
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented polycyclic polyprenylated acylphloroglucinols with anti-Alzheimer's activity from St. John's wort†
Yi
Guo‡
a,
Fang
Huang‡
b,
Weiguang
Sun
a,
Yuan
Zhou
 a,
Chunmei
Chen
a,
Changxing
Qi
a,
Jing
Yang
c,
Xiao-Nian
Li
c,
Zengwei
Luo
a,
Hucheng
Zhu
*a,
Xiaochuan
Wang
*b and
Yonghui
Zhang
*a
a,
Chunmei
Chen
a,
Changxing
Qi
a,
Jing
Yang
c,
Xiao-Nian
Li
c,
Zengwei
Luo
a,
Hucheng
Zhu
*a,
Xiaochuan
Wang
*b and
Yonghui
Zhang
*a
aHubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. E-mail: zhuhucheng@hust.edu.cn; zhangyh@mails.tjmu.edu.cn
bDepartment of Pathophysiology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China. E-mail: wxch@mails.tjmu.edu.cn
cState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650204, China
First published on 26th July 2021
Abstract
Hyperforones A–J (1–10), ten degraded and reconstructed polycyclic polyprenylated acylphloroglucinols (PPAPs) with six different types of unusual architectures, were isolated from Hypericum perforatum (St. John's wort). Compound 1 is characterized by an unprecedented 1,5-epoxyfuro[3′,4′:1,5]cyclopenta[1,2-c]oxecine ring system; compounds 2 and 3 represent the first PPAPs with a contracted B-ring leading to the unique 5/5 core skeletons; compound 4, a proposed biosynthetic precursor of 2, is defined by an oxonane-2,7-dione architecture; compound 5 features an unusual spiro[furo[3′,4′:1,5]cyclopenta[1,2-b]oxepine-3,2′-oxetane] ring system; compounds 6–8 possess a rare macrocyclic lactone ring in addition to the newly formed C-ring; and compounds 9 and 10 contain a newly formed six-membered C-ring, which constructed the unexpected 6/6 scaffold with the B-ring. Hypothetic biosynthetic pathways to generate these scaffolds starting from the classic [3.3.1]-type PPAPs helped to elucidate their origins and validate their structural assignments. Compounds 4 and 6 simultaneously displayed notable activation of PP2A (EC50: 258.8 and 199.0 nM, respectively) and inhibition of BACE1 in cells (IC50: 136.2 and 98.6 nM, respectively), and showed better activities than the positive controls SCR1693 (a PP2A activator, EC50: 413.9 nM) and LY2811376 (a BACE1 inhibitor, IC50: 260.2 nM). Furthermore, compound 6 showed better therapeutic effects with respect to the reduction of pathological and cognitive impairments in 3 × Tg AD mice than LY2811376. Compound 6 represents the first multitargeted natural product that could activate PP2A and simultaneously inhibit BACE1, which highlights compound 6 as a promising lead compound and a versatile scaffold in AD drug development.
Introduction
Since the first polycyclic polyprenylated acylphloroglucinol (PPAP) was isolated from the medicinal plant Hypericum perforatum (St. John's wort) in 1971,1,2 PPAPs have been extensively investigated because of their intricate molecular structures3–5 and diverse biological activities.6–9 Extracts of St. John's wort, one of the top-selling herbal products, have long been used therapeutically for mild to moderate depression in Europe.10 The beneficial effects of extracts of St. John's wort in Alzheimer's disease (AD) have also been continuously reported,11–13 and these reports have led to the question of whether hyperforin-related PPAP derivatives, the major secondary metabolites of H. perforatum, are also bioactive constituents and potential therapeutic agents for AD.Alzheimer's disease is characterized clinically by chronic and progressive dementia and histopathologically by senile plaques made up of β-amyloid (Aβ) and neurofibrillary tangles consisting of hyperphosphorylated tau, which results in a progressive decline in cognitive function and ultimately death.14–16 New approaches to AD treatment have focused on reducing the production of Aβ, in which β-site amyloid precursor protein cleaving enzyme 1 (BACE1) acts as the rate-limiting enzyme17 and the phosphorylation of tau is regulated by a tau-related phosphatase or kinase, such as protein phosphatase-2A (PP2A) or glycogen synthase kinase 3β (GSK-3β).18,19 Recently, Alzheimer's drug candidate aducanumab, a human monoclonal antibody that selectively targets aggregated Aβ, has shocked investors and scientists.20 Biogen's positive analyses of the aducanumab data represent the first time a Phase 3 study has demonstrated that clearance of aggregated Aβ can reduce the clinical decline of Alzheimer's disease.20 The antibody aducanumab now became the first new Alzheimer's disease drug approved in the United States that aims to interfere with the underlying disease process.21 This would provide compelling support for the amyloid hypothesis. However, it may achieve improved results if the strategy of targeting Aβ combined with other strategies, such as tau-targeted therapy. Furthermore, considering the multifactorial nature of AD,16 multi-target-directed ligands (MTDLs) might be synergistically beneficial against several etiopathogenic mechanisms of AD.
Based on our previous research,22–25 we investigated H. perforatum and isolated and characterized ten novel PPAPs, hyperforones A–J (1–10) (Fig. 1). The challenging task of the elucidation of their structures with absolute configurations was completed by spectroscopic (HRESIMS, NMR, ECD, and X-ray crystallography), quantum chemical computations (ECD and 13C NMR calculations), and chemical (modified Mosher's method) methods. These architectures are essentially different from normal PPAPs,26,27 which significantly enrich the structural diversity of PPAPs. Herein, we described the isolation, structural elucidation, and proposed biosynthetic pathways of 1–10 as well as their remarkable anti-AD ability simultaneously targeting PP2A and BACE1.
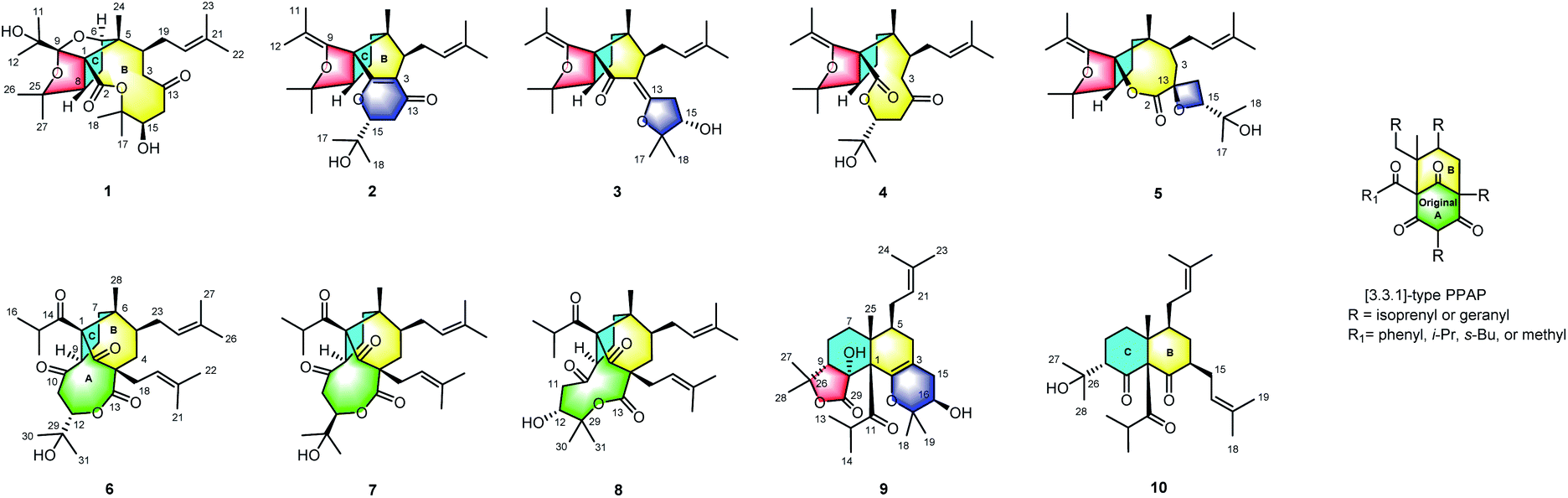 | ||
| Fig. 1 Structures of compounds 1–10. | ||
Results and discussion
Hyperforone A (1), isolated as colorless crystals, had the molecular formula C27H42O7 as determined by its HRESIMS (m/z 501.2824 [M + Na]+, calcd 501.2823) and 13C NMR data. The 1H NMR data (Table S1†) showed signals of an olefinic proton (δH 5.07), two exchangeable protons (δH 4.70 and 3.91), and nine methyl singlets (δH 1.13–1.85, s). The 13C NMR and DEPT spectra (Table S3†) of 1 revealed 27 carbon signals, in which some of the signals could be assigned to an isoprenyl, a propan-2-ol, and two carbonyl groups. The planar structure of 1 was determined by comprehensive interpretations of HSQC, HMBC, and 1H–1H COSY data. The hexahydro-1H-1,5-epoxycyclopenta[c]furan moiety of 1, with a cage-like framework, was determined by 1H–1H COSY correlations of H-6/H-7/H-8 and HMBC correlations (Fig. 2) from Me-24 to C-1, C-5, and C-6, from H-6 to C-1, C-5, and C-9, and from H-8 to C-1 and C-9, assisted by the downfield chemical shifts of C-6, C-9, and C-25. Additionally, HMBC correlations from both Me-11 and Me-12 to C-9 and C-10, from an exchangeable proton (δH 3.91) to C-10, and from both Me-26 and Me-27 to C-25 and C-8 indicated that the propan-2-ol group (C-10–C-12) was located at C-9 and two methyls (Me-26 and Me-27) were located at C-25 of the cage-like framework of 1. Further analysis of its 2D NMR revealed the presence of an oxecane-2,7-dione moiety (ring B), which fused with the cage-like moiety via C-1 and C-5. This conclusion was confirmed by the 1H–1H COSY correlations of H-3/H-4 and H-14/H-15 as well as the HMBC correlations from Me-24 to C-1, C-4, C-5, and C-6, from H-8 to C-2, from both H-3 and H-14 to C-13, and from H-15 to C-16 (Fig. 2). Moreover, 1H–1H COSY interactions of H-4/H-19/H-20 and HMBC correlations from Me-22 and Me-23 to C-20 and C-21 indicated an isoprenyl group at C-4; HMBC correlations from both Me-17 and Me-18 to C-15 and C-16 indicated two methyls at C-16; and the HMBC correlation from an exchangeable proton (δH 4.70) to C-15 suggested the location of a hydroxyl at C-15 of the oxecane-2,7-dione core (Fig. 2). Thus, the planar structure of 1 was established as shown.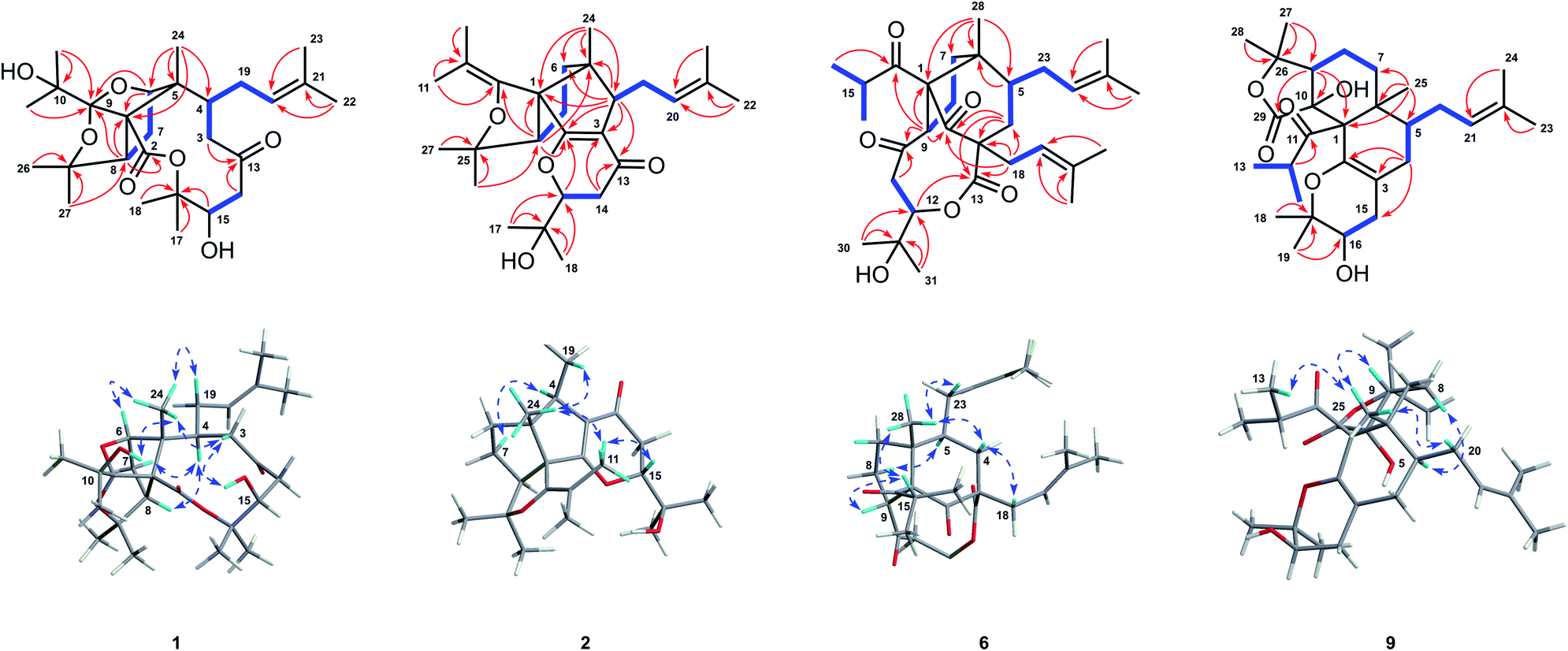 | ||
| Fig. 2 Key 1H–1H COSY (bold), HMBC (arrows), and NOESY (double-headed arrows) correlations of 1, 2, 6, and 9. | ||
The ROESY correlations of Me-24 with H-19, C-10-OH (δH 3.91), and H-3β (δH 3.10), together with the correlation of H-3β with C-15-OH (δH 4.70), implied that the groups of Me-24, the isoprenyl group, the propan-2-ol group, and the hydroxyl group at C-15 were co-facial and assigned as β-oriented (Fig. 2). Furthermore, the ROESY correlation of Me-24 with H-6 confirmed the configuration of C-6, and ROESY correlations of H-4 with H-7 and H-8 determined the configuration of C-8. Finally, the absolute configuration of 1 was unambiguously assigned as 1R,4S,5R,6R,8S,9R,15R by X-ray crystallographic analysis (Fig. 3). This is the first example of PPAP featuring an unusual 1,5-epoxyfuro[3′,4′:1,5]cyclopenta[1,2-c]oxecine skeleton.
