
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBorinostats: solid-phase synthesis of carborane-capped histone deacetylase inhibitors with a tailor-made selectivity profile†
Christoph
Selg
a,
Andrea
Schöler
a,
Julian
Schliehe-Diecks
b,
Maria
Hanl
c,
Laura
Sinatra
a,
Arndt
Borkhardt
b,
Menyhárt B.
Sárosi
 d,
Sanil
Bhatia
b,
Evamarie
Hey-Hawkins
d and
Finn K.
Hansen
*c
d,
Sanil
Bhatia
b,
Evamarie
Hey-Hawkins
d and
Finn K.
Hansen
*c
aInstitute for Drug Discovery, Medical Faculty, Leipzig University, Brüderstraße 34, 04103 Leipzig, Germany
bDepartment of Pediatric Oncology, Hematology and Clinical Immunology, Medical Faculty, Heinrich-Heine University Düsseldorf, Düsseldorf, Germany
cPharmaceutical Institute, Department of Pharmaceutical and Cell Biological Chemistry, University of Bonn, An der Immenburg 4, 53121 Bonn, Germany. E-mail: finn.hansen@uni-bonn.de
dInstitute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Leipzig University, Johannisallee 29, 04103 Leipzig, Germany
First published on 4th August 2021
Abstract
The elevated expression of histone deacetylases (HDACs) in various tumor types renders their inhibition an attractive strategy for epigenetic therapeutics. One key issue in the development of improved HDAC inhibitors (HDACis) is the selectivity for single HDAC isoforms over unspecific pan inhibition to minimize off-target toxicity. Utilizing the carborane moiety as a fine-tuning pharmacophore, we herein present a robust solid phase synthetic approach towards tailor-made HDACis meeting both ends of the selectivity spectrum, namely pan inhibition and highly selective HDAC6 inhibition.
Introduction
Histone deacetylases (HDACs) and their counterpart, the histone acetyl transferases (HATs), comprise two families of enzymes that play a key-role in the regulation of numerous genes and proteins.1,2 By controlling the reversible acetylation status of the ε-amino groups of lysine residues at the N-terminal domain of histone and non-histone proteins, they regulate the state of condensation of the DNA without impinging the DNA sequence itself:3 while HATs will cause chromatin structure to stretch into euchromatin and thus provide access to the DNA for the specific enzyme or other protein complexes involved in transcription and repair, HDACs induce condensed and transcriptionally inactive heterochromatin.1,2,4 As overexpression of HDACs was shown to be linked with several cancer types, their selective inhibition results in several anticancer effects including terminal differentiation, growth arrest and apoptosis in cancer cells.4–7 Therefore, HDAC inhibitors (HDACis) have emerged as valuable epigenetic modulators for treatment of cancer6–8 as well as HIV,9 inflammatory diseases,10 immune disorders, neurodegenerative11–13 and parasitic diseases.15 There are eighteen human HDAC isoforms that have been divided into four classes according to their primary homology to yeast: class I (HDACs 1, 2, 3 and 8), class IIa (HDACs 4, 5, 7, 9), class IIb (HDACs 6 and 10), class III (human sirtuins 1–7) and class IV (HDAC11).24 Within the HDAC family, HDAC6 has drawn major research attention due to occupying a very unique space in the therapeutic spectrum covering intracellular transport, neurotransmitter release, and aggresome formation.13,16 Primarily localized in the cytoplasm it regulates the acetylation of α-tubulin, HSP90,17,18 tau, cortactin, and amyloid β, and influences the microtubule formation and thereby the cell motility and metastatic potential.19 Thus, several research groups have developed small molecule inhibitors with selectivity for this specific isoform mainly following a certain pharmacophore model (Fig. 1).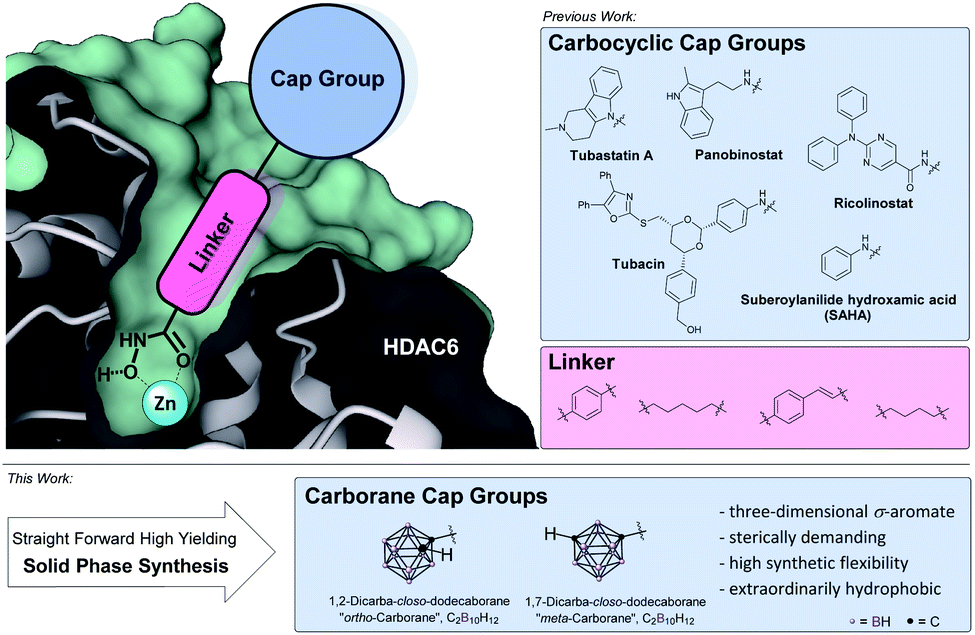 | ||
| Fig. 1 Pharmacophore model of selective HDACis: zinc-binding group (ZBG), hydrophobic alkyl or aryl linker and a sterically demanding, hydrophobic cap group.48,56 | ||
Except for class III HDACs, which are NAD+-dependent, all HDACs are Zn2+-dependent enzymes.20 Thus, the respective inhibitor usually possesses a zinc-binding group (ZBG), a hydrophobic linker and a cap group (Fig. 1, top left). Starting from the rather unselective FDA-approved hydroxamate-based drugs vorinostat, belinostat and panobinostat,20 within this pharmacophore model, HDAC6 isoform selectivity was shown to be connected to steric bulk and hydrophobicity of the cap group owing to the increased size of the outer channel opening of HDAC6 with 17 Å compared to 12 Å in HDAC1.17,21–23 High selectivity was achieved with cap groups bearing extended aromatic residues as in tubacin and tubastatin A as well as bulky cap groups as realized in ricolinostat-type HDACis.14,22,23 In order to increase selectivity, numerous hydrophobic (hetero)aromatic and, to some extent aliphatic cap groups were described in the literature.20,21 These carbon-based systems are for the most part limited to a two-dimensional shape as larger carbocyclic skeletons like adamantyl-, norbornenyl-, bicyclo-[2.2.2]octanyl- or fullerenyl-residues pose problems due to limited modification sites and possible rearrangement reactions or their sheer size.25 Therefore, when it comes to hydrophobic, sterically demanding scaffolds, an emerging building block in drug design is the carborane moiety.26 Replacement of (hetero)aryl moieties by these extremely versatile boron clusters has been widely applied and often resulted in dramatically increased potency, target selectivity and lower toxicity.25,27 Furthermore, carboranes play a key role in the development of boron neutron capture therapy (BNCT) agents and as carriers in nanomedicinal applications.29,30 Carboranes are a family of polyhedral clusters comprised of CH and BH vertices with the icosahedral closo-dicarbadodecaborane (C2B10H12) being the most prominent and well-examined member.27,28 For symmetrical reasons, the two carbon atoms can be arranged within the icosahedron separated by one (ortho-/1,2-), two (meta-/1,7-) or three (para-carborane/closo-1,12-dicarbadodecaborane) bonds (Fig. 1, bottom). The size of the cluster is in between a rotating phenyl ring and adamantane. The cluster fragments CH and BH are interconnected by a strong network of multi-electron-multi-center bonds and thus represent a completely delocalized three-dimensional σ-aromatic compound (deemed “superaromatic”) of very high stability.31 Bearing acidic (carbon-centered) and hydridic (boron-centered) hydrogen atoms, carboranes exhibit a peculiar interaction profile where besides the classic hydrophobic interactions, also hydrogen bonds C–H⋯X (X = O, N, S, F, π-system) and the so-called dihydrogen bonds B–H⋯H–X (with χX > χH > χB) are formed.32,33 As opposed to carbon cage structures, for carboranes manifold achiral, chiral and chiral-at-cage modifications at carbon- as well as at boron-vertices are described in the literature, thus paving the way also for the evaluation and design of target-specific asymmetric drugs.34,35
While boron-containing drugs like bortezomib, crisaborole and tavaborole are established therapeutics, carborane drugs are still scarce in clinical trials.30,36–38 Within this branch, a limited number of boron-containing HDACis have been investigated.39,40 However, only one of them was carborane-based and to the best of our knowledge none of them were tested for their HDAC6 selectivity. In the present publication we therefore seek to establish a systematic approach to carborane-based HDACis to provide a solid foundation for further investigations. By synthesizing a small compound library, we want to enable the direct head-to-head comparison of different carborane cap groups employing a set of known and successfully employed linker groups in combination with the well-investigated aryl or alkyl hydroxamate zinc-binding group. The hit compounds will be further compared to their respective phenyl analogues.
Results and discussion
Despite the readily synthetic availability of aniline-type carborane derivatives we chose the electrophilic carboxylic acid for the formation of the amide connectivity,41 as carboranes with directly bonded electron-withdrawing substituents are prone to degradation by base. Cluster deboronation can also occur in biological environments. Furthermore, as the clusters impose a strong electron-withdrawing effect on the adjacent nitrogen, carboranyl amines exhibit extreme low nucleophilicity.42 To provide different degrees of freedom in movement we decided to evaluate the carborane analogues of phenylacetic acid and benzoic acid (Scheme 1). | ||
| Scheme 1 Synthesis of the carborane building blocks. Reagents: (a) (i) n-BuLi, hexane, 0 °C; (ii) C2H4O, 0 °C; (iii) AcOH, MeOH, rt, 45%; (b) CrO3, acetone, AcOH, H2O, rt, 69%; c) (i) n-BuLi, Et2O, −78 °C; (ii) CO2(g), −78 °C; (iii) H2O, rt, quant. | ||
Following the synthetic protocol of Nekvinda and co-workers,43closo-1,2-dicarbadodecaborane (ortho-1) was monolithiated and treated with oxirane to afford hydroxymethyl carborane 2 which was subsequently oxidized with CrO3 to form the desired carboxymethyl carborane 3. As benzoic acid analogues of ortho-carborane prove problematic in amide couplings, we chose the meta isomer, closo-1,7-dicarbadodecaborane (meta-1), for our second cap group scaffold.44 The respective carboxylic acid 4 was obtained following a slightly modified literature procedure of Kasar and co-workers.46 Monolithiation of meta-1 and subsequent treatment with gaseous CO2 afforded 4 in quantitative yield (Scheme 1).
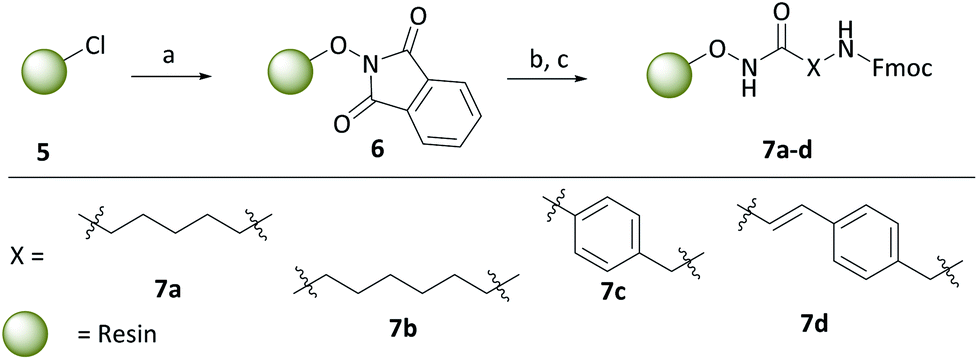
With the carborane cap group precursors 3 and 4 in hand, an elaborate synthetic approach for the linker and hydroxamate moieties was developed. To overcome the problems of the aforementioned possible deboronation that might occur under strongly basic conditions associated with the typical synthesis of hydroxamic acids through hydroxylaminolysis,40 we envisioned that a solid-phase synthesis as recently described by our group would be the suitable tool.47 Beside the prevention of harsh basic conditions, this methodology could offer a fast and efficient carborane-based HDACi library expansion through a parallel synthesis approach. Consequently, we treated in a first step the commercially available 2-chlorotritylchloride resin 5 with N-hydroxyphthalimide to form the modified resin 6. The phthaloyl residue in turn is cleaved using a 5% methanolic hydrazine solution to obtain a free hydroxylamine which can be coupled in a HATU-mediated amide coupling with an N-Fmoc protected linker to form the modified resins 7a–d (Scheme 2).
 | ||
| Scheme 2 Synthesis of the modified resins 7a–d with four different linkers. Reagents: (a) PhthN-OH, NEt3, DMF, 24 h, rt; (b) N2H4·H2O, MeOH, 30 min, rt; (c) Fmoc-linker-COOH, HATU, HOBt·H2O, DIPEA, DMF, 18 h, rt. | ||
To cover a broad spectrum of possible HDACis, we chose linker moieties that were already employed in unselective and selective HDACis described in the literature in resemblance to ricolinostat, vorinostat and tubacin, two different chain lengths, namely C5 and C6 alkyl linkers (7a, 7b), as well as a tubastatin-type benzyl (7c) and a panobinostat-type vinylbenzyl linker (7d) were selected (Scheme 2).
After Fmoc deprotection of the modified resins 7a–d, amide coupling with the respective carboranyl carboxylic acid cap groups 3 and 4 and subsequent cleavage with TFA afforded the desired carborane-capped hydroxamates 8a–d and 9a–d in very good yields (Scheme 3).
 | ||
| Scheme 3 Fmoc deprotection of the modified resins 7a–d, amide coupling with the carboranyl cap groups and cleavage off the resin. Reagents: (a) 20% piperidine in DMF, rt; (b) 3, HATU, DIPEA, DMF, 18 h, rt, (c) 4, COMU, DIPEA, DMF, 18 h, rt; (d) 5% TFA, DCM, 1 h, rt, 74–99%. For definition of X see Scheme 2. | ||
While coupling of ortho-carborane derivative 3 was achieved flawlessly under standard conditions using HATU, DIPEA and DMF, the meta-carboranyl carboxylic acid 4 could only be coupled in moderate yields. However, following the procedure described by Scholz et al., by the replacement of HATU with the more active COMU, excellent yields were also achieved with the sterically demanding and less reactive 4.44b
With the eight hydroxamates 8a–d and 9a–d in hand, we set out to explore their inhibitory activity against HDAC1 and HDAC6 in a fluorogenic assay using ZMAL (Z-Lys(Ac)-AMC) as substrate and vorinostat (suberoylanilide hydroxamic acid, SAHA) as a control (Table 1) following our previously published protocol.45 To our delight, all new compounds showed IC50 values for HDAC6 in the double- or even single-digit nanomolar range comparable to those of SAHA (IC50: 0.031 μM). Two of the compounds (9a and 9d) showed improved HDAC6 inhibition compared to the control. Compound 9d was identified as the most potent HDAC6 inhibitor (IC50: 0.006 μM).
| Structure | Target inhibition IC50a [μM] | MTT IC50b [μM] | Structure | Target inhibition IC50a [μM] | MTT IC50b [μM] | ||||
|---|---|---|---|---|---|---|---|---|---|
| HDAC1 | HDAC6 | SF6/1 | A2780 | HDAC1 | HDAC6 | SF6/1 | A2780 | ||
| a Assays carried out with n ≥ 2 (each in duplicate wells), values are shown as means ± SD. b Assays carried out with n ≥ 3 (each in duplicate wells), values are shown as means ± SD. | |||||||||

|
0.451 ± 0.023 | 0.037 ± 0.006 | 12 | 5.78 ± 1.68 |

|
0.440 ± 0.166 | 0.024 ± 0.005 | 18 | 3.98 ± 0.95 |

|
0.101 ± 0.003 | 0.045 ± 0.011 | 2 | 4.64 ± 0.95 |

|
1.328 ± 0.039 | 0.078 ± 0.0003 | 17 | 5.28 ± 1.25 |

|
0.015 ± 0.002 | 0.031 ± 0.004 | 0.5 | 1.66 ± 0.81 |

|
0.081 ± 0.014 | 0.040 ± 0.008 | 2 | 3.07 ± 1.41 |

|
2.888 ± 0.049 | 0.059 ± 0.007 | 49 | 4.85 ± 1.64 |

|
1.715 ± 0.002 | 0.006 ± 0.0001 | 286 | 4.60 ± 1.67 |

|
0.176 ± 0.007 | 0.095 ± 0.008 | 2 | 1.39 ± 0.30 |

|
0.725 ± 0.041 | 0.025 ± 0.002 | 29 | 5.04 ± 1.21 |

|
0.094 ± 0.014 | 0.031 ± 0.009 | 3 | 1.03 ± 0.27 | |||||
Due to a relatively even distribution of IC50 values no clear structure–activity relationship could be assigned for HDAC6 inhibition. For the activity against HDAC1, however, a clear selectivity trend was observed (Fig. 2).
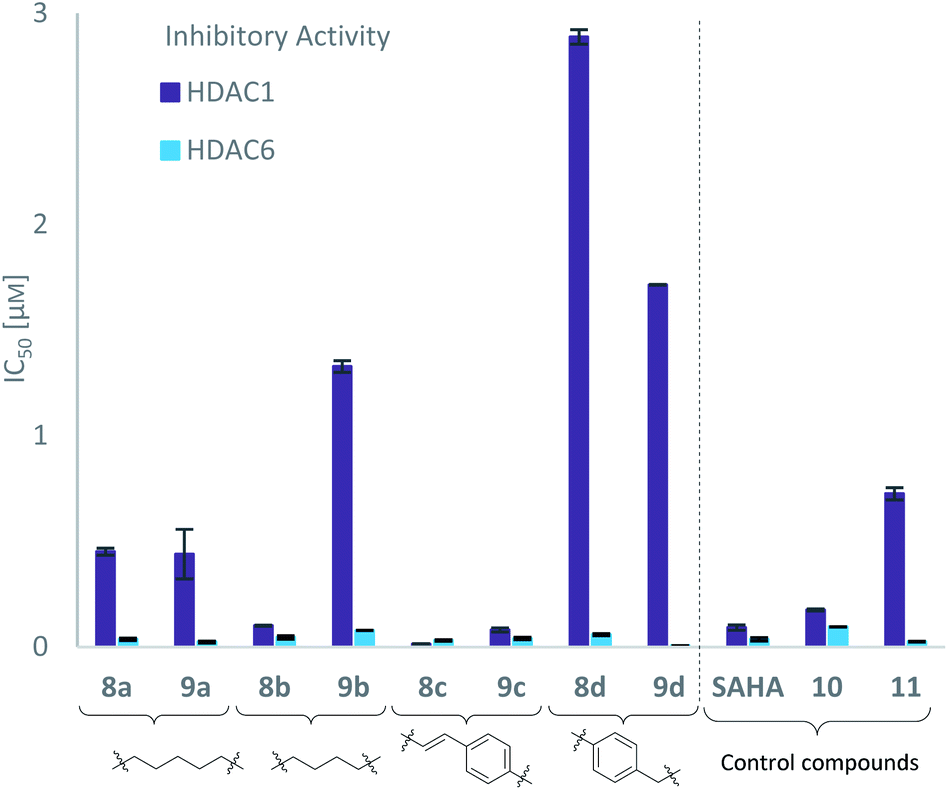 | ||
| Fig. 2 Target inhibition indicated by IC50 values (μM) of carborane-based hydroxamates 8a–d, 9a–d and control compounds SAHA, 10 and 11 grouped by linker against HDAC1 and HDAC6, determined by fluorogenic HDAC inhibition assays. | ||
Three of the compounds bearing an alkyl chain linker (8a, 9a and 9b) showed moderate (12- to 18-fold) preference for HDAC6 with slightly better inhibitory activity for the longer chains. The results of the short chain derivative 8b (SF6/1: 2) whose net chain length and carbonyl group positions match the parent aryl analogue pan inhibitor SAHA (SF6/1: 3) showed no clear preference.
Also, for the vinylbenzyl hydroxamates 8c and 9c, inhibition of both HDACs was very similar. Notably, inverse preference was observed between the two carborane cap groups shifting from either two-fold HDAC1 preference for 8c to two-fold HDAC6 preference for 9c. In contrast to this unselective inhibitory activity, the tubastatin A analogues with a benzyl linker (8d and 9d) showed excellent selectivity for HDAC6 whilst also maintaining very high inhibitory activity. Thus, we identified the meta-carboranyl hydroxamate 9d as the hit compound with an IC50 value of 0.006 μM and a more than 280-fold selectivity for HDAC6 (Table 1 and Fig. 2).
To investigate the influence of the carborane moiety, we synthesized aryl analogues for the best pan inhibitory compound 8c and the best selective HDAC6 inhibitor 9d. The synthesis was carried out employing our solid-phase protocol to afford vinylbenzyl derivative 10 from phenylacetic acid and benzyl derivative 11 using benzoic acid. Both phenyl analogues 10 and 11 showed good inhibitory activity against HDAC6 with IC50 values of 0.095 μM and 0.025 μM, respectively (Table 1). The HDAC6 selectivity (SF6/1: 2 and 29 resp.) was distributed the same way as observed for parent compounds 8c and 9d, yet the carborane compounds clearly outperformed their corresponding phenyl analogue in terms of HDAC6 inhibition and, in the case of 9d, selectivity.
To evaluate the anticancer properties of the carborane-capped HDACis, compounds 8a–d and 9a–d, as well as their phenyl analogues 10 and 11 were tested in MTT assays in the ovarian cancer cell line A2780, again using the FDA-approved HDACi vorinostat (SAHA) as positive control.45 The results are summarized in Table 1. All compounds demonstrated antiproliferative effects in the single-digit micromolar concentration range. The most potent carborane-capped HDACi was compound 8c with an IC50 value of 1.66 μM.
Encouraged by these promising results, we performed additional HDAC isoform profilings examining the HDAC6 selectivity of the two most potent compounds 8c and 9d over all class I isoforms with SAHA as control and in the case of HDAC8, also panobinostat (Table 2).
| Compound | Target inhibition IC50 [μM]a | ||||
|---|---|---|---|---|---|
| HDAC1b | HDAC2b | HDAC3b | HDAC6b | HDAC8c | |
| a Assays carried out with n ≥ 2 (each in duplicate wells), values are shown as means ± SD. b Z-Lys(Ac)-AMC was used as substrate. c Boc-Lys-(Tfa)-AMC was used as substrate. n.d.: not determined. | |||||
| 8c | 0.015 ± 0.002 | 0.020 ± 0.004 | 0.021 ± 0.001 | 0.031 ± 0.004 | 1.661 ± 0.126 |
| 9d | 1.715 ± 0.002 | 1.857 ± 0.167 | 1.346 ± 0.003 | 0.006 ± 0.0001 | 5.780 ± 0.192 |
| SAHA | 0.094 ± 0.014 | 0.163 ± 0.015 | 0.113 ± 0.004 | 0.031 ± 0.009 | 3.410 ± 0.494 |
| Panobinostat | n.d. | n.d. | n.d. | n.d. | 0.166 ± 0.015 |
Considering HDAC2 and HDAC3, again, 8c showed non-selective behavior while compound 9d exhibited substantial selectivity for HDAC6 over HDAC2 and HDAC3 (309- and 224-fold, respectively). The IC50 values for HDAC8 were greater than 1 μM and indicated neglectable inhibition for both compounds.
As already observed for tubastatin A and panobinostat, the potent HDAC6 inhibition may be the result of π–π stacking interactions between the linker aryl groups and the adjacent phenylalanine residues inside the hydrophobic channel.48,54 To further undermine this assumption, compounds 8c and 9d were docked into the human HDAC6 catalytic domain 2 (hHDAC6-CD2, Fig. 3A and B). Hydroxamate inhibitors are known to coordinate to the HDAC Zn2+ ion through mono- or bidentate binding modes.48,54,55 A monodentate Zn2+-binding was predicted for 8c and the amido NH group forms a hydrogen bond with S568. The phenyl ring of 8c forms a π–π stacking interaction with F680. On the other hand, a bidentate Zn2+-binding was predicted for 9d. The phenyl ring of 9d forms a π–π stacking interaction with F620 and the amido CO group can potentially form a hydrogen bond with S568.
 | ||
| Fig. 3 8c (A) and 9d (B) docked into the human HDAC6 catalytic domain 2 (PDB ID: 5EDU).48 HDAC6 is shown in gray. Residue numbering according to PDB ID: 5EDU. 8c (C) docked into human HDAC1 (PDB ID: 5ICN).49 HDAC1 is shown in grey. Residue numbering according to PDB ID: 5ICN. Superimposed HDAC6 backbone trace is shown in light blue for comparison (PDB ID: 5EDU). B: orange, C: gray/green, N: blue, O: red, Zn: purple. Hydrogen atoms are omitted. Ligand–receptor interaction and Zn2+ coordination is shown as dashed lines. | ||
The carborane clusters of both compounds form dihydrogen bonds with H500 and P501, but the ortho-carborane of 8c additionally interacts with F620. The dihydrogen bonds between the ligands and key hHDAC6-CD2 residues H500 and P501 certainly contribute to the observed potent inhibition of HDAC6. The sterically demanding hydrophobic carborane cap groups point outwards the hydrophobic channel and align to HDACs surface binding domain which leaves sufficient space for the carborane to act as isoform selection parameter without decreasing target inhibition.
As an attempt to understand the different selectivity trends, 8c and 9d were docked into human HDAC1.49 74% of the docked poses of 8c contained a monodentate Zn2+-binding. Fig. 3C shows a monodentate binding mode where the ortho-carborane interacts with P29 and the amido NH group forms a hydrogen bond with D99. In the case of 9d, only 39% of the docked poses chelated the zinc ion in the active site. The remaining poses of 9d did not enter the HDAC1 active site (see ESI, Fig. S1†). As a comparison, 100% of the docked HDAC6 poses for both 8c and 9d were chelating the zinc ion. It is reasonable to assume that compound 8c exhibiting a longer linker is capable of chelating the zinc ion of both HDAC1 and HDAC6, thus inhibiting both targets, whereas the shorter benzyl linker realized in 9d in combination with a sterically demanding carborane cap is unable to efficiently engage the zinc ion in HDAC1. With the combination of our linker library and meta- and ortho-carboranes as bioisosteric phenyl mimetics, thus, we could not only significantly improve the potency, but, in the case of 9d also the selectivity of the HDACis compared to their parent phenyl analogues.
In the next step, we investigated the selectivity profile of 8c and 9d in a cellular environment using the corresponding phenyl analogues 10 and 11 as well as vorinostat (SAHA) as control compounds. The acute myeloid leukemia (AML) cell line HL60 was treated for 24 h with 1 μM of each compound and the cell lysates were subsequently immunoblotted with antibodies against acetyl-histone H3 (a marker of HDAC1-3 inhibition), acetyl-α-tubulin (a marker of HDAC6 inhibition), and acetyl-SMC3 (a marker of HDAC8 inhibition). The results are presented in Fig. 4A. None of the compounds induced acetylation of SMC3. The unselective inhibitor 8c caused hyperacetylation of histone H3 indicating inhibition of HDAC1, 2 and/or 3. As expected, the highly selective HDAC6i 9d increased only the protein levels of acetyl-α-tubulin. Consequently, the biochemical and western blot data confirm that 9d is a selective HDAC6i both in a cell-free and cellular setting.
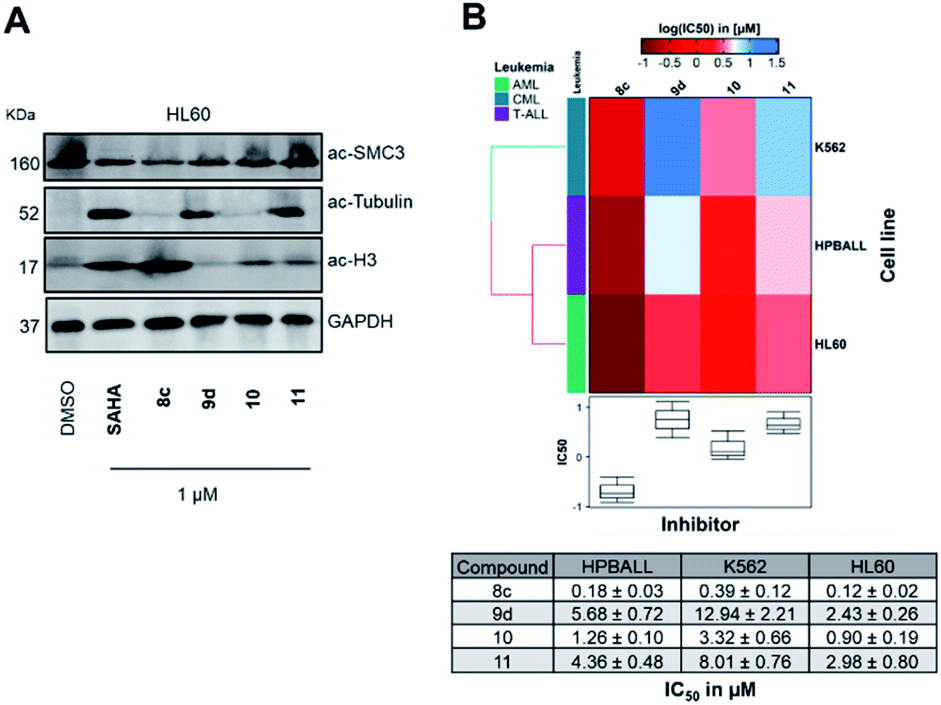 | ||
Fig. 4 (A) Immunoblotting with anti-acetyl SMC3 (HDAC8 inhibition), anti-acetyl-α-tubulin (HDAC6 inhibition) and anti-acetyl-histone H3 (HDAC1 inhibition) antibodies on HL60 cell lysates obtained after 24 h treatment at 1 μM with compounds 8c, 9d, 10, 11 or vorinostat (SAHA) as a control. (B) Comparative cellular viability (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) IC50 μM) of three leukemic cell lines originated from different lineages (K562, HPBALL and HL60) after 72 h treatment with compounds 8c, 9d, 10 or 11. The IC50 data (n = 3) plotted as a heat map with each box of the heat map representing the mean of three independent experiments (n = 3), whereas the color of the individual cell is related to its position along with a logIC50 (μM) gradient. The table below is depicting actual IC50 values used for plotting these heat maps. IC50 μM) of three leukemic cell lines originated from different lineages (K562, HPBALL and HL60) after 72 h treatment with compounds 8c, 9d, 10 or 11. The IC50 data (n = 3) plotted as a heat map with each box of the heat map representing the mean of three independent experiments (n = 3), whereas the color of the individual cell is related to its position along with a logIC50 (μM) gradient. The table below is depicting actual IC50 values used for plotting these heat maps. | ||
To elucidate the antiproliferative effects of compounds 8c and 9d against a selection of cell lines from different leukemia entities, we screened both compounds in a CellTiter Glo assay against the HPBALL (T-cell acute lymphoblastic leukemia, T-ALL), K562 (chronic myeloid leukemia, CML) and HL60 (AML) cell lines. The corresponding phenyl analogues 10 and 11 were used as controls (Fig. 4B).
Compound 9d displayed moderate antiproliferative activity with IC50 values ranging from 2.43 to 12.94 μM, while 8c demonstrated submicromolar activity against all three cell lines with the highest activity observed in HL60 cells (IC50: 0.12 μM). Of note, 8c outperformed its corresponding phenyl analogue 10 in all three cell lines and showed 7–8-fold lower IC50 values.
The relatively low cytotoxicity of the selective HDAC6i 9d in comparison to the unselective HDACi 8c is in good agreement with recent results from literature, which indicate that class I HDAC inhibition is important for single-agent activity.51 HDAC6 together with the motor protein dynein is essential to recruit ubiquitinated proteins to aggresomes. The dual inhibition of the aggresome (via HDAC6 inhibition) and proteasome pathways can consequently lead to accumulation of misfolded, cytotoxic proteins resulting in induction of apoptosis.52 Hence, HDAC6i and proteasome inhibitor exhibit synergistic anticancer properties and this synergism led to the approval of the combination therapy of panobinostat, the proteasome inhibitor bortezomib, and dexamethasone to treat multiple myeloma. This knowledge prompted us to investigate the synergistic effects of 8c, 9d and vorinostat as a control in combination with bortezomib, using 8 × 8 dose–response matrices (Fig. 5). Among the tested combinations, 8c displayed the highest synergy (ZIP synergy score: 10.52) with bortezomib, followed by vorinostat (ZIP synergy score: 7.18) and 9d (5.95). The comparative high synergistic activity of 8c with bortezomib is potentially due to its potent class I HDAC inhibitory activity, as targeting especially class I HDACs are critical targets to overcome bortezomib resistance.53
 | ||
| Fig. 5 (A)–(C) Illustrative synergy map of 8c, 9d and vorinostat (SAHA) after 72 h co-treatment of the acute myeloid leukemia (AML) cell line HL60 with bortezomib (proteasome inhibitor) at depicted concentrations. The mean synergy score calculations were based on the ZIP model and visualizations were performed using Synergy Finder webtool.50 | ||
Conclusions
In summary, we have successfully extended our solid-phase synthesis approach using preloaded resins towards the synthesis of a small compound library of carborane-based HDACis. The syntheses proceeded flawlessly for carborane analogues of benzoic and phenylacetic acid without cluster degradation and intermittent isolation (and detection problems associated with carboranes).44 These carborane derivatives were employed as bioisosteres in HDACi drug design to precisely steer the selectivity of the HDACis between pan inhibition and highly selective HDAC6 inhibition with only minor adjustments to the inhibitor structure. In reference to the successful FDA-approved HDACi vorinostat, we termed the best-in-category cluster compounds borinostat A (9d) and borinostat B (8c). Both borinostats were compared with their respective phenyl analogues 10 and 11 to further support their superiority as a hydrophobic, sterically demanding cap group leading to increased inhibitory activity and, in the case of borinostat A, selectivity for the clinically preferred isoform HDAC6. In vitro cytotoxicity evaluation of the new carborane derivatives showed an antiproliferative effect in the low single-digit micromolar range against human ovarian cancer cells and, in the case of borinostat B, submicromolar activity against three different leukemia cell lines. In synergism studies, both borinostat A and B demonstrated synergistic anticancer activity when combined with the proteasome inhibitor bortezomib.To conclude, due to the tunable selectivity profile, this series of carborane-capped HDACis represents an encouraging starting point to develop unselective HDACis with improved single-agent activity as well as highly selective HDAC6 inhibitors for potential combination therapies.
Author contributions
CS, EH-H, and FKH designed the study. CS performed the synthesis of the carboranes under the supervision of EH-H. CS performed the synthesis of the carborane-capped HDACi under the supervision of FKH. LS provided building blocks and experimental support. AS performed the biochemical HDAC inhibition assays under the supervision of FKH. MH performed the MTT assays under the supervision of FKH. JS-D performed CellTiter Glo assays, western blot experiments and synergy experiments under the supervision of AB and SB. MBS performed the docking studies. CS wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously supported by the Deutsche Forschungsgemeinschaft (MBS: SA 2902/2-1). Funding by the Graduate School Building with Molecules and Nano-objects (BuildMoNa) is gratefully acknowledged. This study was funded in part by the Deutsche Forschungsgemeinschaft – 270650915 (Research Training Group GRK 2158, TP2d to SB). AB acknowledges the financial support from Löwenstern e. V. and from Katharina-Hardt Foundation. We sincerely thank Dr. Matthias Scholz and Linda Schäker-Hübner for advice and fruitful discussion.References
- (a) C. L. Woodcock and R. P. Ghosh, Cold Spring Harbor Perspect. Biol., 2010, 2, a000596 CrossRef PubMed; (b) J. R. Whittle and J. Desai, Cancer, 2015, 121, 1164–1167 CrossRef CAS PubMed.
- M. Grunstein, Nature, 1997, 389, 349–352 CrossRef CAS PubMed.
- (a) S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS; (b) M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- M. New, H. Olzscha and N. B. La Thangue, Mol. Oncol., 2012, 6, 637–656 CrossRef CAS PubMed.
- M. Haberland, R. L. Montgomery and E. N. Olson, Nat. Rev. Genet., 2009, 10, 32–42 CrossRef CAS PubMed.
- Y.-S. Lee, K.-H. Lim, X. Guo, Y. Kawaguchi, Y. Gao, T. Barrientos, P. Ordentlich, X.-F. Wang, C. M. Counter and T.-P. Yao, Cancer Res., 2008, 68, 7562–7569 Search PubMed.
- T. Sakma, K. Uzawa, T. Onda, M. Shiiba, H. Yokoe, T. Shibahara and H. Tanzawa, Int. J. Oncol., 2006, 29, 117–124 Search PubMed.
- M. Paris, M. Porcelloni, M. Bnaschi and D. Fattori, J. Med. Chem., 2008, 52, 1505–1529 CrossRef.
- N. M. Archin, A. L. Liberty, A. D. Kashuba, S. K. Choudhary, J. D. Kuruc, A. M. Crooks, D. C. Parker, E. M. Anderson, M. F. Kearney, M. C. Strain, D. D. Rochman, M. G. Hudgens, R. J. Bosch, J. M. Coffin, J. J. Eron, D. J. Hazuda and D. M. Margolis, Nature, 2012, 487, 482–485 CrossRef CAS PubMed.
- D. Rotilli, G. Simonetti, A. Savarino, A. T. Palamara, A. R. Migliaccio and A. Mai, Curr. Top. Med. Chem., 2009, 9, 272–291 CrossRef PubMed.
- S. Trazzi, C. Fuchs, R. Viggiano, M. de Franceschi, E. Valli, P. Jedynak, F. K. Hansen, G. Perini, R. Rimondini, T. Kurz, R. Bartesaghi and E. Ciani, Hum. Mol. Genet., 2016, 25, 3887–3907 CrossRef CAS.
- K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- P. LoPresti, Cells, 2021, 10, 1–15 Search PubMed.
- (a) J. P. Dompierre, J. D. Godin, B. C. Charrin, F. P. Cordelières, S. J. King, S. Humbert and F. Saudou, J. Neurosci., 2007, 27, 3571–3583 CrossRef CAS PubMed; (b) K. J. Falkenberg and R. W. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673–691 CrossRef CAS PubMed.
- (a) K. T. Andrews, A. Haque and M. K. Jones, Immunol. Cell Biol., 2012, 90, 66–77 CrossRef CAS PubMed; (b) M. K. W. Mackwitz, E. Hesping, K. Eribez, A. Schöler, Y. Antonova-Koch, J. Held, E. A. Winzeler, K. T. Andrews and F. K. Hansen, Eur. J. Med. Chem., 2021, 211, 113065 CrossRef CAS; (c) M. K. W. Mackwitz, E. Hesping, Y. Antonova-Koch, S. Diedrich, T. G. Woldearegai, T. Skinner-Adams, M. Clarke, A. Schöler, L. Limbach, T. Kurz, E. A. Winzeler, J. Held, K. T. Andrews and F. K. Hansen, ChemMedChem, 2019, 14, 912–926 CrossRef CAS; (d) D. Diedrich, K. Stenzel, E. Hesping, Y. Antonova-Koch, T. Gebru, S. Duffy, G. Fisher, A. Schöler, S. Meister, T. Kurz, V. M. Avery, E. A. Winzeler, J. Held, K. T. Andrews and F. K. Hansen, Eur. J. Med. Chem., 2018, 158, 801–813 CrossRef CAS.
- M. Brindisi, A. P. Saraswati, S. Brogi, S. Gemma, S. Butini and G. Campiani, J. Med. Chem., 2020, 63, 23–39 CrossRef CAS.
- B. E. Gryder, Q. H Sodji and A. K. Oyelere, Future Med. Chem., 2012, 4, 505–524 CrossRef CAS.
- O. Witt, H. E. Deubzer, T. Milde and I. Oehme, Cancer Lett., 2009, 277, 8–21 CrossRef CAS PubMed.
- C. Boyault, K. Sadoul, M. Pabion and S. Khochbin, Oncogene, 2007, 26, 5468–5476 CrossRef CAS PubMed.
- (a) C. M. Grozinger and S. L. Schreiber, Chem. Biol., 2002, 9, 3–16 CrossRef CAS PubMed; (b) Y. Li and E. Seto, Cold Spring Harbor Perspect. Med., 2016, 6, a026831 CrossRef PubMed.
- Y. Sixto-López, J. A. Gómez-Vidal, N. de Pedro, M. Bello, M. C. Rosales-Hernández and J. Correa-Basurto, Sci. Rep., 2020, 10, 10462 CrossRef PubMed.
- L. Auzzas, A. Larsson, R. Matera, A. Baraldi, B. Deschênes-Simard, G. Giannini, W. Cabri, G. Battistuzzi, G. Gallo, A. Ciacci, L. Vesci, C. Pisano and S. Hanessian, J. Med. Chem., 2010, 53, 8387–8399 CrossRef CAS PubMed.
- K. V. Butler, J. Kalin, C. Brochier, G. Vistoli, B. Langley and A. P. Kozikowski, J. Am. Chem. Soc., 2010, 132, 10842–10846 CrossRef CAS PubMed.
- X.-X. Wang, R.-Z. Wan and Z.-P. Liu, Eur. J. Med. Chem., 2018, 143, 1406–1418 CrossRef CAS PubMed.
- (a) Y. Endo, in Boron-Based Compounds, Potential and Emerging Applications in Medicine, ed. E. Hey-Hawkins and C. Viñas Teixidor, John Wiley & Sons Ltd, Oxford, 2018, ch. 1.1.2, pp. 3–5 Search PubMed; (b) P. Stockmann, M. Gozzi, R. Kuhnert, M. B. Sárosi and E. Hey-Hawkins, Chem. Soc. Rev., 2019, 48, 3497–3512 RSC.
- (a) M. Scholz, K. Bensdorf, R. Gust and E. Hey-Hawkins, ChemMedChem, 2009, 4, 746–748 CrossRef CAS PubMed; (b) J. F. Valliant, P. Schaffer, K. A. Stephenson and J. F. Britten, J. Org. Chem., 2002, 67, 383–387 CrossRef CAS PubMed.
- (a) R. N. Grimes, in Carboranes, Academic Press, New York, 2nd edn, 2011, pp. 1053–1062 Search PubMed; (b) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS; (c) R. Frank, V. Ahrens, S. Boehnke, S. Hofmann, M. Kellert, S. Saretz, S. Pandey, M. Sárosi, A. Bartók, A. G. Beck-Sickinger and E. Hey-Hawkins, Pure Appl. Chem., 2015, 87, 163–171 CrossRef CAS; (d) A. M. Spokoyny, Pure Appl. Chem., 2013, 85, 903–919 CrossRef CAS PubMed; (e) K. O. Kirlikovali, J. C. Axtell, A. Gonzalez, A. C. Phung, S. I. Khan and A. M. Spokoyny, Chem. Sci., 2016, 7, 5132–5138 RSC; (f) G. R. Kracke, M. R. VanGordon, Y. V. Sevryugina, P. J. Kueffer, K. Kabytaev, S. S. Jalisatgi and M. F. Hawthorne, ChemMedChem, 2015, 10, 62–67 CrossRef CAS PubMed.
- For instructive reviews on carboranes, see: (a) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (b) R. E. Williams, Chem. Rev., 1992, 92, 177–207 CrossRef CAS; (c) R. B. King, Chem. Rev., 2001, 101, 1119–1152 CrossRef CAS PubMed; (d) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC.
- K. Nedunchezhian, N. Aswath, M. Thiruppathy and S. Thirugnanamurthy, J. Clin. Diagn. Res., 2016, 10, ZE01–ZE04 Search PubMed.
- M. Gozzi, B. Schwarze and E. Hey-Hawkins, ChemMedChem, 2021, 16(10), 1533–1565 CrossRef CAS PubMed.
- M. Otsuka, R. Takita, J. Kanazawa, K. Miyamoto, A. Muranaka and M. Uchiyama, J. Am. Chem. Soc., 2015, 137, 15082–15085 CrossRef CAS PubMed.
- M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111(11), 7035–7062 CrossRef CAS PubMed.
- M. Calvaresi and F. Zerbetto, J. Chem. Inf. Model., 2011, 51, 1882–1896 CrossRef CAS PubMed.
- R. Cheng, B. Li, J. Wu, J. Zhang, Z. Qiu, W. Tang, S.-L. You, Y. Tang and Z. Xie, J. Am. Chem. Soc., 2018, 140, 4508–4511 CrossRef CAS PubMed.
- D. V. Smil, S. Manku, Y. A. Chantigny, S. Leit, A. Wahhab, T. P. Yan, M. Fournel, C. Maroun, Z. Li, A.-M. Lemieux, A. Nicolescu, J. Rahil, S. Lefebvre, A. Panetta, J. M. Besterman and R. Déziel, Bioorg. Med. Chem. Lett., 2009, 19, 688–692 CrossRef CAS PubMed.
- N. R. Weinberg, Curr. Opin. Invest. Drugs, 2009, 10, 1236–1242 Search PubMed.
- S. J. Baker, Y.-K. Zhang, T. Akama, A. Lau, H. Zhou, V. Hernandez, W. Mao, M. R. K. Alley, V. Sanders and J. J. Plattner, J. Med. Chem., 2006, 49, 4447–4450 CrossRef CAS PubMed.
- (a) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (b) M. Bello, Curr. Pharm. Des., 2018, 24, 1–10 CrossRef PubMed; (c) G. F. S. Fernandes, W. A. Denny and J. L. Dos Santos, Eur. J. Med. Chem., 2019, 179, 791–804 CrossRef CAS PubMed.
- R. Bakri, A. A. Parikesit, C. P. Satriyanto, D. Kerami and U. S. F. Tambunan, Adv. Bioinf., 2014, 2014, 104823 Search PubMed.
- P. Kavianpour, M. C. M. Gemmell, J. U. Kahlert and L. M. Rendina, ChemBioChem, 2020, 21, 2786–2791 CrossRef CAS PubMed.
- Y. Nie, Y. Wang, J. Miao, Y. Li and Z. Zhang, J. Organomet. Chem., 2015, 798, 182–188 CrossRef CAS.
- (a) R. A. Wiesboeck and M. F. Hawthorne, J. Am. Chem. Soc., 1964, 86, 1642–1643 CrossRef CAS; (b) M. F. Hawthorne, D. C. Young, P. M. Garrett, D. A. Owen, S. G. Schwerin, F. N. Tebbe and P. A. Wegner, J. Am. Chem. Soc., 1968, 90, 862–868 CrossRef CAS; (c) C. Selg, W. Neumann, P. Lönnecke, E. Hey-Hawkins and K. Zeitler, Chem.–Eur. J., 2017, 23, 7932 CrossRef CAS PubMed.
- J. Nekvinda, B. Grüner, D. Gabel, W. M. Nau and K. I. Assaf, Chem.–Eur. J., 2018, 24, 12970–12975 CrossRef CAS PubMed.
- (a) T. Lützenburg, I. Neundorf and M. Scholz, Chem. Phys. Lipids, 2018, 213, 62–67 CrossRef PubMed; (b) M. Scholz and L. M. Wingen, Inorg. Chem., 2017, 56, 5510–5513 CrossRef CAS PubMed.
- N. Erdeljac, K. Bussmann, A. Schöler, F. K. Hansen and R. Gilmour, ACS Med. Chem. Lett., 2019, 10, 1336–1340 CrossRef CAS PubMed.
- R. A. Kasar, G. M. Knudsen and S. B. Kahl, Inorg. Chem., 1999, 38, 2936–2940 CrossRef CAS PubMed.
- L. Sinatra, J. J. Bandolik, M. Roatsch, M. Sönnichsen, C. T. Schoeder, A. Hamacher, A. Schöler, A. Borkhardt, J. Meiler, S. Bhatia, M. U. Kassack and F. K. Hansen, Angew. Chem., Int. Ed., 2020, 59, 22494–22499 CrossRef CAS PubMed.
- (a) Y. Hai and D. W. Christianson, Nat. Chem. Biol., 2016, 12, 741–747 CrossRef CAS PubMed; (b) H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- P. J. Watson, C. J. Millard, A. M. Riley, N. S. Robertson, L. C. Wright, H. Y. Godage, S. M. Cowley, A. G. Jamieson, B. V. Potter and J. W. Schwabe, Nat. Commun., 2016, 7, 11262 CrossRef CAS PubMed.
- A. Ianevski, L. He, T. Aittokallio and J. Tang, Bioinformatics, 2020, 36, 2645 CrossRef PubMed.
- Y. Depetter, S. Geurs, R. De Vreese, S. Goethals, E. Vandoorn, A. Laevens, J. Steenbrugge, E. Meyer, P. de Tullio, M. Bracke, M. D’hooghe and O. De Wever, Int. J. Cancer, 2019, 145, 735–747 CrossRef CAS PubMed.
- (a) T. Hideshima, P. G. Richardson and K. C. Anderson, Mol. Cancer Ther., 2011, 10, 2034–2042 CrossRef CAS PubMed; (b) N. Reßing, M. Sönnichsen, J. D. Osko, A. Schöler, J. Schliehe-Diecks, A. Skerhut, A. Borkhardt, J. Hauer, M. U. Kassack, D. W. Christianson, S. Bhatia and F. K. Hansen, J. Med. Chem., 2020, 63, 10339–10351 CrossRef PubMed.
- J. Kikuchi, T. Wada, R. Shimizu, T. Izumi, M. Akutsu, K. Mitsunaga, K. Noborio-Hatano, M. Nobuyoshi, K. Ozawa, Y. Kano and Y. Furukawa, Blood, 2010, 116, 406–417 CrossRef CAS PubMed.
- S. Shen, M. Svoboda, G. Zhang, M. A. Cavasin, L. Motlova, T. A. McKinsey, J. H. Eubanks, C. Barinka and A. P. Kozikowski, ACS Med. Chem. Lett., 2020, 11, 706–712 CrossRef CAS PubMed.
- N. J. Porter, A. Mahendran, R. Breslow and D. W. Christianson, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13459 CrossRef CAS PubMed.
- D. Sehnal, A. S. Rose, J. Koča, S. K. Burley and S. Velankar, Mol*: towards a common library and tools for web molecular graphics, in MolVA'18: Proceedings of the Workshop on Molecular Graphics and Visual Analysis of Molecular Data, Eurographics Association, Goslar, pp. 29–33 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02268g |
| This journal is © The Royal Society of Chemistry 2021 |