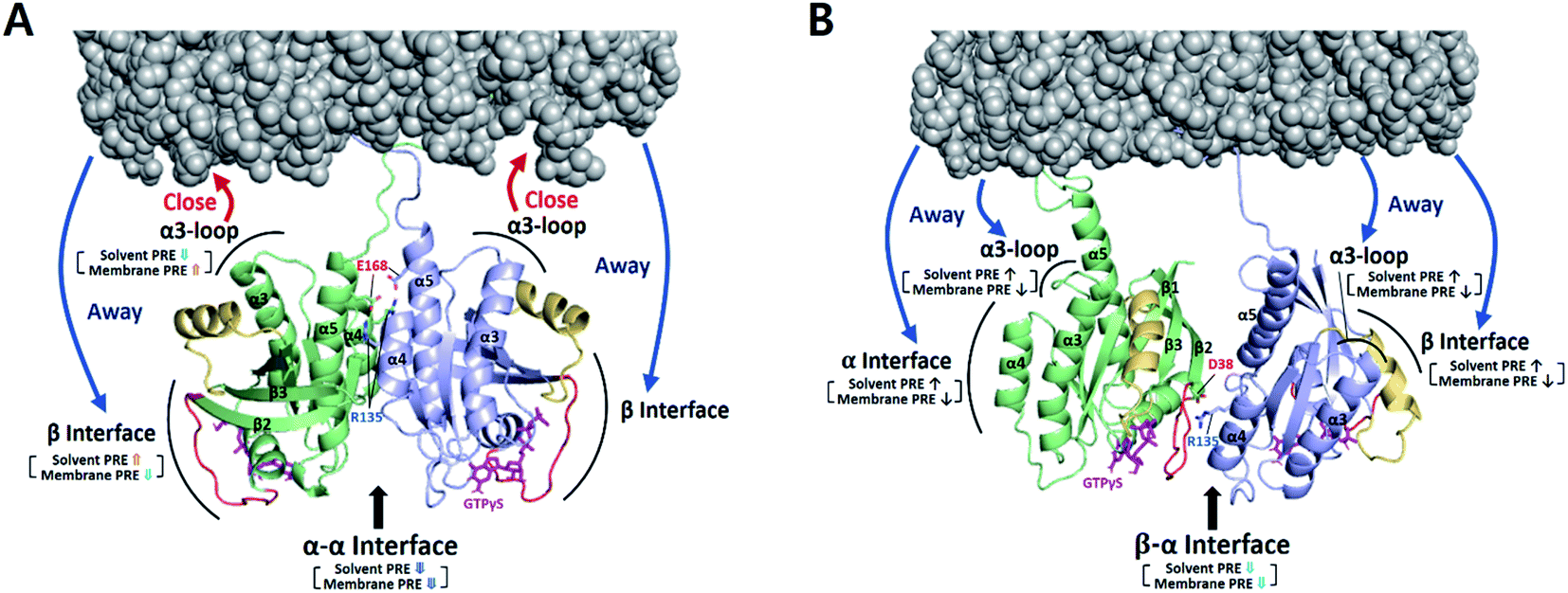 | ||
| Fig. 3 Dimerization-induced changes in the solvent and membrane PRE (based on Δ1H-Γ2,di-mono/1H-Γ2,mono values in Fig. S14b†) for each dimer state mapped onto the NMR-driven structure of the α–α (A) or α–β dimer (B) of KRAS–G12D on the membrane. Dimerization-induced changes in proximity to the membrane are represented by red (closer) and blue (further) arrows. The side chains of key residues for each dimerization interface are depicted. Switch I and II regions are colored red and yellow, respectively. | ||
2.4. NMR-driven structures of membrane-bound α–α and α–β dimers of KRAS–G12D
Based on the two main structural clusters determined by HADDOCK and specific versus nonspecific modes of dimerization, PRE-derived distance restraints were divided into two distinct subsets to separately generate models of each dimer on the membrane. Specifically, probes that exhibit a distance of <22 Å (PRE detection limit) from the corresponding TEMPO-labeled cysteine in the α–α or α–β cluster model (Table S1†) were treated as ambiguous restraints and added to restraints derived from the TEMPO-PRE titration and membrane PRE data for each dimer state observed with the interface mutants in samples (i), (ii), or (iii) (details in ESI Material and Methods†).These restraints were used to generate 200 final HADDOCK models of each dimer on the membrane (Table S2†), each of which formed a single cluster. For the 200 and 20 lowest HADDOCK-score structures of the α–α dimer, the GTPase domains (residues 1–172, excluding the HVR) superimpose very well with average backbone RMSDs of 0.94 ± 0.18 Å and 0.82 ± 0.15 Å, respectively, to the mean structures (Fig. S15†). The RMSDs for the GTPase domains in the structures of the α–β dimer were 1.62 ± 0.55 Å and 1.34 ± 0.47 Å, respectively (Fig. S16†). In both models, lipid head groups of PS (acidic) and/or PC (neutral, zwitterionic) on the membrane surface form electrostatic interactions with the C-terminal HVR residues, primarily the positively charged side chains of lysines (details in Table S3 in the ESI†).
