
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA homoleptic alkynyl-protected [Ag9Cu6(tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) C)12]+ superatom with free electrons: synthesis, structure analysis, and different properties compared with the Au7Ag8 cluster in the M15+ series†
C)12]+ superatom with free electrons: synthesis, structure analysis, and different properties compared with the Au7Ag8 cluster in the M15+ series†
Xiaoshuang
Ma‡
a,
Lin
Xiong‡
b,
Lubing
Qin
a,
Yun
Tang
a,
Guanyu
Ma
a,
Yong
Pei
*b and
Zhenghua
Tang
 *a
*a
aGuangzhou Key Laboratory for Surface Chemistry of Energy Materials, New Energy Research Institute, School of Environment and Energy, South China University of Technology, Guangzhou Higher Education Mega Centre, Guangzhou, Guangdong 510006, P. R. China. E-mail: zhht@scut.edu.cn
bDepartment of Chemistry, Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education, Xiangtan University, Hunan Province, Xiangtan 411105, P. R. China. E-mail: ypei2@xtu.edu.cn
First published on 31st August 2021
Abstract
We report the first homoleptic alkynyl-protected AgCu superatomic nanocluster [Ag9Cu6(tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)12]+ (NC 1, also Ag9Cu6 in short), which has a body-centered-cubic structure with a Ag1@Ag8@Cu6 metal core. Such a configuration is reminiscent of the reported AuAg bimetallic nanocluster [Au1@Ag8@Au6(tBuCC)12]+ (NC 2, also Au7Ag8 in short), which is also synthesized by an anti-galvanic reaction (AGR) approach with a very high yield for the first time in this study. Despite a similar Ag8 cube for both NCs, structural anatomy reveals that there are some subtle differences between NCs 1 and 2. Such differences, plus the different M1 kernel and M6 octahedron, lead to significantly different optical absorbance features for NCs 1 and 2. Density functional theory calculations revealed the LUMO and HOMO energy levels of NCs 1 and 2, where the characteristic absorbance peaks can be correlated with the discrete molecular orbital transitions. Finally, the stability of NCs 1 and 2 at different temperatures, in the presence of an oxidant or Lewis base, was investigated. This study not only enriches the M15+ series, but also sets an example for correlating the structure–property relationship in alkynyl-protected bimetallic superatomic clusters.
C)12]+ (NC 1, also Ag9Cu6 in short), which has a body-centered-cubic structure with a Ag1@Ag8@Cu6 metal core. Such a configuration is reminiscent of the reported AuAg bimetallic nanocluster [Au1@Ag8@Au6(tBuCC)12]+ (NC 2, also Au7Ag8 in short), which is also synthesized by an anti-galvanic reaction (AGR) approach with a very high yield for the first time in this study. Despite a similar Ag8 cube for both NCs, structural anatomy reveals that there are some subtle differences between NCs 1 and 2. Such differences, plus the different M1 kernel and M6 octahedron, lead to significantly different optical absorbance features for NCs 1 and 2. Density functional theory calculations revealed the LUMO and HOMO energy levels of NCs 1 and 2, where the characteristic absorbance peaks can be correlated with the discrete molecular orbital transitions. Finally, the stability of NCs 1 and 2 at different temperatures, in the presence of an oxidant or Lewis base, was investigated. This study not only enriches the M15+ series, but also sets an example for correlating the structure–property relationship in alkynyl-protected bimetallic superatomic clusters.
Introduction
Superatomic coinage bimetallic nanoclusters (NCs) with atomic precision are currently being extensively investigated due to their tunable structure,1,2 enhanced stability,3–5 and significantly modified physicochemical properties,6–8 compared with homonuclear parent clusters.9–13 Among these monolayer-protected clusters, the most studied combination is Au–Ag, as they are fully miscible in bulk.14,15 It is well known that a plethora of stable Au NCs have been synthesized and characterized in the past few decades,16,17 yet stable Ag NCs comprise a rather recent entry in the coinage metal NC field.18,19 In contrast to Au or Ag, they are far fewer examples of Cu-containing coinage alloy NCs, particularly AgCu molecules. As for the capping agent for protection, mixed ligands can yield a more complicated structure and hence obscure the structure–property relationship establishment; therefore, molecules of single structure type particularly thiolate compounds have been widely employed to prepare AgCu NCs. For instance, in 2016, Zheng's group reported the first thiolated chiral three-concentric-shell cluster containing free valence electrons, [Ag28Cu12(SR)24]4− (SR = 2,4-dichlorobenzenethiolate).20 In 2018, Zhu's group showcased the controllable synthesis of the AgCu bimetallic NC [Ag40.13Cu13.87S19(tBuS)20(tBuSO3)12], which consisted of a Cu10Ag2S7 core, a M42(tBuS)20(tBuSO3)12 shell, and another 12 bare S atoms.21 Recently, Bao et al. prepared the [Ag13Cu10(SAdm)12]3+ (Adm = –SC10H15) NC, which has a Ag13 core and a Cu10(SAdm)12 shell.22Recently, alkynyl molecules have been emerging as a new type of ligand for preparing coinage metal NCs,23,24 mainly because alkynyl molecules can generate more diverse surface binding moieties and some undiscovered molecular clusters with magic numbers,25–30 eventually leading to drastically different functionalities.31–33 In terms of AuAg NCs, Wang and Zheng groups documented the fabrication of the Au34Ag28(PhCC)34 NC and its use as a model catalyst to explore the significance of surface ligands in promoting catalysis.34 In another report, Wang et al. discovered that when incorporating alkali metal ions or copper atoms into the alkynyl-protected body-centered cubic (BCC) [Au7Ag8(tBuCC)12]+ NC (which is also NC 2 in this study), site preference can be observed.35 Recently, Yuan et al. found that the alkynyl-protected monomeric (AuAg)34 can be assembled into 1D polymers with Ag–Au–Ag bonds between neighboring clusters through a solvent-mediated approach.36 In the AgCu regime, the alkynyl ligand has also been utilized. For instance, Williams and co-workers reported two halide-ion-templated heterometallic Ag8Cu6 rhombic dodecahedron clusters and investigated the spectroscopic properties and reactivity of these clusters along with those of the parent Ag14 NCs.37 Mak’s group employed the designed tetranuclear precursors [(R–CC–CC)Ag]4 (R = iPr, tBu, and Ch) to construct a series of heteropolynuclear silver(I)–copper(I) diynyl complexes that bear a common trigonal-planar CuAg3 NC core, and such complexes exhibited long-lived emission upon photoexcitation in various media at room temperature and 77 K.38 In a recent study, Zang’s group discovered that the o-carboranealkynyl-protected [Cu6Ag8(C4B10H11)12Cl]NO3 NC can serve as a perfect hypergolic material, as its ignition delay time can be shortened to 15 ms.39 However, in the above examples of AgCu NCs, all the Ag and Cu atoms are present as the +1 charge state, and no free electrons exist in these clusters. According to the superatom theory,40 these molecules are regarded as M(I) clusters or complexes rather than superatoms with free electrons. Due to the presence of free electrons, the superatoms have quite different structures and physicochemical properties, and hence they can find different applications in catalysis,41,42 optoelectronic devices,36 biomedical regimes31 and so on. To the best of our knowledge, no case of a homoleptic alkynyl-protected superatomic AgCu NC with free electrons has been reported so far.
