
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved broad-spectrum antibiotics against Gram-negative pathogens via darobactin biosynthetic pathway engineering†
Sebastian
Gro߇
 abc,
Fabian
Panter‡
abcd,
Domen
Pogorevc‡
abc,
Carsten E.
Seyfert‡
abc,
Selina
Deckarm
abc,
Chantal D.
Bader
abc,
Jennifer
Herrmann
abc and
Rolf
Müller
*abcd
abc,
Fabian
Panter‡
abcd,
Domen
Pogorevc‡
abc,
Carsten E.
Seyfert‡
abc,
Selina
Deckarm
abc,
Chantal D.
Bader
abc,
Jennifer
Herrmann
abc and
Rolf
Müller
*abcd
aHelmholtz Institute for Pharmaceutical Research Saarland (HIPS), Helmholtz Centre for Infection Research, Saarland University Campus, 66123 Saarbrücken, Germany. E-mail: Rolf.Mueller@helmholtz-hips.de
bDepartment of Pharmacy, Saarland University, 66123 Saarbrücken, Germany
cDZIF – German Centre for Infection Research, Partner site Hannover-Braunschweig, Germany
dHelmholtz International Lab for Anti-Infectives, Campus E8 1, 66123 Saarbrücken, Germany
First published on 12th August 2021
Abstract
The development of new antibiotics is imperative to fight increasing mortality rates connected to infections caused by multidrug-resistant (MDR) bacteria. In this context, Gram-negative pathogens listed in the WHO priority list are particularly problematic. Darobactin is a ribosomally produced and post-translationally modified bicyclic heptapeptide antibiotic selectively killing Gram-negative bacteria by targeting the outer membrane protein BamA. The native darobactin A producer Photorhabdus khanii HGB1456 shows very limited production under laboratory cultivation conditions. Herein, we present the design and heterologous expression of a synthetically engineered darobactin biosynthetic gene cluster (BGC) in Escherichia coli to reach an average darobactin A production titre of 13.4 mg L−1. Rational design of darA variants, encoding the darobactin precursor peptide with altered core sequences, resulted in the production of 13 new ‘non-natural’ darobactin derivatives and 4 previously hypothetical natural darobactins. One of the non-natural compounds, darobactin 9, was more potent than darobactin A, and showed significantly improved activity especially against Pseudomonas aeruginosa (0.125 μg mL−1) and Acinetobacter baumannii (1–2 μg mL−1). Importantly, it also displayed superior activity against MDR clinical isolates of E. coli (1–2 μg mL−1) and Klebsiella pneumoniae (1–4 μg mL−1). Independent deletions of genes from the darobactin BGC showed that only darA and darE, encoding a radical forming S-adenosyl-L-methionine-dependent enzyme, are required for darobactin formation. Co-expression of two additional genes associated with the BGCs in hypothetical producer strains identified a proteolytic detoxification mechanism as a potential self-resistance strategy in native producers. Taken together, we describe a versatile heterologous darobactin platform allowing the production of unprecedented active derivatives in good yields, and we provide first experimental evidence for darobactin biosynthesis processes.
Introduction
Increasing levels of antimicrobial resistance (AMR) is a threat for health care systems globally and it leads to increasing morbidity and mortality rates. Notably, multidrug-resistant (MDR) Gram-negative pathogens such as Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and also Enterobacter species cause difficult-to-treat nosocomial infections and were consequently ranked with critical priority by the WHO.1 Some MDR strains developed resistance towards almost all clinically approved antibiotics.2 This exemplifies the need for the discovery and clinical development of new antibiotics to fight AMR-related infections in the future. Most of the recently FDA-approved antibiotics derive from well-known antibiotics classes such as the β-lactam/β-lactamase inhibitor combinations ceftolozane/tazobactam, ceftazidime/avibactam,3 meropenem/vaborbactam,4 imipenem-cilastatin (dipeptidase inhibitor)/relebactam,5 the aminoglycoside plazomicin,6 the tetracycline derivative eravacycline,7 and the siderophore cephalosporin cefiderocol,8 which make rapid resistance development likely because resistance mechanisms against congeners of these antibiotics classes already exist among pathogens.As a result of this challenge, future antibiotics discovery campaigns should focus on antibiotics with novel scaffolds, molecular targets and modes of action,9 such as the cyclic peptidomimetic murepavidin, which targets the outer membrane (OM) biogenesis in Gram-negative bacteria by inhibiting the lipopolysaccharide transport protein D (LptD). Murepavidin is currently in preclinical development as precision antibiotic against P. aeruginosa to treat patients with cystic fibrosis.10 Another example is the benzamide compound TXA709, which is active against Staphylococcus aureus by affecting septum formation during cell division through the inhibition of FtsZ, a protein without homolog in eukaryotic cells.11 Moreover, the streptomyces-derived griselimycins are highly active against Mycobacterium tuberculosis by inhibiting the new antimicrobial target DnaN, the DNA polymerase sliding clamp.12 Other examples are the spiropyrimidintrione zoliflodacin and the linear polyaromatic peptide cystobactamid, which show broad-spectrum antibacterial activity, e.g. against Neisseria gonorrhoeae and MDR Escherichia coli, respectively, by inhibiting DNA gyrase at distinct binding sites compared to other gyrase-targeting antibiotics like the quinolones.13 However, the number of promising antibiotic lead structures with activity against Gram-negative pathogens currently in clinical development is limited. One key reason why it is so difficult to find novel antibiotics active against Gram-negative bacteria is their OM, which constitutes a major additional permeation barrier for most of the candidate compounds under development. Several new antibiotic classes currently under investigation avoid the issue of cell penetration since their molecular target is located in the OM itself. Among these antibiotics classes are the small molecule MRL-494,14 the ribosomally produced and post-translationally modified peptide (RiPP) darobactin,15 and a chimeric peptidomimetic combining the pharmacophores of murepavadin and polymyxin B1.16
The antibiotic darobactin A was isolated from the bacterial nematode symbiont Photorhabdus khanii HGB1456 and it selectively kills Gram-negative bacteria such as P. aeruginosa, K. pneumoniae, E. coli, and A. baumannii.15 In addition, it showed no cytotoxic activity against a variety of human cell lines or symbiotic gut bacteria, including Gram-negative members belonging to the genus Bacteroides. Darobactin targets the β-barrel assembly machinery A protein (BamA)17 located in the OM. It was recently shown that darobactin A binds to the open form BamA lateral gate with high affinity by mimicking a β-strand in its BamA-bound binding pose.18 Consequently, the β-signal binding site of BamA is blocked, cognate substrate binding is inhibited and nascent OM proteins are not inserted into the OM. Darobactin A is a bicyclic heptapeptide and part of the RiPP secondary metabolite class characterized by the precursor peptide amino acid sequence W1N2W3S4K5S6F7 (Fig. 1).15
 | ||
| Fig. 1 (a) The biosynthetic gene cluster (BGC) from Photorhabdus khanii HGB1456 consisting of a homolog of the global translation inhibitor gene relE from E. coli19 (dark grey arrow labelled with asterisk), the darobactin propeptide encoding gene darA (green arrow), the three ABC-type trans-envelope exporter encoding genes darBCD (light grey arrows) and the radical SAM enzyme encoding gene darE (blue arrow). (b) Transcription and translation of darA leads to the synthesis of the darobactin propeptide, which consists of a leader, core (W1N2W3S4K5S6F7) and tail sequence. Two cyclization post-translational modification (PTM) reactions and proteolytic maturation result in formation of darobactin. The proteolytic maturation was recently assumed to be performed by a peptidase from E. coli, which is not encoded by a gene from the darobactin BGC, or by a self-cleavage mechanism catalysed by the propeptide.20 (c) Structural formula of the mature darobactin A. | ||
The darobactin biosynthetic gene cluster (BGC) in P. khanii consists of six open reading frames (ORFs), darABCDE and a homolog of the global translation inhibitor relE from E. coli.15,19 The darA ORF encodes the propeptide sequence and darBCD encode a tripartite ABC-type trans-envelope exporter system, consisting of an ABC transporter permease (DarB), a membrane fusion protein (DarC) and an ATP binding domain (DarD). The darE gene encodes a radical S-adenosyl methionine (SAM) enzyme, which was proposed to catalyse two consecutive aryl-alkyl couplings between the aromatic parts of the tryptophan side chain leading to two ring formations. The first ring is formed by connection of the indole group of W1 with Cβ of W3via an aryl–alkylether bond and the second ring is formed by a carbon–carbon aryl–alkyl bond between the indole group of W3 and Cβ of K5.15 The cyclisations were shown to give darobactin its β-strand mimic structure and are vital for the bioactivity, as the linear W1N2W3S4K5S6F7 peptide did not bind BamA.15,18 A search in the National Centre for Biotechnology Information (NCBI) bacterial genome database using the darobactin propeptide sequence as query led to the identification of additional darobactin BGCs in Photorhabdus, Yersinia, Vibrio, and Pseudoalteromonas species.15 Two additional ORFs, darF and darG, encoding proteins with unknown functions, are located in the BGC from Pseudoalteromonas luteoviolacea.15 Moreover, Imai and co-workers proposed structures of the hypothetical darobactin derivatives B–E based on the darobactin A structure and the propeptide core sequences encoded in the respective darA homologs of those BGCs.15 Due to the low production titre (≤3 mg L−1) of darobactin A in its native producer strain, darABCDE were transferred into E. coli BW25113 and heterologously expressed under the regulation of an arabinose-inducible promoter, resulting in the production of darobactin A.15 However, the corresponding production titre achieved in fermentations of the heterologous producer was not reported and the existence of the hypothesised darobactin derivatives B–E were not experimentally proven.