The α–α dimer interface of KRAS–G12D is nearly identical to our previous wild-type KRAS–GTPγS structure (Fig. S15C†). Like wild type, the specific α–α G12D dimer is stabilized by intermolecular interactions including (i) hydrogen bonds from Q131 to D154 and R161, and (ii) electrostatic interaction between R135 and E168 (Table S4†). In contrast, the nonspecific α–β dimer models exhibit a higher RMSD, and their interface contacts are mainly electrostatic, including (i) D33 and R128, (ii) D33 and R135, and (iii) D38 and R135. These observations are consistent with the notion that electrostatic forces promote the formation of nonspecific ‘encounter complexes’ in which the precise orientation of the protomers is not well defined.25 The higher RMSD of the α–β dimer model also reflects the smaller number of restraints obtained from this dynamic interface.
The mechanism by which the G12D mutation promotes α–β dimerization remains unknown. We speculate that this may involve (i) mutation-induced changes in the conformation or dynamics of the β interface and/or the α interface,8–10 and (ii) the G12D mutation-induced shift of the monomer towards an orientation in which the β interface is exposed.20 Gly12 mutations are known to induce changes in the β interface dynamics that affect its interactions.26 While α–β dimerization was not detected for wild-type KRAS, it is possible that a small, undetectable population exists.
We believe that, along with the two distinct KRAS G12D dimer configurations we detected on the membrane, other conformational states likely co-exist. KRAS samples multiple conformations involving monomer–dimer exchange and multiple preferred membrane interfaces. To date the KRAS α–β interface is unique to the G12D mutant on the membrane, whereas crystalline states have captured snapshots of G12D monomers (PDB IDs: 6GOF, 5USJ) and a distinct β–β dimer (PDB ID: 6QUU).27 A recent crystal structure of KRAS Q61H (PDB ID: 6MNX) also displayed an almost identical β–β interface in the crystalline state.28 We believe that this dynamic nature of KRAS is crucial for its biological function as a molecular switch, and that disease-associated mutations could alter the conformational landscape of KRAS dimerization as well as membrane association.
2.5. KRAS–G12D α–β dimerization depends on phosphatidylserine in the membrane
KRAS nanoclustering has been reported to be enhanced by the presence of PS,21,29 which is the most abundant anionic phospholipid in eukaryotic membranes and is asymmetrically distributed in the inner leaflet of the plasma membrane.30 Other major membrane lipids including phosphocholines, phosphoethanolamines or phosphoinositides exhibited little impact on the formation of KRAS nanoclusters.29 Molecular dynamics (MD) simulations have demonstrated dynamic multiple orientations of KRAS alone or in complex with effectors with respect to a membrane surface, depending on the lipid compositions.31–36To probe the role of PS in KRAS–G12D dimerization on the membrane, we performed TEMPO-PRE experiments with nanodiscs lacking PS (100% phosphocholine, DOPC). Addition of FP-KRAS–G12D bearing a TEMPO spin label at the α interface (Cys118 or Cys169) induced PRE effects in MC-KRAS–G12D consistent with an ‘α–α’ mode of dimerization, however, no PRE effects were induced on the MC-KRAS–G12D β interface (Fig. S17†). Consistently, we did not observe any PRE effects when the TEMPO spin label was attached to the β interface (Cys39), indicating that KRAS–G12D does not form an ‘α–β’ dimer on a PS-deficient membrane.
In membrane PRE experiments using Gd-conjugated lipid without PS (100% DOPC), the nanodisc-bound MC-KRAS–G12D monomer exhibited a higher Gd-induced PRE on the β-interface relative to the α-interface (Fig. S18†), demonstrating that the β interface becomes more occluded by a PS-free membrane, and this trend is reversed in the presence of PS. It is possible that the increased accessibility of the β interface on the PS membrane promotes the ‘α–β’ dimerization as well as effector binding. Upon addition of FP-KRAS–G12D, the α interface in MC-KRAS–G12D exhibited reduced PRE from the membrane lacking PS (Fig. S19†), consistent with protection of this surface by α–α dimerization. At the same time, PRE on the β interface increased, demonstrating that this surface becomes closer to the membrane upon dimerization. Interestingly, this behaviour is opposite to that obtained in the presence of PS (Fig. 3), suggesting that dimerization on a PS-containing membrane would promote binding of effectors to the exposed β interface of KRAS–G12D.
Our membrane PRE data clearly demonstrate that PS is an active participant in modulating the membrane orientations of the KRAS monomer and dimer, as well as the dimerization interface. These effects of the lipid composition are likely related to electrostatic interaction between the C-terminal polybasic region of KRAS and the anionic lipid head groups of PS on the membrane surface. A broad spectrum of KRAS conformations may exist on the membrane and play roles in activating relevant signaling events, which could be affected by various factors including membrane lipid constituents, oncogenic mutations, nucleotide-binding, and post-translational modifications of KRAS. These factors can be addressed using the nanodisc system presented in this study.
2.6. BI-2852 induces β–β dimerization of KRAS–G12D on the membrane
The small molecule BI-2852 was recently developed as an inhibitor of KRAS–G12D.37 The underlying mechanism-of-action was inferred from a crystal structure of KRAS–G12D in complex with BI-2852 (PDB ID: 6GJ8), revealing two inhibitor molecules bound to the β interfaces of two KRAS protomers in a manner that mediates β–β dimerization.38 This compound-stabilized dimer appears to be signaling-incompetent because the effector-binding site is buried in the dimeric interface. However, the KRAS–G12D construct crystallized with the compound lacks the C-terminal membrane anchor, thus it remains unclear whether or how BI-2852 may impact KRAS–G12D dimerization on a membrane. Therefore, we examined the intermolecular PRE between nanodisc-tethered 13C ILV-labeled MC-KRAS–G12D and TEMPO-tagged FP-KRAS–G12D in the presence and absence of BI-2852.A TEMPO spin label was introduced on Cys1 (M1C) in the β interface of FP-KRAS–G12D. In the absence of BI-2852, V45γ was the only probe on MC-KRAS–G12D that exhibited strong PRE effects, consistent with α–β dimerization (Fig. 4A). Addition of BI-2852 induced strong intermolecular PRE effects on several probes centred on the β interface of MC-KRAS–G12D, consistent with the crystal structure of the complex. In addition, BI-2852 binding caused chemical shift changes in and near the β interface (Fig. S20†). These data are consistent with BI-2852 acting as a molecular ‘glue’ to promote β–β dimerization of KRAS–G12D on a membrane.
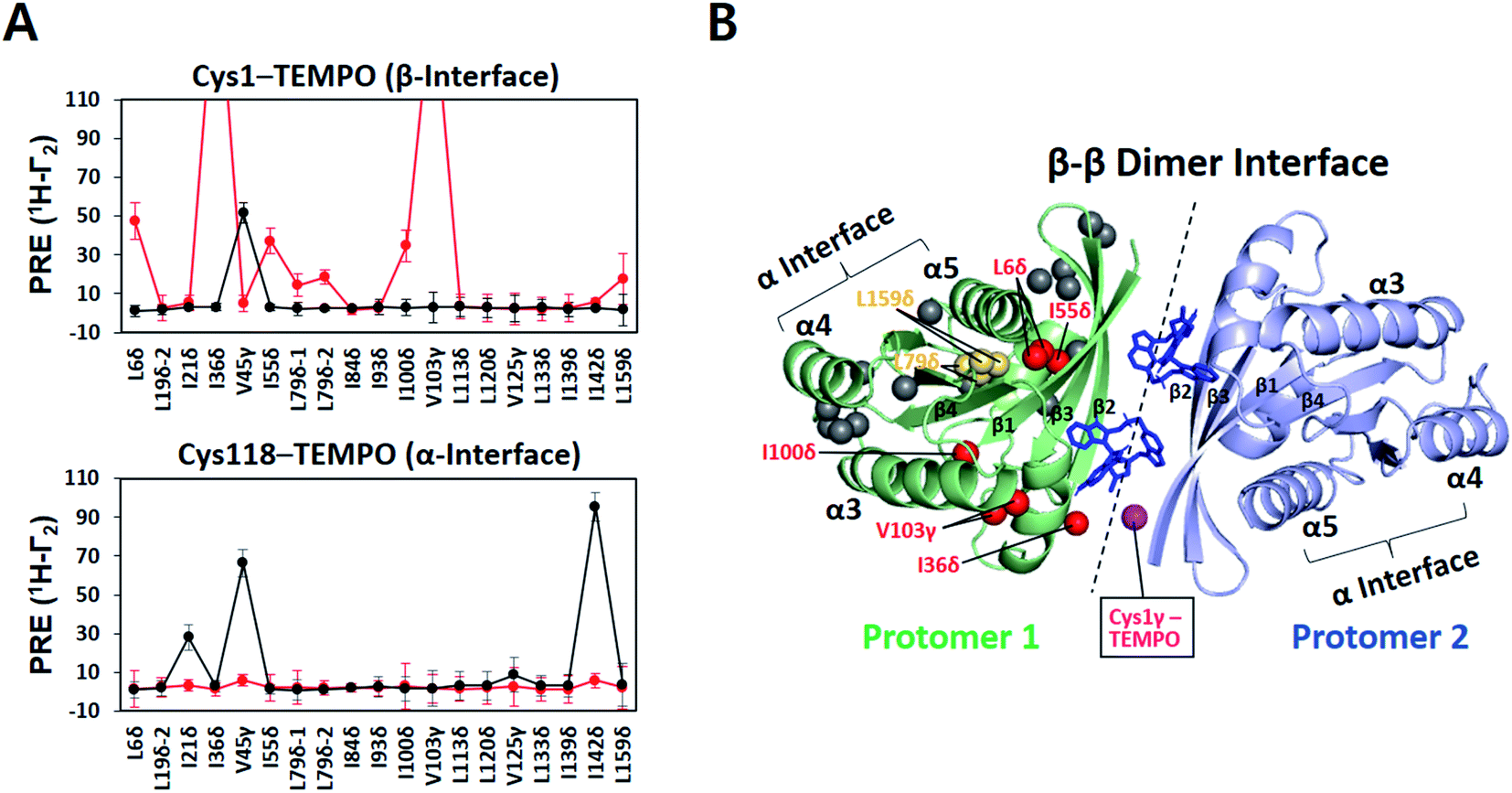 | ||
| Fig. 4 KRAS inhibitor BI-2852 induces KRAS–G12D dimerization through a β–β interface on the membrane surface. (A) Intermolecular 1H-Γ2 PRE rates for ILV 13C-methyls of MC-KRAS–G12D in the presence of equimolar FP-KRAS–G12D bearing a TEMPO spin label at Cys1 in the β interface or Cys118 in the α interface before (black lines) and after addition of BI-2852 (red lines, KRAS–G12D:BI-2852 molar ratio 1:1). (B) MC-KRAS–G12D probes exhibiting PRE effects from Cys1-TEMPO-tagged FP-KRAS–G12D upon addition of BI-2852 mapped onto a NMR-driven structure of a BI-2852-stabilized KRAS–G12D β–β dimer. One protomer is depicted with ILV 13C-methyl probes coloured according to PRE exhibited in red, yellow, and gray, representing strong (>30 s−1), moderate (>10 s−1), and negligible PRE, respectively. Two BI-2852 molecules at the β–β dimer interface are shown in blue. In the opposing protomer, the Cys1 gamma position, the TEMPO conjugation site, is shown in purple. | ||
We generated HADDOCK structures of a BI-2852-stabilized β–β dimer using unambiguous restraints of probes that experience PRE (>10 Hz) from the Cys1 TEMPO spin label in the presence of BI-2852 (Fig. 4B and S21, and Table S5†). The 200 lowest HADDOCK-score structures converged well and this structure is nearly identical to the crystal structure38 (Table S6†), indicating that the BI-2852-stabilized conformation of truncated KRAS–G12D in the crystal structure is preserved in the membrane-bound full-length protein.
In contrast, the intermolecular PRE effects on the α interface of MC-KRAS–G12D from Cys1-TEMPO-tagged FP-KRAS–G12D were substantially reduced by addition of BI-2852 (Fig. 4A), demonstrating that the native α–β dimerization is out-competed by the higher affinity BI-2852-induced β–β dimer. Likewise, PRE effects on the α-interface of MC-KRAS–G12D from TEMPO-tagged Cys118 were also lost upon addition of BI-2852, demonstrating that stabilization of the β–β dimer by this compound also outcompetes α–α dimerization of KRAS–G12D. These observations are consistent with the much higher affinity of KRAS–G12D for BI-2852 (KD of 0.7 μM determined by ITC38), relative to its dimerization affinity (α–α KD 141 μM). The methodologies presented here will be applicable to other potential modulators of RAS dimerization, including small molecules, biologics and endogenous proteins.
2.7. Two dimer interfaces as a mechanism for KRAS–G12D nanoclustering on the membrane
It has been argued that dimerization is required to assemble higher-order multimers (i.e., nanoclusters), which increase the effective local RAS concentration to recruit and activate downstream effectors. Transient signaling from such dynamic complexes has been proposed to yield a switch-like response that improves fidelity by reducing biological noises.39–41 We further emphasize the significance of two dynamic dimer interfaces, which potentially enable assembly of diverse oligomeric states of KRAS–G12D on the membrane. Monomers accumulate in regions enriched in unsaturated PS lipids.21 These clusters would predominantly form specific α–α dimers, which then serve as seeds to assemble transient higher-order species via nonspecific α–β encounter complexes (Fig. 5). The α–β dimer does not appear to be a precursor of the final specific α–α dimer, since disruption of α–β dimers by mutation does not inhibit α–α dimerization. It is apparent from our model that a KRAS–G12D β interface occupied by α–β dimerization would not be accessible for interaction with RAS effectors or GTPase-activating proteins (GAPs) (Fig. S22†). However, it is likely that high-affinity RAS effectors would outcompete the weaker α–β interactions. Previous reports suggest that recruitment and activation of downstream RAF kinases are confined to RAS nanoclusters.42,43 The transient α–β interaction may promote the recruitment and dimerization of RAF by promoting nanoclustering.44 Loosely associated β-interfaces in nanoclusters may be captured effectively by RAF RAS binding domains (RBDs) with fast association rates with the β-interface.45,46 This process may be enhanced by the ‘fly-casting’ mechanism involving both (i) the long flexible linkers (∼163 residues) between the RAF RBD and kinase domain, and (ii) dynamic transition of KRAS between membrane-distal and -proximal states, which was recently characterized through a combination of neutron reflectivity, fast photochemical oxidation of proteins (FPOP), NMR-PRE, and MD simulations.24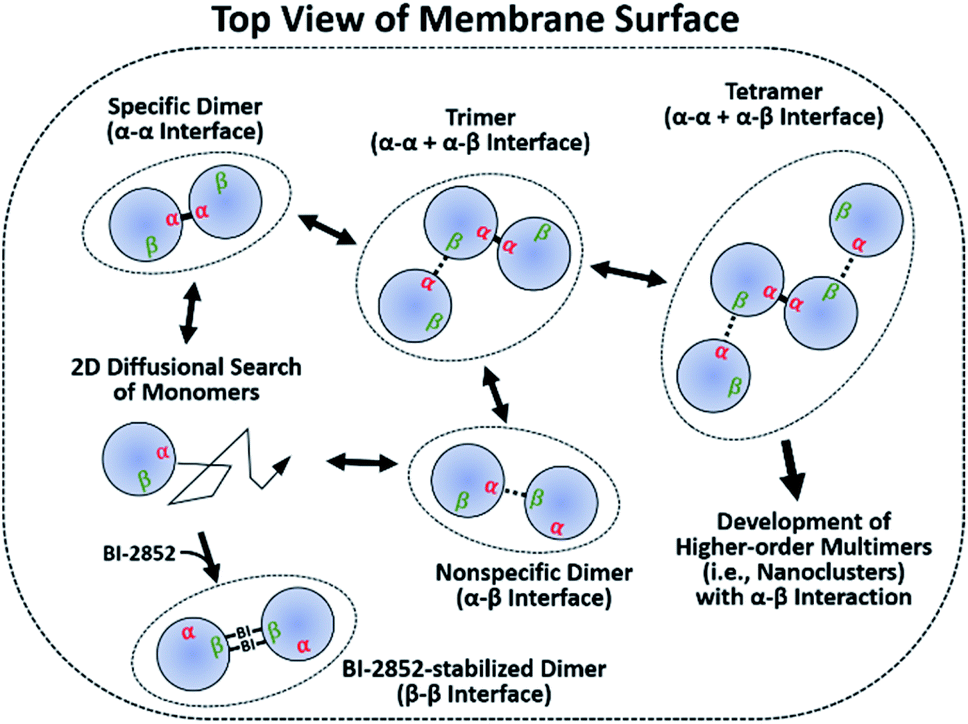 | ||
| Fig. 5 Proposed mechanism for self-assembly of oncogenic KRAS G12D into dimers and nanoclusters on the membrane. Membrane-bound monomers diffuse in two dimensions on the membrane surface to form dimers through a specific α–α or nonspecific α–β interface. α–α dimers serve as seeds for the assembly of higher-order multimers through interactions between their accessible β interfaces and free α interfaces. The small molecule BI-2852 binds to the β interfaces of KRAS–G12D and stabilizes a nonfunctional β–β dimer. | ||
A recent cryo-EM structure of the autoinhibited state of BRAF shows a 14-3-3 dimer bridging phosphorylated sites in conserved region 2 and the C-terminus in a manner that prevents dimerization of the kinase domain and sequesters the CRD.47 KRAS binding to the RBD, which was not resolved in this structure, leads to dephosphorylation of the N-terminal 14-3-3-binding site. This disrupts the autoinhibitory conformation, releasing the CRD and allowing formation of an active kinase domain dimer, which is stabilized by 14-3-3 bridging the C-terminal phosphorylated sites of each protomer. Recruitment of two adjacent RAF molecules by KRAS dimers or nanoclusters at the membrane may synergistically stabilize a signaling-competent KRAS–RAF hetero-tetramer that would propagate mitogen-activated protein kinase (MAPK) signaling. An alternate mechanistic model is that this signaling complex is formed by dimerization of preformed KRAS–RAF hetero-dimeric complexes, as proposed in a recent report that the RAS–RBD binding promotes ‘α–α’ dimerization of RAS on the membrane by allosterically modulating this dimerization interface.48 These MD simulations have revealed that formation of the RAS–RBD hetero-tetramer is coupled with an increase in allosteric connectivity between the galectin-binding sites of the RBDs on the opposite ends of the complex, which is proposed to facilitate binding to the galetin dimer and formation of a supramolecular signaling platform consisting of RAS, RAF and galectin.48 Notably, these mechanistic scenarios for a signaling KRAS–RAF complex may be affected by relative stoichiometry of KRAS, RAF, other effectors, and upstream regulators (i.e., GEF and GAP), which vary in many different types and stages of normal or KRAS mutant cancer cells. For instance, KRAS dimerization or nanoclustering would be promoted by a higher abundance of KRAS than RAF in cells, which was demonstrated to be the case in a proteomics survey of the EGFR-MAPK pathway.49
These structural models raise several questions that remain to be answered. For instance, how association with RAF may impact the orientation of the monomeric and dimeric states of KRAS–GTP on the membrane, whether the kinase domain makes contact with the KRAS:RBD–CRD complex, how the N-terminal RBD/CRD region communicates with the C-terminal kinase domain through a linker that is predicted to be unstructured, and whether or not this linker region becomes more ordered upon assembly of the heterotetrameric complex.
The proposed mechanism for KRAS nanocluster assembly and its role in effector activation remains to be further validated using in vitro experimental setups that enable membrane-assisted KRAS multimerization, as well as cell-based assays to address the effect of KRAS assembly in a biological context.
3 Conclusion
PRE-NMR is a uniquely powerful technique to dissect the structural basis of the dynamic assembly of transient signaling proteins on the biological membrane. In this work, we advanced a nanodisc system by performing paramagnetic NMR titrations and interface-specific mutagenesis to unveil two distinct modes of dimerization of the oncogenic KRAS–G12D mutant on the membrane. On the membrane surface, dimerization of both wild-type KRAS and the G12D mutant is predominantly mediated by the α–α interface. However, KRAS G12D also exhibits dimerization via the asymmetric α–β interface in a nonspecific manner, which is dependent upon the presence of PS in the membrane. This is in contrast to the β–β interface observed in the crystal structure of the same mutant.27 Using our NMR methodology, we also showed that the small molecule BI-2852 (ref. 37) stabilizes a β–β KRAS–G12D dimer in the membrane-associated state, shifting the conformational equilibrium toward a non-functional state on the membrane. These data clearly demonstrate that KRAS dimerization involves multiple interfaces, and mutations and small-molecule binding can significantly alter the conformational landscape of KRAS on the membrane. Our structural data provide mechanistic insight into the self-association of membrane-bound KRAS–G12D, which will aid in the design of new therapeutic strategies specific for KRAS G12D mutant-driven cancers. Moreover, our nanodisc-based assay provides a methodological platform to study assembly of other membrane-bound GTPases and further to screen for candidate inhibitors that disrupt this assembly.Data availability
The 20 final HADDOCK models for both the α–α and α–β dimers on nanodiscs have been deposited in the PDB (http://www.rcsb.org) with the accession codes 7RSC and 7RSE. The assignments used in this study have been deposited in the BMRB (http://www.bmrb.wisc.edu/) with the accession code 51071.Author contributions
K-YL, MI, and CBM designed the research; K-YL and ME prepared the samples; K-YL and GMCG-S performed NMR experiments; TG helped conceptualization; K-YL wrote the paper supervised by MI and CBM; MI and CBM obtained funding.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Canadian Cancer Society Research Institute (Grant # 703209 and 706696) and Canadian Institutes of Health Research (Grant # 410008598) and the Princess Margaret Cancer Foundation. M. I. holds a Canada Research Chair in Cancer Structural Biology. NMR spectrometers were funded by the Canada Foundation for Innovation and the Princess Margaret NMR Core Facility is supported by the Princess Margaret Cancer Foundation. We thank Dominic Esposito, NCI RAS Initiative, for providing plasmids for expression of FP-KRAS. We thank Dr Alexandre Bonvin, Utrecht University, for providing the HADDOCK 2.2 program. We thank Boehringer Ingelheim for providing the compound BI-2852.References
- S. Lu, H. Jang, S. Muratcioglu, A. Gursoy, O. Keskin, R. Nussinov and J. Zhang, Chem. Rev., 2016, 116, 6607–6665 CrossRef CAS PubMed.
- D. K. Simanshu, D. V. Nissley and F. McCormick, Cell, 2017, 170, 17–33 CrossRef CAS PubMed.
- R. Nussinov, C. J. Tsai and H. Jang, Cancer Res., 2018, 78, 593–602 CrossRef CAS PubMed.
- I. Khan, J. M. Rhett and J. P. O'Bryan, Biochim. Biophys. Acta, Mol. Cell Res., 2020, 1867, 118570 CrossRef CAS PubMed.
- C. Ambrogio, J. Kohler, Z. W. Zhou, H. Wang, R. Paranal, J. Li, M. Capelletti, C. Caffarra, S. Li, Q. Lv, S. Gondi, J. C. Hunter, J. Lu, R. Chiarle, D. Santamaria, K. D. Westover and P. A. Janne, Cell, 2018, 172, 857–868 e815 CrossRef CAS PubMed.
- X. Nan, T. M. Tamguney, E. A. Collisson, L. J. Lin, C. Pitt, J. Galeas, S. Lewis, J. W. Gray, F. McCormick and S. Chu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7996–8001 CrossRef CAS PubMed.
- R. Spencer-Smith, A. Koide, Y. Zhou, R. R. Eguchi, F. Sha, P. Gajwani, D. Santana, A. Gupta, M. Jacobs, E. Herrero-Garcia, J. Cobbert, H. Lavoie, M. Smith, T. Rajakulendran, E. Dowdell, M. N. Okur, I. Dementieva, F. Sicheri, M. Therrien, J. F. Hancock, M. Ikura, S. Koide and J. P. O'Bryan, Nat. Chem. Biol., 2017, 13, 62–68 CrossRef CAS PubMed.
- M. B. Cammarata, C. L. Schardon, M. R. Mehaffey, J. Rosenberg, J. Singleton, W. Fast and J. S. Brodbelt, J. Am. Chem. Soc., 2016, 138, 13187–13196 CrossRef CAS PubMed.
- J. A. Parker, A. Y. Volmar, S. Pavlopoulos and C. Mattos, Structure, 2018, 26, 810–820 e814 CrossRef CAS PubMed.
- C. C. Chen, T. K. Er, Y. Y. Liu, J. K. Hwang, M. J. Barrio, M. Rodrigo, E. Garcia-Toro and M. Herreros-Villanueva, PloS One, 2013, 8, e55793 CrossRef CAS PubMed.
- M. R. Mehaffey, C. L. Schardon, E. T. Novelli, M. B. Cammarata, L. J. Webb, W. Fast and J. S. Brodbelt, Chem. Sci., 2019, 10, 8025–8034 RSC.
- J. Guldenhaupt, T. Rudack, P. Bachler, D. Mann, G. Triola, H. Waldmann, C. Kotting and K. Gerwert, Biophys. J., 2012, 103, 1585–1593 CrossRef PubMed.
- H. Jang, S. Muratcioglu, A. Gursoy, O. Keskin and R. Nussinov, Biochem. J., 2016, 473, 1719–1732 CrossRef CAS PubMed.
- S. Muratcioglu, T. S. Chavan, B. C. Freed, H. Jang, L. Khavrutskii, R. N. Freed, M. A. Dyba, K. Stefanisko, S. G. Tarasov, A. Gursoy, O. Keskin, N. I. Tarasova, V. Gaponenko and R. Nussinov, Structure, 2015, 23, 1325–1335 CrossRef CAS PubMed.
- P. Prakash, A. Sayyed-Ahmad, K. J. Cho, D. M. Dolino, W. Chen, H. Li, B. J. Grant, J. F. Hancock and A. A. Gorfe, Sci. Rep., 2017, 7, 40109 CrossRef CAS PubMed.
- A. Sayyed-Ahmad, K. J. Cho, J. F. Hancock and A. A. Gorfe, J. Phys. Chem. B, 2016, 120, 8547–8556 CrossRef CAS PubMed.
- K. Y. Lee, Z. Fang, M. Enomoto, G. Gasmi-Seabrook, L. Zheng, S. Koide, M. Ikura and C. B. Marshall, Angew. Chem., 2020, 59, 11037–11045 CrossRef CAS PubMed.
- F. Hagn, M. L. Nasr and G. Wagner, Nat. Protoc., 2018, 13, 79–98 CrossRef CAS PubMed.
- T. Ravula, N. Z. Hardin and A. Ramamoorthy, Chem. Phys. Lipids, 2019, 219, 45–49 CrossRef CAS PubMed.
- M. T. Mazhab-Jafari, C. B. Marshall, M. J. Smith, G. M. Gasmi-Seabrook, P. B. Stathopulos, F. Inagaki, L. E. Kay, B. G. Neel and M. Ikura, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6625–6630 CrossRef CAS PubMed.
- Y. Zhou, P. Prakash, H. Liang, K. J. Cho, A. A. Gorfe and J. F. Hancock, Cell, 2017, 168, 239–251 e216 CrossRef CAS PubMed.
- M. C. Gregory, M. A. McLean and S. G. Sligar, Biochem. Biophys. Res. Commun., 2017, 487, 351–355 CrossRef CAS PubMed.
- M. A. McLean, M. C. Gregory and S. G. Sligar, Annu. Rev. Biophys., 2018, 47, 107–124 CrossRef CAS PubMed.
- Q. N. Van, C. A. Lopez, M. Tonelli, T. Taylor, B. Niu, C. B. Stanley, D. Bhowmik, T. H. Tran, P. H. Frank, S. Messing, P. Alexander, D. Scott, X. Ye, M. Drew, O. Chertov, M. Losche, A. Ramanathan, M. L. Gross, N. W. Hengartner, W. M. Westler, J. L. Markley, D. K. Simanshu, D. V. Nissley, W. K. Gillette, D. Esposito, F. McCormick, S. Gnanakaran, F. Heinrich and A. G. Stephen, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 24258–24268 CrossRef CAS PubMed.
- J. Y. Suh, C. Tang and G. M. Clore, J. Am. Chem. Soc., 2007, 129, 12954–12955 CrossRef CAS PubMed.
- J. C. Hunter, A. Manandhar, M. A. Carrasco, D. Gurbani, S. Gondi and K. D. Westover, Mol. Cancer. Res., 2015, 13, 1325–1335 CrossRef CAS PubMed.
- A. Bergner, X. Cockcroft, G. Fischer, A. Gollner, W. Hela, R. Kousek, A. Mantoulidis, L. J. Martin, M. Mayer, B. Mullauer, G. Siszler, B. Wolkerstorfer, D. Kessler and D. B. McConnell, Chemistry, 2019, 25, 12037–12041 CrossRef CAS PubMed.
- Z. W. Zhou, C. Ambrogio, A. K. Bera, Q. Li, X. X. Li, L. Li, J. Son, S. Gondi, J. Li, E. Campbell, H. Jin, J. J. Okoro, C. X. Xu, P. A. Janne and K. D. Westover, Cancer Res., 2020, 80, 3719–3731 CAS.
- K. J. Cho, D. van der Hoeven, Y. Zhou, M. Maekawa, X. Ma, W. Chen, G. D. Fairn and J. F. Hancock, Mol. Cell Biol., 2016, 36, 363–374 CrossRef CAS PubMed.
- H. I. Ingolfsson, M. N. Melo, F. J. van Eerden, C. Arnarez, C. A. Lopez, T. A. Wassenaar, X. Periole, A. H. de Vries, D. P. Tieleman and S. J. Marrink, J. Am. Chem. Soc., 2014, 136, 14554–14559 CrossRef CAS PubMed.
- Z. L. Li and M. Buck, Structure, 2017, 25, 679–689 e672 CrossRef CAS PubMed.
- Z. L. Li, P. Prakash and M. Buck, ACS Cent. Sci., 2018, 4, 298–305 CrossRef CAS PubMed.
- P. Prakash, Y. Zhou, H. Liang, J. F. Hancock and A. A. Gorfe, Biophys. J., 2016, 110, 1125–1138 CrossRef CAS PubMed.
- S. Cao, S. Chung, S. Kim, Z. Li, D. Manor and M. Buck, J. Biol. Chem., 2019, 294, 7068–7084 CrossRef CAS PubMed.
- T. Travers, C. A. Lopez, Q. N. Van, C. Neale, M. Tonelli, A. G. Stephen and S. Gnanakaran, Sci. Rep., 2018, 8, 8461 CrossRef PubMed.
- S. Li, H. Jang, J. Zhang and R. Nussinov, Structure, 2018, 26, 513–525 e512 CrossRef CAS PubMed.
- D. Kessler, M. Gmachl, A. Mantoulidis, L. J. Martin, A. Zoephel, M. Mayer, A. Gollner, D. Covini, S. Fischer, T. Gerstberger, T. Gmaschitz, C. Goodwin, P. Greb, D. Haring, W. Hela, J. Hoffmann, J. Karolyi-Oezguer, P. Knesl, S. Kornigg, M. Koegl, R. Kousek, L. Lamarre, F. Moser, S. Munico-Martinez, C. Peinsipp, J. Phan, J. Rinnenthal, J. Sai, C. Salamon, Y. Scherbantin, K. Schipany, R. Schnitzer, A. Schrenk, B. Sharps, G. Siszler, Q. Sun, A. Waterson, B. Wolkerstorfer, M. Zeeb, M. Pearson, S. W. Fesik and D. B. McConnell, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 15823–15829 CrossRef CAS PubMed.
- T. H. Tran, P. Alexander, S. Dharmaiah, C. Agamasu, D. V. Nissley, F. McCormick, D. Esposito, D. K. Simanshu, A. G. Stephen and T. E. Balius, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 3363–3364 CrossRef CAS PubMed.
- Y. Zhou and J. F. Hancock, Biochim. Biophys. Acta, 2015, 1853, 841–849 CrossRef CAS PubMed.
- Y. Zhou, P. Prakash, A. A. Gorfe and J. F. Hancock, Cold Spring Harbor Perspect. Med., 2018, 8, a031831 CrossRef CAS PubMed.
- H. Wu, Cell, 2013, 153, 287–292 CrossRef CAS PubMed.
- T. Tian, A. Harding, K. Inder, S. Plowman, R. G. Parton and J. F. Hancock, Nat. Cell Biol., 2007, 9, 905–914 CrossRef CAS PubMed.
- S. J. Plowman, N. Ariotti, A. Goodall, R. G. Parton and J. F. Hancock, Mol. Cell Biol., 2008, 28, 4377–4385 CrossRef CAS PubMed.
- H. Lavoie and M. Therrien, Nat. Rev. Mol. Cell Biol., 2015, 16, 281–298 CrossRef CAS PubMed.
- J. R. Sydor, M. Engelhard, A. Wittinghofer, R. S. Goody and C. Herrmann, Biochemistry, 1998, 37, 14292–14299 CrossRef CAS PubMed.
- C. Kiel and L. Serrano, Sci. Signaling, 2009, 2, ra38 CrossRef PubMed.
- E. Park, S. Rawson, K. Li, B. W. Kim, S. B. Ficarro, G. G. Pino, H. Sharif, J. A. Marto, H. Jeon and M. J. Eck, Nature, 2019, 575, 545–550 CrossRef CAS PubMed.
- M. R. Packer, J. A. Parker, J. K. Chung, Z. Li, Y. K. Lee, T. Cookis, H. Guterres, S. Alvarez, M. A. Hossain, D. P. Donnelly, J. N. Agar, L. Makowski, M. Buck, J. T. Groves and C. Mattos, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2015648118 CrossRef CAS PubMed.
- T. Shi, M. Niepel, J. E. McDermott, Y. Gao, C. D. Nicora, W. B. Chrisler, L. M. Markillie, V. A. Petyuk, R. D. Smith, K. D. Rodland, P. K. Sorger, W. J. Qian and H. S. Wiley, Sci. Signaling, 2016, 9, rs6 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc03484g |
| This journal is © The Royal Society of Chemistry 2021 |