Herein, we report the first case of a homoleptic alkynyl-protected AgCu superatomic NC, namely [Ag9Cu6(tBuCC)12]+ (NC 1, also Ag9Cu6 in short), which has a BCC-based structure with a three-layered Ag1@Ag8@Cu6 metal core configuration. Such a structure is reminiscent of the reported [Au7Ag8(tBuCC)12]+ (NC 2, also Au7Ag8 in short) molecule with a Au1@Ag8@Ag6 metal architecture, and it is first-time synthesized by an anti-galvanic reaction (AGR) approach with a ultrahigh yield in this study. The structural differences between NC 1 and NC 2 result in significantly different optical absorption properties. Comprehensive DFT calculations disclosed the discrete LUMO and HOMO energy levels of NCs 1 and 2, where the characteristic absorbance peaks of NCs 1 and 2 can be correlated to the specific molecular orbital transitions. Finally, the stability of NCs 1 and 2 at room temperature and 60 °C, in the presence of H2O2 or CH3ONa, was investigated and compared.
Results and discussion
NC 1 was first prepared by following a modified “Two-in-One” method (see experimental details in the ESI†).43 Briefly, in the presence of NaSbF6, tBuCCAg(I) is reduced by (PPh3)2CuBH4 in the mixed solvent of dichloromethane and acetonitrile. The reaction was aged for 12 h during which the solution gradually changed from colorless to yellow, and finally to dark blue. A blue block crystal was obtained by diffusing methanol into the dichloromethane solution containing the crude product. It is worth noting that by introducing one metal in the precursor and another metal in the reducing agent, such a “Two-in-One” method might be universal for synthesizing bimetallic NCs. NC 2 was synthesized by the AGR approach between organometallic tBuCCAg(I) and Au22(tBuCC)18 NC under mild conditions. The synthetic protocols regarding the tBuCCAg(I) precursor and Au22(tBuCC)18 can be found in the ESI.†
The chemical compositions of NCs 1 and 2 were verified by electrospray ionization mass spectrometry (ESI-MS) in positive mode. As shown in Fig. 1, the main peak appears at m/z = 2325.5677 for NC 1 and 3214.8510 for NC 2, corresponding well with [Ag9Cu6(C6H9)12]+ (cal. MW: 2323.5674 Da, deviation: 0.0003 Da) and [Au7Ag8(C6H9)12]+ (cal. MW: 3214.8515 Da, deviation: 0.0005 Da), respectively. Also, the isotopic distributions of the two NCs match perfectly with the simulated results (inset in Fig. 1A and B). One may notice that there are some fragments in the ESI-MS spectra of NC 1, and the peak analysis in Fig. S1† shows that the three peaks at m/z = 2254.6932, 2299.0153, and 2370.0681 Da can be assigned to [Ag7Cu7(C6H9)13]+, [Ag8Cu6(C6H9)13]+, and [Ag10Cu5(C6H9)12]+, respectively. For NC 2, there is a much less pronounced peak, and the peak assignment analysis in Fig. S2† shows that it can be assigned to [Au7Ag8(tBuCC)10]+, which is probably generated by losing two ligands (–tBuCC) from parent NC 2.
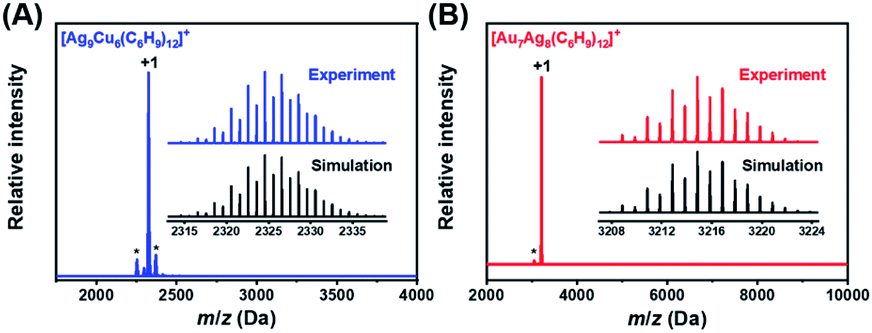 | ||
| Fig. 1 ESI-MS spectra of NCs 1 (A) and 2 (B) in positive mode. Inset: the experimental isotropic pattern (blue for 1, and red for 2) and simulated (black) data of NCs 1 and 2. | ||
Moreover, the fingerprint absorbance peaks of NC 1 are located at 544, 579, and 620 nm, and its characteristic absorbance feature along with the simulated pattern will be discussed next. Nevertheless, we monitored the absorbance change during the formation of NC 1. As shown in Fig. S3A,† upon reduction, an absorbance peak at 477 nm gradually appeared with the maximal value reached at 1 h. After that, such an absorbance peak gradually diminished and the characteristic peak at 579 nm from NC 1 gradually emerged. The 477 nm absorbance peak indicates that some intermediate may exist; however, several attempts to isolate it were not successful. As depicted in Fig. S3B,† the sharp color transition from yellow to dark yellow, slight pink and eventually blue can be clearly visualized. To better understand the AGR process from Au22(tBuCC)18 to NC 2, the reaction process was also monitored using time-resolved UV-visible absorption spectra. As shown in Fig. S4A,† the absorbance features of Au22 NC disappeared immediately upon the addition of tBuCCAg(I), while a new absorption band at ∼537 nm emerged. There are two obvious color changes at the time point of tBuCCAg(I) addition and in the period from 4 to 8 h (Fig. S4B†). After 2 h, the characteristic peak at 487 nm of NC 2 gradually emerged, meanwhile the absorbance peak at 537 nm gradually diminished. It has been postulated but not ascertained that there might be some critical intermediate during the AGR process, which is still under investigation.
