
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short, enantioselective, gram-scale synthesis of (−)-zephyranthine†
Yuxiang
Zhao
 ,
Yanren
Zhu
,
Guolan
Ma
,
Qi
Wei
,
Shaoxiong
Yang
,
Xiaoyu
Zeng
,
Hongbin
Zhang
* and
Jingbo
Chen
*
,
Yanren
Zhu
,
Guolan
Ma
,
Qi
Wei
,
Shaoxiong
Yang
,
Xiaoyu
Zeng
,
Hongbin
Zhang
* and
Jingbo
Chen
*
Key Laboratory of Medicinal Chemistry for Natural Resource, Ministry of Education, Yunnan Provincial Center for Research & Development of Natural Products, School of Chemical Science and Technology, Yunnan University, Kunming, 650091, P. R. China. E-mail: zhanghb@ynu.edu.cn; chenjb@ynu.edu.cn
First published on 21st June 2021
Abstract
A reasonable synthesis design by strategically integrating functional group manipulation into the ring system construction resulted in a short, enantioselective, gram-scale total synthesis of (−)-zephyranthine. The concise route includes a catalytic Michael/Michael cascade for the asymmetric synthesis of a penta-substituted cyclohexane with three contiguous stereogenic centers, a remarkable 8-step one-pot operation to easily assemble the zephyranthine tetracyclic skeleton, the regioselective construction of a double bond in the C ring and an asymmetric dihydroxylation. This synthesis is also flexible and paves a potential path to a variety of cyclohexylamine-fused tricyclic or polycyclic alkaloids.
Introduction
Lycorine-type alkaloids (e.g., 1–4; Fig. 1)1 are members of an Amaryllidaceae alkaloid sub-class2 and display useful biological properties,3 including anticholinergic, antiviral, insect antifeedant, and antineoplastic activities, as well as other pharmacological properties.4 These alkaloids have attracted substantial synthetic attention because of their tetracyclic core structure, multiple chiral centers and bioactivities. As a result, significant effort has been devoted to assembling the tetracyclic skeleton of such alkaloids5 and to the syntheses of the natural products themselves.6–10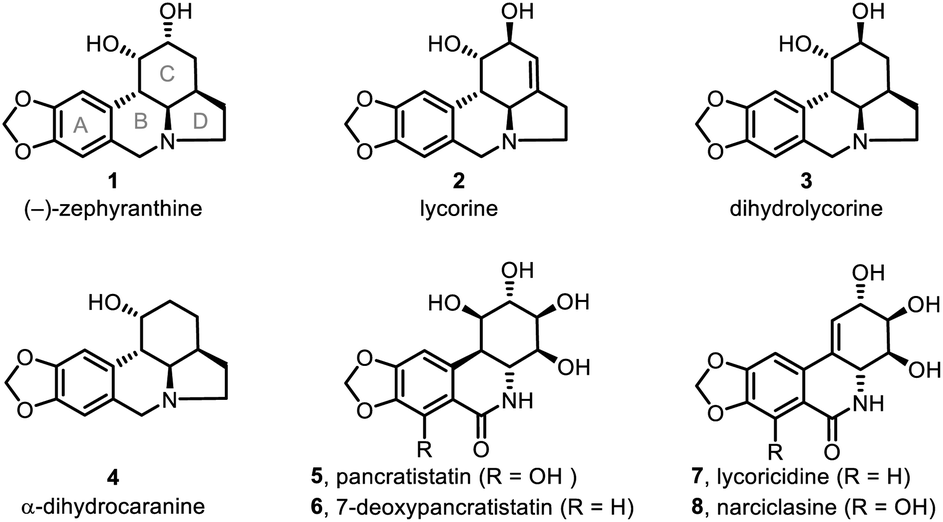 | ||
| Fig. 1 Selected Amaryllidaceae alkaloids. | ||
Unlike other members of the lycorine family, (−)-zephyranthine (1)11 has only a limited number of syntheses reported for its fabrication,6e,12 none of which detail a catalytic asymmetric approach. Herein, we report an efficient, enantioselective, gram-scale protocol for 1 that takes advantage of two one-pot reactions. The first is a catalytic asymmetric double Michael addition to construct the C ring with three consecutive chiral centers. The second is a novel 8-step procedure involving double deacetalyzation, nitro group reduction to its corresponding amine, tandem double ring-closing reductive amination, and then double ester hydrolysis with subsequent tandem decarboxylation to give the tetracyclic skeleton of 1. Although we have successfully developed a remarkably facile route to 1, we encountered obstacles at a later stage. Unfortunately, the crucial regioselective construction of the C1–C2 double bond in the C ring was hindered by mutable substrates containing nitro groups or amine-type nitrogen atoms. However, this failure was counteracted with the successful, kinetically controlled regioselective enolization of the C ring ketone moiety.
Results and discussion
The catalytic double Michael addition of γ,δ-unsaturated-β-ketoester and nitroolefin was previously developed by our group13d for the asymmetric synthesis of multiple-substituted cyclohexanes bearing 3–5 stereogenic centers, which is expected to develop into the key step of a general method to stereoselectively synthesize a variety of cyclohexylamine-fused alkaloids, including (−)-zephyranthine and lepadiformine-type alkaloids.14Our simple retrosynthetic analysis of the target natural product (Scheme 1) revealed that penta-substituted cyclohexane 12, which arose from a catalytic asymmetric double Michael addition of 13 and 14, was likely a key intermediate that would result in the direct formation of tetracyclic ketone 10via a one-pot operation. Subsequent regioselective construction of a double bond in the C ring, followed by dihydroxylation, would lead to 1.
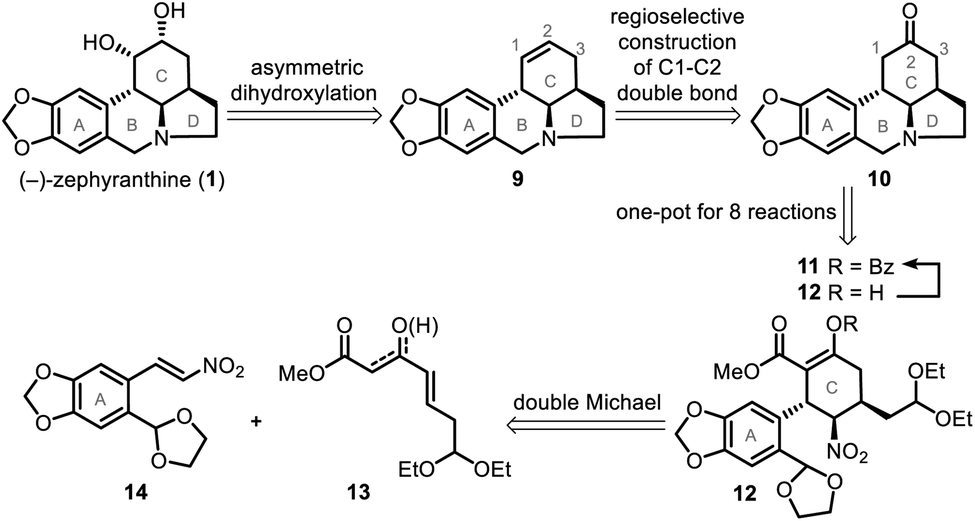 | ||
| Scheme 1 Retrosynthetic analysis of (−)-zephyranthine (1). | ||
γ,δ-Unsaturated-β-ketoester 13 (ref. 13d and 15) and nitroolefin 14 (ref. 5b and 16) were prepared on 10 gram scales using a literature method with minor modifications (Scheme 2, see ESI† for detailed preparation methods).
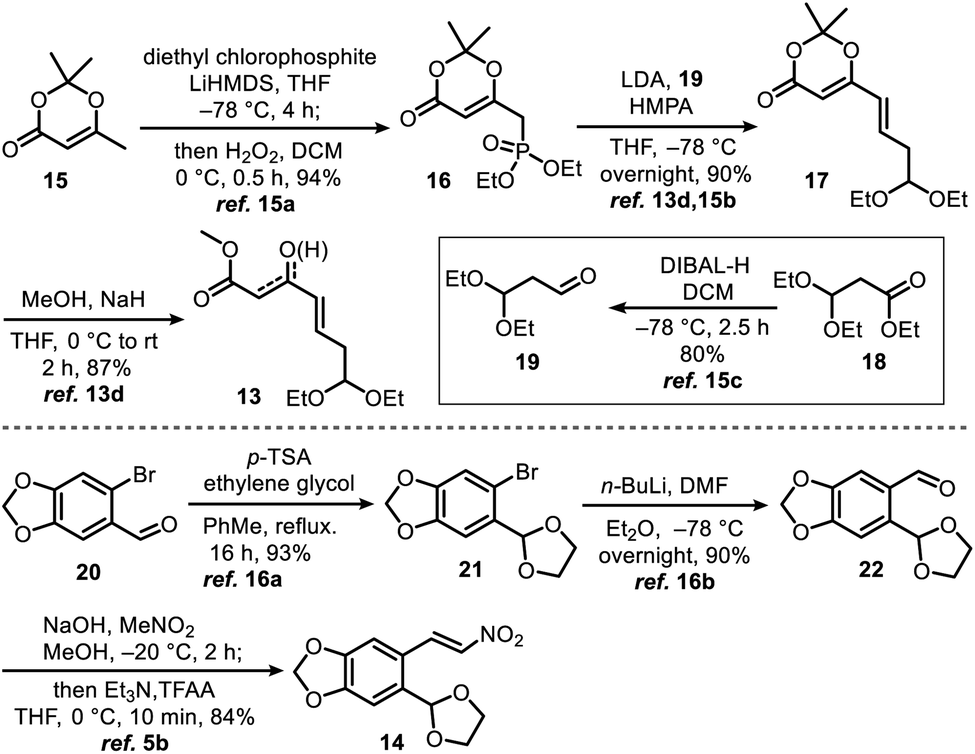 | ||
| Scheme 2 Synthesis of γ,δ-unsaturated-β-ketoester 13 and nitroolefin 14. | ||
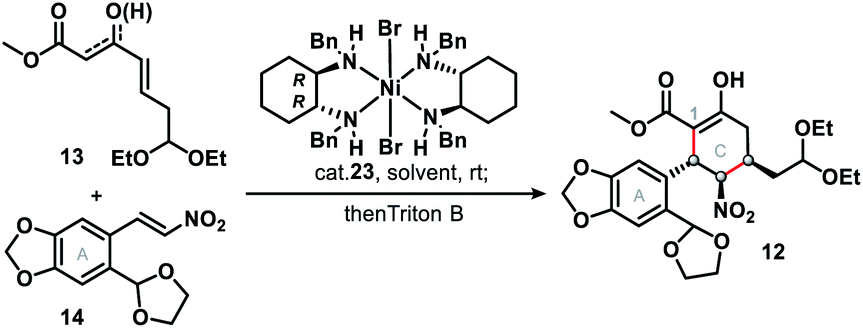
The synthetic journey commenced with a catalytic asymmetric double Michael addition cascade reaction of 13 and 14, which was promoted by Evans' chiral nickel(II) catalyst (23).13 Condition screening (Table 1) revealed that the 1st Michael addition, unlike that in the synthesis of (−)-stenine,13d was very sluggish with the low conversion (<10%) even after 10 days' reaction at room temperature with THF as solvent or in solvent-free conditions; DCM as solvent brought about the fastest reaction that afforded 84% yield of the product with inadequate ee (entry 3) in 48 hours. Finally we found that PhMe as the solvent and Triton B as the base gave both the highest enantioselectivity (90%) and diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), as well as high yield (85%) of penta-substituted cyclohexane 12, which was identified as the single isomer of an enol ester. Furthermore, NMR analysis showed that three consecutive chiral centers (in the C ring) had been correctly constructed so that 1 would be produced in the subsequent steps.
:1), as well as high yield (85%) of penta-substituted cyclohexane 12, which was identified as the single isomer of an enol ester. Furthermore, NMR analysis showed that three consecutive chiral centers (in the C ring) had been correctly constructed so that 1 would be produced in the subsequent steps.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry |
13:14 |
Solvent | Time (h) | Cat. 23 (mol%) | Yieldb (%) | eec (%) |
| a The reaction was performed in the presence of Triton B as a base (1.0 equiv.) at room temperature. b Isolated yields after chromatographic purification. c Enantiomeric excess was determined by high performance liquid chromatography (HPLC), chiracel columns. d Reaction was very sluggish with the low conversion after 10 days. | ||||||
| 1 | 1:1 |
— | 240 | 2 | Traced | ND |
| 2 | 1:1 |
THF | 240 | 2 | Traced | ND |
| 3 | 1:1 |
DCM | 48 | 2 | 84 | 76 |
| 4 | 1:1 |
PhMe | 96 | 2 | 76 | 90 |
| 5 | 1:1 |
PhMe | 96 | 3 | 78 | 90 |
| 6 | 1.1:1 |
PhMe | 96 | 2 | 81 | 90 |
| 7 |
1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1 |
PhMe | 96 | 2 | 85 | 90 |
| 8 | 1.3:1 |
PhMe | 96 | 2 | 85 | 90 |
It can be speculated, as shown in Scheme 3, the steric and electronic effects caused by the γ-ethyl group of the product (R1 = Et, for enantioselective synthesis of stenine)13d of 1st Michael addition make the intermediate 24 (R1 = Et) a less active Michael acceptor in the 2nd Michael addition, therefore, a heterogeneous strong base (KOH/SiO2) condition was required to promote this reaction while avoiding damage to the nitro group. Moreover, an isomerisation phenomenon was observed after the 2nd Michael addition that the keto ester intermediate 25 gradually transformed into its enol ester isomer accompanied by inversion of configuration at the Nα-carbon. Evidently, γ,δ-unsaturated-β-ketoester (R1 = H) in this work is more active, and the 2nd Michael addition as well as the subsequent isomerisation progressed rapidly and completed in 20 minutes after addition of Triton B to the reaction mixture upon completion of 1st Michael addition.
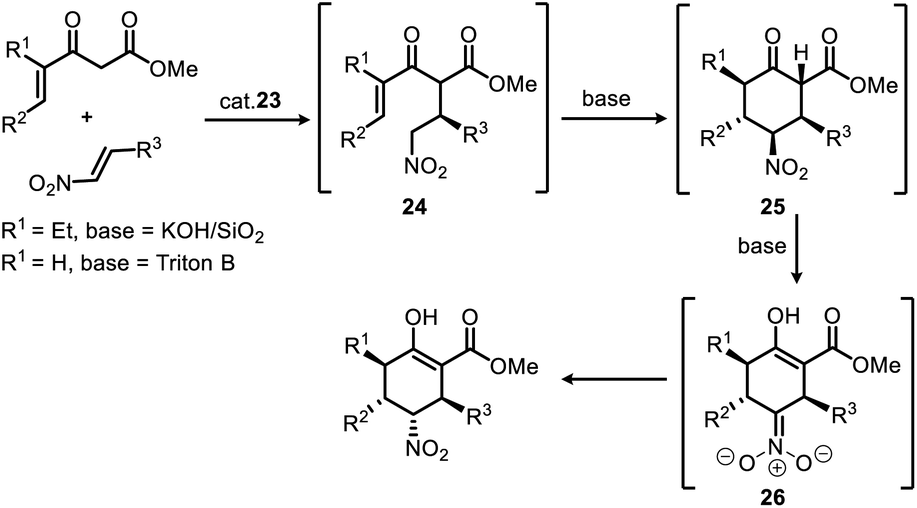 | ||
| Scheme 3 Stereoselective synthesis of multiple-substituted cyclohexanes via a Michael/Michael/isomerization cascade reaction. | ||
To confirm the absolute configuration of compound 12, its enol moiety was either benzyloxycarbonyl (Cbz)- or benzoyl (Bz)-protected to prevent unwanted aldol reactions between the α-carbon (C1) of the β-keto ester and the aldehydes that would form from the acetal moieties upon their subsequent deprotection. Cbz-protection was achieved by treating 12 with benzyl carbonochloridate (CbzCl) in the presence of NaH to give ester 27 in 90% yield. However, 27 was difficult to purify by recrystallization. By replacing CbzCl with benzoyl chloride (BzCl), similar esterification of 12 afforded benzoate 11 in quantitative yield. After recrystallization, the isomeric purity of 11 was greater than 99% ee, as determined by high performance liquid chromatography (HPLC). The absolute configuration of benzoate 11 was confirmed by X-ray crystallography (Scheme 4) with Cu-Kα radiation.
 | ||
| Scheme 4 Protection of the enol hydroxy group of 12 and the absolute configuration of compound 11. | ||
Successful construction of the three contiguous stereogenic centers in the newly formed cyclohexane ring allowed us to begin synthesizing 1. Cyclization of 11 to form tetracyclic ketone 10 was accomplished through a multistep one-pot operation (Scheme 5), which began by treating 11 with HBr (1.0 equiv., 33% in HOAc) in HOAc–THF–H2O (5:1:1) at 50 °C for 2 h to give dialdehyde 28. Subsequent reaction of 28 with zinc powder at room temperature overnight gave 29. After a simple filtration to remove the excess zinc and other solid substances, HCl (8.0 N, 100 equiv.) was added to the reaction mixture to hydrolyze 29 into 30. Tandem decarboxylation of 30 then delivered key intermediate 10 in a total yield of 57% via an eight-step one-pot synthesis.
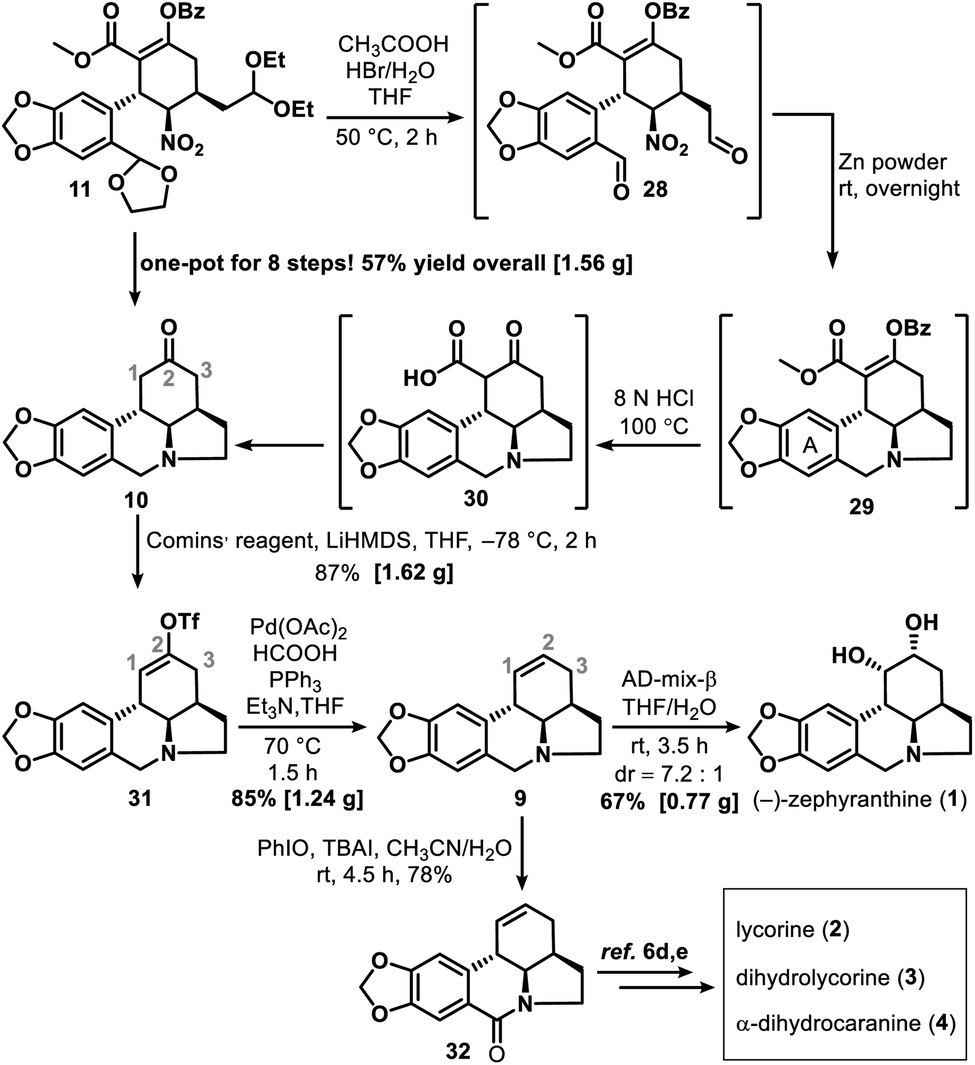 | ||
| Scheme 5 Total synthesis of (−)-zephyranthine (1) and synthesis of 32. | ||
The reaction of ketone 10 with lithium bis(trimethylsilyl)amide and Comins' reagent17 at −78 °C was a kinetically controlled regioselective enolization, which was followed by triflation to afford enol triflate 31 in 87% yield. This then underwent a palladium-promoted hydrogenolysis18 to give 9 in 85% yield. In the last step, an attempt to avoid oxidative damage of the amino nitrogen atom was made by adding some acid to the reaction system; however, this failed owing to the deactivation of AD-mix-β under acidic conditions. Fortunately, the most conventional sharpless asymmetric dihydroxylation19 of 9 with AD-mix-β under acid-free conditions proceeded smoothly and gave 1 in 67% isolated yield (76% yield of 1 and its diastereoisomer in a ratio of 7.2:1). After that, amide 32 was synthesized in 78% yield via a PhIO promoted oxidation20 of 9. Our approach thus provided a formal synthesis of a number of other lycorine-type alkaloids12a (Scheme 5), such as lycorine (2),6d,e dihydrolycorine (3)6d,e and α-dihydrocaranine (4).6d,e
To gain additional insight into the nature of the regioselective enolization of ketone 10, we conducted a theoretical study and the DFT quantum-chemical calculations (Scheme 6, see ESI† for details) revealed that the formation of intermediate 33a is kinetically favored over that of 33b.
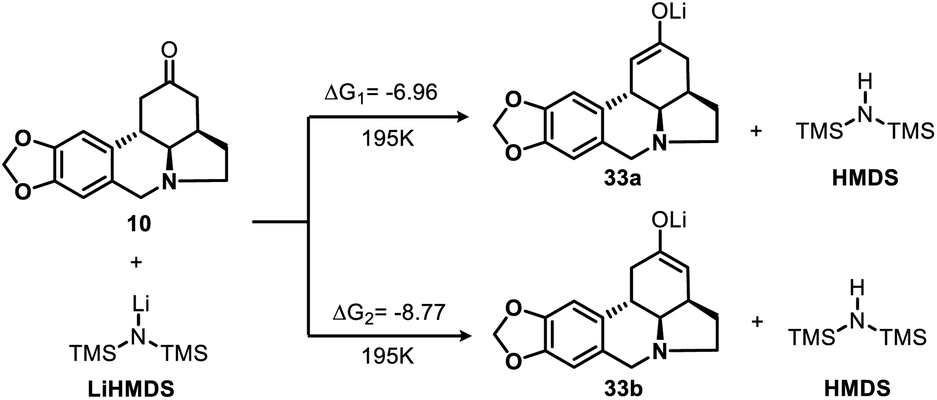 | ||
| Scheme 6 DFT calculations for enolization reaction of 10 (kcal mol−1). | ||
We also obtained 36, the double bond positional isomer of 9, from the same intermediate 10 that gave 9. This was accomplished through a 3-step chemical manipulation of the ketone moiety of the C ring (Scheme 7). Intermediate 10 was reduced with sodium borohydride in methanol to give secondary alcohol 34 (dr = 1.2:1), which underwent mesylation and then DBU-promoted, thermodynamically controlled methanesulfonic acid elimination to afford 36 as a single regioisomer in 57% overall yield.
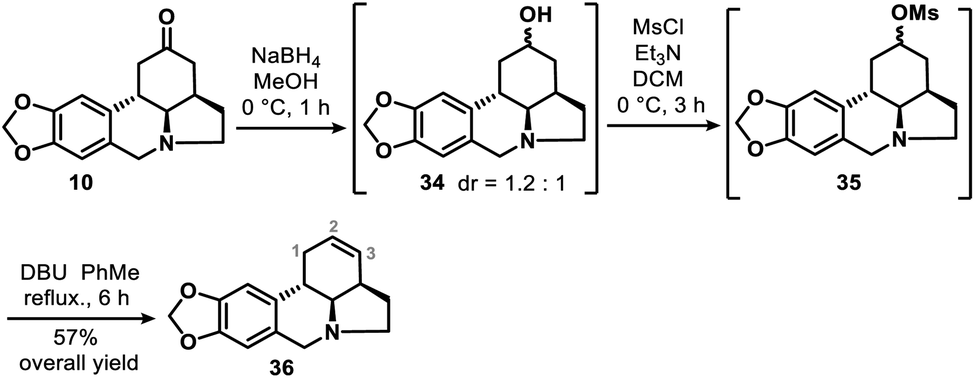 | ||
| Scheme 7 Synthesis of the double bond positional isomer (36) of 9. | ||
To confirm the proposed thermodynamically controlled process, we conducted DFT calculations (see ESI† for details) of elimination reactions of mesylate 35 as indicated in Scheme 8. For both 35a and 35b, the formation of olefin 36 is more favorable than formation of 9 according to the free energy changes. 35a is more likely to undergo elimination than 35b to form compound 36 as less energy required. The calculation results supported our conclusion that the formation of 36 by elimination reaction of 35 (both 35a and 35b) is a thermodynamically controlled process.
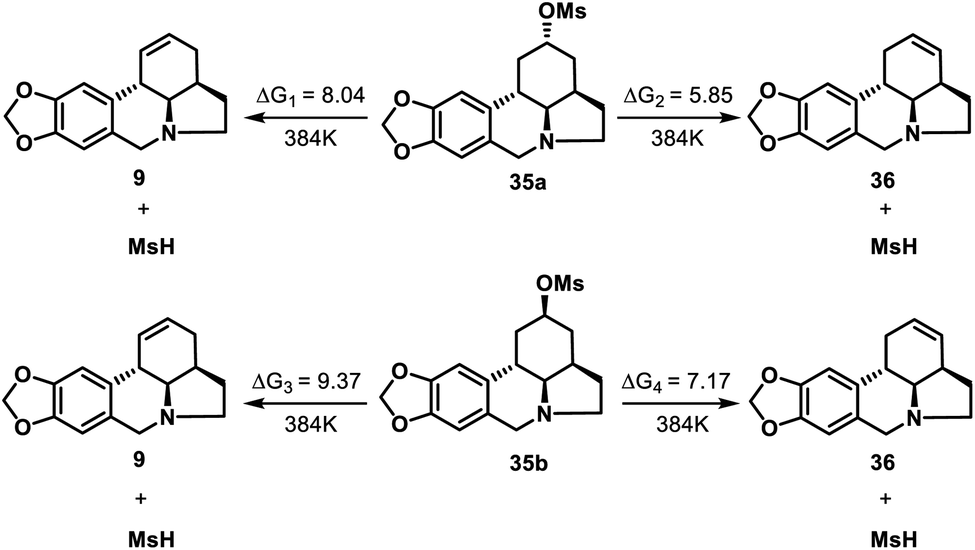 | ||
| Scheme 8 DFT calculations for elimination reaction of 35 (kcal mol−1). | ||
We had also attempted other routes to regioselectively construct the double bond in the C ring, but these did not proceed as we expected (see ESI† for detailed informations). Ideally, deesterification of 37 could efficiently provide 9 (Scheme 9, top) and ensure that the double bond remained in the correct position (C1–C2); however, this reaction was unsuccessful. We did manage to convert the ester group of 37 into a carboxyl or aldehyde group, but the subsequent decarboxylation or deformylation failed. In addition, transformation of 12 to 38 (Scheme 9, bottom) could not be achieved through direct deesterification, and attempts to obtain 39 from 12 with the same one-pot protocol that gave 10 from 11 (Scheme 5) were also unsuccessful due to unwanted aldol reactions.
 | ||
| Scheme 9 Failed alternative routes to produce (top) the C-ring double bond and (bottom) compounds 38 and 39 from 12. | ||
Conclusions
The natural product (−)-zephyranthine (1) was synthesized using a highly efficient and practical approach. Strategically integrating functional group manipulation into the ring system construction resulted in two, multi-step, one-pot reactions that greatly simplified the overall operation and improved its efficiency. From readily available 13 and 14, only six steps (18.7% overall isolated yield) were necessary to acquire 1 g of (−)-1. In addition, regioselective construction of the C ring double bond from 10 delivered 9 or 36 through kinetically or thermodynamically controlled pathways, respectively. This, together with the concise synthesis of amide 32, provided a flexible and practical synthetic pathway for lycorine-type alkaloids and their analogs. The development of multistep one-pot reactions with greater efficiency and further applications in lepadiformine-type alkaloid syntheses are currently underway.Data availability
All computational data associated with this article have been inserted in ESI.Author contributions
H. Z. and J. C. conceived the idea. Y. Z. conducted the most of experiments. Y. Z., G. M., Q. W., S. Y. and X. Z co-synthesized part of substrates. H. Z. and J. C. co-wrote the paper. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21662041, U1702286), the Yunnan Provincial Science & Technology department (2018FA045), the Program for Changjiang Scholars and Innovative Research Teams in Universities (IRT17R94), and the Project of Innovative Research Team of Yunnan Province (202005AE160005). We thank A/Prof. Ruihan Zhang and Dr Chang Xu for their help with the DFT calculations.Notes and references
-
(a) R. D. Harken, C. P. Christensen and W. C. Wildman, J. Org. Chem., 1976, 41, 2450 CrossRef CAS
; (b) S. F. Martin, in The Alkaloids, ed. A. Brossi, Academic Press, New York, 1987, vol. 30, pp. 25−376 Search PubMed
-
(a)
J. W. Cook and J. D. Loudon, in The Alkaloids, ed. R. H. F. Manske and H. L. Holmes, Academic Press, New York, 1952, vol. 2, pp. 331−352 Search PubMed
-
(a) D. B. Fitzgerald, J. L. Hartwell and J. J. Leiter, Nat. Cancer Inst., 1958, 20, 763 CAS
-
(a) G. R. Pettit, S. Freeman, M. J. Simpson, M. A. Thompson, M. R. Boyd, M. D. Williams, G. R. Pettit III and D. L. Doubek, Anti-Cancer Drug Des., 1995, 10, 243 CAS
- For the synthesis of the tetracyclic skeleton of Amaryllidaceae alkaloids, see:
(a) L. D. Miranda and S. Z. Zard, Org. Lett., 2002, 4, 1135 CrossRef CAS PubMed
- For the syntheses of lycorine, dihydrolycorine, and α-dihydrocaranine, see:
(a) E. W. Warnhoff and W. C. Wildman, J. Am. Chem. Soc., 1957, 79, 2192 CrossRef CAS
- For the syntheses of 7-deoxypancratistatin, see:
(a) H. Paulsen and M. Stubbe, Tetrahedron Lett., 1982, 23, 3171 CrossRef CAS
- For the syntheses of pancratistatin, see:
(a) S. Danishefsky and J. Y. Lee, J. Am. Chem. Soc., 1989, 111, 4829 CrossRef CAS
- For the syntheses of lycoricidine, see:
(a) S. Ohta and S. Kimoto, Tetrahedron Lett., 1975, 16, 2279 CrossRef
- For the syntheses of narciclasine, see:
(a) J. H. Rigby and M. E. Mateo, J. Am. Chem. Soc., 1997, 119, 12655 CrossRef CAS
- For the isolation of zephyranthine, see:
(a) S. Ozeki, Chem. Pharm. Bull., 1964, 12, 253 CAS
- For the syntheses of zephyranthine see:
(a) Y. J. Chen, S. L. Cai, C. C. Wang, J. D. Cheng, S. Kramer and X. W. Sun, Chem.–Asian. J., 2017, 12, 1309 CrossRef CAS PubMed
-
(a) This catalyst was selected from a primary screening from chiral thioureas, cinchona alkaloids and Evans' chiral nickel(II) catalyst;;
(b) D. A. Evans and D. Seidel, J. Am. Chem. Soc., 2005, 127, 9958 CrossRef CAS PubMed
- S. M. Weinreb, Chem. Rev., 2006, 106, 2531 CrossRef CAS PubMed
-
(a) J. H. Sahner, H. Sucipto, S. C. Wenzel, M. Groh, R. W. Hartmann and R. Müller, ChemBioChem, 2015, 16, 946 CrossRef CAS PubMed
-
(a) A. E. Gatland, B. S. Pilgrim, P. A. Procopiou and T. J. Donohoe, Angew. Chem., Int. Ed., 2014, 53, 14555 CrossRef CAS PubMed
- D. L. Comins and A. Dehghani, Tetrahedron Lett., 1992, 33, 6299 CrossRef CAS
- S. Cacchi, E. Morera and G. Ortar, Tetrahedron Lett., 1984, 25, 4821 CrossRef CAS
-
(a) S. G. Hentges and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 4263 CrossRef CAS
- W. J. Huang, O. V. Singh, C. H. Chen, S. Y. Chiou and S. S. Lee, Helv. Chim. Acta, 2002, 85, 1069 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2019555. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03147c |
| This journal is © The Royal Society of Chemistry 2021 |