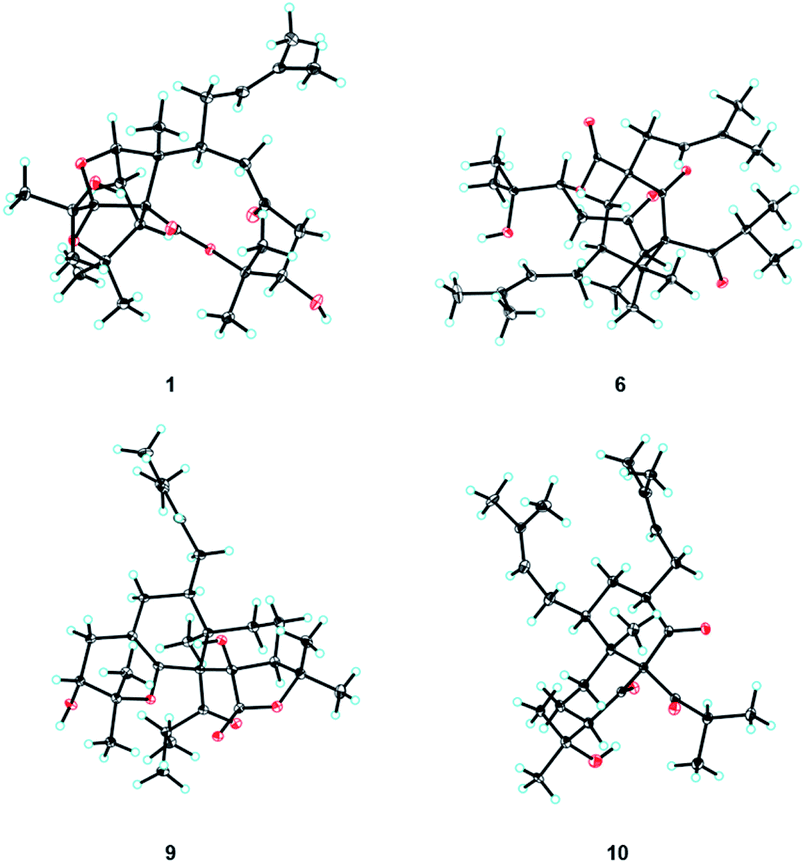 | ||
| Fig. 3 X-ray crystal structures of 1, 6, 9, and 10. | ||
Hyperforone B (2) was obtained as a colorless gum. The molecular formula of 2 was determined to be C27H40O4 based on its HRESIMS data, which included a quasimolecular ion peak at m/z 429.2982 [M + H]+. The 1H NMR spectrum of 2 (Table S1†) displayed signals indicative of an olefinic proton at δH 5.24, an oxygenated methine proton at δH 4.26, and nine methyl singlets. The 13C NMR (Table S3†) and HSQC data showed 27 carbon signals, including an unsaturated ketone carbonyl (δC 193.4), six olefinic carbons (δC 179.7, 146.9, 132.0, 126.1, 117.1, and 108.9), four quaternary carbons, three methines, four methylenes, and nine methyls. These data suggested that 2 is likely a PPAP derivative. The planar structure of 2, which is substantially different from those of regular PPAPs,26,27 was elucidated by extensive analyses of its 2D NMR spectra including its HMBC and 1H–1H COSY spectra (Fig. 2). The 5/5-fused core structure of 2 (rings B and C) was indicated by 1H–1H COSY correlations of H-6/H-7/H-8 and HMBC correlations from Me-24 to C-1, C-4, C-5, and C-6, from H-4 to C-1, C-2, C-3, C-5, and C-6, and from H-8 to C-1, C-2, and C-5. In addition, the 1H–1H COSY cross-peak of H-14/H-15 as well as HMBC correlations from Me-17 and Me-18 to C-15 and C-16 and from H-14 to C-13 and C-3 confirmed the connectivities of C-13/C-14/C-15/C-16/C-17(C-18) and also revealed the location of this fragment at C-3. Moreover, a weak HMBC interaction (recorded in CDCl3) between H-15 and C-2 along with the consideration of the chemical shifts of C-2 and C-15 suggested the linkage of C-2 and C-15 via an oxygen atom to construct the pyranone moiety. HMBC correlations from Me-26 and Me-27 to C-8 and C-25 and from Me-22 and Me-23 to C-20 and C-21, along with a spin system of H-4/H-19/H-20, suggested the presence of a 2-hydroxylisopropyl moiety at C-8 and an isoprenyl group at C-4, respectively. The presence of the isobutenyl at C-1 was revealed by HMBC interactions from Me-11 and Me-12 to C-9 and C-10 and from H-8 to C-9. Finally, in order to satisfy the molecular formula and complete the structure of 2, an enol ether bond was assigned between C-9 and C-25. Thus, the planar structure of 2 was determined.
The relative configuration of 2 was elucidated by a NOESY experiment (Fig. 2) and 13C NMR calculations. Me-24 showed a NOESY interaction with H-19, which revealed their co-facial and ascribed β-orientations. Furthermore, NOESY correlations of Me-24/Me-11 and Me-11/H-15 (recorded in CD3OD) confirmed the configurations of C-1 and C-15, and the NOESY correlation of H-4/H-7 determined the configuration of C-8. Moreover, 13C NMR data calculations were performed for both 2a (8S*) and 2b (8R*) (Fig. S1 and Table S5†), which suggested 2a as the correct structure. Finally, the absolute configuration of 2 was determined by comparing its experimental and calculated ECD spectra (Fig. S2†).
Hyperforone C (3) showed the same molecular formula of C27H40O4 as compound 2. Extensive analyses of its NMR data implied that 3 possesses a similar structure to 2, except for the C-13–C-18 moiety. The 1H–1H COSY interaction of H-14/H-15 and HMBC interactions from H-14 to C-13 and from Me-17, Me-18, and H-15 to C-16, along with the consideration of the chemical shifts of C-15 and C-16, suggested that the fragment of C-13–C-18 is a 2,2-dimethyltetrahydrofuran-3-ol moiety (Fig. S3†). Finally, the remaining degree of unsaturation and the chemical shifts of C-3 and C-13 suggested the presence of a double bond between C-3 and C-13. The relative configurations at C-1, C-4, C-5, and C-8 of 3 were identical to those of 2 according to the similar NOESY correlations (Fig. S3†). The NOESY correlation of H-4 (δH 2.99) with H-14α (δH 3.96) suggested their α-orientation and the Z-configuration of the Δ3(13) double bond in 3. The relative configuration of C-15 was assigned by the coupling constants of H-14α (J = 19.0 Hz) and H-14β (J = 19.0, 6.0, 2.2 Hz) (Fig. S4†). Then, the absolute configuration of 3 was established by comparing its experimental and calculated ECD spectra (Fig. S2†).
Hyperforone D (4) was obtained as a colorless oil, and its molecular formula of C27H42O5 was established based on its HRESIMS and 13C NMR spectra. Comparison of the 1H and 13C NMR data of 4 and 2 (Tables S1 and S3†) showed that the main difference between them was the presence of an additional methylene (δC 49.4) in 4 and the absence of the double bond signal of C-3 in 2, which was speculated to be the result of the cleavage of the C-2–C-3 double bond of 2. The planar structure of 4 was established by detailed analyses of its 2D NMR data, and its structure indicated that it is biosynthetically related to 2. Based on the NOESY correlations of Me-24 with H-19 and H-3, of H-3 with H-15, and of H-8 with H-4 and Me-17, the relative configuration of 4 was similar to that of 2 (Fig. S5†). The absolute configuration of 4 was ultimately determined from its calculated ECD spectrum (Fig. S2†).
The molecular formula of 5, C27H42O5, was deduced by HRESIMS. Preliminary analysis of its 2D NMR data implied that compound 5 has a similar cyclopenta[c]furan moiety as those in 2–4 (Fig. 1). Comprehensive analysis of its NMR spectra indicated the presence of a 1,6-dioxaspiro[3.6]decan-5-one core with an isoprenyl group and propan-2-ol group at C-4 and C-15, respectively (Fig. S3†). Compound 5 bearing an uncommon oxetane ring was further indicated by the presence of a HMBC interaction (recorded in DMSO-d6) from C-16-OH (δH 4.73) to Me-17 and Me-18 and the absence of the HMBC interaction from C-16-OH to C-14, assisted by the consideration of the chemical shifts of C-15 and C-16. The relative configurations at C-1, C-4, and C-5 of 5 were the same as those of 3 according to the NOESY correlations (Fig. S3†). The remaining relative configurations at C-8, C-13, and C-15 were deduced by NOESY correlations (recorded in CD3OD) of H-8/H-14 and H-15/H-3α (δH 1.66). Finally, the absolute configuration of 5 was determined by ECD calculation (Fig. S2†).
The molecular formula of hyperforone F (6) was determined to be C31H46O6 from its HRESIMS spectrum with an ion at m/z 537.3182 [M + Na]+ (calcd for C31H46O6Na, 537.3187). The planar structure of 6 was elucidated by analyses of its NMR spectra (Fig. 2). The 1H NMR and HSQC spectra of 6 suggested the presence of two olefinic protons at δH 5.21 (t, J = 7.6 Hz) and 5.10 (t, J = 7.9 Hz), an oxygenated methine proton at δH 4.74 (dd, J = 11.4, 4.7 Hz), an unshielded methine at δH 4.08 (t, J = 9.7 Hz), and nine methyls (δH 1.73, s; 1.70, s; 1.68, s; 1.65, s; 1.26, d, J = 6.4 Hz; 1.19 × 2, s; 1.12, d, J = 6.3 Hz; and 0.97, s). The 13C NMR and HSQC spectra of 6 revealed 31 carbon resonances, including three carbonyls (δC 214.1, 208.9, and 203.8), one ester carbonyl (δC 170.8), four olefinic carbons (δC 137.0, 133.9, 122.9, and 118.3), three quaternary carbons (δC 81.7, 60.9, and 51.4), an oxygenated tertiary carbon (δC 71.6), four methines, six methylenes, and nine methyl groups. The 5/6-fused core structure of 6 (rings B and C) was elucidated by HMBC correlations from Me-28 to C-1, C-5, C-6, and C-7, from H-9 to C-1, C-2, and C-6, and from H-4 to C-2, C-3, C-5, and C-6 aided by the 1H–1H COSY correlations of H-4/H-5 and H-7/H-8/H-9 (Fig. 2). Additional HMBC correlations from Me-30 and Me-31 to C-12 and C-29, from H-11 and H-12 to C-10 along with the 1H–1H COSY cross-peak of H-11/H-12 revealed the presence of a six-membered fragment (C-10/C-11/C-12/C-29/C-30(C-31)) similar to C-13/C-14/C-15/C-16/C-17(C-18) in 2, which was attached to C-9 as indicated by the HMBC interaction between H-9 and C-10. This fragment further constructed a nine-membered lactone ring by connecting to C-3 via the ester carbonyl (C-13), as revealed by HMBC correlations from H-4 and H-12 to C-13. Moreover, HMBC correlations from Me-16, Me-17, and H-9 to C-14 confirmed the location of the isobutyryl group at C-1. Finally, the remaining two isoprenyl groups were located at C-3 and C-5, respectively, and HMBC correlations from H-18 to C-2, C-3, C-4, and C-13 and from H-23 to C-4, C-5, and C-6 further confirmed the structure of ring B. Consequently, the planar structure of 6 was elucidated.
The relative configuration of 6 was determined through a NOESY experiment (recorded in DMSO-d6) assisted by CS Chem 3D simulations and 13C NMR calculations. NOESY correlations of Me-28/H-23, Me-28/H-4β, and H-4β/H-18 revealed their co-facial and ascribed to β-orientations. In addition, the NOESY correlation between Me-28 and H-15 suggested that rings B and C are cis-fused. An obvious NOESY signal was observed between H-9 and H-15, which determined the configuration of C-9. To determine the configuration of C-12, the NOESY spectrum was further analyzed by the molecular modeling of 6a (12S*) and 6b (12R*) simulated by CS Chem 3D (Fig. 2 and S6†). The NOESY interactions of H-5/H-8α, H-8α/H-11α, and H-11β/H-12 were anticipated for 6a (Fig. 2), and dihedral angles between H2-11 and H-12 in 6a corresponded to their coupling constants. In addition, the calculated 13C NMR were determined for 6a and 6b (Fig. S1 and Table S6†), which also suggested an S* configuration for C-12. Finally, the absolute configuration of 6 was unambiguously confirmed by X-ray crystallography (Fig. 3, CCDC 1812029†).28,29
Hyperforone G (7) has the same molecular formula of C31H46O6 as compound 6. Extensive analyses of the 1D and 2D NMR data of 7 suggested that 7 shares an equivalent planar structure with 6. The only difference between 7 and 6 was the orientation of H-12, which was determined to be α-oriented in 7 by the NOESY correlations of H-9/H-11β, and H-11α/H-12 as well as the 1H–1H coupling constants (J11β,12 = 10.9 Hz and J11α,12 = 2.1 Hz). Thus, compound 7 was determined to be the C-12 epimer of 6.
Hyperforone H (8) also has the same molecular formula as 6 and a closely similar structure based on its HRESIMS and NMR spectra. The absence of the HMBC correlation from H-12 to C-13 as well as the upfield and downfield shifted chemical shifts of C-12 (δC 76.5) and C-29 (δC 87.2) of compound 8 compared with compound 6 (δC 83.8 and 72.2 for C-12 and C-29, respectively) suggested that 8 possesses a 10 membered lactone ring rather than the nine-membered lactone ring in 6 and 7. The relative configurations at C-1, C-3, C-5, C-6 and C-9 of 8 were determined to be the same as those of 6 by the NOESY correlations of Me-28/H-23, Me-28/H-4β, H-4β/H-19, Me-28/H-15, and Me-16/H-9 (Fig. S5†). Meanwhile, the R configuration of C-12 was confirmed by the NOESY correlations of H-8/H-11α, H-11α/Me-31, and Me-30/H-12 as well as a modified Mosher's method (Fig. S7†).30 Finally, the absolute configurations of compounds 7 and 8 were assigned by comparing their experimental ECD spectra with that of compound 6 (Fig. S8†).
Hyperforone I (9) was obtained as colorless crystals. Its molecular formula of C29H44O6 was deduced by HRESIMS data (m/z 489.3190, [M + H]+) assisted by the 13C NMR data indicating eight degrees of unsaturation. The 1H NMR spectrum (Table S2†) showed the presence of nine methyls at δH 0.87, 1.02, 1.17, 1.24, 1.40, 1.42, 1.45, 1.60, and 1.71, one oxygenated methine at δH 3.66 (1H, m, H-16), and one olefinic proton at δH 5.10 (1H, t, J = 7.3 Hz, H-21). The 13C NMR and DEPT spectra of 9 revealed 29 carbon signals (Table S4†), including four quaternary carbons, two carbonyls, four oxygenated tertiary carbons, five methines, five methylenes, and nine methyls. Further analysis of the above data suggested that some of the resonances could be assigned to an unconjugated carbonyl (δC 218.3, C-11), an ester carbonyl (δC 172.0, C-29), two olefinic carbons (δC 109.4, C-3 and 140.3, C-2), a prenylated group (δC 27.6, 123.1, 132.5, 25.8, and 17.8, C-20–C-24), and an isopropyl group (δC 40.5, 23.4, and 20.2, C-11–C-13). The aforementioned NMR data along with the remaining four degrees of unsaturation indicated compound 9 to be a tetracyclic PPAP derivative.
Further analyses of its 2D NMR spectra revealed that compound 9 is quite different from regular PPAPs, and it featured a unique tetradecahydro-2H-isobenzofuro[5,4-h]chromene skeleton (Fig. 2). The 6/6-fused core structure of 9 (rings B and C) was confirmed by 1H–1H COSY correlations of H-7/H-8/H-9 and H-4/H-5 as well as HMBC correlations from Me-25 to C-1, C-5, C-6, and C-7, from H-9 to C-1 and C-10, and from H-4 to C-2 and C-3. Furthermore, 1H–1H COSY interactions of H-5/H-20/H-21 and HMBC cross-peaks from Me-23 and Me-24 to C-21 and C-22 confirmed that the prenylated group (C-20–C-24) was located at C-5 of the 6/6-fused core. Moreover, the 1H–1H COSY interaction of H-15/H-16 and HMBC correlations from Me-18 and Me-19 to C-16 and C-17 and from H-15 to C-2, C-3, and C-4 suggested the connectivities of C-15/C-16/C-17/C-18(C-19), which was located at C-3 of the 6/6-fused core. The degrees of unsaturation and chemical shifts of C-2 and C-17 implied that a pyran ring was constructed by the linkage of C-2–O–C-17. The presence of γ-lactone was supported by HMBC correlations from Me-27 and Me-28 to C-9 and C-26 and from H-9 to C-10 and C-29 as well as the chemical shifts of C-26 (δC 85.8) and C-29 (δC 174.9). The 1H–1H COSY interaction of H-13(H-14)/H-12 and HMBC correlations from H-12, Me-13, and Me-14 to C-11 revealed an isopropyl moiety. Finally, in order to complete the structure of 9, the isopropyl group was located at C-1, and the planar structure of 9 was established.
The relative configuration of 9 was deduced by the NOESY experiment (Fig. 2). The NOESY correlations of Me-25 with H-9 and Me-13 implied that these groups were co-facial and β-oriented. The NOESY correlations of Me-25 with H-20 and of H-5 with H-8 suggested that the C-5 substituent was also β-oriented. Finally, the absolute configuration of compound 9 was unambiguously confirmed as 1R,5S,6R,9R,10R,16R by a single-crystal X-ray diffraction experiment (Fig. 3) with Cu Kα radiation [CCDC 1936569,† Flack parameter, 0.09(6)].28,29
Hyperforone J (10) had the molecular formula of C28H44O4 based on its NMR and HRESIMS data. A further comparison of the NMR data of 10 with those of 9 indicated that 10 also had a 6/6-fused core and had the same substitutions at C-1, C-5, and C-6 as those in 9. The 1H–1H COSY correlations of H-3/H-15/H-16 and HMBC correlations (Fig. S3†) from Me-18 and Me-19 to C-16 and C-17 suggested a prenylated group at C-3. The propan-2-ol moiety at C-9 of the 6/6-fused core was confirmed by HMBC correlations from both Me-27 and Me-28 to C-26 and C-9 along with the chemical shift of C-26 (δC 72.1). Two unconjugated carbonyl groups (δC 211.9, 210.5) were assigned at C-10 and C-2, respectively, on the basis of the HMBC correlations from H-3 to C-2 and from H-9 to C-10 along with the downfield shift of C-1 (δC 87.6).
The relative configuration of 10 was determined by a ROESY experiment (Fig. S3†). The ROESY correlations of Me-25 with H-4β (δH 1.10) and of H-3 with H-4α (δH 2.07) and H-5 demonstrated that Me-25 and substituents at C-3 and C-5 were co-facial and β-oriented. Furthermore, the ROESY correlation of H-20/H-8b (δH 1.73) indicated that H-8b was put in an α-orientation and the ROESY correlations of H-8a (δH 1.98)/H-9/Me-13 implied that the isobutyryl group at C-1 and H-9 were co-facial and β-oriented. Finally, the absolute configuration of compound 10 was defined as 1S,3R,5S,6R,9S by a single-crystal X-ray diffraction experiment (Fig. 3).
Hyperforones A–J (1–10), containing six types of unusual skeletons, are quite different from regular PPAPs.26,27 To illuminate their origins, our hypothetical biosynthetic pathways are outlined in Scheme 1, starting from the classic [3.3.1]-type PPAP hyperforin via a series of retro-Claisen, nucleophilic addition, and oxidative reactions and so on. After the cleavage of the A ring by a retro-Claisen reaction, i underwent further oxidative and nucleophilic addition reactions to construct a five-membered ring fused with a B-ring via C-1 and C-8 (ii). Then, additional retro-Claisen, nucleophilic addition, and oxidative reactions of ii generated the key intermediate iii with a furan ring, which further underwent three different pathways (pathways A–C). Afterward, the C-5 side chain of iii underwent a nucleophilic addition reaction to afford a pyran ring (iv), a furan ring (vi), and an oxetane ring (vii) in paths A, B, and C, respectively. The oxidative cleavage of the double bond between C-5 and C-9 of iv leads to the formation of v, which was followed by a nucleophilic addition and an oxidative reaction generating the core structure of 1 with an unexpected cage-like ring system. The oxidative cleavage of the double bond between C-5 and C-9 of vi leads to the production of 2–4. Compound 5 was formed from vii by Baeyer–Villiger oxidation. Additionally, the nucleophilic addition and decarboxylation of ii leads to intermediates viii and ix, which gave the unique lactone of 6–8 by oxidative cleavage of the double bond between C-3 and C-4. In another pathway, i underwent retro-Claisen, prenylation, oxidation, and nucleophilic addition reactions to afford intermediate x with a 6/6 ring system, which was followed by a decarboxylation reaction generating 10 and by an additional cyclization reaction generating 9. Notably, this is the first report of [3.3.1]-type PPAP derivatives with a contracted B-ring and the original A-ring completely degraded (2 and 3), a cleaved B-ring and the original A-ring completely degraded (4), and nine- (6 and 7) or ten-membered (8) lactone rings formed by the original A-ring via partial degradation, reconstruction, and expansion. Compounds 9 and 10 are also the first examples of PPAPs with a newly formed six-membered C-ring by nucleophilic addition.
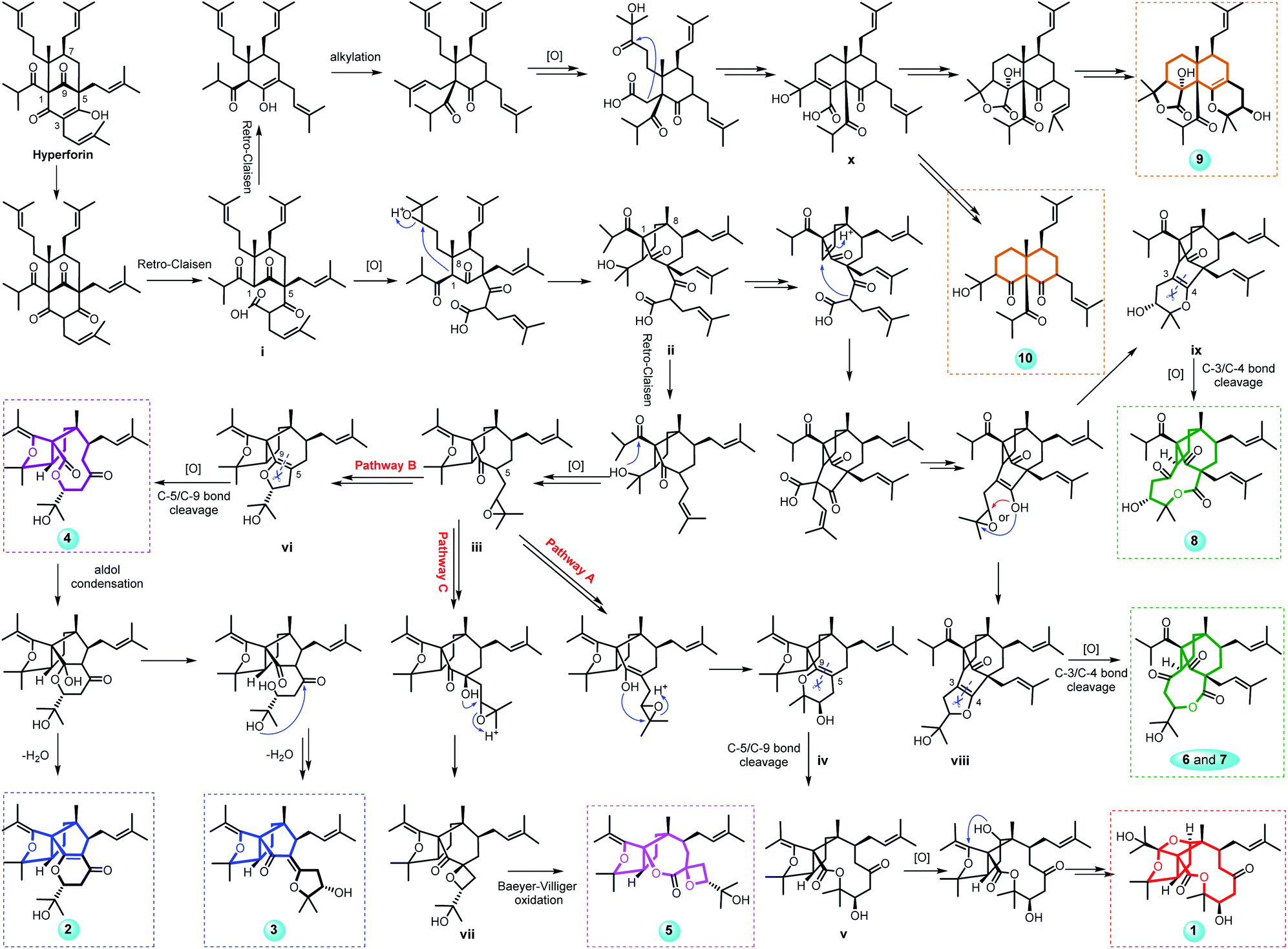 | ||
| Scheme 1 Proposed biosynthetic pathways of compounds 1–10 with six different types of architectures. | ||
Natural products are prone to facile non-enzymatic transformations during extraction, fractionation, analysis, handling, and storage.31 In this study, the air-drying and isolation processes may bring the risk of oxidative transformations. This raises the question of whether compounds 1–10 are handling artifacts. Therefore, comparative LC-MS analyses of authentic standards of 1–10 with a fresh extract of H. perforatum, one where the plant material has not been subjected to air drying or exposure to acidic conditions, were conducted. The results (Fig. S9–S13†) showed that compounds 6–10 could be detected in the fresh extracts, while 1–5 were absent. Therefore, compounds 6–10 were determined as natural products, while compounds 1–5 were not confirmed. In addition, LC-MS method was also used to monitor the changes of compounds 1–10 after exposure to aqueous solution (for detailed experimental procedures and results, please see the “Methods and materials” section and Fig. S14 in the ESI†). The results showed that these compounds possessed good stability in the aqueous solution.
As the two crucial enzymes in the MTDLs strategy for AD, PP2Ac and BACE1 were subjected to molecular docking studies with all compounds, and compounds 6 and 4 fitted well with the major active sites of PP2Ac and BACE1 (Fig. S15 and Table S7†).32 As shown, hydrogen bonds were observed between 6 and two amino acid residues of PP2Ac including Arg214 and Tyr265; meanwhile, hydrophilic interactions were observed for 6 and amino acid residue Gln73 of BACE1. It is interesting that 7, the C-12 epimer of 6, showed much weaker interactions with both PP2Ac and BACE1. Detailed comparison of the molecular docking results of 6 and 7 with these of PP2Ac and BACE1 (Fig. S16†), it is obvious that the different configurations of propan-2-ol group resulted in different conformations, which make it more difficult for 7 to form hydrophilic interactions with both enzymes. Thus, the propan-2-ol group plays a significant role for 6 in the interactions with PP2Ac and BACE1. Similar to that of 6, compound 4 could also form some key hydrogen bonds with the amino acid residues of Tyr127, Arg214, and Tyr265 of PP2Ac and with amino acid residues of Gln73 and Thr231 of BACE1. The enzyme activity assay showed that compounds 1–10 increased PP2A activity with EC50 values in the range of 199.0–7761.0 nM in HEK293/tau cells and inhibited BACE1 activity with IC50 values in the range of 98.6–9553.0 nM in N2a/APP cells (Table S8†). Interestingly, compounds 6 and 4 exhibited superior bioactivities (EC50: 199.0 and 258.8 nM for PP2A; IC50: 98.6 and 136.2 nM for BACE1, respectively) and both compounds showed better activities than the positive controls SCR1693 (Fig. S17†)33 and LY2811376 (Fig. S18†).34,35 All of the compounds showed no cytotoxicity in cell viability assays at a concentration up to 8 μM (Fig. S19 and S20†). In addition, the binding assay of compounds 4 and 6 to proteins BACE1 and PP2A was also performed using the cellular thermal shift assay (CETSA) method to confirm the interaction between compounds and their targets in cell lysates. These results (Fig. S21†) indicated that 4 and 6 could directly interact with BACE1 and PP2A. Subsequently, we treated HEK293/tau cells with 10 nM okadaic acid (OA)36 to evaluate the compounds for their ability to rescue OA-induced PP2A deficiency and detected the effects of the compounds on BACE1 activity, Aβ production and the β-cleavage of APP (APP-β) in N2a/APP cells. The results showed that both compounds 6 and 4, especially 6, exhibited favorable effects toward reducing tau phosphorylation and Aβ production by regulating the activities of PP2A and BACE1 in vitro (Fig. S22†).
As the most effective compound in vitro, three dose escalations of 6 were infused into the cerebral ventricles of 3 × Tg AD mice continuously for one month, and a series of tests were performed to comprehensively evaluate the activities of 6 against AD-like behavioral alterations. First, the open field (OF) test indicated that anxiety and exploratory activity were unchanged (Fig. S23†). Subsequently, the novel object recognition (NOR) test showed a significant increase in the novel recognition index, which was used to evaluate the contextual memory of the mice (Fig. 4A and S24A†). Furthermore, Morris water maze (MWM) tests were carried out to assess the spatial reference learning and memory functions of the mice, and the results showed that 6 clearly attenuated cognitive impairments in 3 × Tg AD mice (Fig. 4B, C and S24B–E†). The results of the therapeutic effects of these compounds on AD pathology showed that middle and high doses of 6 significantly reduced the tau phosphorylation and Aβ production and deposition in vivo by regulating the enzyme activities of PP2A and BACE1, respectively (Fig. 4E–I and S25†). In addition, dendritic spines, mushroom-type spines, and a panel of synaptic proteins were markedly increased in mice treated with 6 (Fig. 4D and S24F–I†), indicating that 6 attenuated cognitive impairments through the recovery of synaptic plasticity.
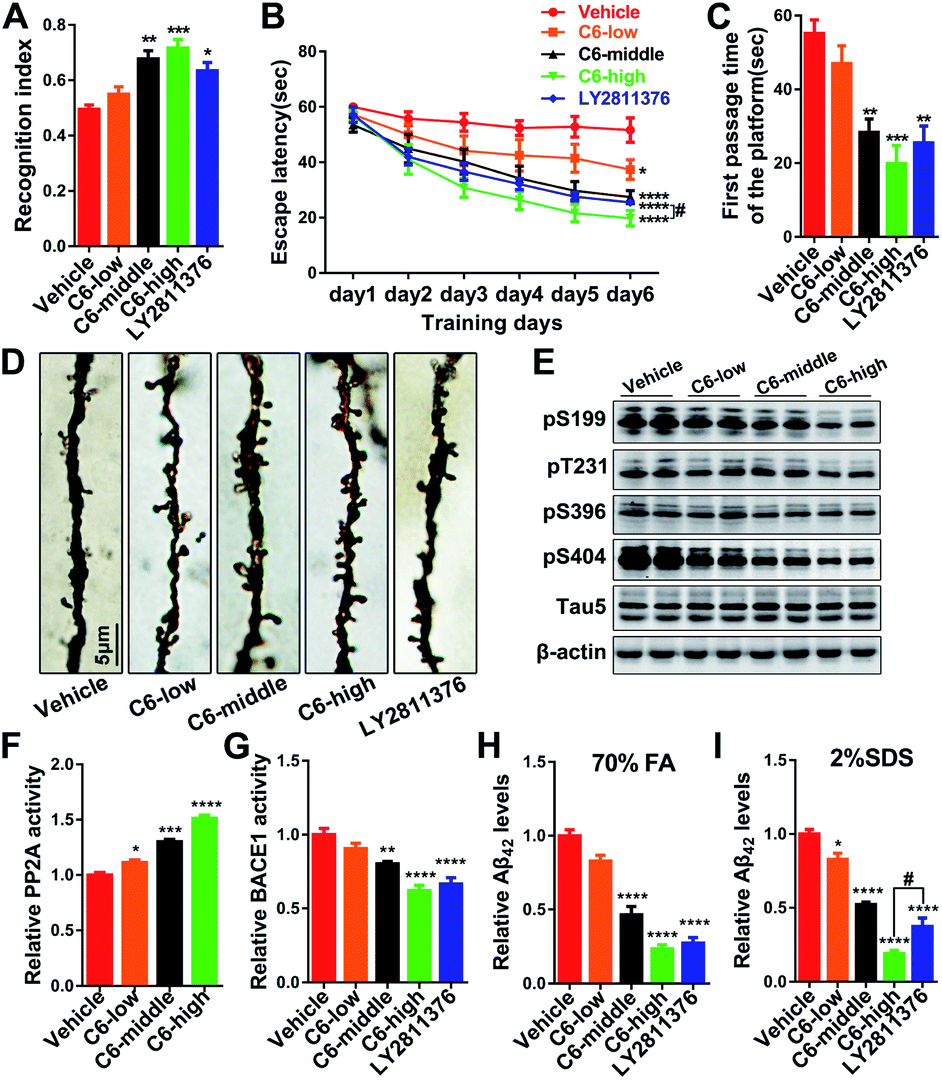 | ||
| Fig. 4 Compound 6 [C6-low (0.725 μg μL−1 × 5 μL), C6-middle (2.9 μg μL−1 × 5 μL), and C6-high (11.6 μg μL−1 × 5 μL)] improved learning and memory by regulating Aβ42 production and tau phosphorylation in 3 × Tg AD mice with LY2811376 (2 μg μL−1 × 5 μL) as the positive control group. (A) Statistical analyses of the NOR results. (B) Escape latencies to find the hidden platform during the MWM training and (C) testing were recorded. (D) Representative Golgi staining photomicrographs of primary dendrites in the hippocampal CA3 region. (E) Western blot showing tau phosphorylation levels at Ser199, Thr231, Ser396 and Ser404, unphosphorylated tau recognized by tau1, and total tau recognized by tau5 in the brains of 3 × Tg mice treated with 6. (F) The PP2A activity was determined using the Activity Assay Kit. (G) The BACE1 activity was determined using a β-secretase activity assay kit. (H) Insoluble Aβ42 (70% FA) and (I) soluble Aβ42 protein levels were determined using a mouse Aβ1–42 ELISA kit. ****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05 versus the vehicle group; #p < 0.05, C6-high versus the positive control group. | ||
Conclusions
In the present study, we identified ten novel PPAPs with six different types of unexpected skeletons from H. perforatum. Due to their complex and variable structures, the challenging task of their structural elucidations was comprehensively established by using spectroscopic (HRESIMS, NMR, ECD, and X-ray crystallography), quantum chemical computational (ECD and 13C NMR calculations), and chemical (modified Mosher's method) methods. Overall, the unique structures and biosynthetic origins of 1–10 help elucidate the structural diversity of the PPAP family. Compound 6 significantly improved learning and memory impairments in AD mice by decreasing tau phosphorylation and Aβ production/deposition and elevating the levels of synapse-associated proteins as well as the number of dendritic spines in vivo. Therefore, 6 could be developed into a promising lead compound with MTDLs for AD therapy targeting multi-etiopathogenic mechanisms. The pharmacological activity of these compounds as well as their challenging structural features will spark great interest among organic chemists and biologists alike.Data availability
Experimental or computational data associated with this article have been provided in the ESI.†Author contributions
Y. G. and F. H. contributed equally to this work. They conducted the main experiments, analyzed the data, and wrote the manuscript; W. S. finished the docking experiments. Y. Z. helped with the LC-MS experiments; C. C. and C. Q. helped with the experimental procedures; J. Y. did the ECD and 13C NMR calculations; X.-N. Li did the X-ray experiments; Z. L. helped with the Mosher experiments; Y. Z., X. W., and H. Z. designed the experiments and revised the manuscript. All of the authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Program for Changjiang Scholars of the Ministry of Education of the People's Republic of China (No. T2016088), the National Natural Science Foundation for Distinguished Young Scholars (No. 81725021), the National Natural Science Foundation for Excellent Young Scholars (81922065), the Innovative Research Groups of the National Natural Science Foundation of China (No. 81721005), the Academic Frontier Youth Team of HUST (2017QYTD19), and the Integrated Innovative Team for Major Human Diseases Program of Tongji Medical College (HUST). We thank Y. Zhou for help with LC-MS experiments and the Analytical and Testing Center at HUST for assistance with the ECD, UV and IR analyses.Notes and references
- A. I. Gurevich, V. N. Dobrynin, M. N. Kolosov, S. A. Popravko and I. D. Riabova, Antibiotiki, 1971, 16, 510–513 CAS.
- N. S. Bystrov, B. K. Chernov, V. N. Dobrynin and M. N. Kolosov, Tetrahedron Lett., 1975, 16, 2791–2794 CrossRef.
- Y. Shimizu, S.-L. Shi, H. Usuda, M. Kanai and M. Shibasaki, Angew. Chem., Int. Ed., 2010, 49, 1103–1106 CrossRef CAS PubMed.
- B. A. Sparling, D. C. Moebius and M. D. Shair, J. Am. Chem. Soc., 2013, 135, 644–647 CrossRef CAS PubMed.
- C. P. Ting and T. J. Maimone, J. Am. Chem. Soc., 2015, 137, 10516–10519 CrossRef CAS PubMed.
- M. Wonnemann, A. Singer and W. E. Müller, Neuropsychopharmacology, 2000, 23, 188–197 CrossRef CAS PubMed.
- C. M. Schempp, K. Pelz, A. Wittmer, E. Schöpf and J. C. Simon, Lancet, 1999, 353, 2129 CrossRef CAS.
- C. M. Schempp, V. Kirkin, B. Simon-haarhaus, A. Kersten, J. Kiss, C. C. Termeer, B. Gilb, T. Kaufmann, C. Borner, J. P. Sleeman and J. C. Simon, Oncogene, 2002, 21, 1242–1250 CrossRef CAS PubMed.
- C. Gey, S. Kyrylenko, L. Hennig, L. H. D. Nguyen, A. Büttner, H. D. Pham and A. Giannis, Angew. Chem., Int. Ed., 2007, 46, 5219–5222 CrossRef CAS PubMed.
- J. Barnes, L. A. Anderson and J. D. Phillipson, J. Pharm. Pharmacol., 2001, 53, 583–600 CrossRef CAS PubMed.
- A. Brenn, M. Grube, G. Jedlitschky, A. Fischer, B. Strohmeier, M. Eiden, M. Keller, M. H. Groschup and S. Vogelgesang, Brain Pathol., 2014, 24, 18–24 CrossRef PubMed.
- J. Hofrichter, M. Krohn, T. Schumacher, C. Lange, B. Feistel, B. Walbroel, H.-J. Heinze, S. Crockett, T. F Sharbel and J. Pahnke, Curr. Alzheimer Res., 2013, 10, 1057–1069 CrossRef CAS PubMed.
- M. C. Dinamarca, W. Cerpa, J. Garrido, J. L. Hancke and N. C. Inestrosa, Mol. Psychiatry, 2006, 11, 1032–1048 CrossRef CAS PubMed.
- K. Iqbal and I. Grundke-Iqbal, J. Cell. Mol. Med., 2008, 12, 38–55 CrossRef CAS PubMed.
- K. L. Viola and W. L. Klein, Acta Neuropathol., 2015, 129, 183–206 CrossRef CAS PubMed.
- A. D. C. Alonso, I. Grundke-Iqbal and K. Iqbal, Nat. Med., 1996, 2, 783–787 CrossRef CAS PubMed.
- R. Vassar, J. Mol. Neurosci., 2004, 23, 105–114 CrossRef CAS PubMed.
- A. Takashima, J. Alzheimer's Dis., 2006, 9, 309–317 CAS.
- C.-X. Gong, T. Lidsky, J. Wegiel, L. Zuck, I. Grundke-Iqbal and K. Iqbal, J. Biol. Chem., 2000, 275, 5535–5544 CrossRef CAS PubMed.
- J. Brainard, News at a glance, Science, 2019, 366, 402–403, DOI:10.1126/science.366.6464.402.
- K. Servick, Science, 2021, 372, 1141 CrossRef CAS PubMed.
- H. Zhu, C. Chen, J. Yang, X. N. Li, J. Liu, B. Sun, S. X. Huang, D. Li, G. Yao, Z. Luo, Y. Li, J. Zhang, Y. Xue and Y. Zhang, Org. Lett., 2014, 16, 6322–6325 CrossRef CAS PubMed.
- L. Hu, Y. Zhang, H. Zhu, J. Liu, H. Li, X. N. Li, W. Sun, J. Zeng, Y. Xue and Y. Zhang, Org. Lett., 2016, 18, 2272–2275 CrossRef CAS PubMed.
- Y. Guo, N. Zhang, C. Chen, J. Huang, X. N. Li, J. Liu, H. Zhu, Q. Tong, J. Zhang, Z. Luo, Y. Xue and Y. Zhang, J. Nat. Prod., 2017, 80, 1493–1504 CrossRef CAS PubMed.
- Y. Guo, Q. Tong, N. Zhang, X. Duan, Y. Cao, H. Zhu, S. Xie, J. Yang, J. Zhang, Y. Liu, Y. Xue and Y. Zhang, Org. Chem. Front., 2019, 6, 817–824 RSC.
- R. Ciochina and R. B. Grossman, Chem. Rev., 2006, 106, 3963–3986 CrossRef CAS PubMed.
- X. W. Yang, R. B. Grossman and G. Xu, Chem. Rev., 2018, 118, 3508–3558 CrossRef CAS PubMed.
- S. Parsons, H. D. Flack and T. Wagner, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 249–259 CrossRef CAS PubMed.
- H. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- R. J. Capon, Nat. Prod. Rep., 2020, 37, 55–79 RSC.
- A. N. Jain, J. Med. Chem., 2003, 46, 499–511 CrossRef CAS PubMed.
- Y. Xia, R. Liu, R. Chen, Q. Tian, K. Zeng, J. Hu, X. Liu, Q. Wang, P. Wang, X. C. Wang and J. Z. Wang, J. Alzheimer's Dis., 2014, 42, 1029–1039 CAS.
- P. C. May, R. A. Dean, S. L. Lowe, F. Martenyi, S. M. Sheehan, L. N. Boggs, S. A. Monk, B. M. Mathes, D. J. Mergott, B. M. Watson, S. L. Stout, D. E. Timm, E. Smith LaBell, C. R. Gonzales, M. Nakano, S. S. Jhee, M. Yen, L. Ereshefsky, T. D. Lindstrom, D. O. Calligaro, P. J. Cocke, D. Greg Hall, S. Friedrich, M. Citron and J. E. Audia, J. Neurosci., 2011, 31, 16507–16516 CrossRef CAS PubMed.
- C. Qi, J. Bao, J. Wang, H. Zhu, Y. Xue, X. Wang, H. Li, W. Sun, W. Gao, Y. Lai, J.-G. Chen and Y. Zhang, Chem. Sci., 2016, 7, 6563–6572 RSC.
- P. K. Kamat, S. Rai, S. Swarnkar, R. Shukla and C. Nath, Mol. Neurobiol., 2014, 50, 852–865 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: HRESIMS, IR, UV, 1D and 2D NMR spectra of all the new compounds. CCDC 1936567, 1812029, 1936569 and 1936570. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03356e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |