
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium(II)-catalyzed regioselective direct C4- and C5-diamidation of indoles and mechanistic studies†
Shreedhar
Devkota
 a,
Suyeon
Kim
bc,
Seok Yeol
Yoo
bc,
Sonaimuthu
Mohandoss
a,
Mu-Hyun
Baik
*cb and
Yong Rok
Lee
*a
a,
Suyeon
Kim
bc,
Seok Yeol
Yoo
bc,
Sonaimuthu
Mohandoss
a,
Mu-Hyun
Baik
*cb and
Yong Rok
Lee
*a
aSchool of Chemical Engineering, Yeungnam University, Gyeongsan 38541, Republic of Korea. E-mail: yrlee@yu.ac.kr
bDepartment of Chemistry, Korea Advanced Institute of Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
First published on 21st July 2021
Abstract
A ruthenium(II)-catalyzed regioselective direct diamidation of 3-carbonylindoles at the C4- and C5-positions using various dioxazolones is described. This novel protocol allows for the effective installation of two amide groups on the benzene ring in indole. A remarkably broad substrate scope, excellent functional group tolerance, and mild reaction conditions are notable features of this protocol. Further explorations reveal that benzo[b]thiophene-3-carboxaldehyde is a viable substrate and affords its corresponding diamidation products. The diamido indoles are further converted into various functionalized products and used as sensors for metal ion detection. Density functional theory studies are also conducted to propose a reaction mechanism and provide a detailed understanding of the regioselectivity observed in the reaction.
Introduction
Indoles and their derivatives are among the most important heteroaromatic skeletons found in a wide range of bioactive natural products,1 functional materials,2 and pharmaceuticals.3 They possess a wide range of pharmacological properties, such as antifungal, antihistaminic, antimicrobial, antioxidant, anticonvulsant, anti-HIV, analgesic, anti-inflammatory, anti-cancer, and antitubercular activity.4 Several substituted indoles are currently utilized as pharmaceuticals.5 Various synthetic approaches to the preparation of substituted indoles have been demonstrated owing to their importance and useful applications.6 C3–H functionalization of indoles has been achieved using electrophilic substitution reactions,7 but functionalization at the C2- and C4-positions remains underdeveloped owing to the inherently poor reactivity at these positions. Recently, significant progress has been made using transition metal-catalyzed C–H functionalization of indoles at the C2- and C4-positions utilizing directing groups.8 Specifically, the use of carbonyl groups at the C3-site as a directing group for C2–H and C4–H functionalization was demonstrated.9 Ru(II)- or Rh(III)-catalyzed regioselective C2- and C4-alkenylation or alkylation of 3-carbonylindoles was reported by the Prabhu,10 Punniyamurthy,11 and Jia groups.12 Regioselective Pd-catalyzed direct arylation of 3-carbonylindoles at the C4- or C5-position was reported by the Shi group.13 For C2- and C4-amination, Ir(III) was shown to be effective and regioselective. C4-Amination of 3-carbonylindoles using sulfonyl azides was described by the Prabhu14 and You groups15 (Scheme 1A). Recently, Wang et al. developed a Co(III)-catalyzed regioselective C2-amidation of 3-acetylindoles using phenyl dioxazolone16 (Scheme 1B).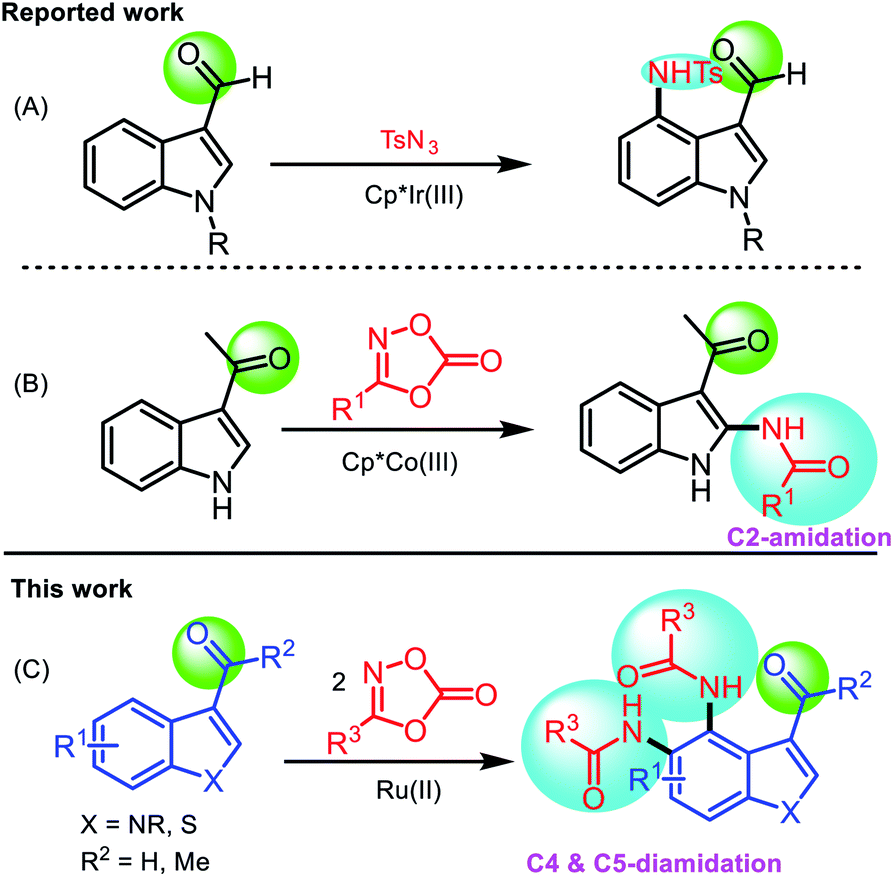 | ||
| Scheme 1 Reported methods (A and B) vs. present strategy (C) for direct amination/amidation of 3-carbonylindoles. | ||
To date, diamidation at both the C4- and C5-positions of indole has not been achieved. Recently, dioxazolones have been increasingly recognized as a versatile class of reagents that serve as an amide source, and are environmentally benign, cheap, readily available, and easy to handle.17 Furthermore, dioxazolones display a potentially useful affinity toward the metal center of metallacyclic intermediates when compared to organic azides, which are considered as conventional amidating reagents.18 Herein, we report a novel Ru(II)-catalyzed direct diamidation of 3-carbonylindoles using dioxazolones to introduce two amide groups at the C4- and C5-positions of indole (Scheme 1C).
Results and discussion
We first examined the reaction of 1-methylindole-3-carboxaldehyde (1a) with 3-phenyl-1,2,4-dioxazol-5-one (2a) using various catalysts, additives, and solvents (Table 1). Initial attempts to react 1a (0.5 mmol) with 2a (0.5 mmol) in the presence of 5 mol% [RhCp*Cl2]2, 15 mol% AgSbF6, and 10 mol% AcOH in 2,2,2-trifluoroethanol (TFE) at 50 °C did not afford the desired product (entry 1, Table 1). When 1a (0.5 mmol) and 2a (0.5 mmol) were reacted in the presence of 5 mol% [Ru(p-cymene)Cl2]2, 15 mol% AgSbF6, and 10 mol% AcOH in TFE at 50 °C for 20 h, diamidation product 5a was obtained in 40% yield (entry 2). Importantly, the putative products of C2– and C4–H functionalization (3a and 4a) were not observed, indicating the intriguing and useful regioselectivity of the reaction. To our delight, the use of 1.1 mmol of 2a increased the yield of 5a to 71% (entry 3). As an alternative to AcOH, the use of 10 mol% 1-AdCO2H, PhCO2H, and PivOH afforded 5a in 75, 70, and 87% yield, respectively (entries 4–6). The yield of 5a was not improved by decreasing or increasing the amount of AgSbF6 or PivOH (entries 7–10). In other solvents, such as 1,2-dichloroethane or hexafluoroisopropanol (HFIP), compound 5a was produced in lower yield (entries 11–12). Increasing the temperature to 70 °C and carrying out the reaction under a N2 atmosphere did not increase the product yield (entries 13–14). The use of other catalysts did not improve the yield of 5a. For example, 5a was isolated in 51% yield when [Cp*IrCl2]2 was used as the catalyst, but no 5a was obtained from [Cp*Co(CO)I2] (entries 15–16). The 1H NMR of 5a showed a singlet corresponding to the aldehyde proton at δ 9.64 ppm, the vinyl proton on the pyrrole ring at δ 8.42 ppm as a singlet, and two amide proton peaks at δ 11.67 and 9.89 ppm as two singlets. The 13C NMR spectrum showed an aldehyde carbon peak at δ 184.9 ppm and two amide carbon peaks at δ 165.9 and 164.2 ppm, respectively. When an excess of 2a (4.4 equivalents) was used to introduce amide substituents at C-4, 5, 6, 7 of the indole ring, only 5a was produced in 86% yield without formation of other products. Further reaction of N-methylindole with 2a under standard reaction conditions did not afford the amidation product.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Additives (mol%) | Yieldb (%) | |
| Solvent | 5a | |||
| a Reaction conditions: 1a (0.5 mmol), 2a (1.1 mmol), solvent (5 mL), and air atmosphere. b Isolated yield. c 1a (0.5 mmol), 2a (0.5 mmol), and solvent (5 mL). d The reaction was performed under a N2 atmosphere. e The reaction was carried out at 70 °C. | ||||
| 1c | [Cp*RhCl2]2 (5) | AgSbF6 (15), AcOH (10) | TFE | 0 |
| 2c | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), AcOH (10) | TFE | 40 |
| 3 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), AcOH (10) | TFE | 71 |
| 4 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), 1-AdCOOH (10) | TFE | 75 |
| 5 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PhCOOH (10) | TFE | 70 |
| 6 | [Ru(p-cymene)Cl 2 ] 2 (5) | AgSbF 6 (15), PivOH (10) | TFE | 87 |
| 7 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (10), PivOH (10) | TFE | 77 |
| 8 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (20), PivOH (10) | TFE | 84 |
| 9 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (5) | TFE | 79 |
| 10 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (20) | TFE | 84 |
| 11 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (10) | 1,2-DCE | 43 |
| 12 | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (10) | HFIP | 67 |
| 13d | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (10) | TFE | 65 |
| 14e | [Ru(p-cymene)Cl2]2 (5) | AgSbF6 (15), PivOH (10) | TFE | 75 |
| 15 | [Cp*IrCl2]2 (5) | AgSbF6 (15), PivOH (10) | TFE | 51 |
| 16 | [Cp*Co(CO)I2] (5) | AgSbF6 (15), PivOH (10) | TFE | 0 |

The scope was explored using the optimized conditions employing various indole-3-carboxaldehyde substrates (1b–1n) and dioxazolone 2a (Table 2). Treatment of unsubstituted indole-3-carboxaldehyde 1b and N-alkyl substituted indole-3-carboxaldehydes 1c–1d bearing n-propyl and n-pentyl groups provided their corresponding products 5b–5d in 80, 85, and 80% yield, respectively (Table 2, A). Similarly, treatment of substituted indole-3-carboxaldehydes 1e–1g bearing N-prenyl, N-benzyl, and N-phenyl groups afforded their corresponding products 5e–5g in 76, 89, and 72% yield, respectively. After exploring the reactions using different N-substituted indoles, we then investigated a series of indoles bearing various substituents on the indole ring. C2-Chloro- or C2-phenyl substituted indole-3-carboxaldehydes 1h and 1i gave compounds 5h and 5i in 74 and 73% yield, respectively (Table 2, B). The structure of 5i was unambiguously confirmed using single-crystal X-ray diffraction (CCDC 2070102†). Interestingly, when C5- and C6-substituted indole-3-carboxaldehydes 1j, 1k, and 1l were used in the reaction, mono amidation products 5j–5l were isolated in 79, 75 and 80% yield, respectively and their corresponding diamidation products were not produced, which was attributed to the steric bulk of these substituents. In addition, the reaction of C7-substituted indole-3-carboxaldehydes 1m and 1n successfully provided their diamidation products 5m and 5n in 78 and 71% yield, respectively.
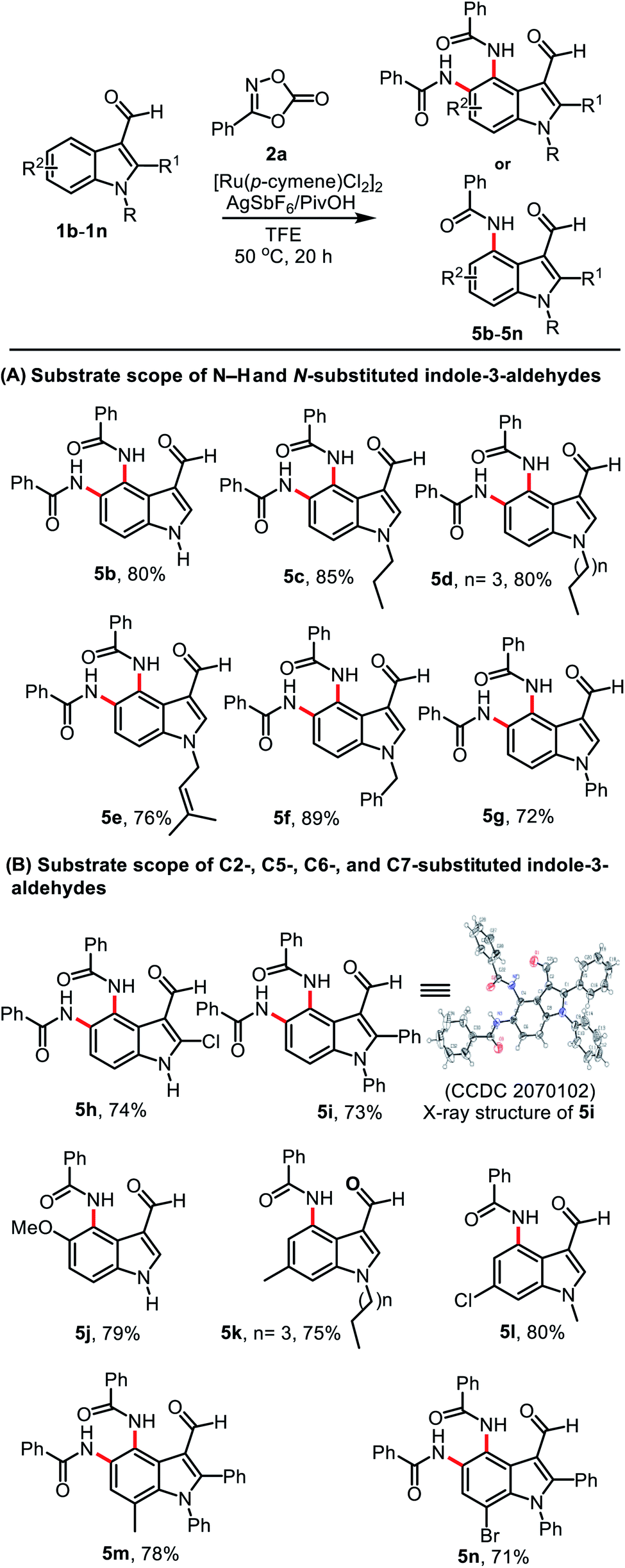
|
To demonstrate the scope of this protocol, various dioxazolones were examined in the reaction under the optimal conditions (Table 3). A combination of 1a or 1b with dioxolones 2b–2c having electron-donating groups of 4-Me or 4-OMe on the aromatic ring showed good reactivity and provided products 6a–6c in 78–81% yield. Similarly, dioxazolones 2d–2f bearing electron-withdrawing groups on the aromatic ring such as 4-Br, 4-F, and 4-Cl were also tolerated in the reaction and provided diamidation products 6d–6f in 75–77% yield (Table 3, A). We found that this reaction was not limited to aryl substrates only; aliphatic dioxazolones 2g and 2h bearing methyl and n-heptyl groups also displayed a good functional group compatibility, affording products 6g and 6h in 74 and 77% yield, respectively (Table 3, B).
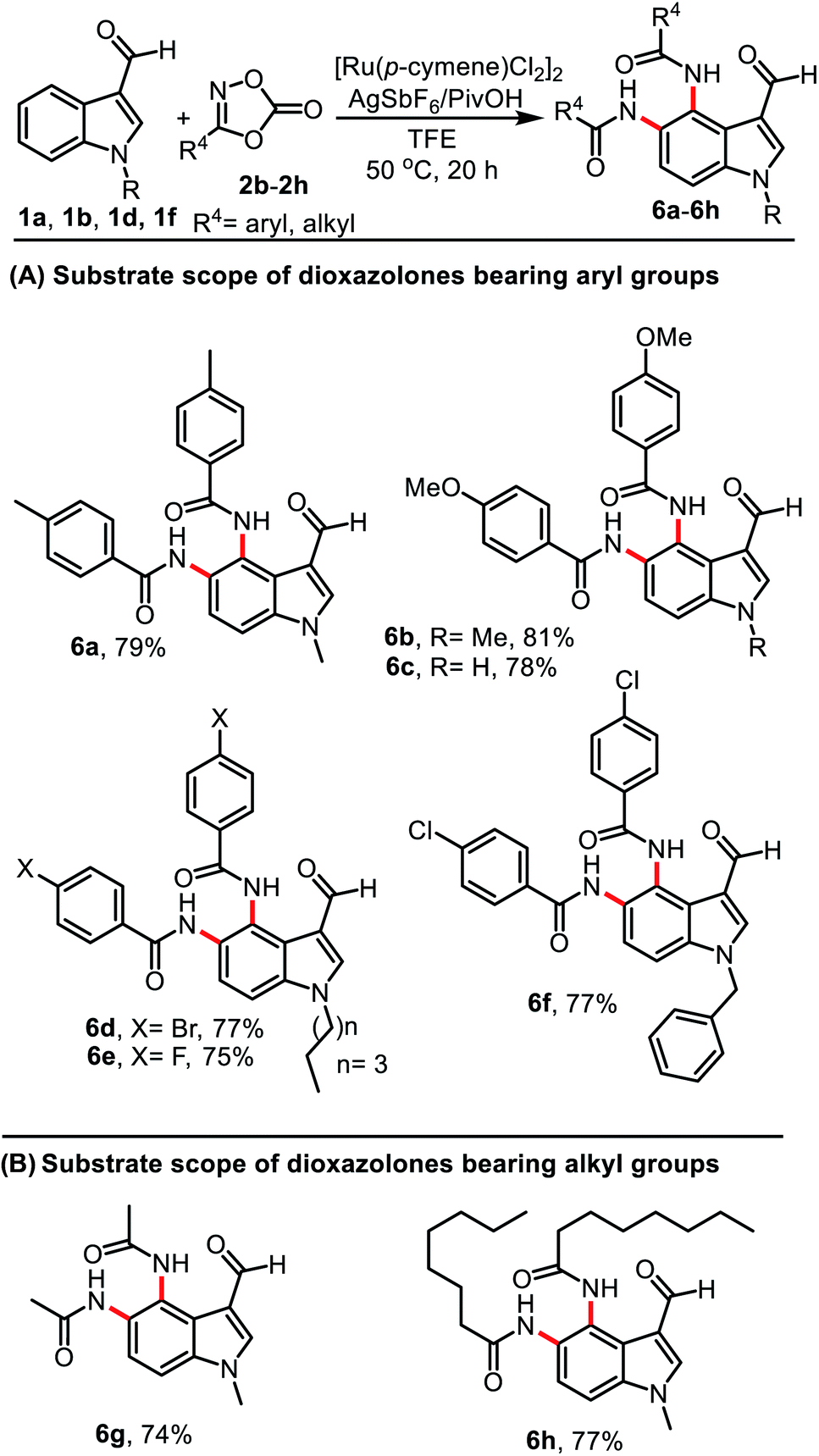
|
We further explored the scope of this reaction using dioxolones 2i–2k bearing heteroaromatic and polyaromatic one (2j) which afforded products 7a and 7b in 75% and 73% yield. When 1a was reacted with 3-(naphthalen-2-yl)-1,4,2-dioxazol-5-one (2k), the desired product 7c was isolated in 83% yield (Table 4, A). To test the versatility of the C–H activation/amidation reaction, which is not restricted to indole-3-formylaldehydes, the possibility of using benzo[b]thiophene-3-carboxaldehyde (1o) and 3-acylindole (1p) was investigated (Table 4, B and C). Gratifyingly, a combination of 1o with 2a, 2b, 2e, or 2k successfully gave products 7d–7g in 65–75% yield. The reactions of 3-acylindole 1p were also successful and provided products 7h–7j in 73–84% yield.

|
Next, we performed the reaction of 1q bearing C3-formyl and N-pyrimidyl directing groups with 2a under the standard reaction conditions (Scheme 2). However, instead of C4/C5-diamidation or C7-amidation products, only C2-amidated product 8 was obtained in 69% yield (Scheme 2).
 | ||
| Scheme 2 Reaction of 1q with 2a. | ||
The compounds 5a and 6g obtained using this new protocol are useful precursors for further synthetic transformations like Wittig olefination, Knoevenagel condensation, and reduction reactions, as summarized in Scheme 3. Treatment of 5a with (triphenylphosphoranylidene)acetonitrile or (t-butoxycarbonylmethylene)triphenylphosphorane in THF led to 9a and 9b in 91 and 88% yield, respectively. The condensation reaction of 5a with dimethyl malonate in the presence of piperidine afforded compound 9c in 83% yield. The chemoselective reduction of 6g using LiAlH4 in THF gave compound 9d in 91% yield.
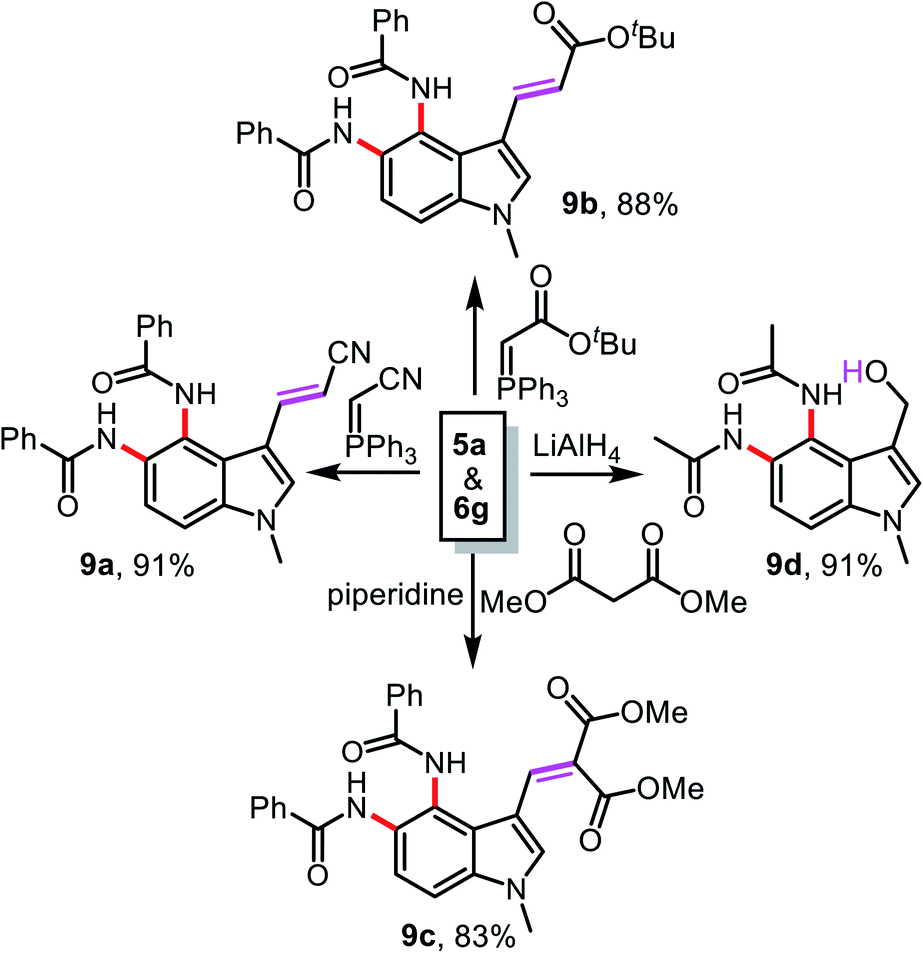 | ||
| Scheme 3 Further functionalization of compounds 5a and 6g. | ||
To gain insight into the mechanism of this reaction, we performed H/D exchange experiments using 1a with 5 mol% [Ru(p-cymene)Cl2]2, 15 mol% AgSbF6, and 5 equiv. of CD3COOD as the deuterium source in TFE at 50 °C. The 1H NMR spectrum of 1a′ showed 11% H/D exchange at the C4-position, while no significant H/D exchange was observed at the C2-position (Scheme 4).
 | ||
| Scheme 4 Control experiment. | ||
Based on our H/D exchange experiments and the observed products, a plausible mechanism for the Ru(II)-catalyzed diamidation of indole was proposed, as shown in Scheme 5, which was explicitly tested using density functional theory (DFT) calculations.10,12 Geometry optimization, vibration, and solvation energy calculations were performed using the B3LYP-D3/LACVP/6-31G** level of theory.19 The electronic energies of all the optimized structures were re-evaluated at the B3LYP-D3/cc-pVTZ(-f) level of theory.20 Details of the computational method can be found in the ESI.† As shown in Fig. 1, our theoretical investigation started with cationic Ru(II) κ2-OPiv complex A as the active catalyst. The dissociation of one neutral ligand allows the metal center to accommodate the indole substrate, 1-methylindole-3-carboxaldehyde (1a), to give B, which was found to be 6.3 kcal mol−1 more stable than the initial species. C4–H ruthenation occurs to form a six-membered ruthenacycle C traversing B-TS located at 15.3 kcal mol−1. The regioisomeric transition state (B′-TS) for C2–H activation demands an additional energy of 4.7 kcal mol−1, indicating a significant kinetic preference for C4-functionalization over C2.
 | ||
| Scheme 5 Plausible mechanism for the formation of 5a. | ||
 | ||
| Fig. 1 DFT-calculated energy profile obtained for the Ru(II)-catalyzed C4-amidation reaction of 1a. | ||
To gain a deeper understanding of the C–H ruthenation step that governs the regioselectivity of the first amidation reaction, the energy difference between B-TS and B′-TS was examined using distortion–interaction analysis.21Fig. 2 shows that the complex was partitioned into the indole substrate and catalyst fragments to obtain the structural distortion energies (Edist) required for each fragment to adopt the structure observed in the transition state. The interaction energy (Eint) was calculated by letting the two distorted fragments interact electronically at the distance observed in the transition state. Substrate 1a exhibits a slightly larger destabilization energy of ∼1 kcal mol−1 for the elongation of C4–H when compared to C2–H despite having almost identical bond lengths of 1.228 and 1.227 Å, respectively, reflecting the lower acidity of C4–H.
Because the catalyst distortions are also nearly identical at ∼31.5 kcal mol−1, the total distortion energies can be considered nearly identical at 56.0 and 55.2 to give an overall electronic energy preference of 3.2 kcal mol−1. Interestingly, the interaction energies show a significant difference with the Eint in the transition states being 4 kcal mol−1, as illustrated in Fig. 2. As the main electronic interactions in these two transition states are related to the Ru–C and O–H bond forming steps, the difference in Eint likely reflects the reactivity at the carbon sites in 1a. Thus, the electronic properties of each carbon site in 1a were evaluated using natural population analysis (NPA) and the Fukui index, indicating that the C4-site was more electron-rich than the C2-site (Fig. S1, see ESI†). This analysis indicates that the relative energies of the two possible transition states were governed by the higher partial negative charge on the C4-site, which leads to a stronger interaction with the electrophilic metal site. This is an unexpected result because the kinetics of conventional concerted metalation–deprotonation (CMD) reactions are thought to be mainly determined by the Brønsted acidity of the C–H bonds. Thus C–H activation in this system is best described as an electrophilic CMD (eCMD), which was recently highlighted as an alternative to the classical CMD mechanism.22 The C4–H activation step affords six-membered ruthenacycle C, which was 2.8 kcal mol−1 lower in energy than the alternative five-membered ruthenacycle (C′).
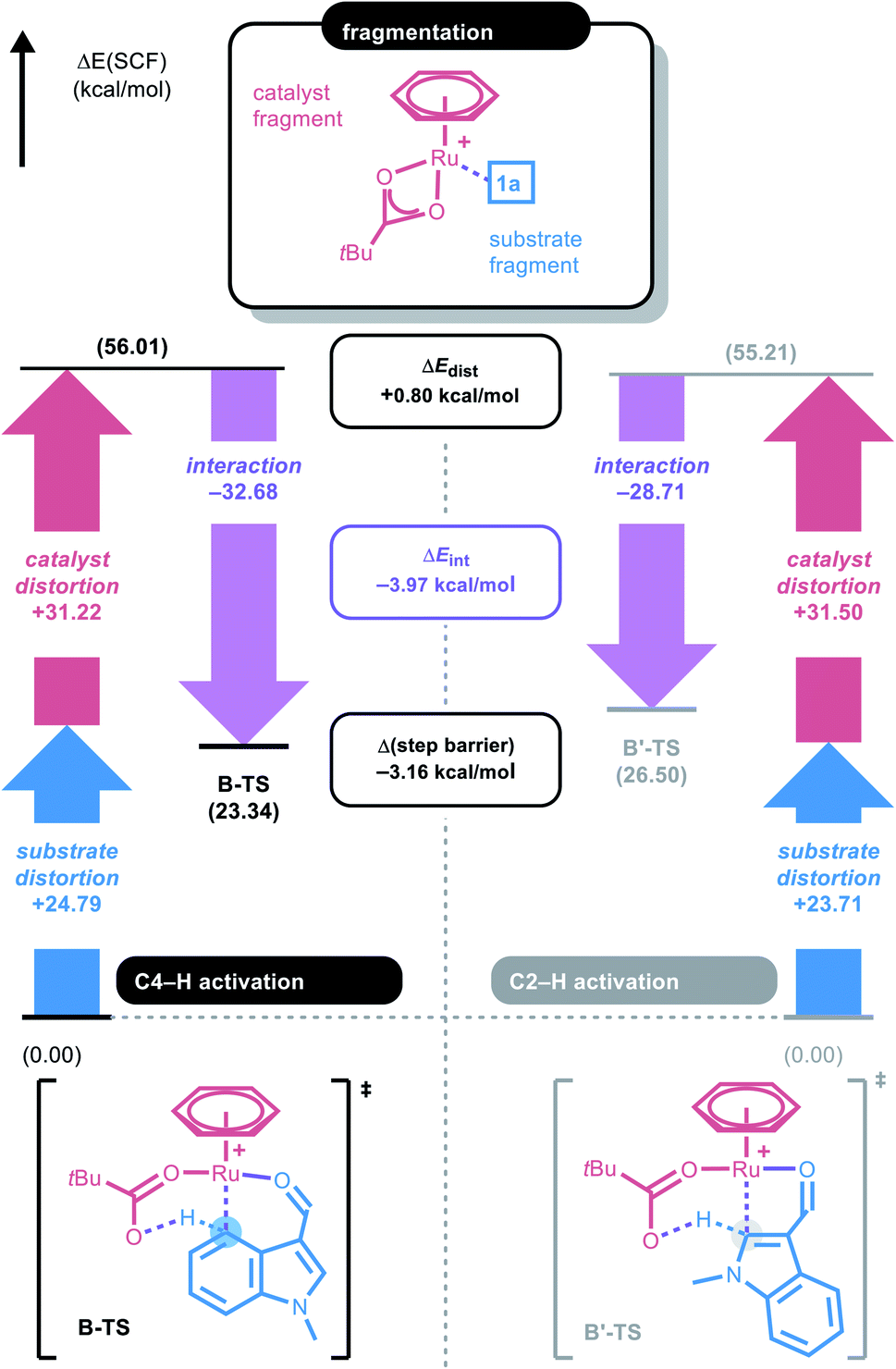 | ||
| Fig. 2 Distortion–interaction analysis diagram for B-TS and B′-TS. | ||
To push the catalytic cycle forward, the PivOH ligand in C must be displaced by 3-phenyl-1,2,4-dioxazol-5-one (2a) to form N-bound complex D located at 2.7 kcal mol−1. CO2 extrusion viaD-TS at 11.5 kcal mol−1 forms N-bound complex D located at 2.7 kcal mol−1. CO2 extrusion viaD-TS at 11.5 kcal mol−1 presented Ru(IV) imido complex E, which can undergo a reductive C–N bond forming reaction. Traversing reductive elimination transition state E-TS demands 12.8 kcal mol−1, which generates cyclic amidato complex F at −51.8 kcal mol−1. To complete the first amidation reaction proto-demetallation occurs with the assistance of PivOH to regenerate the κ2-OPiv moiety in G for the sequential C–H activation of 4a. In principle, mono-amidated indole product 4a may be released from G, but for diamidation to occur, 4a may remain coordinated to the ruthenium center, in line with the experimental observation that 4a was not detected. As summarized in Fig. 3, our calculations show that C5–H ruthenation enhanced reactivity was attributed to the electron-donating and resonance effects of the C4-amide group, which increase the electron density and nucleophilicity of the C5-site. The potential competing C2–H activation of 4a was evaluated and discarded as it requires an insurmountable barrier of 32.0 kcal mol−1 associated with G′-TS. Distortion–interaction analysis revealed that the difference in Eint between G-TS and G′-TS was 20.1 kcal mol−1, which was greater than that found in the first C–H activation step (Fig. S2, see ESI†). Consumption of another equivalent of 2a and CO2 extrusion traversing I-TS located at 18.6 kcal mol−1 gave imido ruthenacycle J. Subsequent C–N bond formation and proto-demetallation regenerate the active catalyst (A) and furnish the desired diamidated product 5a. In principle, C5-amidation may facilitate C6–H activation in a similar way; the increased structural complexity and steric bulk destabilize the resulting ruthenacycle intermediates and further impede the reaction (Fig. S3, see ESI†).23
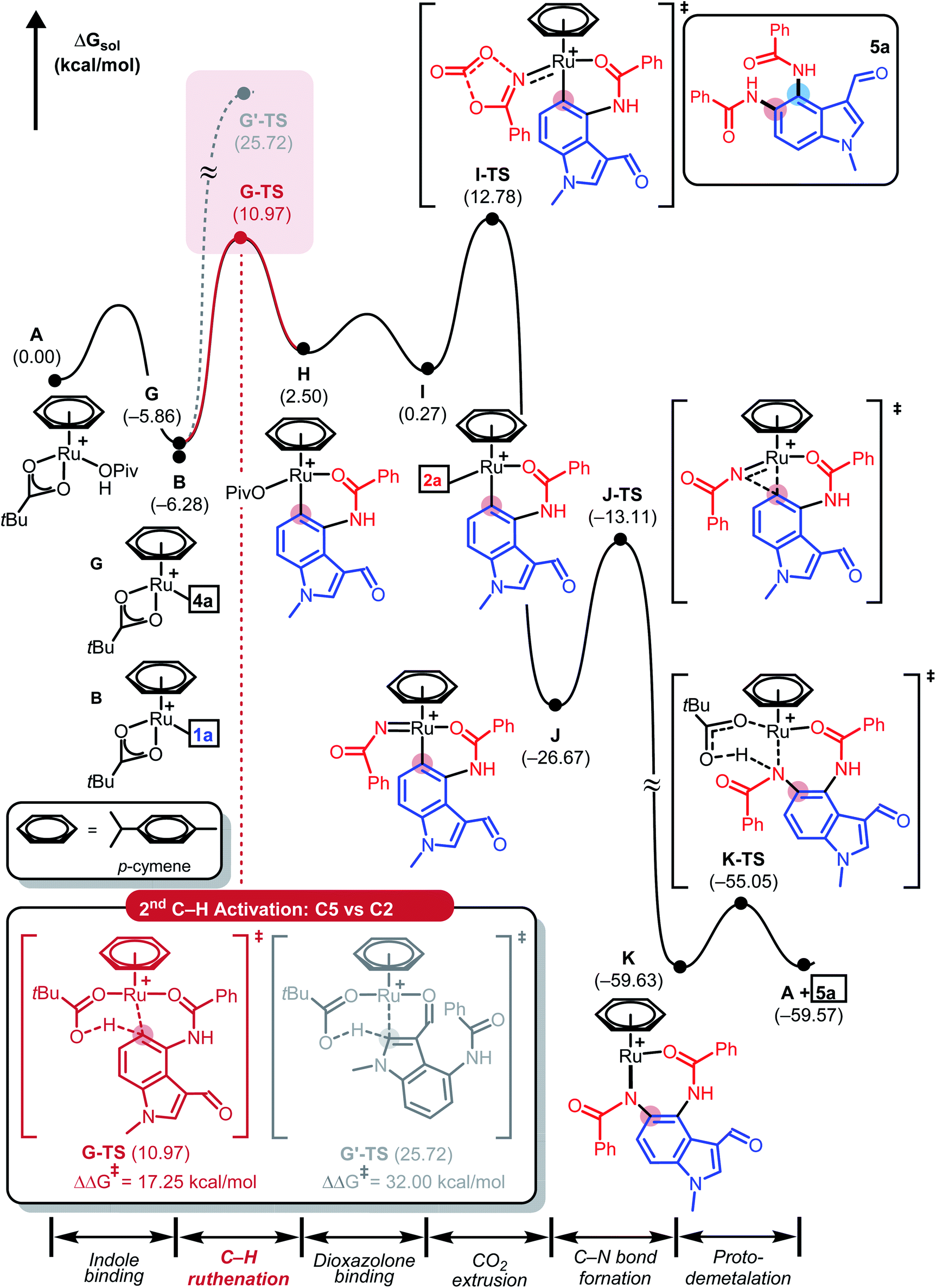 | ||
| Fig. 3 DFT-calculated energy profile for the consecutive Ru(II)-catalysed C5-amidation reaction used to give 5a. | ||
Indole-based receptor systems have attracted significant attention because of the inherent fluorescence properties of indole chromophores.24 The indole fluorophore can transfer an electron to the vacant orbital in transition metals. In addition, the interaction between the indole ring and metal in biological systems has been well documented.25 However, indole derivatives used as potential fluorescent chemosensors for sensing metal cations such as ferric ions (Fe3+) have been less explored.26 Iron is the most abundant element on the Earth and in living cells. Iron is essential for cellular metabolism, enzymatic reactions, and gene expression, and serves as an oxygen carrier in hemoglobin.27 Deficiency or excess Fe3+ in biological systems can cause various disorders. Therefore, the detection, sensing, and monitoring of Fe3+ ions have become a matter of significant concern in environmental and biological samples.
The fluorescence intensities of compounds 5a–5o, 6a–6h, and 7a–7j were examined. These compounds exhibit fluorescence due to the presence of NH groups and other active sites, which can be used to selectively detect and bind to heavy metal ions (Fig. S4, see ESI†). Interestingly, among the screened compounds, 7i showed the most intense fluorescence signal (Fig. S5, see ESI†). Fig. S6† shows the absorbance and fluorescence selectivity response of compound 7i (1.0 × 10−5 mol L−1) dissolved in DMSO towards 10 mM pH 7.4 solutions of different metal ions such as Ag+, Ba2+, Ca2+, Cd2+, Ce3+, Cu2+, Co2+, Fe3+, Hg2+, Mn2+, Na+, Ni2+, Pb2+, Sn2+, Sr2+, Ti3+, and Zn2+. Fig. S6a (see the ESI†) shows that 7i exhibits maximum absorption bands at 306, 294, and 330 nm upon the addition of Ce3+, Cu2+, and Fe3+ ions, respectively. The presence of Ce3+, Cu2+, and Fe3+ ions results in a shift and enhancement of the absorption intensity of 7i, while the response of 7i towards other metal ions was negligible.28 Among the metal ions studied, Fe3+ ions show a maximum tendency to quench the fluorescence intensity of 7i due to the formation of a 7i–Fe3+ complex (Fig. S6b, see ESI†). Upon the addition of Fe3+ (0 to 10 mM), the emission intensity of 7i gradually decreases, accompanied by a red shift of 12 nm from 405 nm to 417 nm, up on excitation at 324 nm (Fig. 4a). The binding of Fe3+ to 7i initiates charge transfer causing 95% fluorescence quenching.29 According to the linear Stern–Volmer equation, the measured fluorescence intensity [F0/F] varied as a function of the concentration of Fe3+, which showed good linearity, confirming the formation of 7i–Fe3+ (Fig. 4b). Based on our fluorescence titration experiments, the detection limit of 7i–Fe3+ was found to be 2.9 μM (Fig. S7, see ESI†).30
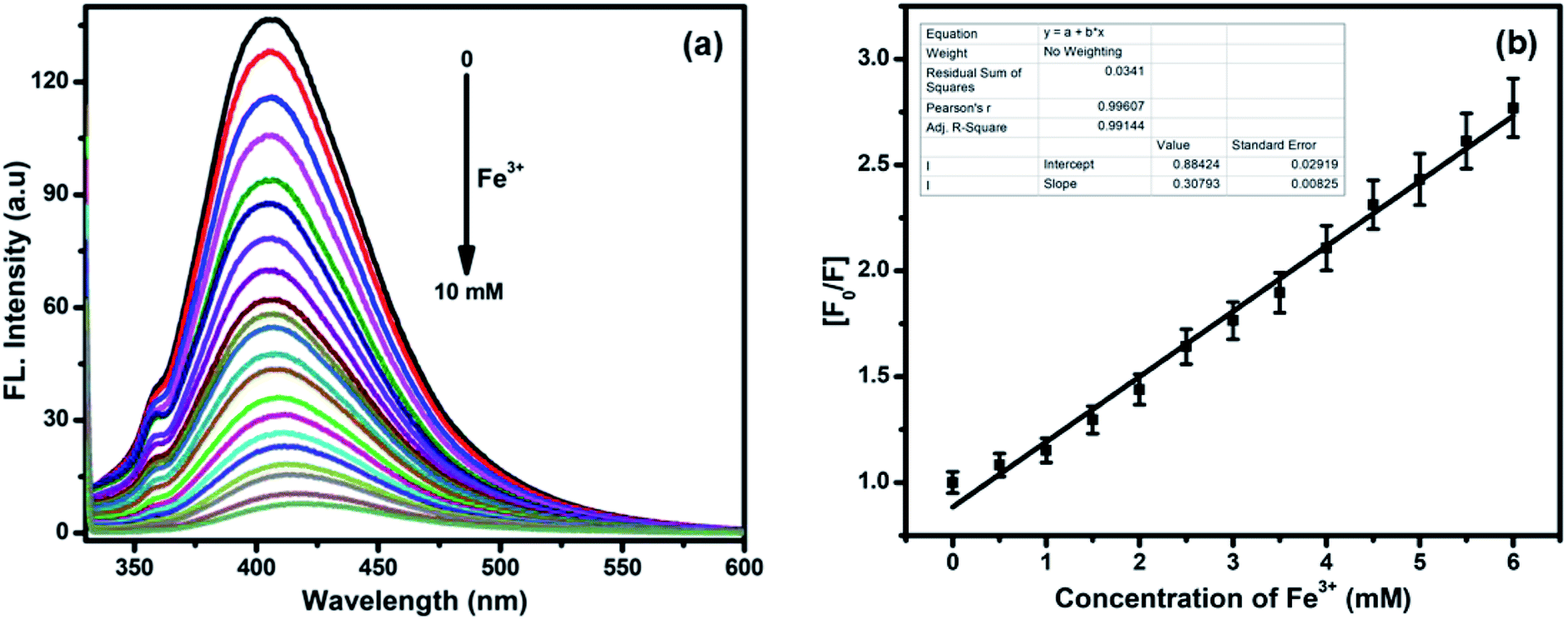 | ||
| Fig. 4 (a) Fluorescence spectra of 7i (1.0 × 10−5 mol L−1) in the presence of various concentrations of Fe3+ (0–10 mM). (b) Stern–Volmer plot based on the relative fluorescence signal [F0/F] and [Fe3+] concentrations. | ||
Conclusions
In conclusion, we have developed a new method to introduce two amide groups at the C4- and C5-positions of indole using a ruthenium(II)-catalyzed regioselective diamidation of 3-carbonylindoles with various dioxazolones. This novel diamidation protocol effectively provides access to a diverse series of functionalized indoles carrying two amide groups on the benzene ring of the indole moiety under mild conditions with excellent functional group tolerance and a broad substrate scope. Density functional theory calculations elucidated the full reaction mechanism and rationalized the regioselectivity observed in the reaction. Further extension of this protocol can also afford the corresponding diamidation products using benzo[b]thiophene-3-carboxaldehyde. To demonstrate the utility of this new method, the synthesized compounds were further elaborated and converted into functionalized molecules, which can be used as sensors for metal ion detection.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. D. performed all the experiments and prepared the ESI.† S. K., S. Y. Y. and M.-H. B. carried out all the computational studies. S. M. performed the chemosensor studies. Y. R. L. conceptualized the work, supervised the project and wrote the manuscript. All the authors discussed and commented to give the final shape of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2021R1A2B5B02002436) and by the Korean Ministry of Education, Science, and Technology (2012M3A7B4049677). We also thank the Institute for Basic Science (IBS-R10-A1) for their financial support.Notes and references
- (a) M. Inman and C. J. Moody, Chem. Sci., 2013, 4, 29–41 RSC; (b) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef CAS PubMed; (c) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC; (d) T. Kawasaki and K. Huguchi, Nat. Prod. Rep., 2005, 22, 761–793 RSC.
- (a) Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed; (b) C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 5450–5454 CrossRef CAS PubMed.
- (a) T. V. Sravanthi and S. L. Manju, Eur. J. Pharm. Sci., 2016, 91, 1–10 CrossRef CAS PubMed; (b) A. Kumari and R. K. Singh, Bioorg. Chem., 2019, 89, 103021–103056 CrossRef CAS PubMed.
- (a) N. Chadha and O. Silakari, Eur. J. Med. Chem., 2017, 134, 159–184 CrossRef CAS PubMed; (b) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed; (c) P. V. Thanikachalam, R. K. Maurya, V. Garg and V. Monga, Eur. J. Med. Chem., 2019, 180, 562–612 CrossRef CAS PubMed.
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- (a) Z. Wang, H. Zeng and C.-J. Li, Org. Lett., 2019, 21, 2302–2306 CrossRef CAS PubMed; (b) S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2010, 132, 11416–11417 CrossRef CAS PubMed; (c) N. Selander, B. T. Worrell, S. Chuprakov, S. Velaparthi and V. V. Fokin, J. Am. Chem. Soc., 2012, 134, 14670–14673 CrossRef CAS PubMed; (d) S. Wagaw, B. H. Yang and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 6621–6622 CrossRef CAS.
- (a) S. Chen, Y. Liao, F. Zhao, H. Qi, S. Liu and G.-J. Deng, Org. Lett., 2014, 16, 1618–1621 CrossRef CAS PubMed; (b) J. Chen, M. Li, J. Zhang, W. Sun and Y. Jiang, Org. Lett., 2020, 22, 3033–3038 CrossRef CAS PubMed; (c) G. Berionni, V. Morozova, M. Heininger, P. Mayer, P. Knochel and H. Mayr, J. Am. Chem. Soc., 2013, 135, 6317–6324 CrossRef CAS PubMed.
- (a) R. Mei, J. Loup and L. Ackermann, ACS Catal., 2016, 6, 793–797 CrossRef CAS; (b) Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 4698–4701 CrossRef CAS PubMed; (c) Q. Liu, Q. Li, Y. Ma and Y. Jia, Org. Lett., 2013, 15, 4528–4531 CrossRef CAS PubMed.
- (a) M. S. Sherikar, R. Kapanaiah, V. Lanke and K. R. Prabhu, Chem. Commun., 2018, 54, 11200–11203 RSC; (b) A. J. Borah and Z. Shi, Chem. Commun., 2017, 53, 3945–3948 RSC; (c) F. Li, Y. Zhou, H. Yang, Z. Wang, Q. Yu and F.-L. Zhang, Org. Lett., 2019, 21, 3692–3995 CrossRef CAS PubMed.
- (a) V. Lanke, K. R. Bettadapur and K. R. Prabhu, Org. Lett., 2016, 18, 5496–5499 CrossRef CAS PubMed; (b) V. Lanke and K. R. Prabhu, Org. Lett., 2013, 15, 6262–6265 CrossRef CAS PubMed.
- S. Pradhan, M. Mishra, P. B. De, S. Banerjee and T. Punniyamurthy, Org. Lett., 2020, 22, 1720–1725 CrossRef CAS PubMed.
- J. Lv, B. Wang, K. Yuan, Y. Wang and Y. Jia, Org. Lett., 2017, 19, 3664–3667 CrossRef CAS PubMed.
- Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 CrossRef CAS PubMed.
- V. Lanke and K. R. Prabhu, Chem. Commun., 2017, 53, 5117–5120 RSC.
- S. Chen, B. Feng, X. Zheng, J. Yin, S. Yang and J. You, Org. Lett., 2017, 19, 2502–2505 CrossRef CAS PubMed.
- X. Shi, W. Xu, R. Wang, X. Zeng, H. Qiu and M. Wang, J. Org. Chem., 2020, 85, 3911–3920 CrossRef CAS PubMed.
- (a) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103–14107 CrossRef CAS PubMed; (b) S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed; (c) B. Sun, T. Yoshino, S. Matsunaga and M. A. Kanai, Chem. Commun., 2015, 51, 4659–4661 RSC; (d) P. W. Tan, A. M. Mak, M. B. Sullivan, D. J. Dixon and J. Seayad, Angew. Chem., Int. Ed., 2017, 56, 16550–16554 CrossRef CAS PubMed.
- Y. Park, J. Heo, M.-H. Baik and S. Chang, J. Am. Chem. Soc., 2016, 138, 14020–14029 CrossRef CAS PubMed.
- (a) J. C. Slater, Phys. Today, 1974, 27, 49 CrossRef; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (d) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (e) S. Grimme, J. Antony, S. Ehrlich and S. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed; (f) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (g) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (h) W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS; (i) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070–10086 CrossRef CAS PubMed.
- (a) L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821–6836 CrossRef CAS PubMed; (b) E. Tan, O. Quinonero, M. E. de Orbe and A. M. Echavarren, ACS Catal., 2018, 8, 2166–2172 CrossRef CAS PubMed; (c) K. Naksomboon, J. Poater, F. M. Bickelhaupt and M. Á. Fernández-Ibáñez, J. Am. Chem. Soc., 2019, 141, 6719–6725 CrossRef CAS PubMed; (d) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378–10382 CrossRef CAS PubMed; (e) T. Rogge, J. C. A. Oliveira, R. Kuniyil, L. Hu and L. Ackermann, ACS Catal., 2020, 10, 10551–10558 CrossRef CAS.
- J. Park, J. Lee and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 4256–4260 CrossRef CAS PubMed.
- H. Shizuka, M. Serizawa, H. Kobayashi, K. Kameta, H. Sugiyama, T. Matsuura and I. Saito, J. Am. Chem. Soc., 1988, 110, 1726–1732 CrossRef CAS.
- (a) Y. L. Sun and A. T. Wu, J. Fluoresc., 2013, 23, 629–634 CrossRef CAS PubMed; (b) Y. Tang, H. Liu, G. Jiang and Z. Gu, J. Appl. Spectrosc., 2017, 84, 911–914 CrossRef CAS.
- (a) Y. C. Hsieh, J. L. Chir, S. T. Yang, S. J. Chen, C. H. Hu and A. T. Wu, Carbohydr. Res., 2011, 346, 978–981 CrossRef CAS PubMed; (b) N. Kaur, P. Kaur, G. Bhatia, K. Singh and J. Singh, RSC Adv., 2016, 6, 82810–82816 RSC; (c) S. Ta, S. Nandi, M. Ghosh, S. Banerjee and D. Das, Spectrochim. Acta, Part A, 2017, 173, 196–200 CrossRef CAS PubMed.
- (a) B. D'Autréaux, N. P. Tucker, R. Dixon and S. Spiro, Nature, 2005, 437, 769–772 CrossRef PubMed; (b) H. Weizman, O. Ardon, B. Mester, J. Libman, O. Dwir, Y. Hadar, Y. Chen and A. Shanzer, J. Am. Chem. Soc., 1996, 118, 12368–12375 CrossRef CAS; (c) D. S. Kalinowski and D. E. S. R Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed; (d) A. Gupta and A. L. Crumbliss, J. Lab. Clin. Med., 2000, 136, 371–378 CrossRef CAS PubMed.
- (a) S. Meghdadi, N. Khodaverdian, A. Amirnasr, P. J. French, M. E. van Royen, E. A. C. Wiemer and M. Amirnasr, J. Photochem. Photobiol., A, 2020, 389, 112193 CrossRef CAS; (b) X. Gong, H. Zhang, N. Jiang, L. Wang and G. Wang, Microchem. J., 2019, 145, 435–443 CrossRef CAS.
- A. Shylaja, S. R. Rubina, S. S. Roja and R. R. Kumar, Dyes Pigm., 2020, 174, 108062–108070 CrossRef CAS.
- S. Mohandoss, R. Atchudan, T. N. J. I Edison, K. Mishra, R. J. I. Tamargo, S. Palanisamy, K. Yelithao, S. G. You and Y. R. Lee, Sens. Actuators, B, 2020, 306, 127581–127594 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data and X-ray crystallographic structure data for 5i. CCDC 2070102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02138a |
| This journal is © The Royal Society of Chemistry 2021 |