Herein, we report the in silico design, chemical synthesis and heterologous expression of a synthetically engineered darobactin BGC in E. coli BL21 (DE3) leading to an improved darobactin A production titre (13.4 (±2.0) mg L−1 in 3 days). In parallel work, Wusain et al. published the heterologous production of darobactin A in E. coli reaching a similar titre.20 We here additionally present the rational in silico design of darA variants with altered core peptide encoding sequences and their heterologous expression. The resulting darobactin derivatives include the previously hypothetical natural darobactins B–E. Most importantly, 13 new ‘non-natural’ derivatives were generated, including darobactin 9, which showed significantly improved antibacterial activity against some of the most problematic pathogens according to the WHO priority list, such as P. aeruginosa, A. baumannii, and K. pneumoniae. Furthermore, in our analysis independent targeted gene deletions of the relE-like gene, darA, darB, darC, darD, and darE revealed that only darA and darE are ultimately required for darobactin formation in E. coli. Moreover, the expression of darF, solely or in combination with darG, as well as purification of DarF and in vitro investigation of the enzyme activity identified the proteolytic degradation of darobactin as potential detoxification mechanism in the respective native producer strain.
Results & discussion
Design of a synthetic darobactin BGC modified for the heterologous production of darobactin A in E. coli BL21 (DE3)
Despite the strong anti-Gram-negative activity of darobactin, heterologous production in E. coli was previously shown to be feasible.15 Aiming for a versatile heterologous darobactin production platform in E. coli, we designed a modified darobactin BGC (GenBank accessions are provided in ESI Data†) in silico based on the BGC sequence originating from P. khanii HGB1456 (GenBank accession WHZZ01000001.1) (see ESI†). The modified BGC includes the five ORFs darABCDE and the relE-like gene. We decided to include the relE-like gene in our initial design as it was co-localized with this darobactin BGC as well as with several other darobactin BGCs in different wild-type strains.15 Thus, we could not exclude that this gene may play a beneficial role in darobactin biosynthesis even though its homolog relE was shown to be a global translation inhibitor in E. coli.19 We placed the isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible T7lac promoter system21 (including the lacI repressor gene and lacO operator) upstream of darA intending to allow time-controlled gene expression and thus the production of darobactin. The constitutive nptII promoter22 was placed upstream of darBCDE, since we assumed that constitutive gene expression of the darBCD-encoded ABC-type trans-envelope exporter system increases the capability of our E. coli production strain to export darobactin prior to induction of the darA gene expression with IPTG and therefore can improve its self-resistance to darobactin. We inserted the tD1 terminator sequence upstream of the nptII promoter to prevent T7lac promoter-dependent gene expression affecting the transcription of the darBCDE operon. Furthermore, we aimed to exclude that a potential native promoter located in the 396 bp long intergenic region between darA and darBCDE influences the gene expression of darBCDE. In addition to the natively present unique restriction endonuclease recognition sites (R-sites), we inserted several more R-sites at strategic locations allowing to remove the relE-like gene, exchange promoters or darA and to facilitate efficient sub cloning of the modified darobactin BGC into different vector systems (see Fig. 2 and ESI Table 2†). | ||
| Fig. 2 (a) Schematic overview of the workflow including the in silico design of the modified darobactin BGC based on the genome sequence from P. khanii HGB1456, chemical DNA synthesis and cloning of the expression construct, transformation of the expression construct into E. coli BL21 (DE3) and heterologous production of darobactin derivatives on the example of darobactin A. (b) Design of the darobactin BGC modified for the heterologous expression in E. coli. The pNOSO-relEdarABCDE expression construct contains the relE homologue (dark grey arrow), lacI repressor gene (black arrow), T7lac promoter (black curved arrow) upstream of darA (green arrow), nptII promoter (black curved arrow) upstream of darBCD (light grey arrows) and darE (blue arrow), tD1 terminator (yellow hexagon), p15A origin of replication (ori; white box) and kanamycin resistance gene (white arrow). Selected unique restriction endonuclease recognition sites (R-sites), which were important for removal of the relE homologue (AgeI), exchange of darA by darA variants with altered propeptide core sequence (NdeI/BamHI) and introduction of darFG (SpeI/PacI), are indicated by dashed lines. (c) Different darA variants were designed for the heterologous production of natural darobactin derivatives (B–E) and non-natural darobactin derivatives (1–21). (d) The darFG fragment was designed to investigate the biosynthetic function of the respective genes upon co-expression with the modified darobactin BGC. | ||
The modified darobactin BGC DNA sequence was chemically synthesized in one part (8.5 kb), delivered in a pUC57 high-copy plasmid and cloned via restriction hydrolysis/DNA ligation into pNOSO (described in ESI†), which features a low-copy p15A origin of replication to allow for genetic stability of the resulting recombinant strain. The expression constructs were transformed into E. coli BL21 (DE3) and the heterologous production of darobactin A was initially confirmed by uHPLC-HRMS and MS2 fragmentation after a small-scale batch cultivation (see ESI†). After initial evaluation of different E. coli expression strains, cultivation media and expression constructs, the highest relative production based on the mass peak intensity was achieved in E. coli BL21 (DE3) pNOSO-relEdarABCDE using FM medium for cultivation (see ESI† and Fig. 3).
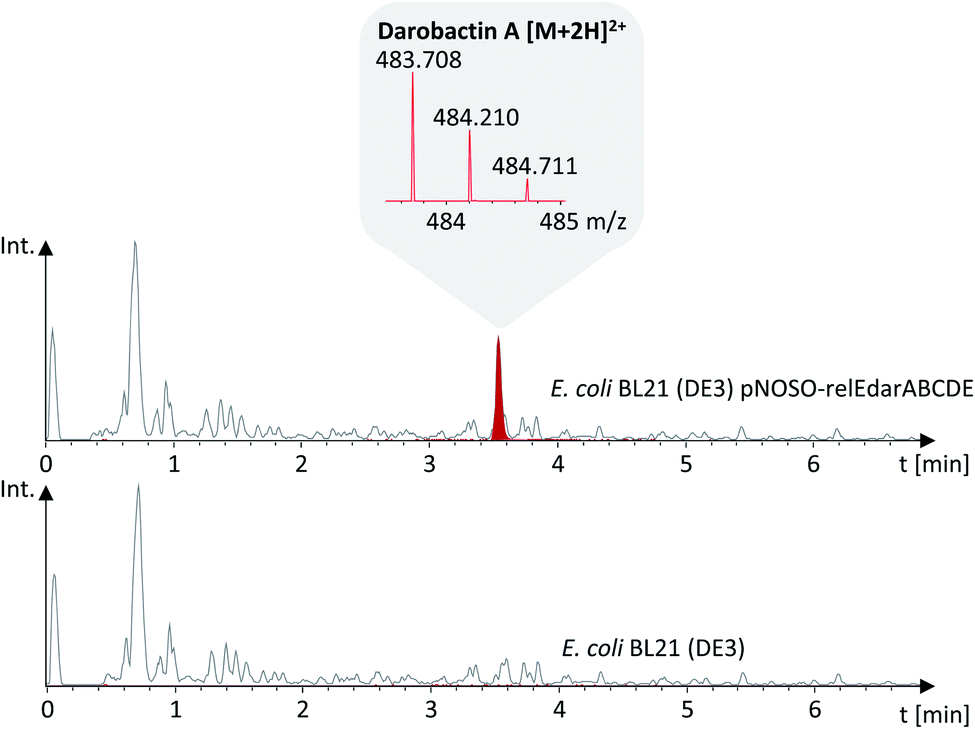 | ||
| Fig. 3 Darobactin A production was confirmed by uHPLC-HRMS analysis of the E. coli pNOSO-relEdarABCDE fermentation broth supernatant. Darobactin A is visualized as extracted ion chromatogram (EIC) at 483.71 Da (red trace) represents the [M + 2H]2+ MS peak, including the corresponding isotope pattern, which is shown in the grey field. The grey trace represents the base peak chromatogram (BPC) of the culture supernatant of E. coli BL21 (DE3) with and without the darobactin expression vector. | ||
Furthermore, we observed peptide impurities in the Base Peak Chromatograms (BPCs) upon expression of high-copy number plasmids (see ESI, Fig. 2†), which would hamper darobactin purification. Thus, we favoured the pNOSO-based expression construct. Since removal of the relE homologue from the plasmid encoded BGC did not affect the darobactin production, we used pNOSO-darABCDE, lacking the relE-like gene, as basis for all following experiments.
Notably, darobactin A production levels were consistently higher if the lac promoter-regulated gene expression of the T7 RNA polymerase, which is encoded in the genome of BL21 (DE3) and related E. coli strains, and the T7lac promoter on the plasmid remained non-induced as compared to induction with 0.1 mM IPTG. We assume that leaky darA gene expression by the lac-repressed T7 promoter leads to a favourable amount of propeptide in the cell for optimal darobactin A production. This leaky expression might be caused by traces of lactose in the complex cultivation medium leading to slight induction of the lac-repressed genes. Higher darA gene expression levels caused by induction of the lac-regulated T7 RNA polymerase expression and induction of the T7lac promoter with IPTG might result in metabolic overburdening of the E. coli cell. This result is not entirely consistent with observations made in parallel in the work by Wuisan et al., in which an increasing darA to darE transcript ratio led to higher darobactin A production.20 We assume that a certain darA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :darE transcript ratio, which has to be determined in future experiments, is required for optimum darobactin A production. To investigate whether slight induction or repression of the T7lac promoter results in higher relative darobactin production, we supplemented the production broth with 10 μM, 1 μM, 100 nM, 10 nM, 1 nM and 0.1 nM concentrations of IPTG or 0.5%, 1.5% and 5% of glucose. However, no production improvement with respect to non-induced T7 polymerase and darA expression was achieved under any of the tested conditions (see ESI, Table 7†).
:darE transcript ratio, which has to be determined in future experiments, is required for optimum darobactin A production. To investigate whether slight induction or repression of the T7lac promoter results in higher relative darobactin production, we supplemented the production broth with 10 μM, 1 μM, 100 nM, 10 nM, 1 nM and 0.1 nM concentrations of IPTG or 0.5%, 1.5% and 5% of glucose. However, no production improvement with respect to non-induced T7 polymerase and darA expression was achieved under any of the tested conditions (see ESI, Table 7†).
Subsequently, we performed a large-scale batch cultivation with E. coli BL21 (DE3) pNOSO-darABCDE omitting any IPTG induction to isolate darobactin A and confirm the structure and minimum inhibitory concentrations (MICs) published by Imai et al. for internal comparison with novel darobactins (see below).
Purification of darobactin A from the culture supernatant was done by adsorption of darobactin on XAD-16N resin followed by elution with 80% MeOH in H2O and several steps of preparative and semi-preparative HPLC (see ESI†). The purified compound was used to calculate a linear regression based on the MS peak surface areas of different stock concentrations (see ESI†). On this basis, an absolute quantification of the darobactin A concentration in the fermentation broth of E. coli BL21 (DE3) pNOSO-darABCDE was performed, providing an average production rate of 13.4 (±2.0) mg L−1.
Production of natural and non-natural darobactin derivatives
Darobactin is a promising candidate for biosynthetic structure engineering attempts due to its excellent antibacterial activity profile, the hypothesised natural variability in its amino acid composition,15 and its presumed insensitivity towards amino acid mutations in its binding site.18 Thus, we investigated the plasticity of our heterologous production system aiming at the production of novel darobactin derivatives with improved antibacterial properties. To this end, we designed four DNA fragments in silico including the entire darA gene with an altered core sequence encoding the propeptides of the hypothetical natural darobactins B–E (Table 1 and Fig. 4).| Darobactin | darA core DNA sequence | Core peptide | Calc. mass [M + 2H]2+ | Obs. mass [M + 2H]2+ | Mass deviation [M + 2H]2+ (ppm) | Mass shift (Da) |
|---|---|---|---|---|---|---|
| A | TGG AAC TGG TCA AAA AGC TTC | W N W S K S F | 483.7089 | 483.7095 | 1.24 | |
| B | TGG AAC TGG ACC AAA CGA TTC | W N W T K R F | 525.2512 | 525.2511 | 0.19 | |
| C | TGG TCA TGG TCA AGA AGC TTC | W S W S R S F | 484.2065 | 484.2066 | 0.21 | |
| D | TGG AAC TGG TCA AGA AGC TTC | W N W S R S F | 497.7119 | 497.7116 | 0.60 | |
| E | TGG TCA TGG TCA AAA AGC TTC | W S W S K S F | 470.2034 | 470.2037 | 0.64 | |
| 1 | TGG TCA TGG ACC AAA CGA TTC | W S W T K R F | 511.7458 | Not observed | ||
| 2 | TGG TCA TGG ACC AGA AGC TTC | W S W T R S F | 491.2143 | 491.2146 | 0.61 | |
| 4 | TGG AAC TGG ACC AAA AGC TTC | W N W T K S F | 490.7167 | 490.7171 | 0.82 | |
| 5 | TGG TGC TGG TCA AAA AGC TTC | W C W S K S F | 478.1920 | Not observed | ||
| 6 | TGG AAC TGG TGC AAA AGC TTC | W N W C K S F | 491.6974 | 587.2104 | +191.026 | |
| 7 | TGG TGC TGG TGC AAA AGC TTC | W C W C K S F | 486.1806 | Not observed | ||
| 8 | TGG AAC TGG TCA AAA TGC TTC | W N W S K C F | 491.6974 | 587.2100 | +191.0252 | |
| 9 | TGG AAC TGG TCA AAA AGC TGG | W N W S K S W | 503.2123 | 503.2139 | 3.18 | |
| 10 | TGG AAC TGG TCA AAA AGC TAC | W N W S K S Y | 491.7063 | 491.7064 | 0.20 | |
| 11 | TGG CAG TGG TCA AAA AGC TTC | W Q W S K S F | 490.7167 | 490.7169 | 0.41 | |
| 12 | TGG ACC TGG TCA AAA AGC TTC | W T W S K S F | 477.2112 | 477.2115 | 0.63 | |
| 13 | TGG GCA TGG TCA AAA AGC TTC | W A W S K S F | 462.2059 | 462.206 | 0.22 | |
| 14 | TGG AAC TGG GCA AAA AGC TTC | W N W A K S F | 475.7114 | 475.7118 | 0.84 | |
| 15 | TGG AAC TGG TCA GCA AGC TTC | W N W S A S F | 455.1799 | 455.1802 | 0.66 | |
| 16 | TGG AAC TGG TCA AAA GCT TTC | W N W S K A F | 475.7114 | 475.7119 | 1.05 | |
| 17 | TGG AAC TGG TCA AAA AGC GCA | W N W S K S A | 445.6932 | 445.6936 | 0.90 | |
| 18 | TGG AAC TGG TCA AAA AGC CAG | W N W S K S Q | 410.1746/474.2039 | 474.2043 | 0.84 | +128.0594 |
| 19 | TGG AAC TGG TCA AAA | W N W S K | 366.6586 | Not observed | ||
| 20 | TGG AAC TGG TCA | W N W S | 303.6190 | Not observed | ||
| 21 | TGG AAC TGG | W N W | 260.1023 | Not observed |
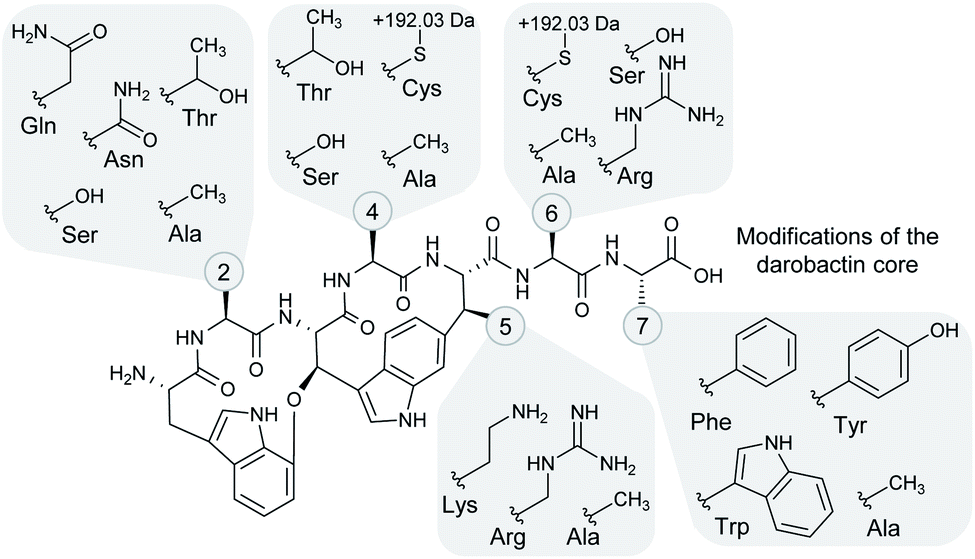 | ||
| Fig. 4 Structural variation among the heterologously produced darobactin derivatives. The side chain moieties of the different amino acids in positions 2, 4–6 and 7 (see numbered circles) that were observed in the heterologously produced darobactin derivatives (see also Table 1) are shown in the grey-shaded areas. Cysteine side chains in positions 4 and 6 led to formation of an adduct with a mass shift of +191.026 Da and a potential adduct sum formula of C6H9NO4S based on ChemCalc prediction (discussed in more detail in the text). Amino acid side chain moieties of derivatives, which were not observed upon expression of the respective pNOSO construct with altered darA sequence in E. coli BL21 (DE3), are not shown (darobactins 1, 5 and 7). | ||
The existence of these darobactin congeners was previously assumed based on the in silico analysis of darobactin propeptide sequences in the respective BGCs but has not yet been confirmed.15
After chemical synthesis of the DNA fragments containing the chimeric darA genes (named darA-B to darA-E), the native darA sequence was replaced with darA-B to -E, independently, in pNOSO-darABCDE using restriction hydrolysis/DNA ligation cloning to generate pNOSO-darABCDE-B to -E. Heterologous expression of the respective constructs in E. coli BL21 (DE3) resulted in the production of the previously in silico predicted native darobactins B–E, as confirmed by uHPLC-HRMS and MS2 fragmentation analysis (see Table 1, ESI Fig. 4–7 and 23–26†).
Based on the amino acid tolerability found at positions 2 and 4–6 in the natural darobactins A–E, we designed and cloned three new expression constructs (pNOSO-darABCDE-1, -2, and -4) with altered core sequences, in which naturally occurring amino acids were combined in a non-natural manner.
For example, in darobactins B–E position 2 is L-serine or L-asparagine, position 4 is L-serine or L-threonine, position 5 is L-lysine or L-arginine and position 6 is L-serine or L-arginine. However, natural darobactins A–E do not cover all possible combinations. The darA core sequence in pNOSO-darABCDE-1, on the other hand, encodes the core peptide W1S2W3T4K5R6F7 (named darobactin 1 here), representing one of the possible non-natural combinations of natural occurring amino acids. After heterologous expression of pNOSO-darABCDE-1, -2, and -4 in E. coli BL21 (DE3), production of the respective non-natural darobactins was confirmed by uHPLC-HRMS (see Table 1, ESI Fig. 8–10 and 27–29†).
Next, we aimed to investigate whether our heterologous production system also tolerates the exchange of natively occurring amino acids in positions 2 and 4–6 of the darobactin core peptide by amino acids natively not found in the respective positions of the darobactins A–E. Therefore, numerous darA variants (5–8 and 11–17) were designed in silico, in which the core sequences encode non-natural darobactin A derivatives containing L-glutamine (darobactin 11) or L-threonine (darobactin 12) in position 2, L-cysteine in positions 2, 4, 2 and 4, or 6 (darobactins 5, 6, 7, and 8, respectively), and L-alanine in positions 2 and 4–7 (darobactins 13–17, respectively). After chemical synthesis and subsequent cloning of the darA variants into pNOSO-darABCDE, generating pNOSO-darABCDE-5, -6 to -8, and -11 to -17, heterologous expression of the respective constructs in E. coli BL21 resulted in the production of darobactins 11–17 (see Table 1, ESI Fig. 14–20 and 33–39†), whereas darobactin-related but unexpected masses were detected after analysis via GNPS-based spectral networking23 of the corresponding tandem MS upon expression of pNOSO-darABCDE-6 and -8 (m/z 587.209 and 587.212 instead of 491.697 [M + 2H]2+) (see Table 1, ESI Fig. 10, 11, 29 and 30†). Darobactin derivative production was confirmed by observation of the corresponding exact mass followed by analysis of the corresponding MS2 spectrum that shows characteristic fragments for darobactin scaffolds (see ESI†). In case of expression of pNOSO-darABCDE-5, -7 and -13, no expected darobactins nor darobactin-related compounds were detected. This clearly shows that the chemical space to exchange amino acids at certain positions in darobactin is limited. For example, in position 2 the exchange of the naturally occurring amino acids L-asparagine and L-serine by the sterically and physicochemically similar amino acids L-glutamine and L-threonine was tolerated, whereas L-alanine was not accepted in this position. The attempt to introduce one L-cysteine at different positions either resulted in production of darobactin-related compounds with unexpected masses or, similar to our attempt involving two L-cysteine residues at the same time, to complete abolishment of the production. Since the unexpected masses were higher than the expected masses, we assume that the reactive cysteine side chain thiol group underwent chemical reactions leading to adduct formation. Based on the measured exact mass we observe a mass shift of +191.02 Da with respect to the expected darobactin 6 mass. ChemCalc24 analysis predicted a potential sum formula of C6H9NO4S based on the exact mass of the adduct, taking into account the seven golden rules for heuristic sum formula prediction.25 We assume that the presence of a sulphur makes adduct formation via a disulphide bond likely, but purification of darobactins 6 and 8 with subsequent structure elucidation is required to make a precise statement about the respective structures. However, we de-prioritized these compounds as they were produced in significantly lower amounts based on comparison of the MS peak area under the curve with respect to darobactin A (see Table 2).
:15–1:240 dilutions applied in the MIC assay) where growth inhibition was last observed. The lower the crude extract concentration factor at which growth inhibition of a certain pathogen was last observed, the stronger the antimicrobial activity. Weak activity is highlighted in light green colors (e.g. growth inhibition at 6.67× and 3.34× concentration of the crude extract) and strong activity is highlighted in green to dark-green colors (e.g. 1.67×–0.42× concentration), whereas “-” means no activity even at 6.67× concentration. Crude extract concentrations of 0.83× and 0.42× are synonymous with a total crude extract dilution of 1:1.2 and 1:2.4, respectively. The assignment of a concentration factor as measure of the antimicrobial activity is described in more detail in ESI (see ESI Fig. 41). The area under curve (AUC) ratio serves as measure of the production titre of the respective derivative in the crude extract based on the MS peak surface compared to the AUC of darobactin A. “Traces” means an AUC ratio below 0.01. It should be noted, however, that without purification of all congeners an exact quantification is not possible

|
Furthermore, L-alanine was successfully incorporated into the darobactin scaffold in case of darobactins 14–17 (see Table 1, ESI Fig. 17–20 and 36–39†). This was surprising, since the C-terminal amino acid (position 7) occurring in natural darobactins A–E was always L-phenylalanine. Still, replacement with L-alanine in case of darobactin 17 was found possible. The L-phenylalanine moiety is conserved in natural darobactins A–E at this position15 and was shown to bind BamA exactly where a conserved aromatic peptide side chain in the β-signal sequences of the cognate BamA substrates bind.18 Consequently, we hypothesise that an aromatic side chain moiety in position 7 is crucial for the bioactivity of darobactins and that further amino acid exchanges in this position against other aromatic amino acids might result in active darobactin variants. We therefore designed two new darA variants (9 and 10), encoding L-tryptophan and L-tyrosine in position 7, respectively. After cloning of darA-9 and darA-10 into pNOSO-darABCDE and heterologous expression of the respective constructs in E. coli BL21 (DE3), the production of darobactins 9 and 10 exhibiting a terminal L-tryptophan and L-tyrosine moiety, respectively, was confirmed by uHPLC-HRMS (see Table 1, ESI Fig. 12, 13, 31 and 32†).
As the heterologous production system tolerated the majority of amino acid exchanges in the darobactin precursor peptide, we subsequently aimed for a truncation of the core structure. This experiment might provide first insights into a possible structure–activity-relationship, which will be investigated in more detail in the future, the mechanism of tail sequence cleavage and pave the way for semi-synthesis on darobactin scaffolds. We designed four additional darA variants (18 to 21) in which the propeptide sequence length was reduced by one (darA-18) up to four (darA-21) amino acids, respectively, without removing the darobactin tail sequence. However, after cloning and heterologous expression of the generated constructs in E. coli BL21(DE3), we only observed production of one unexpected darobactin derivative (darobactin 18) upon expression of pNOSO-darABCDE-18, which was shown to carry a L-glutamine at position 7 (see Table 1, ESI Fig. 21 and 40†). We had expected the production of a truncated darobactin derivative, thus having a terminal L-serine in position 6. Interestingly, the L-glutamine found in position 7 is the first amino acid of the tail sequence. Truncation of the core sequence therefore seems to lead to a maturation process in which the first amino acid of the tail sequence is recognized as part of the core and is thus not cleaved off in the proteolytic process. The attempt to truncate the darobactin core sequence by two or more amino acids resulted in a complete loss of production (see Table 1). Consequently, we assume that a core sequence of seven amino acids length is recognized by the so far unidentified protease (see below). Notably, in our experiment we did not remove the tail sequence from darA, but only removed one or several of the darobactin core sequence codons. In the contemporaneously performed work of Wuisan and co-workers a darobactin BGC with a darA variant without the tail sequence was heterologously expressed in E. coli, showing that darobactin A was still produced.20 Future experiments, in which the entire tail encoding sequence plus additional core amino acid encoding sequences are removed from darA might thus result in the formation of truncated darobactin congeners.
In summary, 18 natural and non-natural darobactin derivatives were successfully heterologously produced in E. coli BL21 (DE3) after expression of the darobactin BGC with different darA core sequences. Five out of the 18 produced derivatives were native darobactins (A–E), four of which (darobactins B–E) were not previously observed, and 11/19 were non-natural derivatives (1, 2, 4, 9–12, and 14–17), whereas the remaining three showed unexpected masses (6, 8, and 18).
Purification and antibacterial activity profiling of darobactin 9
Since the purification of highly water-soluble darobactin A was a time- and resource-intensive process, we assumed that purification of the newly produced derivatives to be similarly challenging. Thus, we devised a prioritization and preselection strategy enabling us to categorize the newly produced derivatives over others according their anti-Gram-negative activity. Therefore, we performed MIC assays using a small panel of Gram-negative pathogens (K. pneumoniae DSM-30104, P. aeruginosa PAO1, E. coli ATCC25922 and A. baumannii DSM-30008) using crude extracts of all heterologous producer strains (Table 2). Each crude extract was tested in serial dilutions and the given values are the concentration factor for which no visible growth was observed. The assignment of the concentration factor and the interpretation of the estimated antimicrobial activity of the crude extract is described in more detail in the legend of Table 2 and in ESI (see ESI Fig. 41†).However, we could not assign absolute quantities and exact concentrations of novel darobactins without isolating all of them. Thus, the concentration of the darobactin derivatives differed in each crude extract and this experiment does not allow direct comparison of the bioactivity observed for a certain darobactin derivative with the antibiotic activity of darobactin A. Nevertheless, we were able to de-prioritise darobactin derivatives for which no bioactivity was observed (darobactins C, E, 5, 7, 13, 15 and 17), even though this might be caused by low concentration of the respective compound in the crude extract. In order to further narrow down the pool of the remaining darobactin derivatives of interest and select a candidate for isolation and biological assessment based on the pure compound, we calculated the area under curve (AUC) of the respective darobactin derivative MS peaks (Table 2). As the darobactin derivatives differ in their ionisability in the ESI process, this analysis can only serve as a rough estimate of the amount of compound in each crude extract. For instance, darobactin B, in which the L-serine at amino acid position 6 was replaced by the well-ionisable L-arginine, showed a higher MS signal provided from the [M + 3H]3+ ions than the MS signal provided from [M + 2H]2+ ions, whereas [M + 3H]3+ ions were only barely observed for darobactin A. Since we cannot predict the ionisation behaviour of the derivatives we cannot provide an absolute quantification of each congener. We therefore refrained from calculating mg L−1 values for each analog based on the absolute quantification of the darobactin A production titre. The largest AUC values were calculated for darobactins 4, 9, 10, 14 and 16. Due to the challenges associated with isolation and purification of compounds for follow up experiments by NMR and bioactivity assays we therefore prioritized compounds likely to exhibit good activity at reasonable productivity.
Since L-phenylalanine is conserved in position 7 in native darobactins and an essential structural feature to mimic the β-signal of native BamA substrates,18 we were surprised that the extract containing darobactin 9, in which the L-phenylalanine was replaced by L-tryptophan, showed similar antibacterial activity to the darobactin A-containing sample (Table 2). On the contrary, the extract containing darobactin 10 with L-tyrosine in position 7 showed only weak activity despite its structural similarity to darobactin A and similar production level based on the AUC of the respective MS peaks. This indicates that the additional OH-group in L-tyrosine compared to L-phenylalanine negatively affects the interaction of darobactin with BamA, whereas expansion of the side chain π-system by introducing an indole moiety, as it is the case for L-tryptophan, potentially enables stronger aromatic interaction with the BamA target site. Since darobactin 9 was produced in similar quantities to darobactin A based on comparison of the respective AUCs, we classified darobactin 9 as promising starting point for further investigations and purified the compound from a large-scale production culture (see ESI†). The purified compound was used to determine the production titre of darobactin 9 in the fermentation broth of E. coli BL21 (DE3) pNOSO-darABCDE-9 (see ESI†), providing an average production rate of 2.9 (±0.6) mg L−1. The predicted structure (Fig. 5) was verified by acquisition of tandem MS data followed by acquisition of multidimensional NMR experiments. Next, we assessed the MICs of pure darobactin 9, showing that this derivative exhibits superior antibacterial activity against the strains of a larger pathogen panel being on average two-fold more potent than darobactin A (Table 3).
 | ||
| Fig. 5 Structural formula of darobactin 9 carrying a C-terminal L-tryptophan. | ||
| Strain | Dar. 9 | Dar. A | |
|---|---|---|---|
| a MDR clinical isolates (see ESI). | |||
| E. coli | BL21 (DE3) | 0.25 | 0.5–1 |
| BL21 (DE3) + BGC | 2–4 | 8–16 | |
| ATCC-25922 | 1–2 | 2 | |
| JW0451-2 (ΔacrB) | 0.125 | 0.5 | |
| K12 ΔtolC | 0.125 | 0.5 | |
| ECO24a | 1–2 | 8 | |
| ECO25a | 1–2 | 8 | |
| A. baumannii | DSM-30007 | 2 | 16 |
| DSM-30008 | 1–2 | 4 | |
| C. freundii | DSM-30039 | 0.5 | 1 |
| E. cloacae | DSM-30054 | 1 | 2 |
| K. pneumoniae | DSM-30104 | 1–2 | 2–4 |
| KPN12a | 4 | 8 | |
| KPN19a | 1 | 2 | |
| KPN62a | 2 | 4 | |
| P. aeruginosa | PAO1 | 0.125 | 0.5–1 |
| PA14 | 2 | 16 | |
| PA14 ΔmexAB | 1 | 4 | |
| P. mirabilis | DSM-4479 | 32–64 | 64 |
| P. vulgaris | DSM-2140 | 8 | 16 |
| S. marcescens | DSM-30121 | 64 | >64 |
| E. faecalis | ATCC-29212 | >64 | >64 |
| S. aureus | ATCC-29213 | >64 | >64 |
Encouragingly, most Gram-negative bacterial strains were inhibited by sub to low μg mL−1 concentrations of darobactin 9, and the new derivative was particularly active against E. coli (0.125–2 μg mL−1), Citrobacter freundii (0.5 μg mL−1), Enterobacter cloacae (1 μg mL−1), and K. pneumoniae (1–2 μg mL−1). Among the species displaying high MICs for darobactin A, we observed significant improvements for darobactin 9, where MICs on A. baumannii of 1–2 μg mL−1 and MICs on P. aeruginosa of 0.125–2 μg mL−1 were obtained, which corresponds to an approximately 8-fold improvement over darobactin A. Darobactin 9 also gained some activity against Proteus spp. (8–64 μg mL−1) and Serratia marcescens (64 μg mL−1) both being gaps in the antibacterial spectrum of darobactin A. Intriguingly, darobactin 9 was equally active against recent clinical MDR isolates (see ESI†) and MICs were as low as 1–2 μg mL−1 and 1–4 μg mL−1 on E. coli and K. pneumoniae, respectively, further highlighting the potential of darobactins as new broad-spectrum and resistance-breaking antibiotics. Thus, darobactin 9 is a very promising starting point for further structure engineering attempts in the future in which we attempt to further engineer darobactin 9. Especially with respect to difficult-to-treat A. baumannii and P. aeruginosa infections, the improvement in MICs is highly encouraging for such efforts.
Targeted gene deletions and co-expression of darF and darG
Even though hypothetical biosynthetic functions were previously assigned to darBCD, encoding a tripartite ABC-type trans-envelope exporter, and darE, encoding a radical SAM enzyme proposed to catalyse both ring formations in darobactins, experimental confirmation has yet to be performed on a molecular basis. Thus, we conducted targeted, seamless gene deletions of darB, darC, darD, and darE, independently, with subsequent heterologous expression of the deletion constructs in E. coli BL21 (DE3). UHPLC-HRMS analysis of the fermentation broth supernatant (described in ESI†) showed that deletions of darB, darC and darD led to a two- to four-fold decrease in darobactin A production with titres of ca. 6 mg L−1, 5 mg L−1 and 3 mg L−1, respectively, compared to the control.Given that the gene products of darBCD encode an efflux transporter machinery, which is presumably non-functional after deletion of one of its components, the removal of darobactin A from the cell is likely impaired, leading to lower extracellular concentrations as detected by mass spectrometry. Similar observations were made by Wuisan and co-workers, who measured 50% reduced extracellular darobactin A concentration upon heterologous expression of only darA and darE in E. coli, compared to the control strain expressing darABCDE.20 Interestingly, the authors could rule out that darobactin A accumulated in the cell, since intracellular concentrations were also reduced when darBCD was not expressed.20 Since darobactin production was still observed despite deletion of the efflux transporter co-located with the BGC, E. coli BL21 (DE3) might express a darBCD homolog located elsewhere in the genome that partly compensates for the loss of function. Consequently, we searched for darBCD homologs in the genome of E. coli BL21 (DE3) (GenBank accession CP001509) and found only darD homologs (ABC-transporter ATP-binding protein; approx. 30 hits ≥70% pairwise identity), whereas no close darB and darC homologs were found. Nevertheless, we cannot exclude that another efflux system, such as AcrAB-TolC,26 YdhE,27 and MdtABC-TolC28 MDR efflux transporters encoded in the E. coli BL21 (DE3) genome, is involved in the export of darobactin from the cell. Since E. coli strains with deletion of the acrB or tolC were significantly more sensitive towards darobactin A and darobactin 9 as compared to strains harbouring those genes (Table 3), we assume that involvement of both of these broad-spectrum efflux systems in darobactin export is likely. In parallel work, a cooperation of DarBCD with TolC, both being involved in the export of darobactin, was discussed.20 However, deletion of tolC from the genome of the heterologous darobactin producer E. coli BAP-1 did not affect the ratio between intra- and extracellular darobactin concentrations.20
Deletion of darE in our heterologous expression construct resulted in complete abolishment of darobactin A production, confirming its essential function in darobactin biosynthesis. To identify potential darobactin-related compounds in the fermentation broth, we performed a MS2 spectral similarity analysis, but no darobactin-related compounds, such as truncated derivatives, monocyclised, or linear darobactin, were detected. Even though we are not able to confirm the involvement of DarE in the cyclisation reactions on an enzyme level in vitro, it is safe to assume that the previously hypothesised function is correct, because bicyclised darobactin was still produced after darBCD deletion and taking into account that darA encodes the darobactin propeptide, which was proven by Wuisan and coworkers.20 Furthermore, alkyl–arylic couplings like the ones observed for darobactin are usually formed in radical reactions catalysed by radical-SAM-type enzymes or Cytochrome P450 enzymes, which supports the previous hypothesis.29 As no further genes are located within the BGC, we assume the involvement of other genes in the cyclization reactions to be unlikely since those genes would need to be located elsewhere in the genomes of both the native and heterologous producer strains.
Some of the darobactin BGCs found by genome mining in potential producer strains like Pseudoalteromonas luteoviolacea S4054 include two additional ORFs of unknown function, darF and darG. A BLAST (Basic Local Alignment Search Tool)30 search in the non-redundant protein database (nr) using the protein sequence of DarF and DarG as query provided only hypothetical proteins of unknown function and ABC transporters as respective hits (see ESI Data†). HHpred31 analysis of DarF however, revealed homology to zinc-dependent metalloproteases (see ESI Data†), making it a potential candidate protease involved in the darobactin maturation process which was not yet described. Since darF is not part of the darobactin BGC in P. khanii HGB1456, many other native producer strains and the heterologous producer, a homolog would have to be located elsewhere in these genomes. In parallel work, Wuisan and coworkers speculated that the proteolytic maturation of the darobactin propeptide is indeed performed by a protease elsewhere in the genome of E. coli even though they could not rule out an autoproteolytic function of DarA itself.20 Thus, we performed a BLAST search in the genomes of P. khanii HGB1456 and E. coli BL21 (DE3) using the DarF protein sequence as query, however without identification of potential homologs. To investigate the function of darF and darG, we co-expressed both genes under the regulation of the nptII promoter in combination with darABCDE in E. coli BL21 (DE3) and observed complete abolishment of darobactin A production. In order to investigate whether this effect is caused by combined co-expression of darFG or whether it can be related to darF or darG alone, we co-expressed both genes independently with darABCDE. While co-expression of darG only slightly decreased darobactin yields, co-expression of darF led to complete production abolishment. Since a spectral networking analysis did not identify any darobactin-related compounds upon expression of darF, we hypothesised that DarF acts as a protease degrading either the mature darobactin A itself or the corresponding propeptide, thus impeding formation of mature darobactin. To investigate this hypothesis, we heterlogously expressed and purified DarF from E. coli BL21 (DE3) and incubated the enzyme with darobactin A or darobactin 9 in vitro (see ESI†). We indeed observed degradation of darobactins A and 9 after 12 h of incubation with DarF, whereas no degradation was seen in the control experiments without DarF (see ESI Fig. 49 and 50†). These observations supported that DarF is a darobactin-degrading enzyme. A possible function of DarF in P. luteoviolacea S4054 might be a feedback-regulated self-detoxification or self-resistance mechanism, in which high darobactin A concentrations lead to darF gene expression and consequently the degradation of darobactin A, thereby setting a limit to darobactin concentrations not harmful for the native producer. In our heterologous expression system, however, the potential feedback-regulated gene expression might be impaired, because darF is constitutively overexpressed under the regulation of the nptII promoter, which seems to lead to complete degradation of darobactin.
In summary, darA and darE are the only genes essential for darobactin production in E. coli BL21 (DE3), which was also shown in parallel work by Wuisan et al.20 Co-expression of darBCD increases the production level of darobactin A by a factor of 2–4, whereas darG co-expression reduces the production level, as judged by comparison of the darobactin A mass peak. Since co-expression of darF led to complete abolishment of darobactin production and no homologs were identified in the genomes of P. khanii HGB1456 and E. coli BL21 (DE3), we rule out that DarF is the protease involved in the proteolytic maturation process of the compound, in which the leader peptide or tail sequence are removed. Both the mechanism of action of DarF and the proteolytic maturation mechanism of darobactin, performed by a so far unidentified protease enzyme in the native producer strain and a similar enzyme in E. coli BL21 (DE3) have to be investigated in future experiments.
Conclusions
In this work, we developed a versatile heterologous production platform for darobactin derivatives based on a darobactin BGC from P. khanii HGB1456 modified for the expression in E. coli BL21 (DE3). Excluding the previously published darobactin A,15 17 new darobactin derivatives were produced after expression of darA variants encoding different core peptide sequences. Every position in the molecule was shown to be reasonably flexible for structure engineering by incorporation of different amino acids, except of the two L-tryptophans in positions 1 and 3, which are required for formation of the bicyclic darobactin core structure, proving the versatility of the presented system. Likewise, high substrate tolerance has been observed for biosynthetic enzymes in other RiPP pathways, e.g. in lanthipeptide, thiopeptide, lasso peptide and sactipeptide biosynthesis,32 underlining the potential of RiPP structure engineering to generate new antibiotics.However, due to difficulties in the isolation and compound purification process that will have to be addressed in the future, we were so far only able to purify our most promising congener darobactin 9, which showed favourable activity against MDR clinical pathogens and significantly improved antibacterial activity towards P. aeruginosa and K. pneumoniae strains with respect to darobactin A. For the other darobactin derivatives, we relied on appearance of the corresponding mass peak after heterologous expression of the respective plasmid-based BGC, followed by comparison of the calculated to the measured exact mass and corresponding MS2 fragmentation pattern to assign the predicted structures for the remaining derivatives not isolated so far. However, for the L-cysteine-containing derivatives we observed an unexpected mass shift of +191.02 Da that we were not able to account for, while the MS2 spectrum indicates the molecule contains the darobactin core structure including the L-cysteine. It therefore seems likely that the sulphur nucleophile in the respective darobactin derivatives has reacted with a metabolite in the cell in order to detoxify the sulphur nucleophile. Nevertheless, future experiments will address the optimization of cultivation conditions and downstream purification steps, finally allowing characterization of the remaining derivatives, which showed promising antibacterial activity in the crude extract-based MIC assay.
Even though the production titre of darobactin A in our heterologous host (>13 mg L−1 in 3 days) was improved compared to the native producer strain P. khanii HGB1456, parallel work of Wusain et al. reached a titre of >30 mg L−1 in 2 days.20 Nevertheless, the production titres of darobactin A and of a few derivatives achieved with our system is sufficient for isolation and purification, with the production resulted from leaky expression of darA by an uninduced T7lac promoter. Furthermore, it is unlikely that the codon usage in the biosynthetic genes responsible for darobactin production are optimal for translation in E. coli, since the GC content of the darobactin BGC is only 32% as compared to E. coli genome with about 50% GC content. As we did not perform codon optimization so far, it is likely that production improvement can be achieved on both transcriptional and translational level after further optimizations of the darobactin expression vector.
Moreover, determination of the MIC of darobactin A and darobactin 9 against the heterologous producer strain E. coli BL21 (DE3) pNOSO-darABCDE showed that growth inhibition occurred at concentrations of 8–16 and 2–4 μg mL−1 (see Table 3), respectively, indicating that the maximum production titres of both compounds (>13 mg L−1 darobactin A and <3 mg L−1 darobactin 9) might be limited by self-resistance of the producer. In order to achieve industrial scale production titres, the self-resistance of the producer might be improved through targeted mutation of BamA in the genome, since it was previously shown that certain BamA mutations confer resistance towards darobactin A.15 As experiments that occurred in parallel to this work showed BamA mutations to lead to a significant loss in fitness and darobactin A productivity,20 the co-expression of mutated BamA contemporaneously with native BamA might be an alternative route to be explored. Furthermore, utilizing a phylogenetically more distant heterologous producer strain, e.g. of Gram-positive or eukaryotic origin, could circumvent the self-resistance issue completely as these organisms do not rely on BamA.
Targeted gene deletions and the attempts to produce truncated darobactins provided a first insight into the biosynthesis of this compound class. Interestingly, only two genes, darA and darE, are required for the formation of darobactin, a result also observed in parallel work by Wuisan and coworkers.20 Deletion of the relE-like gene, which is associated with the darobactin BGC in some of the native producer strains, had no impact on the darobactin A production in E. coli BL21 (DE3). However, as the gene expression of the relE homologue was still regulated by its native promotor, its expression might be weak or totally absent in E. coli BL21, which prevented us to elucidate its actual function and influence on darobactin biosynthesis. In future experiments, the relE homologue might be re-introduced into pNOSO-darABCDE downstream of darBCDE under the regulation of the nptII promoter for further investigation into its biological role. Moreover, despite the identification of darF and darG as genes associated with the darobactin BGC in several native producer strains, their role in darobactin biosynthesis was previously not discussed. Co-expression of darF and darG in the heterologous producer and the in vitro investigation of purified DarF showed that DarF is a protease leading to degradation of darobactin, whereas the function of DarG remains unclear. We hypothesise that the expression of darF in the native producer strain might be feedback-regulated, thereby limiting the darobactin production. However, experimental evidence has to be provided in future experiments. The proteolytic maturation process of darobactin however, including the enzymes that are potentially involved, still remains unclear.
Taken together, this work provided experimental evidence for darobactin biosynthesis processes, which was in part also confirmed by parallel work by Wuisan et al.20 Importantly, we achieved efficient structure engineering of darobactin and heterologous production of new and superior bioactive derivatives like darobactin 9 in good yields. Therefore, the work reported further advances the darobactin natural product class towards a potential use as an antibiotic drug.
Data availability
The acession data is referred to in the results section and the accession numbers are provided in ESI.†Author contributions
Sebastian Groß: conceptualization (equal); methodology (supporting); project administration (equal); investigation (supporting); visualization (equal); writing – original draft (lead); writing – review & editing (equal). Fabian Panter: methodology (supporting); investigation (equal); formal analysis (lead); visualization (equal), writing – original draft (supporting); writing – review & editing (equal). Domen Pogorevc: conceptualization (equal), methodology (lead), project administration (equal), investigation (supporting), formal analysis (supporting). Carsten E. Seyfert: investigation (equal), formal analysis (supporting). Selina Deckarm: investigation (equal). Chantal D. Bader: methodology (supporting), investigation (supporting), formal analysis (supporting) Jennifer Herrmann: formal analysis (supporting), writing – original draft (supporting). Rolf Müller: conceptualization (equal); supervision; funding acquisition; writing – review & editing (equal).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Stefanie Schmidt for performing the MIC assays and Dr F. P. Jake Haeckl for helpful proofreading of the manuscript. The authors would also like to thank Dr Patrick Chhatwal, Leonard Knegendorf, and Prof. Dr Dirk Schlüter from the Institute for Medical Microbiology and Hospital Epidemiology of Hannover Medical School (MHH), Germany, for providing clinical isolates.Notes and references
- (a) E. Tacconelli, E. Carrara, A. Savoldi, S. Harbarth, M. Mendelson, D. L. Monnet, C. Pulcini, G. Kahlmeter, J. Kluytmans, Y. Carmeli, M. Ouellette, K. Outterson, J. Patel, M. Cavaleri, E. M. Cox, C. R. Houchens, M. L. Grayson, P. Hansen, N. Singh, U. Theuretzbacher, N. Magrini, A. O. Aboderin, S. S. Al-Abri, N. Awang Jalil, N. Benzonana, S. Bhattacharya, A. J. Brink, F. R. Burkert, O. Cars, G. Cornaglia, O. J. Dyar, A. W. Friedrich, A. C. Gales, S. Gandra, C. G. Giske, D. A. Goff, H. Goossens, T. Gottlieb, M. Guzman Blanco, W. Hryniewicz, D. Kattula, T. Jinks, S. S. Kanj, L. Kerr, M.-P. Kieny, Y. S. Kim, R. S. Kozlov, J. Labarca, R. Laxminarayan, K. Leder, L. Leibovici, G. Levy-Hara, J. Littman, S. Malhotra-Kumar, V. Manchanda, L. Moja, B. Ndoye, A. Pan, D. L. Paterson, M. Paul, H. Qiu, P. Ramon-Pardo, J. Rodríguez-Baño, M. Sanguinetti, S. Sengupta, M. Sharland, M. Si-Mehand, L. L. Silver, W. Song, M. Steinbakk, J. Thomsen, G. E. Thwaites, J. W. M. van der Meer, N. van Kinh, S. Vega, M. V. Villegas, A. Wechsler-Fördös, H. F. L. Wertheim, E. Wesangula, N. Woodford, F. O. Yilmaz and A. Zorzet, Lancet Infect. Dis., 2018, 18, 318 CrossRef PubMed; (b) Global Antimicrobial Surveillance System (GLASS) Report 2016-2017, https://apps.who.int/iris/bitstream/handle/10665/279656/9789241515061-eng.pdf?ua=1, accessed 27 November 2020 Search PubMed.
- K. Kupferschmidt, Science, 2016, 352, 758 CrossRef CAS PubMed.
- D. van Duin and R. A. Bonomo, Clin. Infect. Dis., 2016, 63, 234 CrossRef CAS PubMed.
- R. G. Wunderink, E. J. Giamarellos-Bourboulis, G. Rahav, A. J. Mathers, M. Bassetti, J. Vazquez, O. A. Cornely, J. Solomkin, T. Bhowmick, J. Bishara, G. L. Daikos, T. Felton, M. J. L. Furst, E. J. Kwak, F. Menichetti, I. Oren, E. L. Alexander, D. Griffith, O. Lomovskaya, J. Loutit, S. Zhang, M. N. Dudley and K. S. Kaye, Infect. Dis. Ther., 2018, 7, 439 CrossRef PubMed.
- J. R. Smith, J. M. Rybak and K. C. Claeys, Pharmacotherapy, 2020, 40, 343 CrossRef CAS PubMed.
- L. D. Saravolatz and G. E. Stein, Clin. Infect. Dis., 2020, 70, 704 CAS.
- J. A. Sutcliffe, W. O'Brien, C. Fyfe and T. H. Grossman, Antimicrob. Agents Chemother., 2013, 57, 5548 CrossRef CAS PubMed.
- A. Ito, T. Sato, M. Ota, M. Takemura, T. Nishikawa, S. Toba, N. Kohira, S. Miyagawa, N. Ishibashi, S. Matsumoto, R. Nakamura, M. Tsuji and Y. Yamano, Antimicrob. Agents Chemother., 2018, 62, e01454-17 CrossRef PubMed.
- U. Theuretzbacher, K. Bush, S. Harbarth, M. Paul, J. H. Rex, E. Tacconelli and G. E. Thwaites, Nat. Rev. Microbiol., 2020, 18, 286 CrossRef CAS PubMed.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, M. B. van der, F. Bernardini, A. Lederer, R. L. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Kach, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010 CrossRef CAS PubMed.
- M. Kaul, L. Mark, Y. Zhang, A. K. Parhi, Y. L. Lyu, J. Pawlak, S. Saravolatz, L. D. Saravolatz, M. P. Weinstein, E. J. LaVoie and D. S. Pilch, Antimicrob. Agents Chemother., 2015, 59, 4845 CrossRef CAS PubMed.
- A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel, C. König, N. C. Ammerman, M. B. Barrio, K. Borchers, F. Bordon-Pallier, M. Brönstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. W. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. H. Grosset, S. Lagrange and R. Müller, Science, 2015, 348, 1106 CrossRef CAS PubMed.
- (a) G. S. Basarab, G. H. Kern, J. McNulty, J. P. Mueller, K. Lawrence, K. Vishwanathan, R. A. Alm, K. Barvian, P. Doig, V. Galullo, H. Gardner, M. Gowravaram, M. Huband, A. Kimzey, M. Morningstar, A. Kutschke, S. D. Lahiri, M. Perros, R. Singh, V. J. A. Schuck, R. Tommasi, G. Walkup and J. V. Newman, Sci. Rep., 2015, 5, 11827 CrossRef CAS PubMed; (b) S. Baumann, J. Herrmann, R. Raju, H. Steinmetz, K. I. Mohr, S. Hüttel, K. Harmrolfs, M. Stadler and R. Müller, Angew. Chem., Int. Ed. Engl., 2014, 53, 14605 CrossRef CAS PubMed.
- E. M. Hart, A. M. Mitchell, A. Konovalova, M. Grabowicz, J. Sheng, X. Han, F. P. Rodriguez-Rivera, A. G. Schwaid, J. C. Malinverni, C. J. Balibar, S. Bodea, Q. Si, H. Wang, M. F. Homsher, R. E. Painter, A. K. Ogawa, H. Sutterlin, T. Roemer, T. A. Black, D. M. Rothman, S. S. Walker and T. J. Silhavy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21748 CrossRef CAS PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576, 459 CrossRef CAS PubMed.
- A. Luther, M. Urfer, M. Zahn, M. Müller, S.-Y. Wang, M. Mondal, A. Vitale, J.-B. Hartmann, T. Sharpe, F. Lo Monte, H. Kocherla, E. Cline, G. Pessi, P. Rath, S. M. Modaresi, P. Chiquet, S. Stiegeler, C. Verbree, T. Remus, M. Schmitt, C. Kolopp, M.-A. Westwood, N. Desjonquères, E. Brabet, S. Hell, K. LePoupon, A. Vermeulen, R. Jaisson, V. Rithié, G. Upert, A. Lederer, Z. Peter, A. Wach, K. Moehle, K. Zerbe, H. H. Locher, F. Bernardini, G. E. Dale, L. Eberl, B. Wollscheid, S. Hiller, J. A. Robinson and D. Obrecht, Nature, 2019, 576, 452 CrossRef CAS PubMed.
- A. Konovalova, D. E. Kahne and T. J. Silhavy, Annu. Rev. Microbiol., 2017, 71, 539 CrossRef CAS PubMed.
- H. Kaur, R. P. Jakob, J. K. Marzinek, R. Green, Y. Imai, J. R. Bolla, E. Agustoni, C. V. Robinson, P. J. Bond, K. Lewis, T. Maier and S. Hiller, Nature, 2021, 1, 125–129 CrossRef PubMed.
- S. K. Christensen, M. Mikkelsen, K. Pedersen and K. Gerdes, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14328 CrossRef CAS PubMed.
- Z. G. Wuisan, I. D. M. Kresna, N. Böhringer, K. Lewis and T. F. Schäberle, Metab. Eng., 2021, 66, 123 CrossRef CAS PubMed.
- J. W. Dubendorff and F. W. Studier, J. Mol. Biol., 1991, 219, 45 CrossRef CAS.
- D. Pogorevc, F. Panter, C. Schillinger, R. Jansen, S. C. Wenzel and R. Müller, Metab. Eng., 2019, 55, 201 CrossRef CAS PubMed.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng, D. D. Nguyen, J. Watrous, C. A. Kapono, T. Luzzatto-Knaan, C. Porto, A. Bouslimani, A. V. Melnik, M. J. Meehan, W.-T. Liu, M. Crusemann, P. D. Boudreau, E. Esquenazi, M. Sandoval-Calderon, R. D. Kersten, L. A. Pace, R. A. Quinn, K. R. Duncan, C.-C. Hsu, D. J. Floros, R. G. Gavilan, K. Kleigrewe, T. Northen, R. J. Dutton, D. Parrot, E. E. Carlson, B. Aigle, C. F. Michelsen, L. Jelsbak, C. Sohlenkamp, P. Pevzner, A. Edlund, J. McLean, J. Piel, B. T. Murphy, L. Gerwick, C.-C. Liaw, Y.-L. Yang, H.-U. Humpf, M. Maansson, R. A. Keyzers, A. C. Sims, A. R. Johnson, A. M. Sidebottom, B. E. Sedio, A. Klitgaard, C. B. Larson, C. A. Boya P, D. Torres-Mendoza, D. J. Gonzalez, D. B. Silva, L. M. Marques, D. P. Demarque, E. Pociute, E. C. O'Neill, E. Briand, E. J. N. Helfrich, E. A. Granatosky, E. Glukhov, F. Ryffel, H. Houson, H. Mohimani, J. J. Kharbush, Y. Zeng, J. A. Vorholt, K. L. Kurita, P. Charusanti, K. L. McPhail, K. F. Nielsen, L. Vuong, M. Elfeki, M. F. Traxler, N. Engene, N. Koyama, O. B. Vining, R. Baric, R. R. Silva, S. J. Mascuch, S. Tomasi, S. Jenkins, V. Macherla, T. Hoffman, V. Agarwal, P. G. Williams, J. Dai, R. Neupane, J. Gurr, A. M. C. Rodriguez, A. Lamsa, C. Zhang, K. Dorrestein, B. M. Duggan, J. Almaliti, P.-M. Allard, P. Phapale, L.-F. Nothias, T. Alexandrov, M. Litaudon, J.-L. Wolfender, J. E. Kyle, T. O. Metz, T. Peryea, D.-T. Nguyen, D. VanLeer, P. Shinn, A. Jadhav, R. Muller, K. M. Waters, W. Shi, X. Liu, L. Zhang, R. Knight, P. R. Jensen, B. O. Palsson, K. Pogliano, R. G. Linington, M. Gutierrez, N. P. Lopes, W. H. Gerwick, B. S. Moore, P. C. Dorrestein and N. Bandeira, Nat. Biotechnol., 2016, 34, 828 CrossRef CAS PubMed.
- L. Patiny and A. Borel, J. Chem. Inf. Model., 2013, 53, 1223 CrossRef CAS PubMed.
- T. Kind and O. Fiehn, BMC Bioinf., 2007, 8, 105 CrossRef PubMed.
- N. Weston, P. Sharma, V. Ricci and L. J. V. Piddock, Res. Microbiol., 2017, 425–431 Search PubMed.
- F. Long, C. Rouquette-Loughlin, W. M. Shafer and E. W. Yu, Antimicrob. Agents Chemother., 2008, 52, 3052 CrossRef CAS PubMed.
- S. Nagakubo, K. Nishino, T. Hirata and A. Yamaguchi, J. Bacteriol., 2002, 184, 4161 CrossRef CAS PubMed.
- (a) J. J. Hug, J. Dastbaz, S. Adam, O. Revermann, J. Koehnke, D. Krug and R. Müller, ACS Chem. Biol., 2020, 15, 2221 CrossRef CAS PubMed; (b) Y. Ye, H. Fu and T. K. Hyster, J. Ind. Microbiol. Biotechnol., 2021, 48, 3–4 Search PubMed.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403 CrossRef CAS PubMed.
- L. Zimmermann, A. Stephens, S.-Z. Nam, D. Rau, J. Kübler, M. Lozajic, F. Gabler, J. Söding, A. N. Lupas and V. Alva, J. Mol. Biol., 2018, 430, 2237 CrossRef CAS PubMed.
- G. A. Hudson and D. A. Mitchell, Curr. Opin. Microbiol., 2018, 45, 61 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02725e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |