
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical investigations into the biosynthesis of the gymnastatin and dankastatin alkaloids†
Bingqi
Tong
ab,
Bridget P.
Belcher
ab,
Daniel K.
Nomura
 abc and
Thomas J.
Maimone
*ab
abc and
Thomas J.
Maimone
*ab
aDepartment of Chemistry, University of California–Berkeley, Berkeley, CA 94720, USA. E-mail: maimone@berkeley.edu
bNovartis-Berkeley Center for Proteomics and Chemistry Technologies, University of California–Berkeley, Berkeley, CA 94720, USA
cDepartments of Nutritional Science and Toxicology, Cell and Molecular Biology, The Innovative Genomics Institute, University of California–Berkeley, Berkeley, CA 94720, USA
First published on 31st May 2021
Abstract
Electrophilic natural products have provided fertile ground for understanding how nature inhibits protein function using covalent bond formation. The fungal strain Gymnascella dankaliensis has provided an especially interesting collection of halogenated cytotoxic agents derived from tyrosine which feature an array of reactive functional groups. Herein we explore chemical and potentially biosynthetic relationships between architecturally complex gymnastatin and dankastatin members, finding conditions that favor formation of a given scaffold from a common intermediate. Additionally, we find that multiple natural products can also be formed from aranorosin, a non-halogenated natural product also produced by Gymnascella sp. fungi, using simple chloride salts thus offering an alternative hypothesis for the origins of these compounds in nature. Finally, growth inhibitory activity of multiple members against human triple negative breast cancer cells is reported.
Introduction
Natural products have long been a rich source of medicinal agents and sources of inspiration for the design of numerous clinical candidates and FDA-approved drugs.1 Among these, members that interact with protein targets through covalent bond formation have the potential to open up new areas of druggable space, provide sustained target engagement, and confer unique selectivity as a result of architectural complementarity to many fully synthetic small molecules.2 Moreover, natural products featuring more than one covalent warhead offer the prospect of engaging several nucleophilic protein residues and potentially multiple protein partners.3Against this backdrop, we became interested in the fascinating array of chlorinated gymnastatin and dankastatin alkaloids first disclosed in 1997 from the fungal strain Gymnascella dankaliensis isolated from the sponge Halichondria japonica (Fig. 1).4 Presumably produced through the merger of tyrosine and a 14-carbon polyketide fragment (see 1) to first generate gymnastatin N (2), electrophilic halogenation and various oxidative cyclization reactions create a small library of architecturally complex natural products from a common and simple precursor. Gymnastatin and dankastatin alkaloids possess a veritable treasure trove of distinctive electrophilic functional groups, including chloroenone, α-chloroketone, epoxyketone, lactol, and α,β,γ,δ-unsaturated amide moieties; indeed, some members contain as many as five potential electrophilic sites.5 While detailed target identification studies are lacking, many of these tyrosine-derived alkaloids are reported to possess significant anti-cancer activity.4
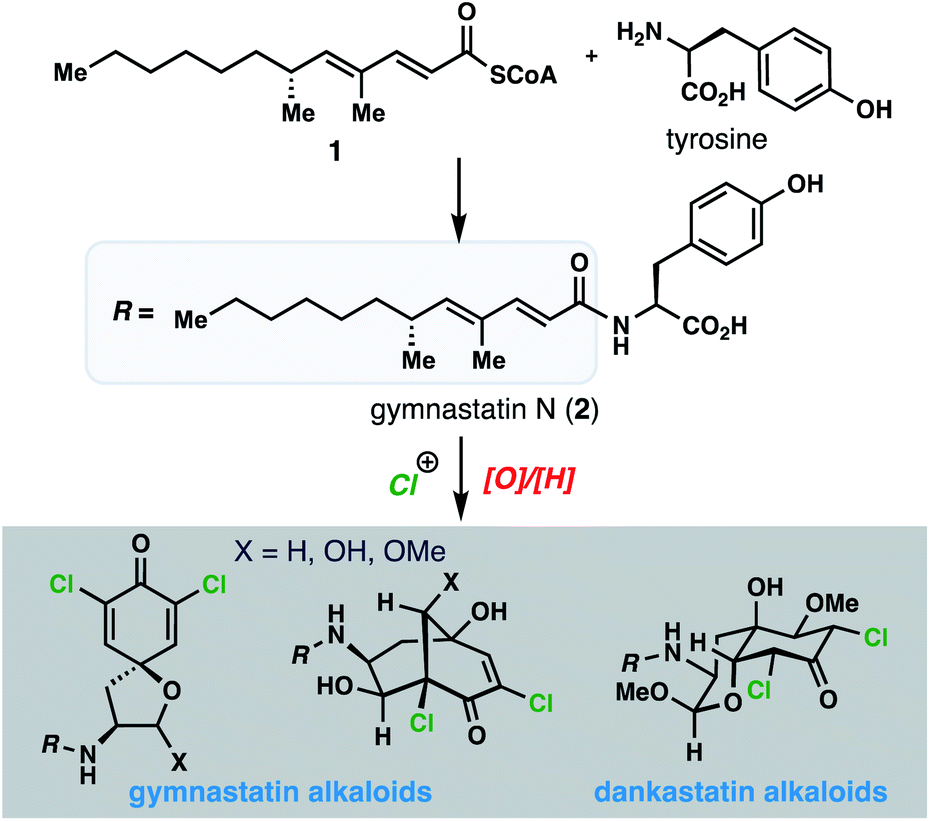 | ||
| Fig. 1 Tyrosine-derived alkaloids from Gymnascella sp. fungi. | ||
With over 20 members isolated possessing varying degrees of oxygenation, halogenation, and cyclization, it is not unreasonable to suspect that gymnastatin A (3) plays a central role in the biosynthesis of other tyrosine-derived alkaloids (see 4–14), possibly through chemistry which can be replicated without enzymes (Fig. 2). Indeed, biosynthetic logic has guided synthetic routes to various gymnastatin members and related alkaloids.6,7 Despite this, detailed chemical insight regarding the formation, stereochemistry, and interconversion of various members is lacking. Given our interest in bicyclo[3.3.1]nonane-containing natural products and covalently binding natural products, especially those containing similar lipophilic amide side chains,3 we sought to develop a unified synthetic platform to these alkaloids as a gateway into studying their biological targets.8,9 Herein we report simple synthetic solutions to multiple chlorinated gymnastatin and dankastatin metabolites, uncovering very subtle factors which favor the formation of a given skeletal type. We also provide an unappreciated link between this natural product family and the well-known fungal natural product aranorosin (15) which has also been isolated from a terrestrial variant of G. dankaliensis. Finally, we report growth inhibitory activity of six members spanning all three scaffold types (spirocyclic dienone, bicyclo[3.1.1]nonane, and oxo-decalin) against human triple negative breast cancer cells.
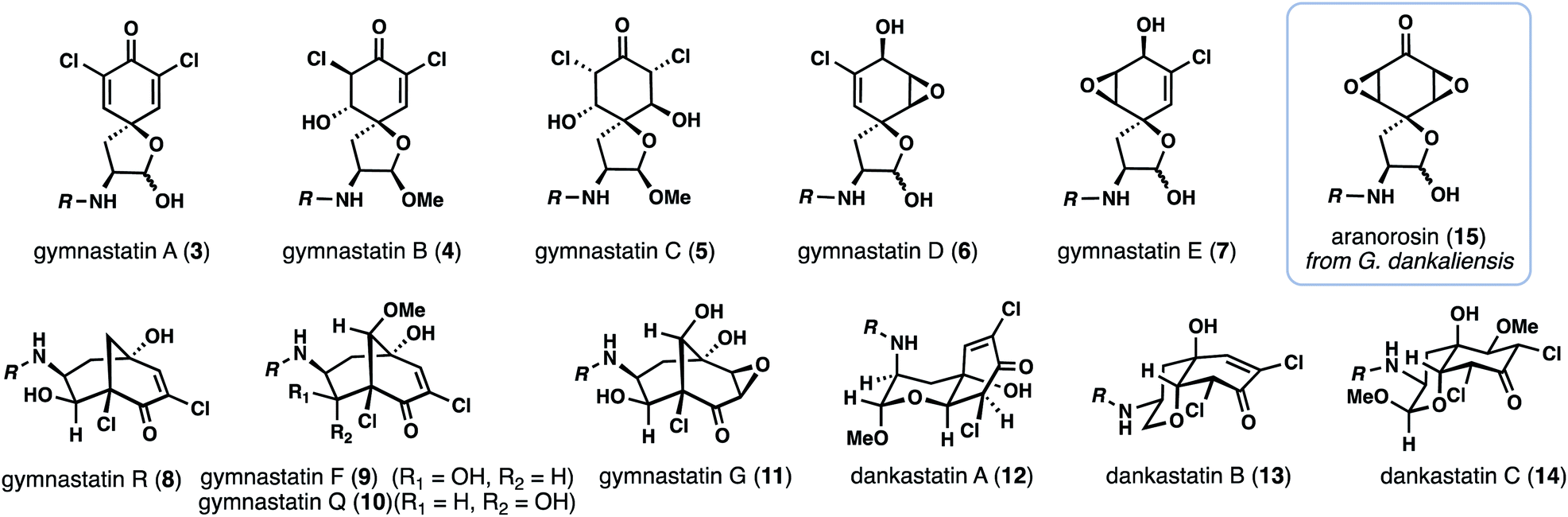 | ||
| Fig. 2 Selected chlorinated gymnastatin and dankastatin members and related natural product aranorosin. | ||
Results and discussion
Bicyclo[3.3.1]nonane-containing gymnastatins and the oxo-decalin-containing dankastatins are proposed to arise from 3via aldol (see 16) and oxa-Michael (see 17) pathways respectively (Fig. 3A).4 The presence of a C-9 methyl ether in gymnastatins F (9) and Q (10) relative to a secondary hydroxyl group in gymnastatins G (11) raises questions regarding the identity of the “OR” group that can trigger this process (i.e. H2O vs. MeOH), in addition to stereochemical concerns arising from inter- vs. intramolecular delivery of the oxygen nucleophile. Additionally, dankastatins exists as two different sets of oxo-cis-decalin diastereomers (compare 12vs.13/14); how (or if) nature controls the formation of a given isomer is an intriguing question.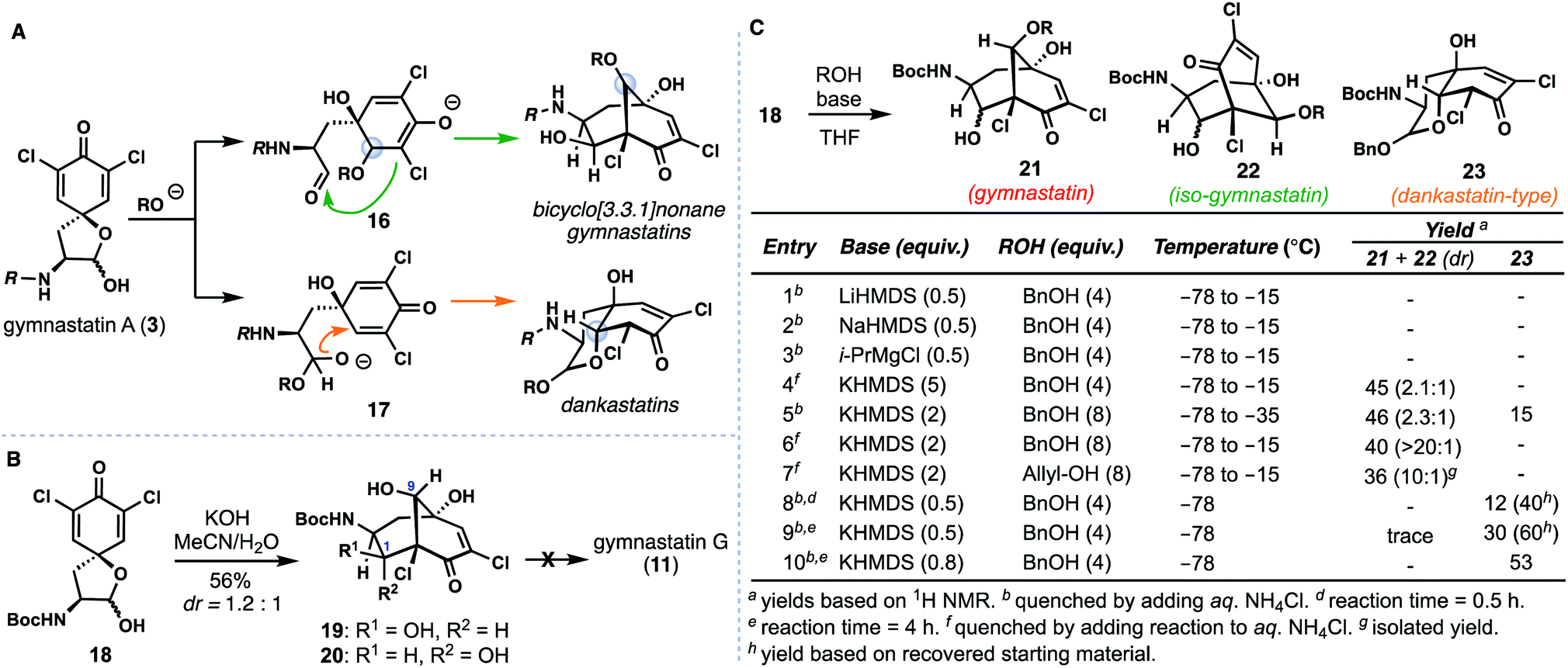 | ||
| Fig. 3 Understanding ring formation in the biosynthesis of gymnastatin and dankastatin alkaloids. (A) Chemical and stereochemical possibilities. (B) Stereochemical problems encountered when employing water as an oxygen nucleophile (C) optimization studies. | ||
We initially targeted gymnastatin G (11) owing to its potent reported activity against the P388 lymphocytic leukemia cell line, and reactive epoxyketone functionality.4a Inspired by the work of Nishiyama on ether-containing gymnastatins F/Q, we had hoped that simply substituting methanol with water would forge the bicyclo[3.3.1]nonane core of 11 in a biomimetic cascade (Fig. 3B).6d Known compound 18, derived from (L)-tyrosine,6 was treated with aqueous KOH in MeCN yielding two bicyclo[3.3.1]nonane-containing products, 19 and 20 in a 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Surprisingly, however, the C-9 stereocenter was incorrectly set during this process.10 Isomer 20 could be converted to 19 by treatment with catalytic amounts of base, suggesting that diastereomers at C-1 are formed in a reversible aldol reaction step, and that, the oxa-Michael addition step, albeit producing an undesired outcome, was stereoselective. Presumably this outcome arises from fast intramolecular oxa-Michael addition, wherein a hydrated aldehyde intermediate (see 17, R = H) serves to deliver the oxygen nucleophile internally forming the cis-6,6-fused (dankastatin-type) bicyclic lactol. Subsequent lactol ring-opening then generates an aldehyde which participates in the aldol process. This observed reactivity questions the strategy nature employs in setting the correct C-9 stereocenter if water is used as a nucleophile. From a synthetic standpoint, we were also not successful in advancing 19/20 into gymnastatin G (11) (vide infra).11
:1 ratio. Surprisingly, however, the C-9 stereocenter was incorrectly set during this process.10 Isomer 20 could be converted to 19 by treatment with catalytic amounts of base, suggesting that diastereomers at C-1 are formed in a reversible aldol reaction step, and that, the oxa-Michael addition step, albeit producing an undesired outcome, was stereoselective. Presumably this outcome arises from fast intramolecular oxa-Michael addition, wherein a hydrated aldehyde intermediate (see 17, R = H) serves to deliver the oxygen nucleophile internally forming the cis-6,6-fused (dankastatin-type) bicyclic lactol. Subsequent lactol ring-opening then generates an aldehyde which participates in the aldol process. This observed reactivity questions the strategy nature employs in setting the correct C-9 stereocenter if water is used as a nucleophile. From a synthetic standpoint, we were also not successful in advancing 19/20 into gymnastatin G (11) (vide infra).11
Given these results, we examined alternative alcohol-based nucleophiles in order to prevent the proposed reaction pathway that leads to undesired C-9 stereochemistry; the resulting alkyl ethers formed could in principle be deprotected and ultimately processed to 11 which we desired for biological testing (Fig. 3C). Dienone 18 was reacted with various quantities of either allyl or benzyl alcohol using a variety of bases and subsequently quenched at various temperatures. Employing sub stoichiometric quantities of Li-, Mg-, and Na-based bases was ineffective at low temperatures (entries 1–3), but potassium bases employed in excess afforded substantial amounts of the desired bicyclo [3.3.1]nonane 21 and isomeric counterpart 22 (entries 4–7). The gymnastatin-type scaffold was favored under these conditions, and optimal ratios of 21 were obtained using two equivalents of base (entries 6 and 7).12 Of note, in entry 5, wherein the reaction was kept colder, we observed formation of small amounts of the dankastatin scaffold (see 23) in addition to 21/22. Finally, maintaining a −78 °C reaction temperature (entries 8–10) led to substantial quantities of 23 showing that under carefully controlled conditions either scaffold can be generated from 18.
With conditions identified for construction of the key bicyclo[3.3.1]nonane core with the correct C-9 stereocenter, we reinvestigated the synthesis of gymnastatin G (11) (Scheme 1). While the epoxide found in 11 could be envisioned to arise from the chloroenone motif of gymnastatin F/Q, we had been unable to realize this process using previously prepared isomer 19/20.11 Given these observations, we proceeded to investigate a monochlorinated tyrosine derivative as a means to synthesize 11.
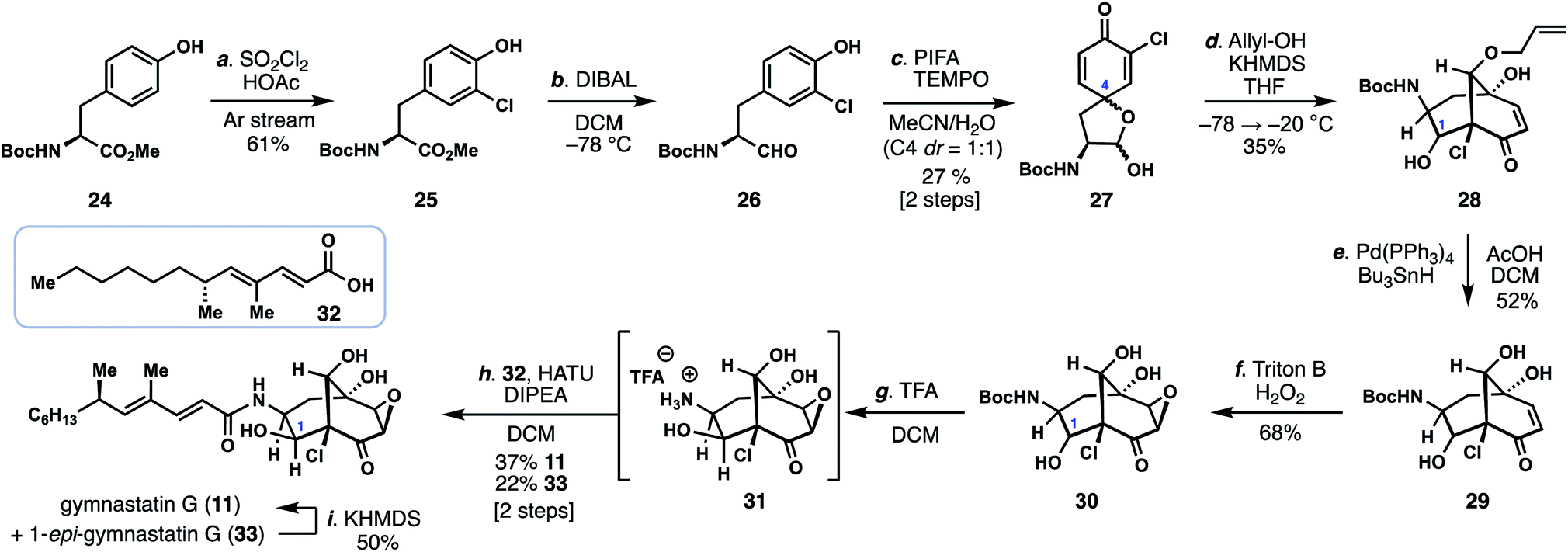 | ||
| Scheme 1 Total synthesis of gymnastatin G. | ||
Carefully controlled mono-chlorination of Boc-tyrosine methyl ester (24) was achieved using sulfuryl chloride under a stream of argon, by which the produced HCl could be removed thus preventing undesired removal of the Boc group under acidic conditions. The resulting ester (25) was then reduced with DIBAL providing aldehyde 26, which was subsequently dearomatized with PIFA in the presence of TEMPO to provide spirolactol 27 as a mixture of four diastereomers, two of which are inconsequential. Using conditions discovered previously (vide supra), treatment of 27 with allyl alcohol and KHMDS led to the bicyclo[3.3.1]nonane-containing product 28 in 35% yield as a mixture of diastereomers. Of note, the C-1 hydroxyl group, formed in the aldol step, was α-disposed in the major product. Moreover, no isomeric bicyclo[3.3.1]nonane-containing products possessing an α-chloroenone motif were formed. The carefully-chosen allyl protecting group was removed under reductive palladium-catalysis (Pd(PPh3)4, Bu3SnH) providing triol 29. Diastereoselective epoxidation of 29 with H2O2 and Triton B afforded epoxyketone 30; notably the basic conditions employed also partially epimerized the C-1 stereocenter (presumably via a retro-aldol/aldol reaction) to now favor β-stereochemistry as found in 11. The mixture of epoxides were then exposed to TFA, removing the Boc group, and the free amine (31) coupled to known acid 32 (HATU, DIPEA) thus delivering gymnastatin G (11) and its C1-epimer (33) in 60% combined yield.13 Epimer 33 could also be converted into 11 in 50% yield by treatment with KHMDS.
With access to the most complex gymnastatin member secured for biological testing, we turned our attention toward the dankastatin family given that our initial screening of cyclization conditions turned up conditions to favor this scaffold. Chlorinated dankastatin members (see 12–14), however, are produced with two different isomeric cis-decalin frameworks. Notably, in dankastatin A (12) the tertiary alcohol and neighboring proton (see C-4 and C-9) are on opposite faces as compared to dankastatins B (13) and C (14). In analogy to work in Fig. 3C, treating 18 with KHMDS/MeOH generated compound 37 and not the dankastatin A-type cis-fused skeleton 35 (Fig. 4A). We presume that in the cyclization of 18, an axial configuration of the amide side chain (see 34vs.36), disfavors formation of 35.14 Again, this raises the question as to how the dankastatin A-type skeleton is prepared in nature. Fortunately, isomer 37 does however, bear resemblance to dankastatins B and C, thus offering a potential pathway to these targets (Fig. 4B).
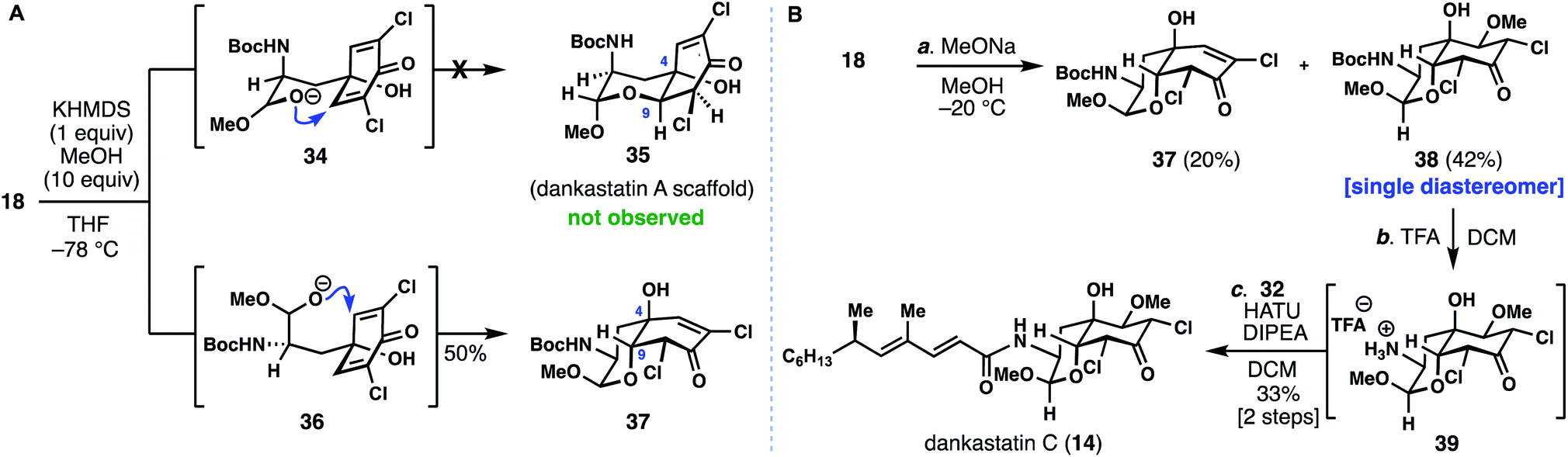 | ||
| Fig. 4 Studies towards the dankastatins. (A) Challenges in forming dankastatin A. (B) Total synthesis of dankastatin C. | ||
Dankastatin C, a more recently isolated member of the dankastatin family,4c possesses a structure suggestive of a hydration event on a biosynthetic intermediate akin to chloroenone 37. In order to synthesize this structure, subtle adjustments were made to the conditions for the intramolecular oxa-Michael addition. Sodium methoxide was utilized as base with MeOH as solvent and the reaction mixture maintained at −20 °C for a prolonged period—long enough for the double MeOH adduct (38) to be the major product but without significant bicyclo[3.3.1]nonane formation. If KHMDS was used as base, bicyclo[3.3.1]nonane-containing products predominated. Through this process, 38 was formed as a single diastereomer in 42% yield along with 20% of 37. Finally, Boc deprotection of 38 with TFA, followed by coupling with side chain 32 (HATU, DIPEA) forged dankastatin C (14).
Unlike dankastatin C, and in fact the majority of other tyrosine-derived alkaloids from Gymnascella, dankastatin B (14) features an alcohol, rather than aldehyde, oxidation state at C-1. To access this natural product, Boc-tyrosine methyl ester (24) was dichlorinated (SOCl2, HOAc) and reduced with DIBAL to yield 40 (Fig. 5). With 40 in hand, we sought to find suitable oxidative dearomatization conditions that were compatible with the free hydroxy group. After some exploration, success was realized using singlet oxygen-based conditions (O2, TPP, hν) in the presence of cesium carbonate (see inset). The yields of this process were initially quite low (entries 1–3), but in the presence of PPh3 the dankastatin core (see 42) could be formed directly, albeit in low yield (entry 4). Interestingly, in addition to 42, we observed a minor isomer (iso-42) which corresponds to the dankastatin A scaffold (dr ∼ 1.6:1). Through reductant and temperature optimization (entries 5–11), we found that high yields of dienone 41 (70%) could be obtained using an electron-deficient phosphine (P(3,5-(CF3)2C6H3)3) at low temperature. Isolated 41 could then be converted to 42 under basic conditions (KHMDS) in 78% yield. While this sequence requires two steps, the yield (78%) and diastereoselectivity (dr = 8:1) were substantially higher than the one-pot transformation. Deprotection of 42 (TFA) and coupling with acid 32 yield dankastatin B (13).
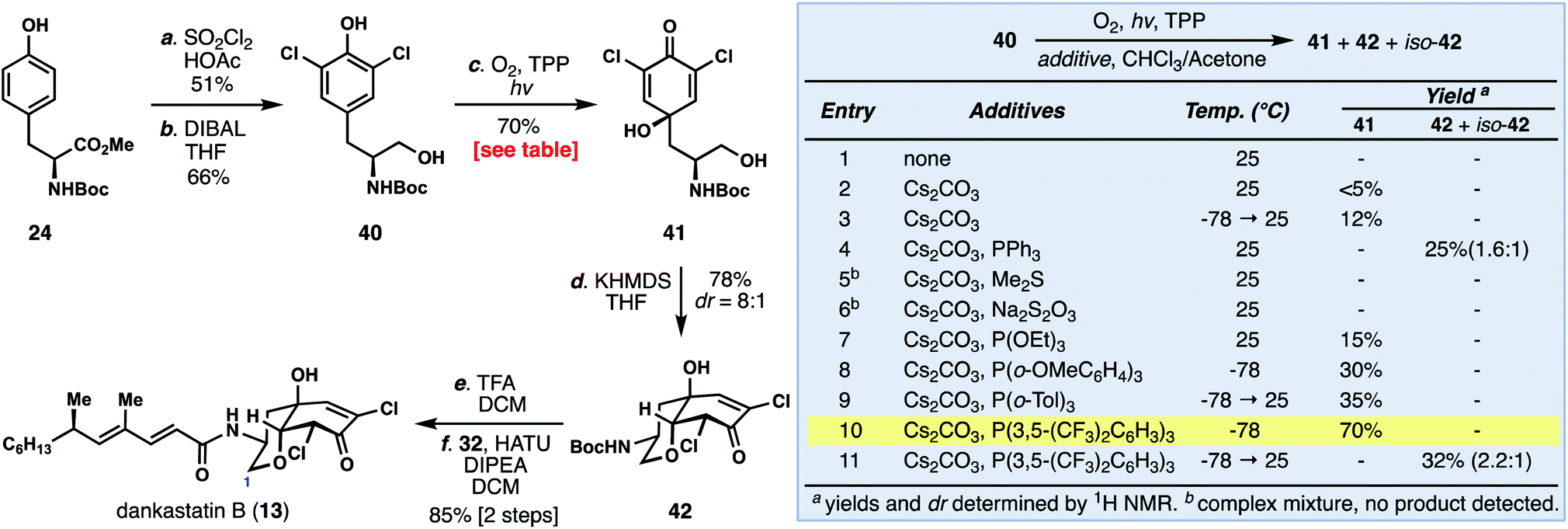 | ||
| Fig. 5 Total synthesis of dankastatin B. | ||
The successful application of proposed biomimetic strategies in the synthesis of dankastatin and gymnastatin alkaloids sheds some light on how nature might make these natural products and the challenges it faces and/or solves in doing so. Yet problems we encountered in our pursuit of gymnastatin G and dankastatin A led us to consider alternative hypotheses for the origins of these chlorinated alkaloids derived from tyrosine. Specifically, we were drawn to the bis-epoxyketone-containing natural product aranorosin (15), which is not halogenated, but bears clear structural, and likely biosynthetic, similarities to 3–14.4d Notably, the α-chloroenone in gymnastatin A and the epoxyketone in aranorosin are of the same oxidation level and we wondered if nature might use nucleophilic, chloride-mediated chemistry and not electrophilic chloronium-induced reactions in the construction of this alkaloid family.15,16
Commercially available aranorosin reacted with LiCl (1.5 equiv.) at room temperature in THF, forming a variety of chlorinated products under very mild conditions (Fig. 6).17 Notably, gymnastatin G (11) and 1-epi-gymnastatin G (33) were isolated from the reaction mixture in 23% combined yield, presumably through an aldol reaction of intermediate 46. In addition, two more natural products, namely aranochlor A (44) and aranochlor B (43), which are oxidized variants of gymnastatins D (6) and E (7) respectively, were also formed in the reaction (in 12%) and can be viewed as links between gymnastatin A and aranorosin.18 Notably, the two diastereomeric natural products (presumably generated via E1cB reactions of 45 and 46) were generated in nearly a 1:1 ratio—an apparent result of nonselective epoxide opening; this observation echoes back to the two diastereomeric skeletons of dankastatins found in nature. Also detected was a small amount of unsaturated aldehyde 47, a structure reminiscent of prior C–C cleavage products.12 While we are unaware of 47 being a real natural product, it is interesting to consider whether Gymnascella dankaliensis might employ oxidized tyrosines as precursors to electrophilic small molecules containing dehydroalanine-like motifs. In any event, investigations into the enzymology surrounding gymnastatin and dankastatin alkaloid biosynthesis is appealing.
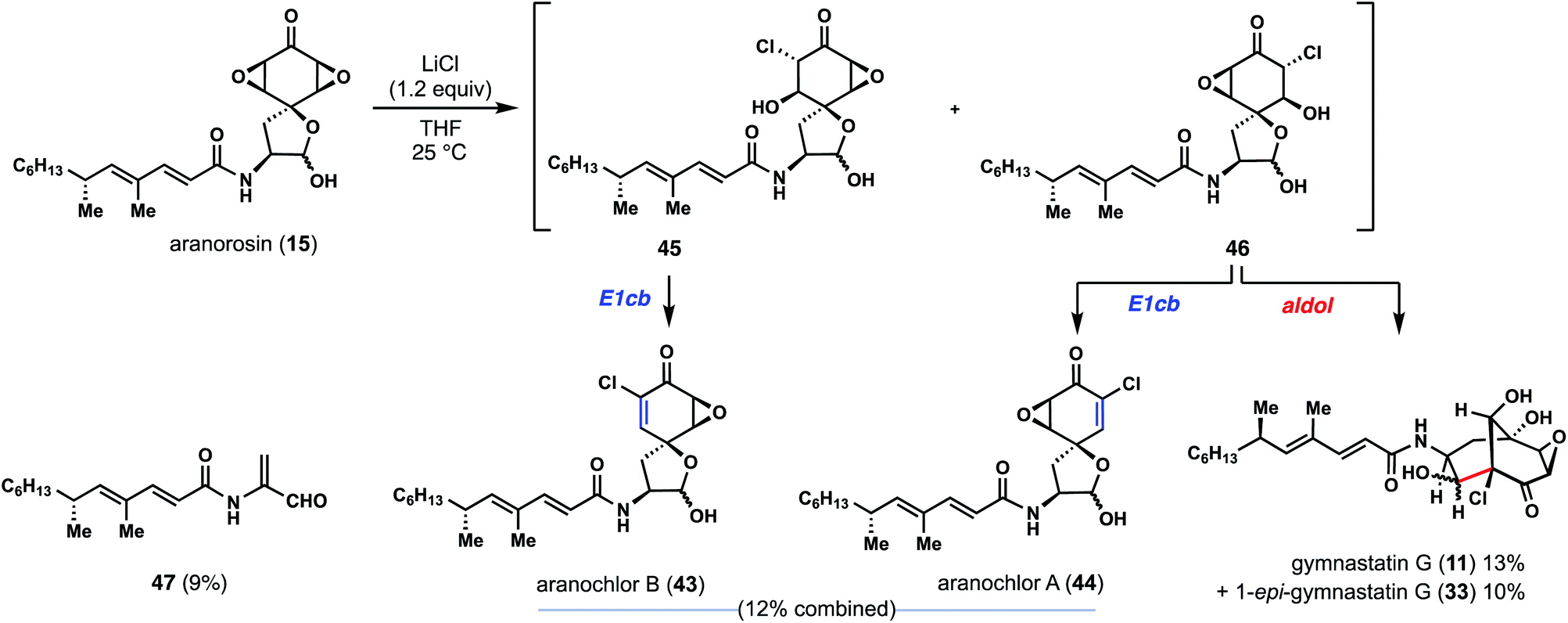 | ||
| Fig. 6 Aranorosin as a possible biosynthetic precursor to chlorinated alkaloids from Gymnascella sp. | ||
With access to these natural products, which contain all of the relevant structural types found in this family, we evaluated their cytotoxicity against aggressive human triple negative breast cancer cells (231MFP cell line) (Fig. 7).19 As noted, many chlorinated tyrosine-derived alkaloids have shown strong anticancer activity, although many of these studies have been conducted in murine tumor cell lines.4 Dankastatin B exhibited the highest potency (EC50 = 0.6 μM) followed closely by aranorosin (EC50 = 1.6 μM), gymnastatin A (EC50 = 2.1 μM), and finally dankastatin C (EC50 = 5 μM). Interestingly, bicyclo[3.3.1]nonane-containing gymnastatins Q and G, which notably also contain only a single electrophilic site in the oxidized tyrosine core, were far less active in this cellular context. Whether these natural products have the same (or related) biological target profiles remains to be determined – work to address these questions is currently underway and will be reported in due course.
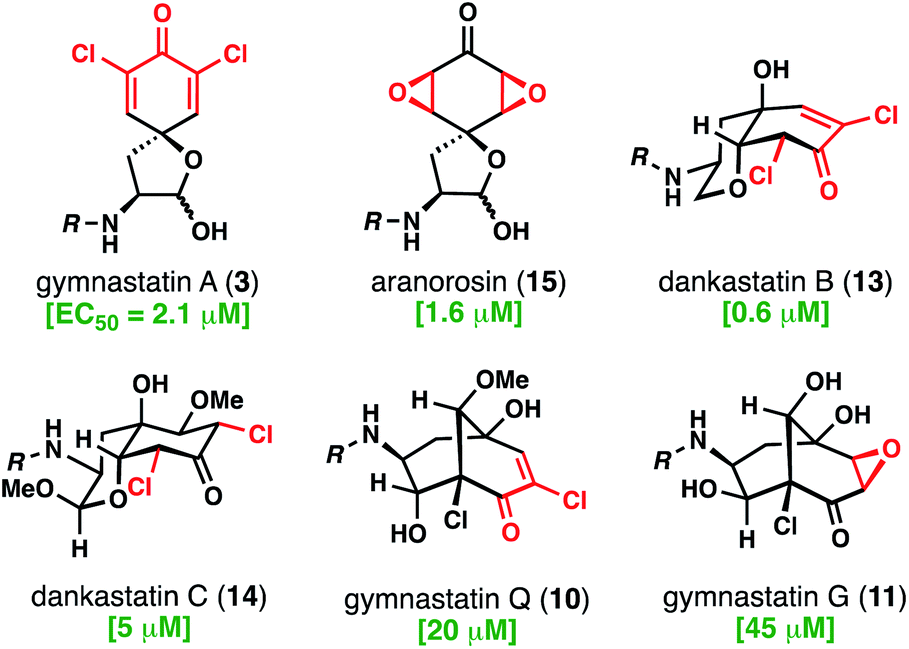 | ||
| Fig. 7 Anti-triple negative breast cancer (231 MFP) activity of select electrophilic alkaloids from Gymnascella sp. | ||
Conclusion
In summary, we have completed the first total syntheses of gymnastatin G and dankastatins B and C, and in the process, explored stereochemical and structural questions surrounding the origins of chlorinated, tyrosine-derived alkaloids. During our studies, we discovered that very small and subtle changes to abiotic reaction conditions could be leveraged to promote the formation of either oxo-decalin or bicyclo[3.1.1]nonane motifs; how nature modulates these product ratios remains an open and interesting question. Additionally, an alternative biosynthetic hypothesis for the origins of chlorinated gymnastatin alkaloids from the well-known fungal metabolite aranorosin was also presented; notably this pathway can circumvent certain stereochemical problems associated with the abiotic Michael/aldol cascade approach. Finally, as a result of these investigations, dankastatin B has emerged as a potent, and easily synthesized, small molecule hit against triple negative breast cancer.Author contributions
T. J. M. and B. T. initiated the project and B. T. conducted all of the synthetic experiments. B. P. B. and D. K. N. performed biological assays. T. J. M. wrote the paper with the assistance of B. T. All authors provided feedback and contributed to editing the manuscript.Conflicts of interest
This study was funded in part by the Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies. Daniel K. Nomura is a co-founder, shareholder, and adviser for Frontier Medicines.Acknowledgements
Financial support is acknowledged from the NIH NIGMS (R01GM136945) and the Novartis Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT). T. J. M. acknowledges unrestricted financial support from Novartis, Bristol-Myers Squibb, Amgen, and Eli Lilly. We are grateful to Dr Hasan Celik and Dr Jeffrey Pelton for NMR spectroscopic assistance and NIH grants GM68933 and S10OD024998. Dr Nicholas Settineri is acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged.Notes and references
- (a) D. J. Newman and G. M. Cragg, Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019, J. Nat. Prod., 2020, 83, 770 CrossRef CAS PubMed; (b) S. E. Kearney, et al., Canvass: A Crowd-Sourced, Natural-Product Screening Library for Exploring Biological Space, ACS Cent. Sci., 2018, 4, 1727 CrossRef CAS PubMed.
- D. K. Nomura and T. J. Maimone. Target Identification of Bioactive Covalently-Acting Natural Products, in Activity-Based Protein Profiling. Curr. Topics in Microbiol. Immunol., ed. B. F. Cravatt, K.-L. Hsu and E. Weerapana, Springer, Switzerland, 2019, vol. 420, pp. 351–374 Search PubMed.
- Y. Isobe, M. Okumura, L. M. McGreggor, S. M. Brittain, M. D. Jones, X. Liang, R. White, W. Forrester, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, Manumycin polyketides act as molecular glues between UBR7 and P53, Nat. Chem. Biol., 2020, 16, 1189 CrossRef CAS PubMed.
- (a) T. Amagata, K. Minoura and A. Numata, Gymnastatins F–H, Cytostatic Metabolites from the Sponge-Derived Fungus Gymnascella dankaliensis, J. Nat. Prod., 2006, 69, 1384 CrossRef CAS PubMed; (b) T. Amagata, M. Tanaka, T. Yamada, K. Minoura and A. Numata, Gymnastatins and Dankastatins, Growth Inhibitory Metabolites of a Gymnascella Species from a Halichondria Sponge, J. Nat. Prod., 2008, 71, 340 CrossRef CAS PubMed; (c) T. Amagata, T. Makoto, T. Yamada, Y.-P. Chen, K. Minoura and A. Numata, Additional cytotoxic substances isolated from the sponge-derived Gymnascella dankaliensis, Tetrahedron Lett., 2013, 54, 5960 CrossRef CAS; (d) L. Hammerschmidt, A. H. Aly, M. Abdel-Aziz, W. E. G. Müller, W. Lin, G. Daletos and P. Proksch, Cytotoxic acyl amides from the soil fungus Gymnascella dankaliensis, Bioorg. Med. Chem., 2015, 23, 712 CrossRef CAS PubMed; (e) H. Wang, H. Dai, C. Heering, C. Janiak, W. Lin, R. S. Orfali, W. E. G. Müller, Z. Liu and P. Proksch, Targeted solid phase fermentation of the soil dwelling fungus Gymnascella dankaliensis yields new brominated tyrosine-derived alkaloids, RSC Adv., 2016, 6, 81685 RSC; (f) D. Xu, M. Luo, F. Liu, D. Wang, X. Pang, T. Zhao, L. Xu, X. Wu, M. Xia and X. Yang, Cytochalasan and Tyrosine-Derived Alkaloids from the Marine Sediment-Derived Fungus Westerdykella dispersa and their Bioactivities, Sci. Rep., 2017, 7, 11956 CrossRef PubMed.
- Notably, many brominated variants of these alkaloids also exist, see: ref. 4d and e.
- For syntheses of various gymnastatins, see: (a) M. K. Gurjar and P. Bhaket, Total Synthesis of a Novel Cytotoxic Metabolite Gymnastatin A, Heterocycles, 2000, 53, 143 CrossRef CAS; (b) C. W. Phoon, B. Somanadhan, S. C. H. Heng, A. Ngo, S. B. Ng, M. S. Butler, A. D. Buss and M. M. Sim, Isolation and total synthesis of gymnastatin N, a POLO-like kinase 1 active constituent from the fungus Arachniotus punctatus, Tetrahedron, 2004, 60, 11619 CrossRef CAS; (c) T. Ogamino, S. Ohnishi, Y. Ishikawa, T. Sugai, R. Obata and S. Nishiyama, Synthesis and biological assessment of hemiacetal spiro derivatives towards development of efficient chemotherapeutic agent, Sci. Technol. Adv. Mater., 2006, 7, 175 CrossRef CAS; (d) K. Murayama, T. Tanabe, Y. Ishikawa, K. Nakamura and S. Nishiyama, A synthetic study on gymnastatins F and Q: the tandem Michael and aldol reaction approach, Tetrahedron Lett., 2009, 50, 3191 CrossRef CAS; (e) L. Raffier and O. Piva, Application of the diastereoselective photodeconjugation of α,β-unsaturated esters to the synthesis of gymnastatin H, Beilstein J. Org. Chem., 2011, 7, 151 CrossRef CAS PubMed.
- For selected syntheses of biogenetically related alkaloids, see: (a) P. Wipf, Y. Kim and P. C. Fritch, Total synthesis and structure assignment of the antitumor antibiotic aranorosin, J. Org. Chem., 1993, 58, 7195 CrossRef CAS; (b) A. McKillop, L. McLaren, R. J. K. Taylor, R. J. Watson and N. J. Lewis, The total synthesis of the diepoxycyclohexanone antibiotic aranorosin and novel synthetic analogues, J. Chem. Soc., Perkin Trans. 1, 1996, 1385 RSC; (c) A. McKillop, L. McLaren, R. J. Watson, R. J. K. Taylor and N. Lewis, A concise synthesis of the novel antibiotic aranorosin, Tetrahedron Lett., 1993, 34, 5519 CrossRef CAS; (d) H. Yu., Y. Zong and T. Xu, Total synthesis of (−)-penicimutanin a and related congeners, Chem. Sci., 2020, 11, 656 RSC; (e) H. Yu, Y. Tian, Y. Zong, J. Zhang and T. Xu, Total Synthesis and Structural Reassignment of Aranorosinol A, Aranorosinol B, and EI-2128-1, J. Org. Chem., 2020, 85, 4335 CrossRef CAS PubMed.
- (a) C. P. Ting and T. J. Maimone, Total Synthesis of Hyperforin, J. Am. Chem. Soc., 2015, 137, 10516 CrossRef CAS PubMed; (b) C. P. Ting, G. Xu, X. Zeng and T. J. Maimone, Annulative Methods Enable a Total Synthesis of the Complex Meroterpene Berkeleyone A, J. Am. Chem. Soc., 2016, 138, 14868 CrossRef CAS PubMed; (c) G. Xu, M. Elkin, D. J. Tantillo, T. R. Newhouse and T. J. Maimone, Traversing Biosynthetic Carbocation Landscapes in the Total Synthesis of Andrastin and Terretonin Meroterpenes, Angew. Chem., Int. Ed., 2017, 56, 12498 CrossRef CAS PubMed; (d) X. Shen, C. P. Ting, G. Xu and T. J. Maimone, Programmable Meroterpene Synthesis,, Nat. Commun., 2020, 11, 508 CrossRef CAS PubMed.
- For recent studies from our group, see: (a) C. A. Berdan, R. Ho, H. S. Lehtola, M. To, X. Hu, T. R. Huffman, Y. Petri, C. R. Altobelli, S. G. Demeulenaere, J. A. Olzmann, T. J. Maimone and D. K. Nomura, Parthenolide Covalently Targets and Inhibits Focal Adhesion Kinase in Breast Cancer Cells, Cell Chem. Biol., 2019, 26, 1027 CrossRef CAS PubMed; (b) J. N. Spradlin, X. Hu, C. C. Ward, S. M. Brittain, M. D. Jones, L. Ou, M. To, A. Proudfoot, E. Ornelas, M. Woldegiorgis, J. A. Olzmann, D. E. Bussiere, J. R. Thomas, J. A. Tallarico, J. M. McKenna, M. Schirle, T. J. Maimone and D. K. Nomura, Harnessing the Anti-Cancer Natural Product Nimbolide for Targeted Protein Degradation, Nat. Chem. Biol., 2019, 15, 747 CrossRef CAS PubMed; (c) B. Tong, J. N. Spradlin, L. F. T. Novaes, E. Zhang, X. Hu, M. Moeller, S. M. Brittain, L. M. McGregor, J. M. McKenna, J. A. Tallarico, M. Schirle, T. J. Maimone and D. K. Nomura, A Nimbolide-Based Kinase Degrader Preferentially Degrades Oncogenic BCR-ABL, ACS Chem. Biol., 2020, 15, 1788 CrossRef CAS PubMed; (d) B. Tong, M. Luo, Y. Xie, J. N. Spradlin, J. A. Tallarico, J. M. Mckenna, M. Shirle, T. J. Maimone and D. K. Nomura, Bardoxolone Conjugation Enables Targeted Protein Degradation of BRD4, Sci. Rep., 2020, 10, 15543 CrossRef CAS PubMed.
- X-ray crystallography confirmed the incorrect stereochemistry at C-9.
- In principle, the epoxide motif found in gymnastatin G could arise from the chloroenone found in gymnastatins F/Q via conjugate addition of water and halide displacement. In our hands, this reaction did not occur using 19/20. Additionally, chemoselective hydrodechlorination of the vinyl chloride in the presence of the tertiary chloride was not possible.
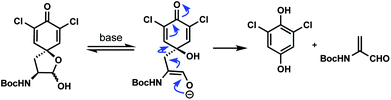
- A somewhat significant (up to 20%) side reaction in many of these reactions was the cleavage reaction shown below. A related process in proteins involving tyrosine has been observed during thyroid hormone synthesis, see: J.-M. Gavaret, H. J. Cahnmann and J. Nunez, Thyroid hormone synthesis in thyroglobulin. The mechanism of the coupling reaction, J. Biol. Chem., 1981, 256, 9167 CrossRef CAS
 .
. - While gymnastatin G was reported as quite labile, we found our synthetic material was stable enough for full characterization. Nevertheless, the synthetic gymnastatin G was acylated with Ac2O, and the NMR resonances of the resulting diacetate matched those reported by the Numata group.
- The same result was obtained using gymnastatin A indicating this outcome is not unique to the Boc protecting group.
- While there is no evidence that such a transformation is related to the actual biosynthesis of these alkaloids, we note that chloroperoxidases can also carry out P450-like transformations. Given that an oxidase likely constructs aranorosin's bis epoxide motif through double epoxidation of a dienone (see ref. 4d), it is not inconceivable to consider that outer sphere attack by chloride on an initially oxidized product could be relevant to the biosynthesis of these metabolites.
- Studies by the Wipf lab on the reactivity of aranorosin with sulfur nucleophiles has shown that bicyclo[3.3.1]nonane-containing products can be formed, see: (a) P. Wipf, P. Jeger and Y. Kim, Thiophilic ring-opening and rearrangement reactions of epoxyketone natural products, Bioorg. Med. Chem. Lett., 1998, 8, 351 CrossRef CAS PubMed; (b) J. T. Hammill, J. Contreras-García, A. M. Virshup, D. N. Beratan, W. Yang and P. Wipf, Synthesis and chemical diversity analysis of bicyclo[3.3.1]non-3-en-2-ones, Tetrahedron, 2010, 66, 5852 CrossRef CAS PubMed.
- Notably, these conditions are unusually mild for chloride-induced opening/elimination of an epoxyketone, which typically require acid activation or elevated temperatures. Presumably the lithium cation is able to fulfill this role given the highly activated aranorosin bis-epoxide system. Importantly, when tetrabutyl ammonium chloride was used, no conversion occurred under the same conditions.
- T. Mukhopadhyay, R. G. Bhat, K. Roy, E. K. S. Vijayakumar and B. N. Ganguli, Aranochlor A and Aranochlor B, Two New Metabolites from Pseudoarachniotus roseus: Production, Isolation, Structure Elucidation and Biological Properties, J. Antibiot., 1998, 51, 439 CrossRef CAS PubMed.
- N. Jessani, M. Humphrey, W. H. Mcdonald, S. Niessen, K. Masuda, B. Gangadharan, J. R. Yates III, B. M. Mueller and B. F. Cravatt, Carcinoma and stromal enzyme activity profiles associated with breast tumor growth in vivo, Proc. Natl. Acad. Sci., 2004, 101, 13756 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data. See DOI: 10.1039/d1sc02613e |
| This journal is © The Royal Society of Chemistry 2021 |