The electronic structures of NCs 1 and 2 were subsequently probed by X-ray photoelectron spectroscopy (XPS), and the results are presented in Fig. S5, S6 and Table S1.† As depicted in Fig. S5A,† the XPS survey scan profile confirmed the co-existence of Ag and Cu elements. The Ag/Cu atomic ratio is estimated as 10.33/6.90, in good agreement with the theoretical value (9/6). It can be noted that the binding energy of the Ag 3d5/2 electrons is located at 368.41 eV, higher than that of bulk Ag (367.9 eV) and lower than that of Ag(I) (368.87 eV) (Fig. S5B†).44 It suggests that the valence state of Ag in NC 1 is between 0 and +1. Meanwhile, the binding energy of Cu 2p3/2 (933.12 eV) agrees well with that of Cu(I) (932.45–933.48 eV), implying that six Cu atoms are present as Cu(I) (Fig. S5C†).45,46 For NC 2, from the XPS survey scan profile (Fig. S6A†), the Ag/Au atomic ratio can be estimated as 8.27/7.28, in good agreement with the theoretical value (8/7). As shown in Fig. S6B,† the binding energy of the Au 4f7/2 electrons is located at 84.50 eV, in between those of bulk Au (84.0 eV) and Au(I) (86.0 eV),47 suggesting that the M1 core in NC 2 is Au(0). In addition, the binding energy of the Ag 3d5/2 electrons is located at 368.83 eV, indicating that the valence state of Ag in NC 2 is +1 (Fig. S6C†).45
Subsequently, the atomic packing structure of NC 1 was examined using a single-crystal X-ray diffractometer. As illustrated in Fig. S7,† NC 1 crystallizes in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) , and each unit cell has a SbF6− counterion, indicating that NC 1 possessed a +1 charge state. More detailed structural parameters are summarized in Table S2.† The structural anatomy of NC 1 is shown in Fig. 2A, which contains nine silver atoms, six copper atoms and twelve tert-butylacetylene ligands, and hence the molecule can be formulated as [Ag9Cu6(tBuCC)12]SbF6. The six copper atoms and twelve alkynyl ligands form six tBuCC–Cu–CCtBu motifs on the surface of this quasi-spherical structure of NC 1 (Fig. 2B). Interestingly, all tert-butylacetylene ligands bind with Cu atoms via σ bonding, and with Ag atoms in the π manner (Fig. 2C). Note that such linear motifs were observed for the first time in the alkynyl-protected AgCu bimetallic NCs, even though similar linear motifs have been previously documented in several alkynyl-protected AuAg NCs, including Au7Ag8,35 Au24Ag20,48 Au34Ag28,34 (AuAg)34,36 Au57Ag53,49 and Au80Ag30.50 As illustrated in the space-filling structure (Fig. 2D), six Cu sites and eight Ag sites are exposed, which might serve as open active sites for catalysis. Despite the orientation of the rigid ligand on the surface of NC 1 being similar to that of NC 2, the exposure extent of Ag atoms in NC 2 is somehow more than that in NC 1 (Fig. S8A and B†). However, for NC 2, even with the same ligand, it is more expansive, probably owing to the much larger radius of the Au atom (Fig. S8C and D†) than that of the Cu atom.
, and each unit cell has a SbF6− counterion, indicating that NC 1 possessed a +1 charge state. More detailed structural parameters are summarized in Table S2.† The structural anatomy of NC 1 is shown in Fig. 2A, which contains nine silver atoms, six copper atoms and twelve tert-butylacetylene ligands, and hence the molecule can be formulated as [Ag9Cu6(tBuCC)12]SbF6. The six copper atoms and twelve alkynyl ligands form six tBuCC–Cu–CCtBu motifs on the surface of this quasi-spherical structure of NC 1 (Fig. 2B). Interestingly, all tert-butylacetylene ligands bind with Cu atoms via σ bonding, and with Ag atoms in the π manner (Fig. 2C). Note that such linear motifs were observed for the first time in the alkynyl-protected AgCu bimetallic NCs, even though similar linear motifs have been previously documented in several alkynyl-protected AuAg NCs, including Au7Ag8,35 Au24Ag20,48 Au34Ag28,34 (AuAg)34,36 Au57Ag53,49 and Au80Ag30.50 As illustrated in the space-filling structure (Fig. 2D), six Cu sites and eight Ag sites are exposed, which might serve as open active sites for catalysis. Despite the orientation of the rigid ligand on the surface of NC 1 being similar to that of NC 2, the exposure extent of Ag atoms in NC 2 is somehow more than that in NC 1 (Fig. S8A and B†). However, for NC 2, even with the same ligand, it is more expansive, probably owing to the much larger radius of the Au atom (Fig. S8C and D†) than that of the Cu atom.
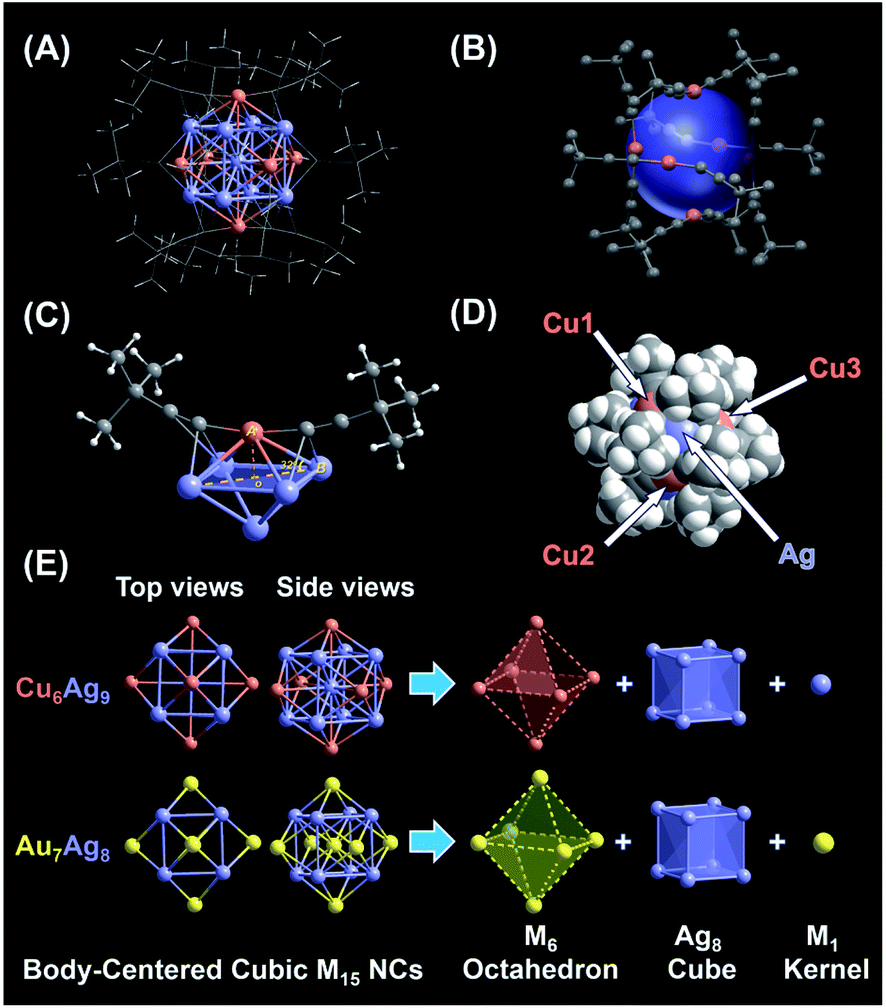 | ||
| Fig. 2 Structural analysis of body-centered cubic (BCC) M15 NCs. (A) Overall structure of monocationic NC 1. (B) The arrangement of six linear tBuCC–Cu–CCtBu staple motifs on the metal surface. (C) Coordination mode of tBuCC– ligands: μ2-η1 (Ag) and η1 (Cu). (D) The space-filling structure of NC 1. (E) Anatomy of NCs 1 and 2. Color legend: Au, yellow; Ag, purple; Cu, orange; C, light gray; hydrogen, white. | ||
Next, the anatomical structure of NC 1 is compared with that of NC 2 (the detailed structural parameters of NC 2 are summarized in Table S3†). As shown in Fig. 2E, NC 1 adopts a core–shell–shell configuration of Ag1@Ag8@Cu6, similar to that of NC 2, as both of them can be classified as the BCC-based M15+ series with an M1 kernel@Ag8 cube@M6 octahedron architecture. However, the AuAg core in NC 2 is slightly constricted compared to the AgCu core in NC 1, as the average adjacent Ag–Ag bond length is 3.333 Å and 3.271 Å in the Ag8 cube for NC 1 and 2, respectively (Fig. S9†). In addition, the average bond length from the central Ag to the Ag atoms in the Ag8 cube is 2.887 Å, comparable with that of bulk Ag (2.889 Å). Note that, compared with the reported Ag–Ag bond lengths in alkynyl-protected Ag NCs listed in Table S4,† there is argentophilic Ag–Ag interaction in the Ag@Ag8 cube for NC 1. Moreover, as illustrated in Fig. S10A,† the capped Cu atom isn't located exactly above the centre of the Ag4 plane, which is different from the surface of NC 2 (Fig. S10B†). That means, besides the atom differences in the M1 kernel and M6 octahedron, NC 1 and NC 2 have some subtle structural differences, and such subtle differences may affect their physicochemical properties as well. In addition, the structure of the thiolate-protected M15 NC Au15(SR)13 also has been theoretically proposed, but it is quite different from that of NC 1 and NC 2, as it consists of a tetrahedral Au4 core, a [Au7(SR)7] ring, and two [Au2(SR)3] “staple” motifs.51
Next, we first compared the optical absorbance properties of NC 1 and NC 2. For NC 1, as shown in Fig. 3A and S11,† there are four prominent peaks at 333 nm (3.72 eV), 544 nm (2.28 eV), 579 nm (2.14 eV), and 620 nm (2.00 eV), a broad absorption peak at 422 nm (2.94 eV), and a weak shoulder at 357 nm (3.47 eV). The energy bandgap derived from the absorbance spectrum is ∼1.80 eV. For NC 2, as presented in Fig. 3B and S11,† there are also four prominent peaks at 3.13 nm (3.96 eV), 422 nm (2.80 eV), 477 nm (2.60 eV), and 506 nm (2.45 eV), and a weak shoulder at 339 nm (3.65 eV), while the energy bandgap is ∼2.22 eV. It is worth noting that, despite some similar absorbance patterns, the absorbance features are drastically different for NC 1 and NC 2 in terms of the peak position and optical bandgap. Such huge discrepancies can be probably attributed to the structural differences. As both NCs adopt an M1@Ag8@M6 metal configuration, the M1 kernel (Ag vs. Au) can make a dramatically different contribution to the absorbance, and the outer M6 octahedron (Ag6vs. Au6) not only modulates the geometrical configuration, but also influences the electronic structure (discussed next). Given the standard absorbance curve (Fig. S12A and B†) of the two NCs, according to Lambert–Beer's law, the molecular absorptivity (ε) of NC 1 (ε = 0.35 × 104 M−1 cm−1) and NC 2 (ε = 0.78 × 104 M−1 cm−1) can be determined, as summarized in Table S5.† Therefore, through calculation, the yield of NC 1 was 41.05% (based on Ag, and the yield is 41.74% based on Cu), and the yield of NC 2 was up to 86.72% (based on Au). The details of the calculation process can be found in the ESI† (Fig. S13 and Tables S6, S7†). It is worth noting that the yield of NC 2 here is much higher than that of the previously reported method. This is mainly due to the fact that the reported method is a “bottom-up” direct reduction approach, in which other polydisperse clusters are also produced, while the AGR method here can yield more homogeneous products, and NC 2 is the main oxidation product (from 4e of Au22 NC to 2e of Au7Ag8 NC). In addition, we also studied the photo-luminescence properties of the two M15 NCs. As shown in Fig. S14,† NC 2 strongly emits in the near-IR region (λmax = 818 nm), in good agreement with the previous report.35 However, there is no obvious emission peak for NC 1.
To elucidate the relationship between the electronic structure and optical properties of NCs 1 and 2, we carried out time dependent-density functional theory (TD-DFT) calculations. The optimized structure based on the crystal structure is used as a model for TD-DFT calculation. As shown in Fig. 3E and F, there is no change in structure after optimization, except for a slightly distorted orientation of the ligands. The distribution of the electronic cloud map of the highest occupied molecular orbital (HOMO) of NC 1 is quite similar to that of NC 2, whereas the cloud density position in the lowest unoccupied molecular orbital (LUMO) of NC 1 is quite different from that of NC 2. From the cloud density distribution, it can be clearly noted that charge transfer occurs from ligands to the metal core for NC 2, resulting in free electron localization in the metal core. While in stark contrast, charge transfer happens from the metal core to ligands for NC 1, which leads to a non-radiative loss of excited state electron energy. Such different charge transfer behaviors are probably the main cause of the different luminescence properties. From Fig. 3E, the molecular orbitals of NC 1 revealed a jelliumatic shell closing at the HOMO state of 1S with two electrons and the LUMO state of 1P, in which there was conversion from S to P. It is also in good accord with the electron counting results, as the metal core offers fifteen delocalized electrons; while twelve electrons are delocalized at the metal-core bonds, one electron has to be deducted to form a cation, and hence NC 1 can be literally considered as a two-electron jelliumatic molecule. The absorption feature of NCs 1 and 2 was also theoretically simulated. As shown in Fig. 3C and D, four prominent peaks namely α, β, γ, and δ can be clearly recognized for both NCs, and the detailed transitions corresponding to the significant peaks are listed in Tables S8 and S9† for NCs 1 and 2, respectively. For NC 1, an optical bandgap of ca. 1.80 eV is extrapolated according to the value of the absorption edge, which is close to the absorption peak at 1.72 eV (α) in the simulated spectrum. Note that the band α could not be merely considered as a HOMO to LUMO transition, but might be resulted from three transition modes (HOMO to LUMO, HOMO to LUMO + 1, and HOMO to LUMO + 2) with nearly equal contribution values (96.2%, 96.1%, and 95.6%) (Fig. S15A†). Similar transition modes are observed in NC 2 (Fig. 3D and S16A†), and the excitation energy (ΔE = 2.04 eV) is also close to the optical bandgap (exp. 2.22 eV). The β peak at 2.22 eV of NC 1 can also be attributed to three transition modes (HOMO to LUMO + 3, HOMO to LUMO + 4, and HOMO to LUMO + 5), whereas the β peak at 2.89 eV in NC 2 can be assigned to two transition modes (HOMO − 8 to LUMO and HOMO − 8 to LUMO + 1) with the contribution of 42.3% and 12.5%, respectively (Fig. S15B and S16B†). In addition, the γ peak of NC 1 comprises fifteen transition modes (e.g., HOMO − 6 (67.7%), HOMO − 7 (55.9%), HOMO − 1 (43.5%) to LUMO + 2 and so on) (Fig. S15C†). In contrast, the γ peak of NC 2 can be attributed to six transition modes, including HOMO to LUMO + 4 (69.0%), LUMO + 8 (59.4%), and LUMO + 9 (56.7%) (Fig. S16C†). The δ peak of NC 1 is mainly contributed by the HOMO − 28 to LUMO + 2 (48.2%) transitions (Fig. S15D†), while the δ peak of NC 2 is predominantly contributed by two transition modes (i.e., HOMO − 6 to LUMO + 4, 21.4%, and HOMO − 19 to LUMO + 2, 24.1%) (see Fig. S16D†). One can conclude that even if NCs 1 and 2bear a similar M15+ geometrical configuration, the optical absorption properties are quite different, mainly owing to the differences in the M1 kernel (Ag vs. Au) and M6 octahedron (Cu vs. Au), and probably the subtle difference in the Ag8 cube as well.
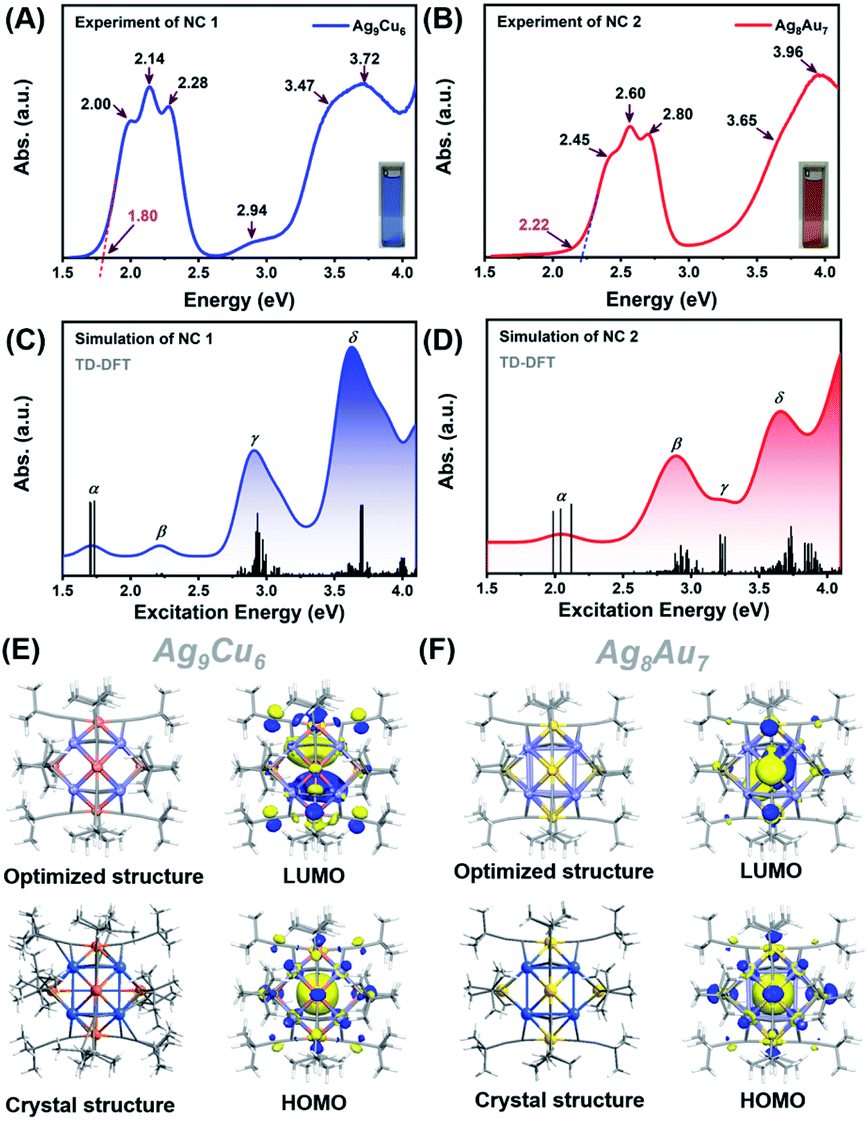 | ||
| Fig. 3 Experimental absorption spectrum plotted in the energy axis of NCs 1 (A) and 2 (B). Insets show the digital photographs of the NCs in CH2Cl2. Simulated absorption spectra of NCs 1 (C) and 2 (D). The electron density diagrams of the HOMO and LUMO orbitals for NCs 1 (E) and 2 (F). | ||
Furthermore, we also calculated the electronic structures of NCs 1 and 2 to unravel the relationship between the electronic structure and the optical absorption properties of M15+ NCs. According to the Kohn–Sham (KS) molecular orbital energy level diagram (Fig. 4A and B), there is an obvious energy gap between the HOMO and the remaining occupied orbitals for the two NCs, which may be because the two electrons at the HOMO orbital with higher energy tend to relax to the HOMO – 1 orbital. It is worth noting that for the frontier unoccupied molecular orbitals (including the LUMO), the Ag(sp) atomic orbital makes the most significant contribution in both NCs. That means, for both NCs, Ag8 cubic atoms are the major contributor to the unoccupied molecular orbitals. It indicates that the unoccupied molecular orbitals of the M15+ clusters might be localized at some specific position (i.e., M8 cube), while the M1 kernel and M6 octahedron make less contribution, reminiscent of the case of M21(SCH3)15.52 This explains that there are triplet peaks located at high wavelength for both NCs. However, for the remaining occupied molecular orbitals (not including the HOMO) particularly the deep occupied orbitals, the Cu(d) atomic orbital makes the most important contribution in NC 1, while Au(d) and Ag(d) both contributed significantly in NC 2. For the HOMO, drastic differences can be observed, Ag(sp) and Au(sp) atomic orbitals make the most significant contribution to the HOMO orbital in NCs 1 and 2, respectively. The absorption band α in NC 1 is primarily attributed to intraband Ag(sp) to Ag(sp) transitions, while the interband Au(d) to Ag(sp) transitions lead to the α band in NC 2. This finding reveals that the central M(0) atom of M15 NCs plays a significant role in the HOMO energy. Moreover, the HOMO energy of NC 1 is higher than that of NC 2, probably due to the fact that, compared with Ag, the central Au atom has higher cohesive force thus causing NC 2 to have lower HOMO energy than NC 1. Similarly, band β at 2.22 eV in NC 1 and that at 2.89 eV in NC 2 originate from Ag(sp) to Ag(sp), and Au(d) to Ag(sp) transitions, respectively. Notably, the band γ of NC 1 comprises Cu(d) to Ag(sp) transitions, suggesting that the Cu6 octahedron is the major contributor to the deep occupied orbitals. The absorption band δ (i.e., 3.63 eV for NC 1 and 3.66 eV for NC 2) has a large portion of C(p) character of alkynyl ligands, which greatly affects their high-energy absorption bands around ∼350 nm. It can be noted that the same tBuCC– ligand makes almost equal yet predominant contribution to the δ absorption band for both NCs, suggesting the important role of the surface ligand in the optical properties.
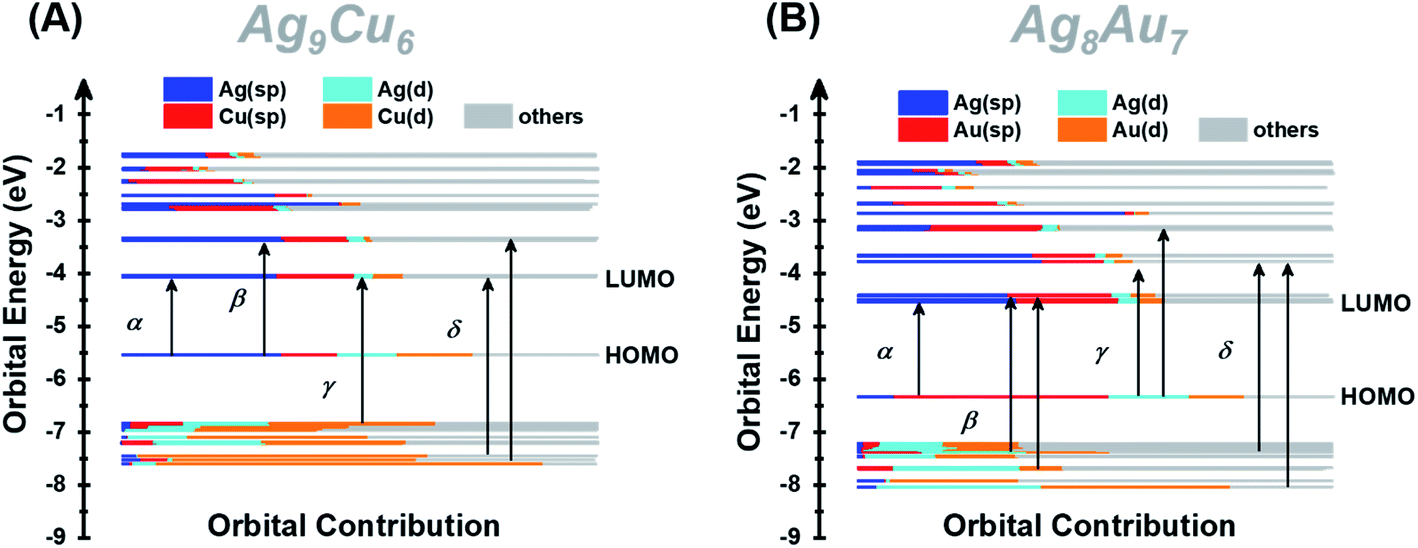 | ||
| Fig. 4 Kohn–Sham (KS) molecular orbital energy level diagram and the associated populations of atomic orbitals in each K–S molecular orbital for NCs 1 (A) and 2 (B). | ||
Despite the similar structural scaffold of the two NCs, the subtle differences in metal composition and structural coordination mode might result in a vast difference of their stability. The stability of NCs 1 and 2 was investigated by monitoring the time-resolved UV-vis absorbance spectra at different temperatures, in the presence of an oxidant (e.g., H2O2) or Lewis base (e.g., CH3ONa). Fig. 5 shows the intensity of the peaks at 579 nm for NC 1 and 487 nm for NC 2versus time. At room temperature, the absorbance of NCs 1 and 2 remained almost unchanged for 24 h (Fig. S17A and B†), indicating that the two NCs can be stable under ambient conditions. As shown in Fig. 5A, the relative absorbance intensity of NC 2 preserved 81% of its initial value in 24 h, however, NC 1 retained 94%, indicating much less decomposition. It was because both NCs are situated at the lowest energy state, as confirmed by DFT calculations of the vibration frequency of NCs 1 and 2 (Table S10†). When incubated at 60 °C (Fig. 5B), the relative intensity of NC 1 remained 81% at 24 h, slightly higher than that of NC 2 (78%), indicating excellent thermal stability for both NCs. However, the absorption intensity of NC 1 dropped much faster than that of NC 2 in the first 5 h (Fig. S17C and D†), indicating that it is more susceptible to heat. However, in the following 5–24 h, the relative intensity of NC 1 remained almost unchanged. Previous studies have documented that the thermal stability of bimetallic NCs not only depends on the extent of free electron centralization,4,53 but also the metal–ligand interaction needs to be considered,54 specifically, the interaction between the surface metal atoms (Ag, Au or Cu) and the alkynyl ligand must be taken into account for these two title NCs. As shown in Fig. S9,† the σ and π bonds between the metal and carbon atoms of the surface binding motif in NC 1 (Cu1–C1: 1.856 Å; Cu1–C2: 1.884 Å; Ag1–C1: 2.395 Å; Ag2–C2: 2.355 Å) are slightly stronger than those in NC 2 (Au1–C1: 1.983 Å; Au1–C2: 1.980 Å; Ag1–C1: 2.495 Å; Ag2–C2: 2.505 Å). Therefore, the more compact structure of NC 1 can prevent decomposition at higher temperature. Furthermore, NC 1 is more stable than NC 2 in the presence of the Lewis base (1 wt% CH3ONa in EtOH), but rapidly decomposed upon the addition of the oxidant (30 wt% H2O2). As shown in Fig. 5C, NC 1 decomposed slightly faster than NC 2 in the first 5 h, and both reached the same intensity at 8 h, after that, NC 2 decomposed slightly faster. The higher relative intensity (84% vs. 80%) indicates that NC 1 possessed slightly superior stability in the presence of the Lewis base. During this process, the color of NC 1 showed no change, while the NC 2 solution gradually turned from orange to light orange, as visualized in Fig. S18A and B.† Note that the cohesive force between Ag and Au in NC 2 is higher than that between Cu and Ag in NC 1, making NC 2 more vulnerable to the Lewis base, as CH3ONa is a nucleophilic agent, and hence it can attack the surface of NC 2 more favorably. However, NC 2 is more robust than NC 1 upon adding H2O2 aqueous solution (Fig. S18C and D†). As shown in Fig. 5D, NC 1 decomposed completely in 1 h, suggesting that NC 1 is sensitive to H2O2, probably because the Cu(I) atoms on the surface of NC 1 can be easily oxidized into Cu(II). In contrast, the corresponding Au(I) atoms in NC 2 possessed strong antioxidation capacity. However, it still lost 35% of the initial value, and such a decomposition can be presumably attributed to the fact that the Ag atoms in the Ag8 cube are easily attacked by the lone pair electrons of the peroxy radical (O22−).55 Such a phenomenon occurs even more seriously for NC 1, leading to an accelerated and complete decomposition in 1 h.
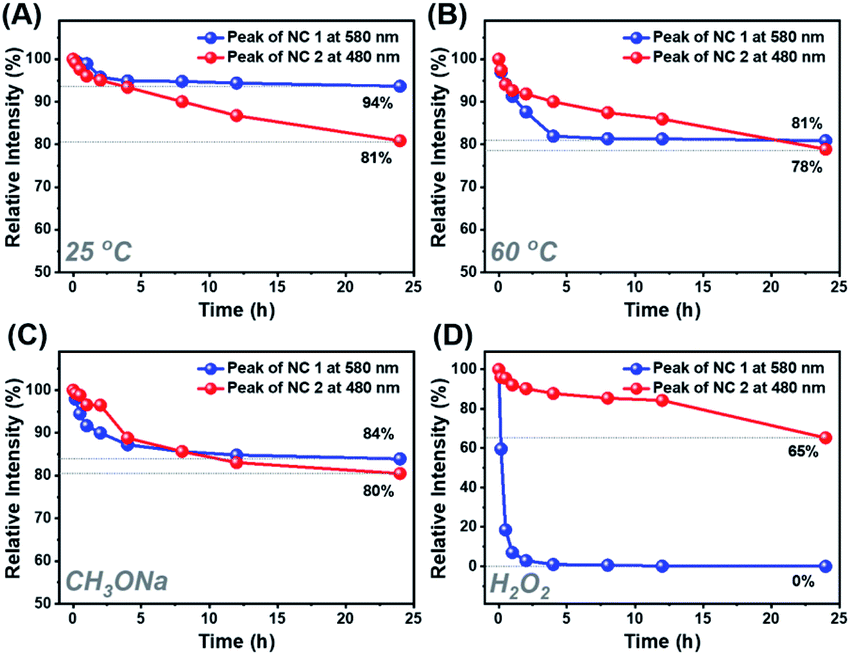 | ||
| Fig. 5 Stability comparison of NCs 1 and 2 under different conditions. The relative absorption intensity value at specific wavelengths is used (NC 1, 579 nm; NC 2, 487 nm) in the time resolved UV-vis absorption spectra. The thermal stability of NCs 1 and 2 in 1,2-dichloroethane solution under (A) ambient conditions and (B) heating to 60 °C. The stability of NCs 1 and 2 in CH2Cl2 solution in the presence of (C) an oxidant (H2O2) and (D) a Lewis base (CH3ONa). | ||
Conclusions
In conclusion, a novel homoleptic alkynyl-protected AgCu superatom [Ag9Cu6(tBuCC)12]+ was synthesized for the first time via a one-pot reaction with high yield (>40%). X-ray crystallographic analysis revealed that it possesses a BCC-based Ag1@Ag8@Cu6 configuration. Also, the BCC-based [Au7Ag8(tBuCC)12]+ cluster with a Au1@Ag8@Au6 metal core was also prepared by an AGR approach with a ultrahigh yield (>86%). DFT calculations revealed that the different absorption features of the two NCs can be attributed to the differences in the M1 kernel (Ag vs. Au), M6 octahedron (Cu vs. Au), and the subtle differences in the Ag8 cube. The characteristic absorbance peaks of NCs 1 and 2 are successfully correlated with the specific molecular orbital transitions. NC 1 possessed superior stability to NC 2 at both room temperature and elevated temperature, and NC 1 also showed better tolerance to the Lewis base but is much more sensitive to the oxidant. We envision that this study can stimulate more research efforts on Cu-containing bimetallic superatomic NCs in terms of their synthesis, structural analysis, property exploration and beyond.
Data availability
All the data in this study are provided in the main text and ESI.†Author contributions
X. M. and Z. T. conceived the idea, X. M. conducted most of the experiments, L. Q., Y. T., and G. M. helped the characterization, L. X. and Y. P. conducted the DFT calculations, X. M. and Z. T. wrote up the draft, Y. P. and Z. T. provided the funding support, and all the authors contributed to the final proof of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. T. acknowledges the financial support from Guangzhou Science and Technology Plan Projects (No. 201804010323). Y. P acknowledges the financial support from the National Natural Science Foundation of China (Grant No. 91961121 and 21773201).Notes and references
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed.
- X. Kang, S. Jin, L. Xiong, X. Wei, M. Zhou, C. Qin, Y. Pei, S. Wang and M. Zhu, Chem. Sci., 2020, 11, 1691–1697 RSC.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- X. Kang, H. Abroshan, S. Wang and M. Zhu, Inorg. Chem., 2019, 58, 11000–11009 CrossRef CAS PubMed.
- X. Kang, X. Wei, P. Xiang, X. H. Tian, Z. W. Zuo, F. Q. Song, S. X. Wang and M. Z. Zhu, Chem. Sci., 2020, 11, 4808–4816 RSC.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- M. Suyama, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2020, 124, 23923–23929 CrossRef CAS.
- X. Zou, S. He, X. Kang, S. Chen, H. Yu, S. Jin, D. Astruc and M. Zhu, Chem. Sci., 2021, 12, 3660–3667 RSC.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem.–Eur. J., 2020, 26, 16150–16193 CrossRef CAS PubMed.
- H. Hirai, S. Ito, S. Takano, K. Koyasu and T. Tsukuda, Chem. Sci., 2020, 11, 12233–12248 RSC.
- S. Wang, Q. Li, X. Kang and M. Zhu, Acc. Chem. Res., 2018, 51, 2784–2792 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- N. Yan, N. Xia, L. Liao, M. Zhu, F. Jin, R. Jin and Z. Wu, Sci. Adv., 2018, 4, eaat7259 CrossRef CAS PubMed.
- Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 32, e1905488 CrossRef PubMed.
- J. Yang and R. Jin, ACS Mater. Lett., 2019, 1, 482–489 CrossRef CAS.
- Q. F. Yao, Z. N. Wu, Z. H. Liu, Y. Z. Lin, X. Yuan and J. P. Xie, Chem. Sci., 2021, 12, 99–127 RSC.
- J. Yan, H. Su, H. Yang, C. Hu, S. Malola, S. Lin, B. K. Teo, H. Hakkinen and N. Zheng, J. Am. Chem. Soc., 2016, 138, 12751–12754 CrossRef CAS PubMed.
- S. Yang, J. Chai, H. Chong, Y. Song, H. Yu and M. Zhu, Chem. Commun., 2018, 54, 4314–4316 RSC.
- Y. Bao, X. Wu, H. Gao, M. Zhou, S. Chen, S. Jin, H. Yu and M. Zhu, Dalton Trans., 2020, 49, 17164–17168 RSC.
- X. Ma, Y. Tang, G. Ma, L. Qin and Z. Tang, Nanoscale, 2021, 13, 602–614 RSC.
- L. L. Zhang, G. Zhou, G. Zhou, H. K. Lee, N. Zhao, O. V. Prezhdo and T. C. W. Mak, Chem. Sci., 2019, 10, 10122–10128 RSC.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS PubMed.
- X. Ma, Z. Tang, L. Qin, J. Peng, L. Li and S. Chen, Nanoscale, 2020, 12, 2980–2986 RSC.
- Z. Lei, X. K. Wan, S. F. Yuan, J. Q. Wang and Q. M. Wang, Dalton Trans., 2017, 46, 3427–3434 RSC.
- Z. Lei, X. K. Wan, S. F. Yuan, Z. J. Guan and Q. M. Wang, Acc. Chem. Res., 2018, 51, 2465–2474 CrossRef CAS PubMed.
- X. Ma, G. Ma, L. Qin, G. Chen, S. Chen and Z. Tang, Sci. China: Chem., 2020, 63, 1777–1784 CrossRef CAS.
- K. K. Chakrahari, R. P. B. Silalahi, J. H. Liao, S. Kahlal, Y. C. Liu, J. F. Lee, M. H. Chiang, J. Y. Saillard and C. W. Liu, Chem. Sci., 2018, 9, 6785–6795 RSC.
- T. T. Jia, G. Yang, S. J. Mo, Z. Y. Wang, B. J. Li, W. Ma, Y. X. Guo, X. Chen, X. Zhao, J. Q. Liu and S. Q. Zang, ACS Nano, 2019, 13, 8320–8328 CrossRef CAS PubMed.
- X. K. Wan, J. Q. Wang, Z. A. Nan and Q. M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- X. Y. Chang, G. T. Xu, B. Cao, J. Y. Wang, J. S. Huang and C. M. Che, Chem. Sci., 2017, 8, 7815–7820 RSC.
- Y. Wang, X. K. Wan, L. Ren, H. Su, G. Li, S. Malola, S. Lin, Z. Tang, H. Hakkinen, B. K. Teo, Q. M. Wang and N. Zheng, J. Am. Chem. Soc., 2016, 138, 3278–3281 CrossRef CAS PubMed.
- Y. Wang, H. Su, L. Ren, S. Malola, S. Lin, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2016, 55, 15152–15156 CrossRef CAS PubMed.
- P. Yuan, R. Zhang, E. Selenius, P. Ruan, Y. Yao, Y. Zhou, S. Malola, H. Hakkinen, B. K. Teo, Y. Cao and N. Zheng, Nat. Commun., 2020, 11, 2229 CrossRef CAS PubMed.
- T. U. Connell, S. Sandanayake, G. N. Khairallah, J. M. White, R. A. O'Hair, P. S. Donnelly and S. J. Williams, Dalton Trans., 2013, 42, 4903–4907 RSC.
- S. C. K. Hau, M. C. Yeung, V. W. Yam and T. C. W. Mak, J. Am. Chem. Soc., 2016, 138, 13732–13739 CrossRef CAS PubMed.
- Q. Y. Wang, J. Wang, S. Wang, Z. Y. Wang, M. Cao, C. L. He, J. Q. Yang, S. Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2020, 142, 12010–12014 CrossRef CAS PubMed.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS PubMed.
- C. Yin, S. Liu, Z. Qin, Y. Zhang, G. Li and Z. Zhao, Eur. J. Inorg. Chem., 2020, 2021, 392–397 CrossRef.
- Z. Qin, S. Sharma, C. Q. Wan, S. Malola, W. W. Xu, H. Hakkinen and G. Li, Angew. Chem., Int. Ed., 2021, 60, 970–975 CrossRef CAS PubMed.
- H. Shen, Y. Z. Han, Q. Wu, J. Peng, B. K. Teo and N. Zheng, Small Methods, 2020, 5, 2000603 CrossRef.
- W. Du, S. Jin, L. Xiong, M. Chen, J. Zhang, X. Zou, Y. Pei, S. Wang and M. Zhu, J. Am. Chem. Soc., 2017, 139, 1618–1624 CrossRef CAS PubMed.
- X. Zou, Y. Li, S. Jin, X. Kang, X. Wei, S. Wang, X. Meng and M. Zhu, J. Phys. Chem. Lett., 2020, 11, 2272–2276 CrossRef CAS PubMed.
- X. Gao, S. He, C. Zhang, C. Du, X. Chen, W. Xing, S. Chen, A. Clayborne and W. Chen, Adv. Sci., 2016, 3, 1600126 CrossRef PubMed.
- Z. Tang, D. A. Robinson, N. Bokossa, B. Xu, S. Wang and G. Wang, J. Am. Chem. Soc., 2011, 133, 16037–16044 CrossRef CAS PubMed.
- Y. Wang, H. Su, C. Xu, G. Li, L. Gell, S. Lin, Z. Tang, H. Hakkinen and N. Zheng, J. Am. Chem. Soc., 2015, 137, 4324–4327 CrossRef CAS PubMed.
- Z. J. Guan, J. L. Zeng, S. F. Yuan, F. Hu, Y. M. Lin and Q. M. Wang, Angew. Chem., Int. Ed., 2018, 57, 5703–5707 CrossRef CAS PubMed.
- J. L. Zeng, Z. J. Guan, Y. Du, Z. A. Nan, Y. M. Lin and Q. M. Wang, J. Am. Chem. Soc., 2016, 138, 7848–7851 CrossRef CAS PubMed.
- P. Liu, W. Han, M. Zheng, W. Li, J. Ren, A. Tlahuice-Flores and W. W. Xu, J. Phys. Chem. A, 2021, 125, 5933–5938 CrossRef CAS PubMed.
- Y. Li, M. G. Taylor, T. Y. Luo, Y. Song, N. L. Rosi, G. Mpourmpakis and R. Jin, J. Phys. Chem. Lett., 2020, 11, 7307–7312 CrossRef CAS PubMed.
- X. Kang, X. Wei, S. Jin, Q. Yuan, X. Luan, Y. Pei, S. Wang, M. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18834–18840 CrossRef CAS PubMed.
- Q. Tang, G. Hu, V. Fung and D. E. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- L. Han, P. Xing and B. Jiang, Org. Lett., 2014, 16, 3428–3431 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization, supporting figures and tables. Details and crystal data of Au7Ag8(CCtBu)12SbF6 and Ag9Cu6(CCtBu)12SbF6 (cif). CCDC 2072510 and 2072663. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03679c |
| ‡ X. Ma and L. Xiong contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |