DOI:
10.1039/D1SC01952J
(Edge Article)
Chem. Sci., 2021,
12, 8872-8883
Proton-coupled electron transfer reactivities of electronically divergent heme superoxide intermediates: a kinetic, thermodynamic, and theoretical study†
Received
8th April 2021
, Accepted 26th May 2021
First published on 27th May 2021
Abstract
Heme superoxides are one of the most versatile metallo-intermediates in biology, and they mediate a vast variety of oxidation and oxygenation reactions involving O2(g). Overall proton-coupled electron transfer (PCET) processes they facilitate may proceed via several different mechanistic pathways, attributes of which are not yet fully understood. Herein we present a detailed investigation into concerted PCET events of a series of geometrically similar, but electronically disparate synthetic heme superoxide mimics, where unprecedented, PCET feasibility-determining electronic effects of the heme center have been identified. These electronic factors firmly modulate both thermodynamic and kinetic parameters that are central to PCET, as supported by our experimental and theoretical observations. Consistently, the most electron-deficient superoxide adduct shows the strongest driving force for PCET, whereas the most electron-rich system remains unreactive. The pivotal role of these findings in understanding significant heme systems in biology, as well as in alternative energy applications is also discussed.
Introduction
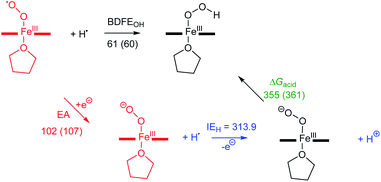
Activation of dioxygen by heme proteins plays a pivotal role in metalloenzyme-mediated oxidation, oxygenation, and dioxygen reduction reactions in biology.1 The central paradigm of this dioxygen binding and activation process by heme centers embodies a distinctive panel of intermediates, where the stepwise reduction of O2 leading up to O–O bond cleavage occurs in parallel to oxidation of the heme iron center. The initial heme–dioxygen adduct (i.e., heme–superoxo (FeIII–O2−˙) or heme–oxy (FeII–O2) species) is common to all dioxygen activating heme enzymes, and exhibits a remarkably divergent reactivity profile mainly depending upon the intricate structural tuning within a given active site. Specifically, the identity and properties of the amino acid side chain ligating at the heme proximal site, distal and proximal non-covalent interactions about the heme center, and electronic properties of the heme ligand itself,1a,2 all of which, in concert choreograph the specific biological role of a heme superoxide intermediate. These include implications in (Chart 1): (i) reversible dioxygen binding in hemoglobin (Hb) and myoglobin (Mb);3 (ii) indole dioxygenation reactivity in tryptophan and indoleamine 2,3-dioxygenases (TDO/IDO);4 (iii) indole monooxygenation by MarE;5 (iv) interaction with physiologically present nitric oxide (˙NO(g)) to generate heme peroxynitrite (FeIII–OONO) species in tryptophan nitrating TxtE6 and other proteins;7 (v) reactivity with electrons and/or protons in native mechanisms of heme oxygenase (HO),1b,8 cytochrome P450 (Cyt P450),9 aromatase,10 and nitric oxide synthase (NOS);11 (vi) abstraction of a hydrogen atom in one of the proposed mechanisms of nitric oxide synthase.1a,12 Particularly, heme superoxide reactivities with exogenous electrons and/or electrons and protons are central to an array of heme enzymes, where the site of reduction and/or protonation is critical for the overall outcome of the biological function. For example, reduction followed by protonation of the distal oxygen atom with respect to the iron center gives rise to the corresponding heme hydroperoxide species (e.g., Cyt. P450),9d whereas the protonation at the proximal oxygen has been often shown to liberate protonated superoxide, as in the case of unproductive degradation of oxyhemoglobin to met-hemoglobin.13
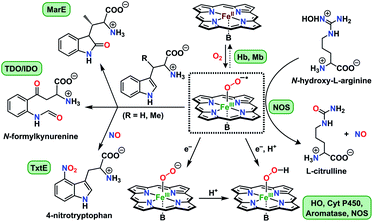 |
| | Chart 1 Versatile biological reactivity pathways of heme superoxo/heme oxy intermediates (shown in the center); B = axially coordinating amino acid side chain at the heme proximal site. | |
Detailed physicochemical elucidation of reduction–protonation chemistries of heme superoxide adducts is also of supreme interest with regard to the oxygen reduction reaction (ORR), a cornerstone in numerous critical biological (e.g., cellular respiration and oxidative phosphorylation) and industrial (e.g., synthetic catalysis and batteries) processes.14 In that, whether the heme superoxide abstracts a hydrogen atom (H˙ = H+ + e−) in a single mechanistic step (i.e., concerted), or an electron and a proton (or vice versa) in two consecutive steps (i.e., stepwise) is salient in dictating the overall thermodynamics of the reaction landscape. All of these concerted and stepwise mechanistic possibilities fall under the umbrella of proton-coupled electron transfer (PCET) reactions,15 of which, the precise mechanistic details are solely dependent on a few key thermodynamic parameters (vide infra).16 Recent thorough investigations by Mayer and coworkers have underscored the importance of PCET processes of heme superoxide adducts with relevance to the cathodic reaction in fuel cells, where efficient, cheap dioxygen reducing metallocatalysts could be of momentous importance.17 Specifically, the proton affinity of the heme-bound superoxide moiety is critical as it dictates the overpotential barrier of the ORR catalyst, thereby determining the rate/efficiency of the catalytic process.17b,18 Essentially, the pKa of the protonating acid is climacteric for the overall catalytic outcome as it should deliver weakly interacting protons that enhance the susceptibility of heme superoxide toward reduction, while preventing dissociation of protonated superoxide; protonated superoxide radicals are highly unstable, and have long known to decay giving O2 and H2O2 (2HO2−˙ → H2O2 + O2).19 Such subtleties related to pKa of the proton source are also critical further downstream in the ORR pathway, where protonation of proximal or distal O-atom of the heme hydroperoxide dictates whether 2-electron (O2(g) + 2H+ + 2e− → H2O2) or 4-electron (O2(g) + 4H+ + 4e− → 2H2O) reduction of oxygen is accomplished, respectively.20 Patently, the latter process is of preference for fuel cell applications, where the complete 4-electron reduction of O2(g) to H2O engender an efficient cathodic reaction, preventing the generation of partially reduced reactive oxygen species. Suitably, the detailed body of work by Dey and coworkers on bio-inspired ORR electrocatalysis have offered classic examples of how to adapt inexpensive, environmentally benign metallosystems for the efficient reduction of O2(g) to H2O.20c,21
Despite the widespread significance of heme superoxide mediated PCET pathways, a comprehensive fundamental understanding of the exact mechanistic, structural, and thermochemical parameters governing these processes is still lacking. Synthetic small molecule models can be useful tools in this endeavor, where in-depth thermodynamic and kinetic investigations into systematically varied heme structures are more feasible. Nonetheless, heme superoxide mimics are abundantly known as incompetent oxidants, and their directed reactivities with organic substrates are extremely scarce. Indeed, our recent work marks the first report where synthetic heme superoxide intermediates were shown to react with exogenously added indole substrates in the efficient modelling of tryptophan dioxygenation chemistry of indoleamine and tryptophan 2,3-dioxygenases.22 Similarly, heme superoxide adducts that efficiently react with added acids (i.e., protons (H+)), reductants (i.e., electrons(e−)), and/or H˙ donors are only a handful.23 Intriguingly, Naruta,24 Dey,25 and their coworkers have presented unique examples of heme superoxide intermediates that react with intramolecular H+ or H˙ donors, ultimately giving rise to the corresponding heme hydroperoxo (FeIII–OOH) adduct. To the best of our knowledge, recent work by Karlin and coworkers is the only instance where proton-coupled electron transfer reactivities of heme superoxide intermediates have been shown, where H˙ abstraction from an exogenous, weak O–H bond substrate generates the corresponding heme hydroperoxo species in a single kinetic step.26
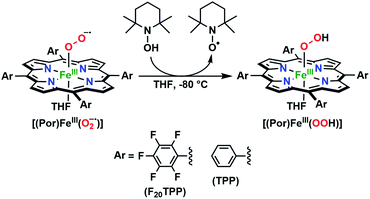
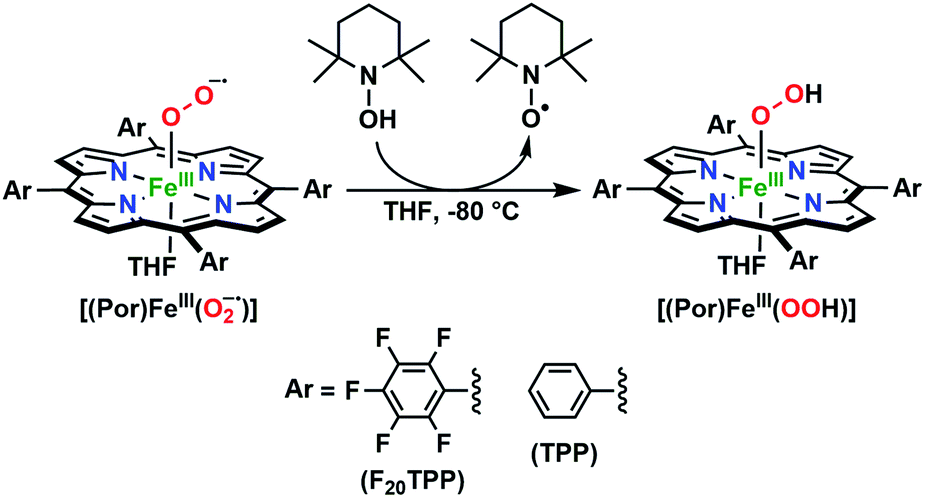
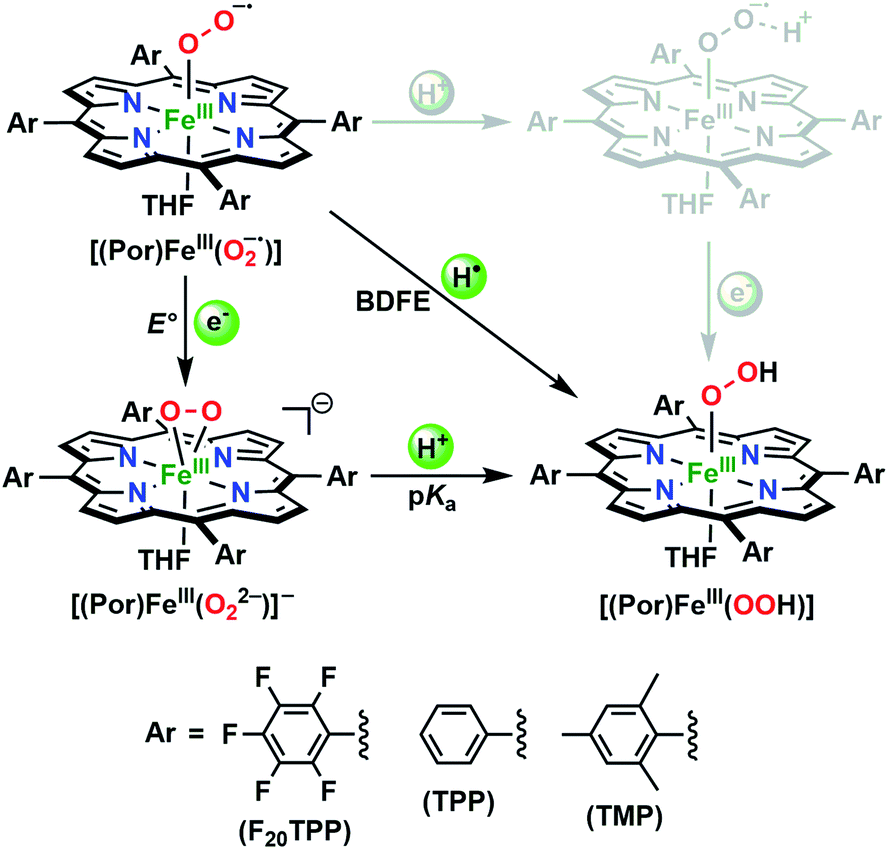
In the present investigation, we have interrogated the concerted proton–electron (i.e., H˙) abstraction reactivities of three electronically different, geometrically similar heme iron superoxo complexes, [(Por)FeIII(O2−˙)] (where Por = porphyrin supporting ligand; Chart 2), using the weak O–H bond substrate TEMPO–H (TEMPO–H = 1-hydroxy-2,2,6,6-tetramethylpiperidine; BDFE = 66.5 kcal mol−1 in THF16a), resulting in the corresponding heme hydroperoxo adduct, [(Por)FeIII(OOH)], under cryogenic conditions. In detail experimental and theoretical analyses into key thermodynamic and kinetic parameters are also presented, revealing intriguing insights into how subtle alterations in electronic atmospheres about heme centers can effect profound outcomes in their physical properties and related reactivities. In that, the most electron-deficient superoxide adduct reacts the fastest with TEMPO–H, while the most electron-rich superoxo species remains unreactive. This reactivity pattern is further corroborated by the experimentally determined (i.e., by means of the Bordwell relationship; eqn (1)27) O–H bond dissociation free energies (BDFEs) of the heme hydroperoxo products. This study marks the first report where detailed thermodynamic (elucidation of pKa, E°, and BDFE values) and kinetic (rate comparisons, kinetic isotope effects and activation parameters) investigations are described for a series of structurally similar, electronically divergent heme superoxide intermediates, along with strong theoretical justification. These findings are crucial in the unequivocal comprehension of both biological systems that are indispensable in human therapeutic applications, as well as processes central to alternative energy sources such as fuel cells.
| | | BDFE = 1.37(pKa) + 23.06E° + CG,solv | (1) |
 |
| | Chart 2 The series of meso-substituted heme superoxide intermediates with varied electronic properties discussed in this work. | |
Results and discussion
Formation and PCET reactivities of [(Por)FeIII(O2−˙)] complexes
The series of superoxo adducts utilized for this study offer disparate electronic atmospheres about the heme center (Chart 2): F20TPP (F20TPP = 5,10,15,20-tetrakis(pentafluorophenyl)-porphyrin), TPP (TPP = 5,10,15,20-tetraphenylporphyrin) and TMP (TMP = 5,10,15,20-tetramesitylporphyrin). Upon bubbling of dry O2(g) into a THF solution of [(THF)2(F20TPP)FeII] (419 nm (Soret; ε = 2.5 × 105 M−1 cm−1) and 539 nm (ε = 1.2 × 104 M−1 cm−1)) at −80 °C, new electronic absorption spectral features centered at 413 nm (Soret; ε = 1.58 × 105 M−1 cm−1) and 532 nm (ε = 1.3 × 104 M−1 cm−1) were generated, indicating the formation of the EPR-silent ferric superoxo species, [(F20TPP)FeIII(O2−˙)] (Fig. S1†). In consistence, the isotopically shifted resonance Raman frequencies for ν(Fe–O) and ν(O–O) features were observed at 577 (Δ18O2 = −23) and 1137 (Δ18O2 = −22) cm−1, respectively (Fig. S2†). The rest of the series of heme superoxo adducts (i.e., [(TPP)FeIII(O2−˙)] and [(TMP)FeIII(O2−˙)]) were prepared similarly (Fig. S1†), and their spectroscopic signatures are in strong agreement with previous reports.1b,22,28
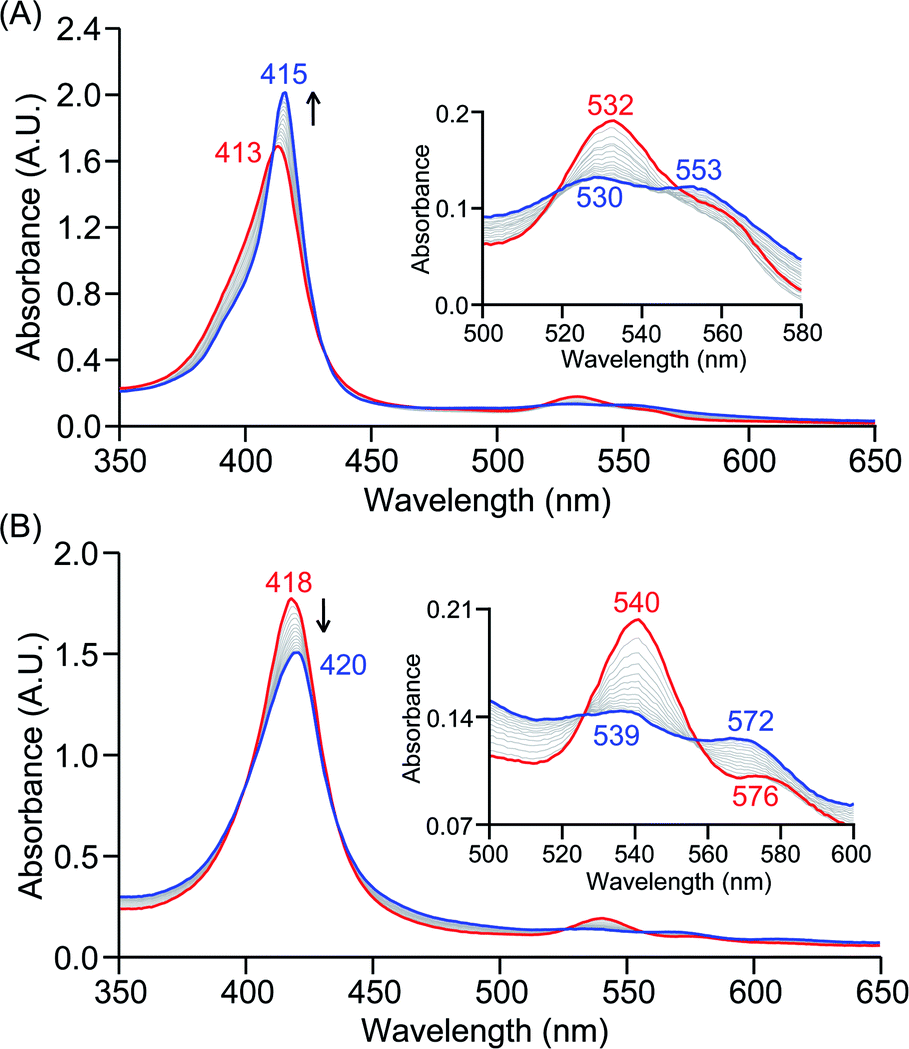
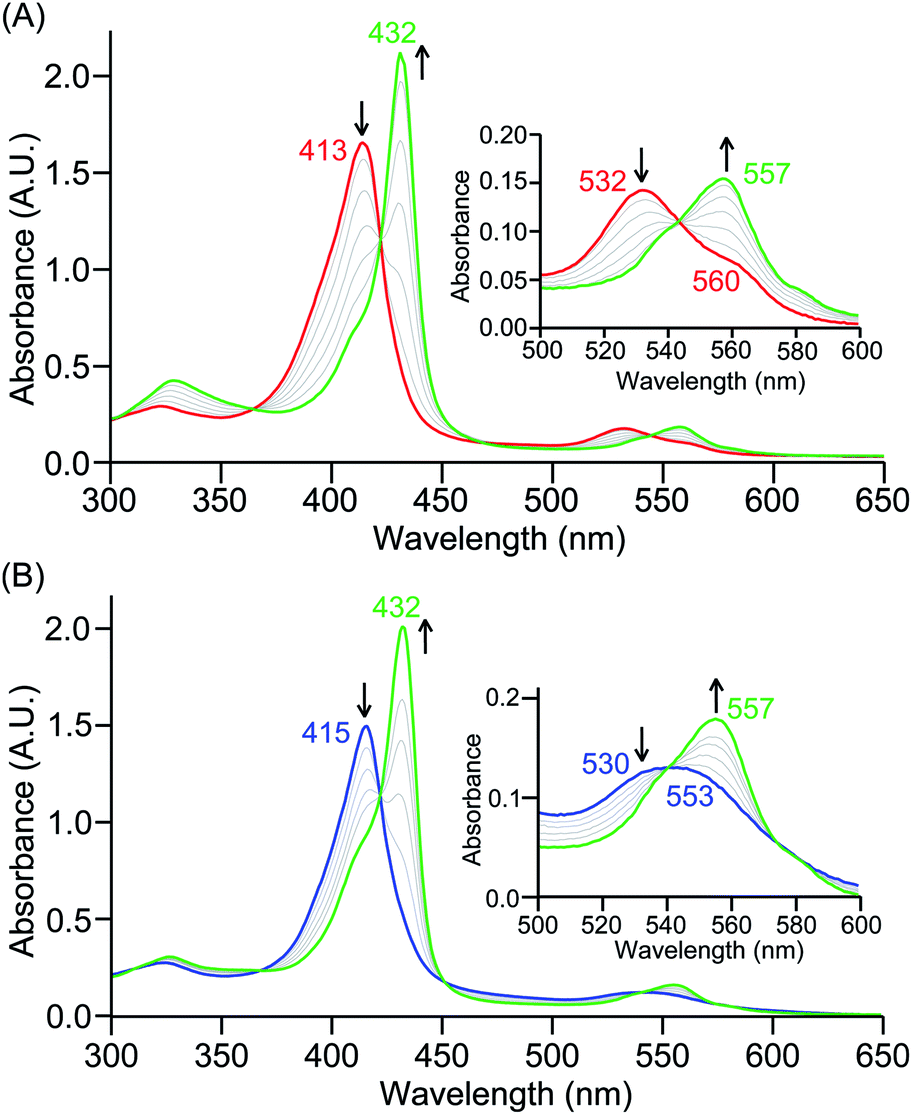
The PCET reactivities of these heme superoxide oxidants were then evaluated against variable concentrations of the TEMPO–H substrate (BDFE = 66.5 kcal mol−1 in THF). When 100 equiv. of TEMPO–H was added into a solution of [(F20TPP)FeIII(O2−˙)] in THF at −80 °C, patent changes in absorption features (Soret: 413 to 415 nm; Q-band: 532 to 530 and 553 nm) were evidenced (Fig. 1A), indicating the H˙ abstraction reactivity of [(F20TPP)FeIII(O2−˙)] with TEMPO–H (Scheme 1). Importantly, the H˙ here is transferred in a single mechanistic step (i.e., concerted), rather than a H+ and an e− in two separate steps. This is due to only H+ or e− transfer from TEMPO–H substrate being extremely thermodynamically uphill compared to H˙ transfer.16a,c[(TPP)FeIII(O2−˙)] also exhibited a similar PCET reactivity with TEMPO–H (Soret: 418 to 420 nm; Q-band: 540 and 576 to 539 and 572 nm) under identical experimental conditions (Fig. 1B). On the contrary however, no reactivity was observed between [(TMP)FeIII(O2−˙)] and TEMPO–H. Moreover, several other PCET substrates with weak C–H (i.e., xanthene (BDFE = 73.3 kcal mol−1 in DMSO29), 9,10 dihydroanthracene (BDFE = 76 kcal mol−1 in DMSO16a)), and O–H (i.e., 5,6-isopropylidene ascorbic acid (BDFE = 70.5 kcal mol−1 in MeCN30), p-OMe-2,6-di-tert-butylphenol (BDFE = 77.4 kcal mol−1 in DMSO29)) bonds were tested against the above series of heme superoxo adducts, where no evidence of any reactivity could be observed.
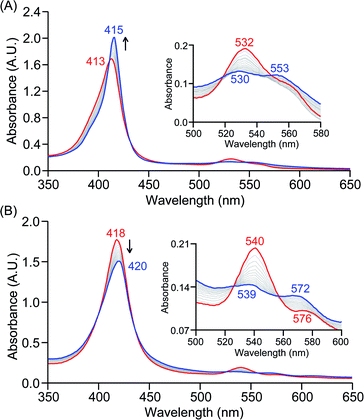 |
| | Fig. 1 Electronic absorption spectral changes observed (in THF at −80 °C) during the reaction of a 50 μM solution of (A) [(F20TPP)FeIII(O2−˙)] and (B) [(TPP)FeIII(O2−˙)] with 100 equiv. of TEMPO–H (red = initial ferric superoxo complex; blue = final ferric product). Insets show the expanded Q-band regions, and arrows indicate the direction of peak transition. | |
 |
| | Scheme 1 Generalized reaction scheme depicting hydrogen atom abstraction reactivities involving heme superoxide adducts and TEMPO–H substrate. | |
Spectroscopic characterization of [(Por)FeIII(OOH)] complexes
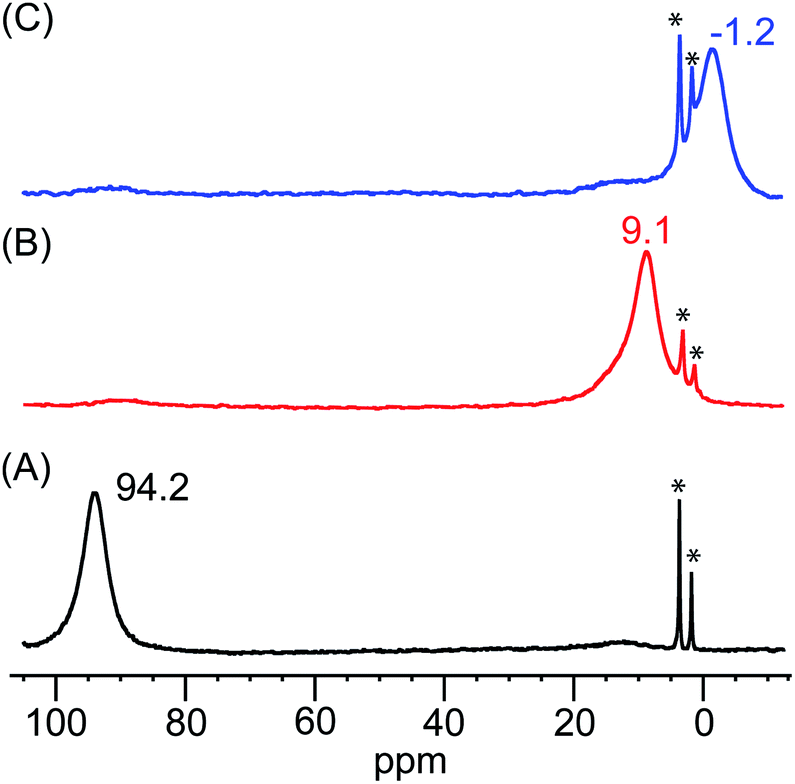
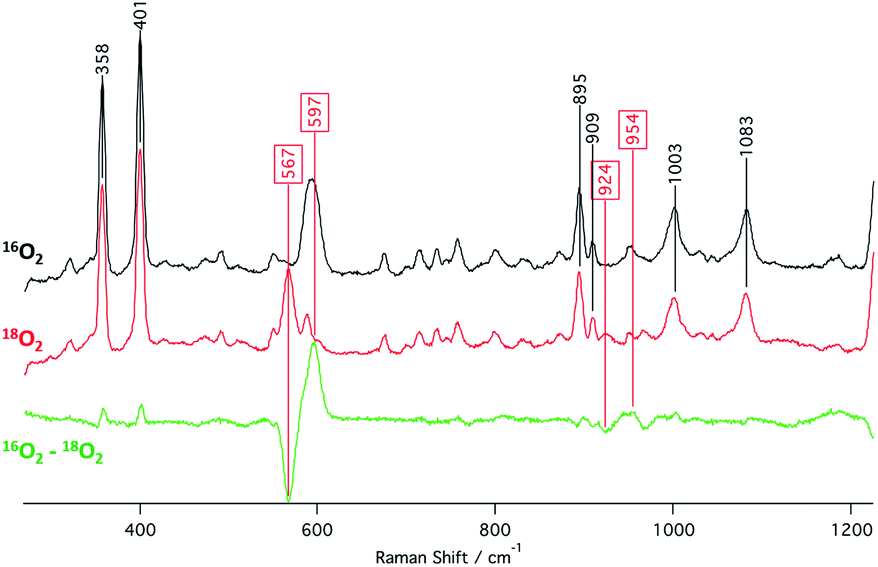
The identities of the expected heme hydroperoxo products (i.e., [(Por)FeIII(OOH)]) from PCET reactivities between heme superoxo adducts and TEMPO–H substrate were established as follows: low-temperature 2H NMR analyses were carried out on heme complexes supported by the pyrrole-position deuterated F20TPP-d8 porphyrinate. [(THF)2(F20TPP-d8)FeII] and [(F20TPP-d8)FeIII(O2−˙)] exhibited a single 2H NMR resonance at δpyrrole = 94.2 and δpyrrole = 9.1 ppm, respectively (Fig. 2). The addition of TEMPO–H induced an upfield shift in the pyrrole resonances to δpyrrole = −1.2 ppm, which is indicative of the formation of a low-spin (S = 1/2) ferric heme system (Fig. 2).31 This is in excellent agreement with the δpyrrole2H NMR resonances observed for the low-spin [(F8TPP-d8)FeIII(OOH)] species by Karlin and coworkers (δpyrrole = −0.63 ppm).26a The EPR spectrum of the final reaction mixture of [(F20TPP)FeIII(O2−˙)] and TEMPO–H predominantly consists of an organic radical signal (g = 2.0; attributed to the TEMPO˙ radical; yield = 82%), which is overlapped with the S = 1/2 FeIII features of the low-spin heme hydroperoxo product complex (Fig. S3 and S4†). Notably, all synthetic heme hydroperoxo adducts reported to-date consist of low-spin ferric centers,23a,c,24a,b,25,26,32 which parallels our aforementioned 2H NMR and EPR characterizations. Moreover, the putative [(F20TPP)FeIII(OOH)] product complex exhibited the isotopic-sensitive resonance Raman frequency for ν(Fe–O) at 597 (Δ18O2 = −30) cm−1 (Fig. 3), which is in-line with other heme hydroperoxo species reported thus far.23c,24a,b,25,26,33 The same feature for [(TPP)FeIII(OOH)] was observed at 589 (Δ18O2 = −26) cm−1 (Fig. S5†). The identities and yields of the [(Por)FeIII(OOH)] products were further substantiated by H2O2 quantification following acidification, where [(F20TPP)FeIII(OOH)] and [(TPP)FeIII(OOH)] produced 92% and 88% yields of H2O2, respectively (Fig. S6†).
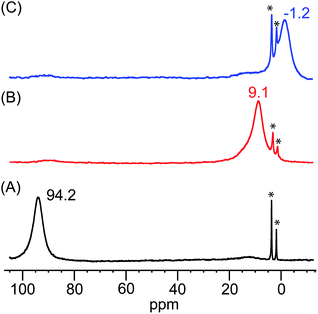 |
| | Fig. 2
2H NMR spectra (in THF at −80 °C) of (A) [(THF)2(F20TPP-d8)FeII] (black), (B) [(F20TPP-d8)FeIII(O2−˙)] (red), and (C) final heme product of the reaction between [(F20TPP-d8)FeIII(O2−˙)] and TEMPO–H (blue) (* peaks at 3.58 and 1.73 ppm correspond to the THF solvent). | |
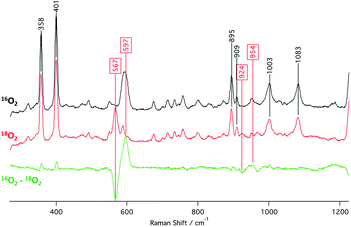 |
| | Fig. 3 Resonance Raman spectra (λex = 413.1 nm) collected from a 2 mM frozen THF solution of the final heme product from the reaction between TEMPO–H and [(F20TPP)FeIII(O2−˙)] prepared with 16O2(g) (black) and 18O2(g) (red). | |
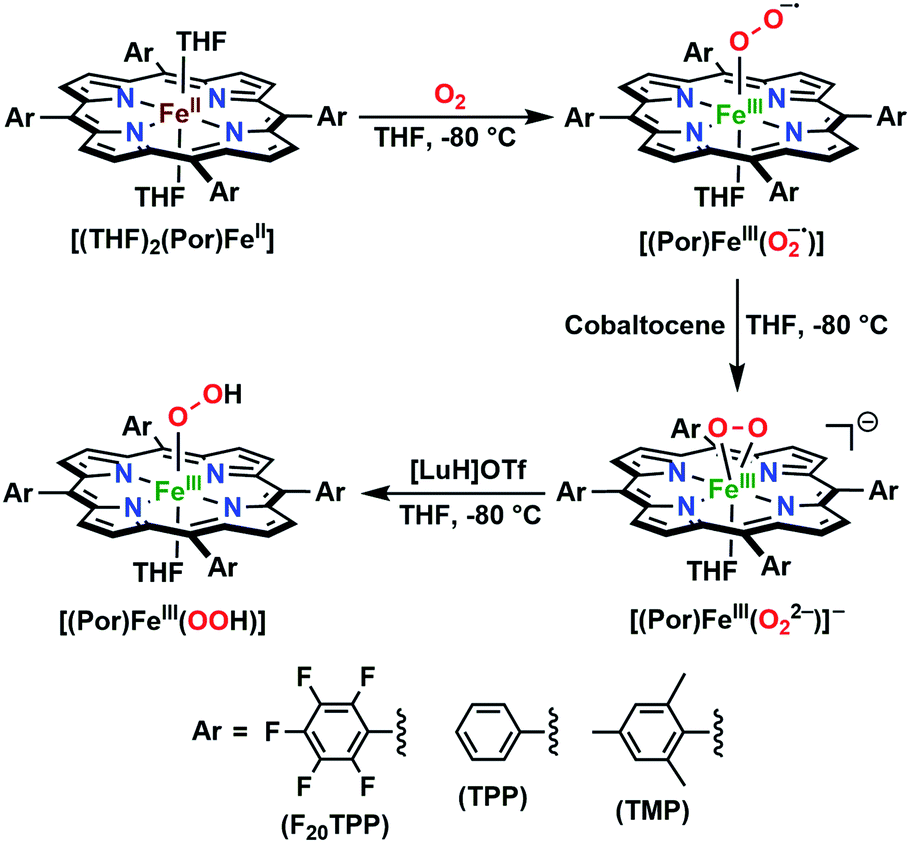
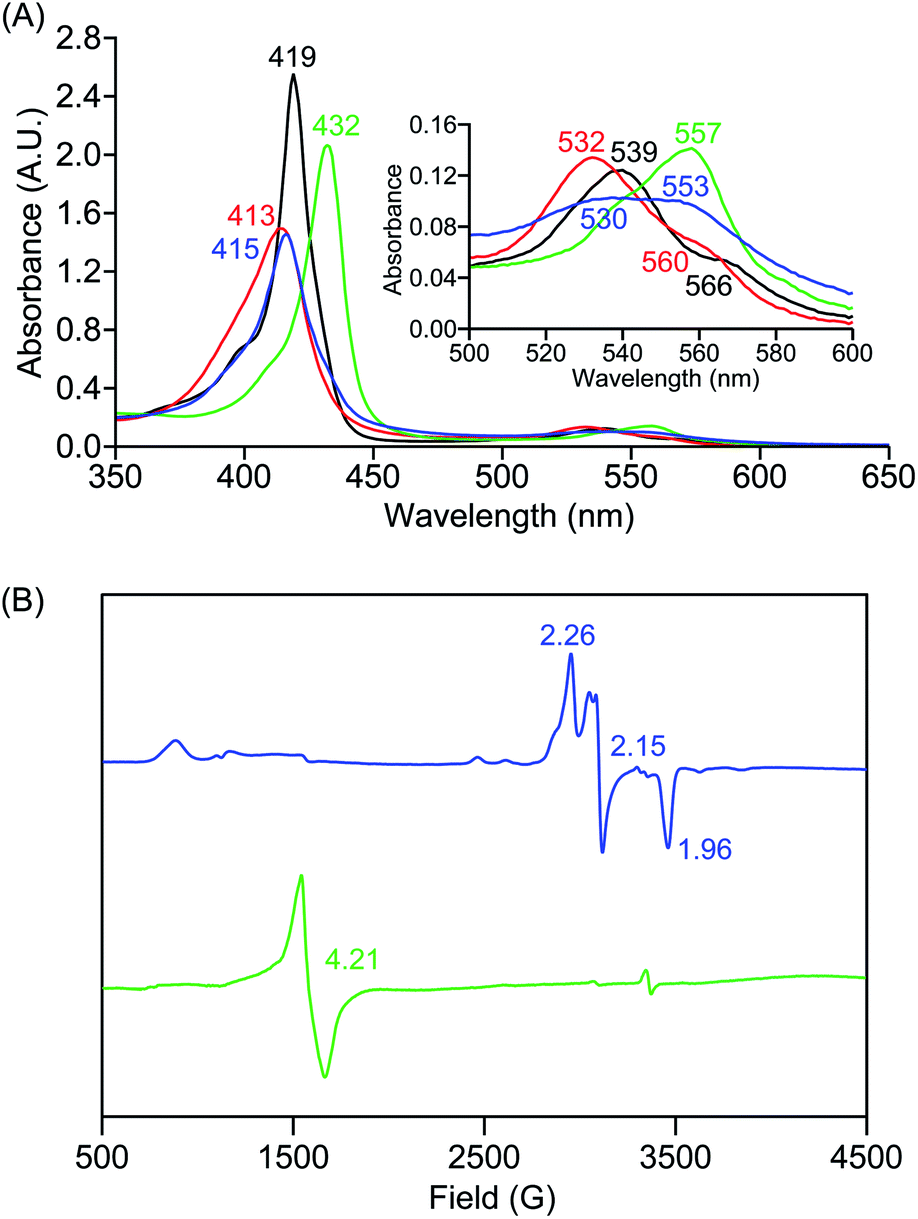
In further effort to characterize the final [(Por)FeIII(OOH)] products, we have generated authentic [(Por)FeIII(OOH)] complexes for all three heme systems by reduction, followed by protonation of each superoxo complex (Scheme 2). In that, the one-electron reduction of the [(Por)FeIII(O2−˙)] complex was achieved with 1 equiv. of cobaltocene, leading to the corresponding side-on ferric peroxo species, [(Por)FeIII(O22−)]− (as would be expected for a five-coordinate or solvent-ligated heme superoxide complex1b). The peroxo complex was subsequently protonated with 1 equiv. of 2,6-lutidinium triflate ([LuH]OTf) affording the end-on ferric hydroperoxo complex, [(Por)FeIII(OOH)]. For example, when 1 equiv. of cobaltocene is added into [(F20TPP)FeIII(O2−˙)] in THF at −80 °C, the formation of the side-on ferric heme–peroxo complex, [(F20TPP)FeIII(O22−)]− was evidenced by the formation of new electronic absorption features at 432 nm (Soret, ε = 2.0 × 105 M−1 cm−1) and 557 nm (ε = 1.5 × 104 M−1 cm−1) (Fig. 4A; for [(TPP)FeIII(O2−˙)] and [(TMP)FeIII(O2−˙)] see Fig. S7†).23b,c,34 Characteristic resonance Raman features of [(F20TPP)FeIII(O22−)]− were observed at ν(Fe–O) = 469 (Δ18O2 = −15) and ν(O–O) = 808 (Δ18O2 = −41) cm−1 (Fig. S8†). Successive addition of 1 equiv. of [LuH]OTf produced new electronic absorption features at 415 nm (Soret, ε = 1.5 × 105 M−1 cm−1), and 553 nm (ε = 1.0 × 104 M−1 cm−1) (Fig. 4A; also see Fig. S7†); the EPR features corresponding to authentic [(F20TPP)FeIII(O22−)]− and [(F20TPP)FeIII(OOH)] adducts were observed at g = 4.21, and g = 2.26, 2.15, and 1.96, respectively (Fig. 4B and Table S1†). These electronic absorption and EPR characteristics of our authentic [(Por)FeIII(OOH)] species closely resemble those of previously reported heme ferric hydroperoxo adducts,24b,25,26,32 and manifest unequivocal similarities to those of the PCET reaction products of [(Por)FeIII(O2−˙)] and TEMPO–H (Fig. S9 and Table S1†). In summation, the close resemblance of spectroscopic properties of PCET reaction products and authentic hydroperoxo compounds strongly suggest the near stoichiometric conversion of [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] to [(F20TPP)FeIII(OOH)] and [(TPP)FeIII(OOH)], respectively, upon abstracting a H˙ from TEMPO–H under the aforementioned reaction conditions.
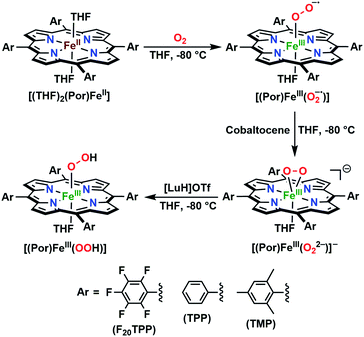 |
| | Scheme 2 Synthetic protocol for the generation of authentic [(Por)FeIII(OOH)] adducts by reduction, followed by protonation of the [(Por)FeIII(O2−˙)] complexes. All reactions were carried out in THF at −80 °C. | |
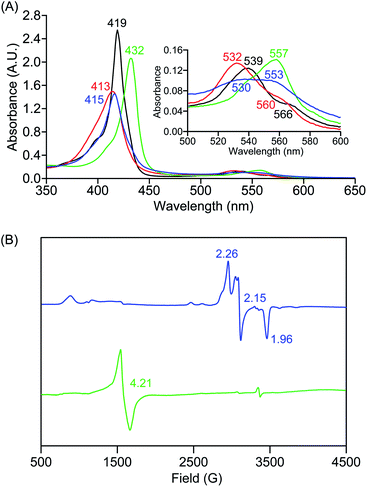 |
| | Fig. 4 (A) UV-vis spectra for 50 μM solutions of [(THF)2(F20TPP)FeII] (black), [(F20TPP)FeIII(O2−˙)] (red), [(F20TPP)FeIII(O22−)]− (green) and [(F20TPP)FeIII(OOH)] (blue) collected in THF at −80 °C, and (B) EPR spectral features (in frozen THF at 7 K) for 2 mM solutions of [(F20TPP)FeIII(O22−)]− (green) and [(F20TPP)FeIII(OOH)] (blue) complexes. | |
Mechanistic investigations into PCET reactivities of [(Por)FeIII(O2−˙)] complexes
Precise physicochemical signatures of proton coupled electron transfer reactions mediated by heme superoxide intermediates are gravely understudied; yet could be of momentous importance in a broad combination of applications. We have carried out thorough thermodynamic and kinetic examinations into the entire series of heme superoxide adducts, which exert firm support for the overall experimental findings presented in this study. Pseudo first-order kinetic analyses were carried out by reacting the superoxo complexes with 50−200 equiv. of TEMPO–H in THF at −80 °C (Fig. S10†). All reactions were followed by electronic absorption spectroscopy, and in all cases, the pseudo-first-order rate constants (kobs) increased linearly as a function of the TEMPO–H substrate concentration (Fig. 5A and S11†), leading to second order rate constants (k2) of 2.23 M−1 s−1 and 1.24 M−1 s−1 for [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)], respectively (Table 1). As well, kinetic isotope effects (KIE (kH/kD)) for PCET mediated by [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] were found to be 11.7 and 6.7 (Fig. 5A, S11† and Table 1), respectively, using the isotopically labelled substrate, TEMPO–D. It is noteworthy that Karlin and coworkers have observed a similar KIE (6) for [(F8TPP)FeIII(O2−˙)], and values of the same magnitude (4.8–12.1) have been reported for PCET effected by various other non-heme superoxo complexes of Fe, Co, and Cu (Table 1).29,35,36a These large (i.e., >2) KIE values present compelling evidence into the rate limiting nature of the homolytic O–H bond cleavage process (and thus, concerted H˙ transfer from the substrate), rather than only proton or electron transfer being the slowest step (where the reaction rate would linearly correlate with either pKa or E° of the substrate, respectively, rather than the BDFE).36b Activation parameters for TEMPO–H oxidation by [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] were determined via Eyring analyses of reaction rates from −70 °C to −100 °C in THF (Fig. 5B and S11†). The experimental ΔH‡ and ΔS‡ activation parameter values, and the activation energies (ΔG‡) derived therefrom are listed in Table 1. Similar Eyring analyses for PCET reactivities of a limited, yet diverse group of non-heme superoxo complexes have been reported, all of which are in the same order of magnitude as the heme superoxo values reported herein.35a–c,36a Notably, ΔH‡ and ΔS‡ for most non-heme superoxides are larger than those of heme superoxide adducts; substantially negative ΔS‡ values of the latter suggest highly ordered transition states as one of the possible contributing factors to their sluggish PCET capabilities. Interestingly, Shaik and coworkers found higher activation barriers for Cyt P450 superoxide mediated hydrogen atom abstraction when compared with the corresponding compound-I oxidant.37 These activation parameters also divulge the remarkable influence of the electronic properties of a heme center on its ability to execute successful H˙ atom abstraction. In that, the most electrophilic superoxide adduct, [(F20TPP)FeIII(O2−˙)], exhibits the smallest activation barrier, and thus, reacts the fastest. To the best of our knowledge, this study marks the first report where activation parameters are presented for any substrate reactivity mediated by synthetic heme superoxide intermediates, hence impeding any detailed comparisons with like systems. Nonetheless, these findings impart substantial insights into methodologies for further improvement of ORR efficiencies in fuel cells, and mechanistic understanding of human heme proteins with significant pathological and/or therapeutic value. For example, these activation parameters could shine light on important structural properties of heme ORR catalysts that could be modulated in order to optimally adjust the activation barriers for precise ORR-related reduction and/or protonation events.
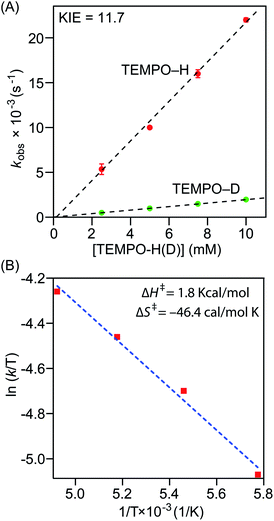 |
| | Fig. 5 (A) Plot of pseudo-first-order rate constants (kobs) versus [TEMPO–H] (red) or [TEMPO–D] (green) for a 50 μM solution of [(F20TPP)FeIII(O2−˙)] in THF at −80 °C. (B) Eyring plot showing ln(k/T) versus 1/T for the reaction between a 50 μM THF solution of [(F20TPP)FeIII(O2−˙)] and TEMPO–H at −70, −80, −90 and −100 °C. | |
Table 1 Kinetic and thermodynamic parameters for PCET reactivities of heme and nonheme–superoxo complexes with H-atom donor substrates
| Identity of the heme/nonheme superoxo complexa |
Kinetic parameters |
Thermodynamic parameters |
Ref.g |
|
k
2 (M−1 s−1) |
KIE (kH/kD) |
ΔH‡ (kcal mol−1) |
ΔS‡ (cal mol−1 K−1) |
ΔG‡ (kcal mol−1) |
E° vs. Fc+/0 (V)e |
pKaf |
BDFEO–Hf (kcal mol−1) |
|
See Chart 2 for structures related to this work.
At −80 °C.
In THF.
No reactivity was observed.
Fc+/0 = ferrocene/ferrocenium redox couple.
Determined for the corresponding hydroperoxo species.
See reference for experimental conditions.
|
|
[(F20TPP)FeIII(O2−˙)]
|
2.23b ± 0.09 |
11.7b |
1.8 ± 0.1 |
−46.4 ± 0.3 |
10.7 ± 0.2 |
−1.17b,c ± 0.01 |
25.6b,c ± 0.4 |
69.1c ± 0.4 |
This work |
|
[(TPP)FeIII(O2−˙)]
|
1.24b ± 0.09 |
6.7b |
2.9 ± 0.2 |
−42.0 ± 0.4 |
11.0 ± 0.2 |
−1.18b,c ± 0.01 |
24.6b,c ± 0.1 |
67.5c ± 0.2 |
This work |
|
[(TMP)FeIII(O2−˙)]
|
NRd |
NRd |
NRd |
NRd |
NRd |
−1.20b,c ± 0.01 |
24.2b,c ± 0.1 |
66.5c ± 0.2 |
This work |
|
[(F8TPP)FeIII(O2−˙)]
|
0.5 |
6.0 |
— |
— |
— |
−1.17 ± 0.01 |
28.8 ± 0.5 |
73.5 ± 0.9 |
26a
|
|
[(PIm)FeIII(O2−˙)]
|
— |
— |
— |
— |
— |
−1.33 ± 0.01 |
28.6 ± 0.5 |
69.5 |
26b
|
|
(TMPImOH)FeIII(O2−˙)
|
— |
— |
— |
— |
— |
−1.32 |
25.1 |
— |
24a
|
|
(TMPImOEt)FeIII(O2−˙)
|
— |
— |
— |
— |
— |
−1.75 |
32.3 |
— |
24a
|
|
[CoIII(py)(O2)(TBP8Cz)]−
|
3.6 ± 0.8 |
9.2 |
6.7 ± 0.1 |
−23 ± 0.4 |
— |
— |
— |
— |
35a
|
|
Co(O2)(Me3TACN)(S2SiMe2)
|
0.87 ± 0.03 |
8.8 |
3.6 |
−36.4 |
— |
— |
— |
— |
35b
|
|
[(DMM-tmpa)CuII(O2−˙)]+
|
23 |
11 |
3.6 ± 0.6 |
−32 ± 3 |
— |
— |
— |
— |
35c
|
|
(TMC)FeIII superoxo complex |
1.2 |
6.3 |
— |
— |
— |
— |
— |
— |
35d
|
|
FeIII(BDPP) superoxo complex |
|
7.0 |
|
|
|
|
|
|
35e
|
|
[LCuII-(O2−˙)]+
|
1.9 |
12.1 |
— |
— |
— |
— |
— |
— |
35f
|
|
[FeIII(S2Me2N3(Pr,Pr))(O2)]
|
— |
4.8 |
— |
— |
— |
— |
— |
— |
35g
|
| μ-1,2-Superoxo CuII2 complex |
0.13 |
— |
9.03 ± 0.4 |
26.7 ± 1.6 |
16.8 ± 0.9 |
−0.59 |
22.2 |
71.7 ± 1.1 |
36a
|
|
[Cu
II
2
(XYLO)(O
2
−
˙)]
2+
|
5.6 |
— |
— |
— |
— |
−0.525 ± 0.01 |
24 ± 0.6 |
81.8 ± 1.5 |
29
|
Comprehending these observed reactivity disparities on thermodynamic grounds calls for experimental examination of O–H bond dissociation free energies (BDFEs) of the [(Por)FeIII(OO–H)] complexes. That is, since the concerted homolytic O–H bond cleavage is the slowest step of the reaction (vide supra), O–H bond strength of the [(Por)FeIII(OO–H)] product complex predominantly dictates the thermodynamic driving force for the overall reaction. These BDFE values can be deduced in a fairly straightforward fashion using the Bordwell equation (eqn (1)),27 if one could determine the redox potential (E°) and acidity (pKa) of the metal oxidants that make up the corresponding thermodynamic cycle/square scheme (Scheme 3).16a,29,36a,38 In addition to shedding light on the perceived reactivities, BDFEs also divulge reactivity limitations to be expected based on the thermodynamic portrayal. All of the heme superoxide complexes employed in this study can be quantitatively converted to the corresponding heme hydroperoxo adducts by stepwise reduction-protonation (by cobaltocene and [LuH]OTf, respectively) reactivity as illustrated in Scheme 2. However, to elucidate the relevant E° and pKa values experimentally, the equilibrium constants must be determined for each of the reduction and protonation steps.
 |
| | Scheme 3 Thermodynamic square scheme used to determine the O–H bond dissociation free energies of the [(Por)FeIII(OOH)] complexes in THF, along with the relevant thermodynamic parameters. | |
The reduction potential (E°) of the [(Por)FeIII(O2−˙)]/[(Por)FeIII(O22−)]− redox couple for each system was elucidated by titrating the [(Por)FeIII(O2−˙)] complexes in THF at −80 °C (Fig. 6A and S12†) with the weak reductant, Cr(η6-C6H6)2 (E1/2 = −1.15 V vs. Fc+/0 in CH2Cl2;39Scheme 4), to afford equilibrium mixtures of [(Por)FeIII(O2−˙)] and [(Por)FeIII(O22−)]−. The array of calculated equilibrium constants (Table S2†) yielded corresponding reduction potentials via Nernst equation (Table S3†), and were found to be −1.17, −1.18, −1.20 V (vs. Fc+/0) for [(F20TPP)FeIII(O2−˙)], [(TPP)FeIII(O2−˙)], and [(TMP)FeIII(O2−˙)], respectively (Table 1). Naruta and co-workers have recently reported theoretical reduction potential values for two tethered axial imidazole coordinated TMP-based heme superoxide systems, which are more negative compared to our values (−1.32 and −1.75 V (vs. Fc+/0 in EtCN))24a (Table 1). This trend is consistent with recently reported values for two F8TPP based systems by Karlin and coworkers, where the superoxo adduct with tethered axial imidazole coordination displayed a lower reduction potential (−1.33 V)26b compared to the parent complex (−1.17 V)26a (Table 1). These differences most likely reflect the enrichment of electron density in the dz2 orbital of the iron center upon imidazole ligation, making it much less susceptible for reduction. Notably, our reduction potential for [(F20TPP)FeIII(O2−˙)] is in excellent agreement with the electrochemically determined values (−1.09 vs. Fc+/0 in dimethylformamide at −30 °C) by Anxolabéhère-Mallart and co-workers for the same system.18,32b,40 Finally, the reversibility of the redox couple, [(Por)FeIII(O2−˙)]/[(Por)FeIII(O22−)]−, was also established: the [(Por)FeIII(O22−)]− complexes generated by one-electron reduction of [(Por)FeIII(O2−˙)] adducts by Cr(η6-C6H6)2 can be reoxidized stoichiometrically by tris(4-bromophenyl)ammoniumyl hexachloroantimonate ([(4-BrC6H4)3N]SbCl6; E1/2 = 0.67 V vs. Fc+/0 in MeCN;39Scheme 4) giving the starting superoxide complex, which can then be re-reduced to [(Por)FeIII(O22−)]− by the addition of 5 equiv. of Cr(η6-C6H6)2 (Fig. S13†).
 |
| | Fig. 6 Electronic absorption spectral changes observed (in THF at −80 °C) upon incremental addition of (A) Cr(η6-C6H6)2 into [(F20TPP)FeIII(O2−˙)] (red) inducing its one-electron reduction to [(F20TPP)FeIII(O22−)]− (green), and (B) tBuP2(dma) into [(F20TPP)FeIII(OOH)] (blue) effecting its deprotonation giving [(F20TPP)FeIII(O22−)]− (green). | |
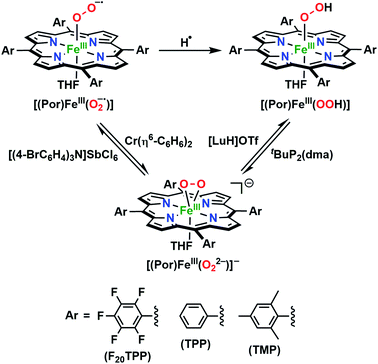 |
| | Scheme 4 Interconversion of [(Por)FeIII(O2−˙)], [(Por)FeIII(O22−)]− and [(Por)FeIII(OOH)] complexes in THF at −80 °C, resulting in equilibrium mixtures that allowed the determination of corresponding reduction potential (E°) and pKa values. | |
In order to complete the thermodynamic square scheme analysis (Scheme 3), the pKa values for the [(Por)FeIII(OOH)] complexes were deduced utilizing the derivatized phosphazene weak base, tBuP2(dma) (pKa of conjugate acid = 24.9 in THF at RT41), in THF at −80 °C.42 Gradual addition of the base afforded equilibrium mixtures of [(Por)FeIII(OOH)] and [(Por)FeIII(O22−)]− (Fig. 6B and S14†), yielding the corresponding equilibrium constants (Table S4†). Accordingly, pKa values of 25.6, 24.6, and 24.2 were computed for [(F20TPP)FeIII(OOH)], [(TPP)FeIII(OOH)], and [(TMP)FeIII(OOH)], respectively (Tables 1 and S5†). These experimentally determined pKa's are generally smaller (i.e., more acidic), as would be expected, than those of tethered axial imidazole coordinated TMP-based heme superoxide systems (≈25.1–32.3) previously reported from theoretical studies performed in propionitrile solvent24a (Table 1). Moreover, the reversibility of the acid–base interconversion of the [(Por)FeIII(OOH)]/[(Por)FeIII(O22−)]− couple was established by re-protonation of the aforementioned [(Por)FeIII(O22−)]− complexes by [LuH]OTf giving stoichiometric quantities of [(Por)FeIII(OOH)] species (Fig. S15†). Finally, our thermochemical findings also parallel those recently reported for [(F8TPP)FeIII(O2−˙)] (pKa = 28.8; E° = −1.17 V) and [(PIm)FeIII(O2−˙)] (pKa = 28.6; E° = −1.33 V) by Karlin and coworkers (Table 1), where similarly reversible protonation and reduction processes were observed.26
The substitution of experimental thermodynamic parameters, E° and pKa (Table 1), in the Bordwell equation (CG(THF) = 61 kcal mol; a solvent-dependent constant)26a,43 lead to BDFE's of 69.1, 67.5, and 66.5 kcal mol−1 for the O–H bonds of [(F20TPP)FeIII(OOH)], [(TPP)FeIII(OOH)], and [(TMP)FeIII(OOH)], respectively (Table 1). It is noteworthy that these results are in great accord with previously reported experimental O–H BDFE's by Karlin and coworkers (73.5, 69.5 kcal mol; Table 1), as well as computationally calculated BDEs of several heme hydroperoxide complexes by Morokuma and coworkers (64–66 kcal mol−1),44 Moreover, our BDFE values are in excellent agreement with the observed PCET reactivity pattern, where [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] facilitate hydrogen atom abstraction from TEMPO–H (BDFEheme hydroperoxo > BDFETEMPOH), while [(TMP)FeIII(O2−˙)] remained unreactive (BDFEheme hydroperoxo ≈ BDFETEMPOH; i.e., thermodynamically neutral). Finally, these BDFEs also unveil the thermodynamic basis for the incapacity of hydrogen atom abstraction by [(Por)FeIII(O2−˙)] complexes from other substrates with stronger (i.e., BDFE ≥ 69.1 kcal mol−1) C–H, N–H, or O–H bonds.
Computational studies
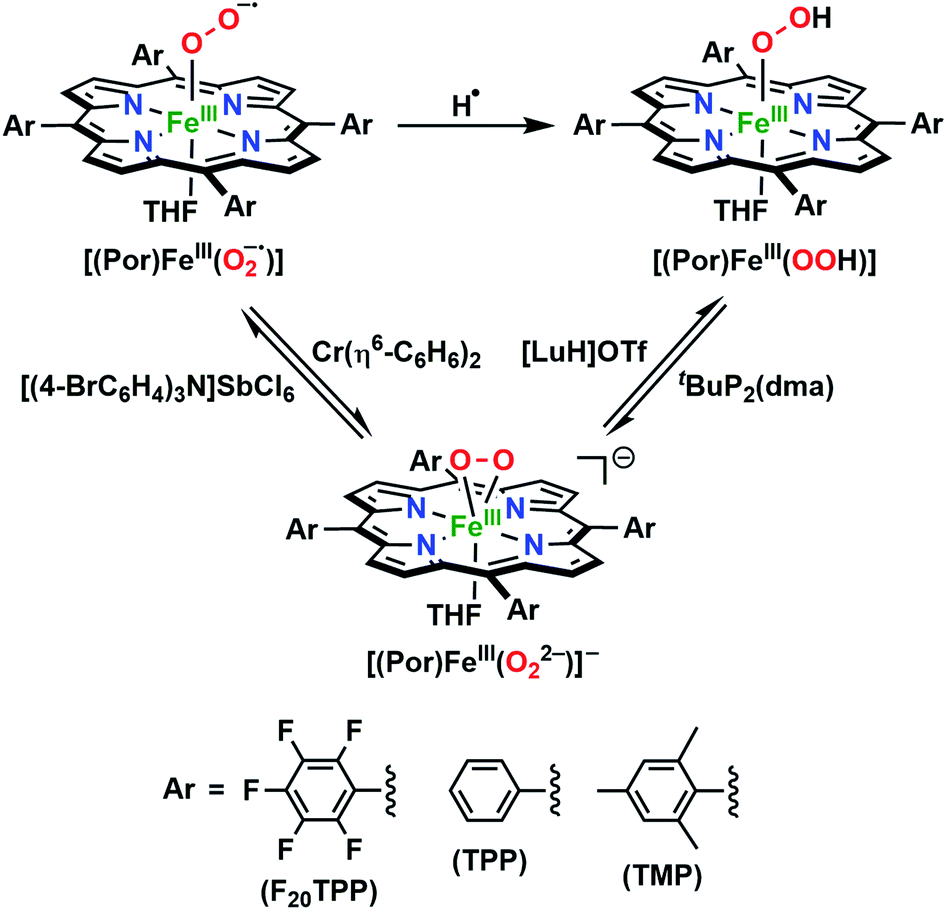
To gain further insight into the intricate electrochemical properties of the iron(III)–superoxo complexes, we did a computational study on both [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] systems and studied their reactivities with TEMPO–H. The [(TPP)FeIII(O2−˙)] complex is calculated as an end-on superoxo configuration with an open-shell singlet spin ground state with two unpaired electrons antiferromagnetically coupled in 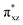 and
and 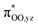 orbitals. The former orbital is a dominant 3dxz atomic orbital on iron in the plane of the FeOO group, while the latter is the antibonding interaction along the O–O bond that also interacts with the 3dyz orbital on iron. The triplet spin state has the same electron configuration but ferromagnetically coupled and its energy with zero-point energy correction is ΔE + ZPE = 5.6 kcal mol−1 higher in energy. Our calculated ground state; therefore, matches experimental assignments (see above) that the system is EPR silent. Subsequently, we calculated the hydrogen atom abstraction transition states (TSHA) for the reaction of 1,3[(TPP)FeIII(O2−˙)] from TEMPO–H and the optimized geometries are shown in Fig. 7. The reactions are concerted with a single hydrogen atom transfer leading to an iron(III)–hydroperoxo product. The transition states are early with short TEMPO–H distances of 1.075 and 1.057 Å in the singlet and triplet spin states, respectively. At the same time, the accepting O–H distance is long: 1.419 Å in 1TSHA and 1.473 Å in 3TSHA. Generally, early transition states correspond with low-energy hydrogen atom abstraction reactions, while later transition states have much higher energy barriers.45
orbitals. The former orbital is a dominant 3dxz atomic orbital on iron in the plane of the FeOO group, while the latter is the antibonding interaction along the O–O bond that also interacts with the 3dyz orbital on iron. The triplet spin state has the same electron configuration but ferromagnetically coupled and its energy with zero-point energy correction is ΔE + ZPE = 5.6 kcal mol−1 higher in energy. Our calculated ground state; therefore, matches experimental assignments (see above) that the system is EPR silent. Subsequently, we calculated the hydrogen atom abstraction transition states (TSHA) for the reaction of 1,3[(TPP)FeIII(O2−˙)] from TEMPO–H and the optimized geometries are shown in Fig. 7. The reactions are concerted with a single hydrogen atom transfer leading to an iron(III)–hydroperoxo product. The transition states are early with short TEMPO–H distances of 1.075 and 1.057 Å in the singlet and triplet spin states, respectively. At the same time, the accepting O–H distance is long: 1.419 Å in 1TSHA and 1.473 Å in 3TSHA. Generally, early transition states correspond with low-energy hydrogen atom abstraction reactions, while later transition states have much higher energy barriers.45
 |
| | Fig. 7 Optimized geometries of the hydrogen atom abstraction transition states from TEMPO–H by [(TPP)FeIII(O2−˙)] in the singlet and triplet spin states. Bond lengths are in angstroms, the imaginary frequency in cm−1 and group spin densities (ρ) in atomic units. The right-hand-side shows the singly occupied molecular orbitals in the singlet and triplet reactants. | |
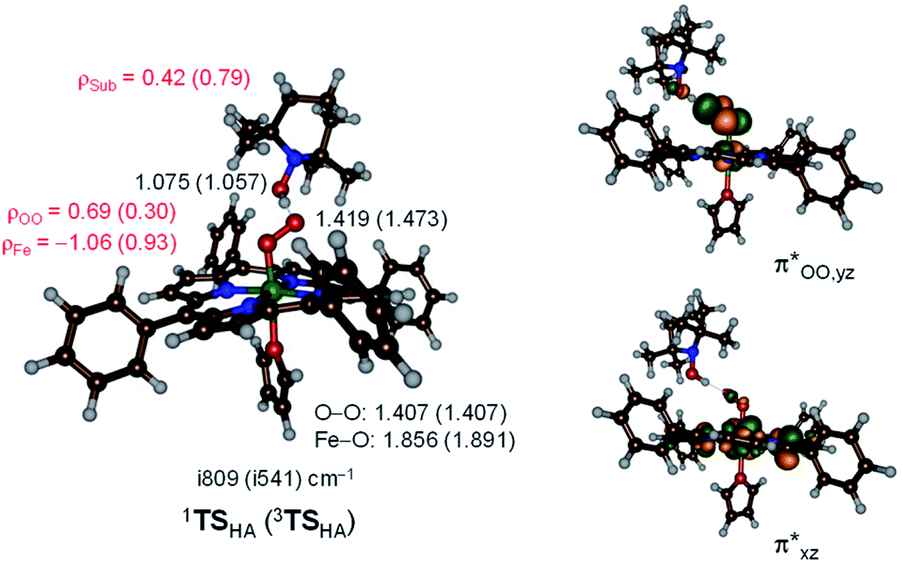
Hydrogen atom abstraction by 1,3[(TPP)FeIII(O2−˙)] leads to electron transfer into the 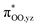 orbital and generates a doublet spin [(TPP)FeIII(OOH)] coupled to a TEMPO radical. This electron transfer is confirmed by the group spin densities that show an increase of spin on the TEMPO group to ρSub = 0.42, while at the same time spin is lost on the dioxygen moiety ρOO = 0.69. The hydrogen atom abstraction barrier is ΔE‡ + ZPE = 3.2 kcal mol−1 in the singlet spin state and 9.9 kcal mol−1 for the triplet spin state barrier. The low-spin barrier matches the experimentally determined ΔH‡ = 2.9 kcal mol−1 excellently. The experimentally determined entropy contribution is relatively large, probably due to a solvent cage surrounding the molecular complex. As shown by previous work of ours, reactions in solution often have a solvent cage around the active complex that affect entropies and particularly reduces vibrational contributions.46 Because of the fact that the solvent cage was not included in the model, the computational free energy of activation is relatively low and much lower than that observed in experiment as the gas-phase entropy is overestimated.
orbital and generates a doublet spin [(TPP)FeIII(OOH)] coupled to a TEMPO radical. This electron transfer is confirmed by the group spin densities that show an increase of spin on the TEMPO group to ρSub = 0.42, while at the same time spin is lost on the dioxygen moiety ρOO = 0.69. The hydrogen atom abstraction barrier is ΔE‡ + ZPE = 3.2 kcal mol−1 in the singlet spin state and 9.9 kcal mol−1 for the triplet spin state barrier. The low-spin barrier matches the experimentally determined ΔH‡ = 2.9 kcal mol−1 excellently. The experimentally determined entropy contribution is relatively large, probably due to a solvent cage surrounding the molecular complex. As shown by previous work of ours, reactions in solution often have a solvent cage around the active complex that affect entropies and particularly reduces vibrational contributions.46 Because of the fact that the solvent cage was not included in the model, the computational free energy of activation is relatively low and much lower than that observed in experiment as the gas-phase entropy is overestimated.
The imaginary frequency in the transition state is modest: i809 cm−1 in 1TSHA and i541 cm−1 in 3TSHA. Typical values for hydrogen atom abstraction imaginary frequencies are of the order of i1200–i1800 cm−1 and implicate a narrow and sharp peak on the potential energy surface.47 The smaller values seen here, also imply that quantum mechanical tunnelling will be less. Indeed, we calculated a KIE = 3.6 using the Eyring model and KIE = 4.0 with the Wigner tunnelling model for the reaction that passes 1TSHA. By contrast, for hydrogen atom abstraction of aliphatic substrates by P450 compound I or non-heme iron dioxygenases typically values well larger than 12 are calculated.48 Our obtained KIE value is in good agreement with the experimentally determined value of 6.7 (Table 1) and hence the calculations give a similar potential energy landscape and curvature as derived from the experimental work.
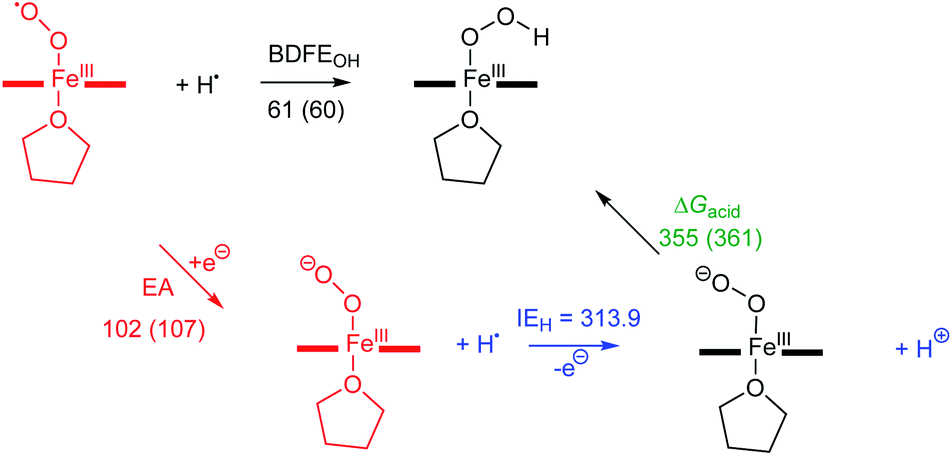
Next, we calculated the BDFE of [(TPP)FeIII(OOH)] as the energy difference of its optimized geometry with that of a hydrogen atom and the [(TPP)FeIII(O2−˙)] complex and find a value of 61 kcal mol−1 as an energy difference between the singlet spin iron–superoxo and the doublet spin iron(III)–hydroperoxo complex and a hydrogen atom (Fig. 8). Our calculated value matches the experimental derivation from redox potential and pKa values well and shows the hydroperoxo O–H bond is relatively weak. Thus, the BDFE of porphyrin/heme ligated iron(III)–hydroxo complexes were calculated for several systems previously. A value of 80.8 kcal mol−1 was obtained for model of horseradish peroxidase that has a porphyrin equatorial ligand and imidazole as axial ligand, while a value of 88.9 kcal mol−1 was found for a P450 model with a thiolate axial ligand.49 As often the BDFE represents a measurement of the ability of an oxidant to abstract hydrogen atoms efficiently, this means that [(TPP)FeIII(O2−˙)] will be a weak oxidant and only able to activate substrates with weak C–H or O–H bonds. Indeed, as reported above, our experimental studies show that only reactions with TEMPO–H led to hydrogen atom transfer, while the system is inactive with other aliphatic substrates. These results; therefore, support previous computational studies on the oxidative properties of the iron(III)–hydroperoxo and iron(III)–superoxo intermediates in P450 enzymes that found them to be sluggish oxidants.37 In addition to the BDFE values, we also calculated the one-electron reduction potential (or electron affinity, EA) of the iron(III)–superoxo complex and find values of 102 and 107 kcal mol−1 for [(TPP)FeIII(O2−˙)] and [(F20TPP)FeIII(O2−˙)], respectively. Finally, we estimated the gas-phase acidity of the iron(III)–hydroperoxo complex (ΔGacid) from the experimentally determined ionization energy of a hydrogen atom (IEH = 313.9 kcal mol−1)50 and the difference between EA and BDFEOH.
 |
| | Fig. 8 Thermochemical cycle for [(TPP)FeIII(O2−˙)] (and [(F20TPP)FeIII(O2−˙)] in parenthesis) with calculated reaction energies for electron transfer (EA), O–H bond dissociation and gas-phase acidity. All data in kcal mol−1. | |
Conclusions
Despite the ubiquitous nature of proton-coupled electron transfer processes mediated by heme superoxo adducts in both biology and alternative energy applications, understanding of precise reactivity limitations in terms of key physicochemical properties of the heme oxidant is still in its infancy. To this end, we present a detailed thermodynamic, kinetic, and theoretical analysis of PCET reactivities of a series of electronically disparate, structurally equivalent heme superoxo model systems. These heme ferric superoxo adducts, [(Por)FeIII(O2−˙)], abstract a hydrogen atom (H˙; i.e., H+ + e−) from the weak O–H bond substrate TEMPO–H, in a single kinetic step (i.e., concerted) leading to the stoichiometric generation of the corresponding heme ferric hydroperoxo, [(Por)FeIII(OOH)], species and TEMPO˙ radical, where the substrate O–H bond cleavage is the overall rate-determining process. Accordingly, H/D KIE's were found to be 11.7 and 6.7 (Table 1) for [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] adducts (Chart 2), respectively, which are in excellent agreement with our theoretical findings. All heme reactants and products have been fully characterized using electronic absorption, EPR, resonance Raman, and 2H NMR spectroscopies under cryogenic conditions. Besides, the identities and yields of the resultant [(Por)FeIII(OOH)] complexes have been probed by the comparison of their spectroscopic features with those of the independently prepared (i.e., by reduction–protonation of the [(Por)FeIII(O2−˙)] counterpart) complexes, and by quantification of hydrogen peroxide liberated upon acidification, respectively.
Variable temperature kinetic (Eyring) studies have allowed the ascertainment of activation parameters (i.e., ΔH‡, ΔS‡, and ΔG‡) that dictate the aforementioned PCET reactivities of [(Por)FeIII(O2−˙)] complexes, which are in strong support of their second-order PCET rates. Indeed, computational findings are in great agreement within the limitations of the employed solvation model. Notably, this study marks the first report with both experimental and theoretical insights into activation parameters of any substrate reactivity of synthetic heme superoxo systems, which, in this case, could be monumental in the rational design of oxygen reduction catalysts for fuel cell or similar applications. To gain further understanding into the strong influence of heme center electronics on the competency to abstract an H˙ by the ligated superoxide unit, we have determined O–H BDFE's for the entire series of [(Por)FeIII(OOH)] complexes utilizing experimental E° and pKa values deduced from redox and acid–base titrations (Table 1), respectively. The O–H BDFE's of 69.1, 67.5, and 66.5 kcal mol−1 found for [(F20TPP)FeIII(OOH)], [(TPP)FeIII(OOH)], and [(TMP)FeIII(OOH)], respectively, provide strong thermodynamic evidence in support of the observed limitations in reactivity. That is, [(F20TPP)FeIII(O2−˙)] and [(TPP)FeIII(O2−˙)] successfully abstracted an H˙ from TEMPO–H, while [(TMP)FeIII(O2−˙)] did not react due to the nullified thermodynamic driving force (i.e., BDFEheme hydroperoxo ≈ BDFETEMPOH). Moreover, the trends in experimentally observed O–H BDFE's are unequivocally supported by our computational results. Lastly, this work reveals previously unknown, critical aspects surrounding electronically driven feasibilities of substrate reactivities facilitated by heme superoxo intermediates. This knowledge will broaden the current understanding of long overlooked reactivity properties of mid-valent heme–oxygen intermediates in biology, while offering novel avenues for the design of better ORR catalysts leading to enhanced efficiencies (e.g., fine-tuning of (1) heme systems steered by electronic properties, (2) acidities of proton sources utilized in ORR catalysis, and (3) structure-based thermodynamic (e.g., pKa, E°, BDFE) and kinetic (e.g., reaction rates and feasibilities, rate limiting events) properties of heme ORR catalysts to further the overall efficacy etc.). If geometric, electronic, and/or secondary sphere properties of heme superoxide intermediates can be optimized to promote the oxidation of stronger organic substrates is an intriguing unknown, and will be a focus of our future interrogations.
Data availability
The datasets supporting this article have been uploaded as part of the supplementary material.
Author contributions
G. B. W. conceived the project idea and supervised the investigation; P. M. and C. L. synthesized all compounds, and generated and analyzed the experimental data; I. I. and S.-R. Y. conducted and analyzed the resonance Raman characterization experiments; E. F. G. and S. d. V. carried out theoretical computations and interpreted the data. G. B. W., S. d. V. and P. M. wrote and edited the manuscript with input from all other co-authors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The University of Alabama at Birmingham (UAB) is gratefully acknowledged for startup funds and the Faculty Development Grant provided to G. B. W. G. B. W, P. M., and C. L. thank Prof. Mary Ellen Zvanut (UAB) and Dr Ken Belmore (University of Alabama, Tuscaloosa) for assistance with EPR and low temperature NMR experiments. E. F. G and S. d. V. thank the BBSRC for funding for a studentship under grant number BB/J014478/1. This work was supported by National Institutes of Health Grants GM115773 and GM126297 to S.-R. Y.
References
-
(a) T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed;
(b) S. M. Adam, G. B. Wijeratne, P. J. Rogler, D. E. Diaz, D. A. Quist, J. J. Liu and K. D. Karlin, Chem. Rev., 2018, 118, 10840–11022 CrossRef CAS;
(c) X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS;
(d) J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3569 CrossRef CAS PubMed;
(e) A. Decker and E. I. Solomon, Curr. Opin. Chem. Biol., 2005, 9, 152–163 CrossRef CAS PubMed.
-
(a) Y. Watanabe, H. Nakajima and T. Ueno, Acc. Chem. Res., 2007, 40, 554–562 CrossRef CAS;
(b) L. J. Smith, A. Kahraman and J. M. Thornton, Proteins: Struct., Funct., Bioinf., 2010, 78, 2349–2368 CrossRef CAS PubMed;
(c) S. Amanullah, A. Singha and A. Dey, Coord. Chem. Rev., 2019, 386, 183–208 CrossRef CAS.
-
(a) M. F. Perutz, M. G. Rossmann, A. F. Cullis, H. Muirhead, G. Will and A. C. T. North, Nature, 1960, 185, 416–422 CrossRef CAS PubMed;
(b) A. F. Riggs, Curr. Opin. Struct. Biol., 1991, 1, 915–921 CrossRef CAS;
(c) J. P. Collman, R. Boulatov, C. J. Sunderland and L. Fu, Chem. Rev., 2004, 104, 561–588 CrossRef CAS PubMed.
-
(a)
S. J. Thackray, I. Efimov, E. L. Raven and C. G. Mowat, in Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature, The Royal Society of Chemistry, 2011, pp. 400–426 Search PubMed;
(b) I. Efimov, J. Basran, S. J. Thackray, S. Handa, C. G. Mowat and E. L. Raven, Biochemistry, 2011, 50, 2717–2724 CrossRef CAS PubMed;
(c)
A. Lewis-Ballester, K. N. Pham, M. Liao, M. A. Correia and S.-R. Yeh, in Dioxygen-Dependent Heme Enzymes, The Royal Society of Chemistry, 2019, pp. 181–221 Search PubMed;
(d) A. Lewis-Ballester, D. Batabyal, T. Egawa, C. Lu, Y. Lin, M. A. Marti, L. Capece, D. A. Estrin and S.-R. Yeh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17371–17376 CrossRef CAS;
(e) E. L. Raven, J. Biol. Inorg Chem., 2016, 1–9 Search PubMed.
- Y. Zhang, Y. Zou, N. L. Brock, T. Huang, Y. Lan, X. Wang, Z. Deng, Y. Tang and S. Lin, J. Am. Chem. Soc., 2017, 139, 11887–11894 CrossRef CAS PubMed.
-
(a) J. D. Caranto, Curr. Opin. Chem. Biol., 2019, 49, 130–138 CrossRef CAS PubMed;
(b) S. M. Barry, J. A. Kers, E. G. Johnson, L. Song, P. R. Aston, B. Patel, S. B. Krasnoff, B. R. Crane, D. M. Gibson, R. Loria and G. L. Challis, Nat. Chem. Biol., 2012, 8, 814–816 CrossRef CAS PubMed;
(c) S. C. Dodani, J. K. B. Cahn, T. Heinisch, S. Brinkmann-Chen, J. A. McIntosh and F. H. Arnold, ChemBioChem, 2014, 15, 2259–2267 CrossRef CAS PubMed;
(d) F. Yu, M. Li, C. Xu, Z. Wang, H. Zhou, M. Yang, Y. Chen, L. Tang and J. He, PLoS One, 2013, 8, e81526 CrossRef PubMed;
(e) S. Louka, S. M. Barry, D. J. Heyes, M. Q. E. Mubarak, H. S. Ali, L. M. Alkhalaf, A. W. Munro, N. S. Scrutton, G. L. Challis and S. P. de Visser, J. Am. Chem. Soc., 2020, 142, 15764–15779 CrossRef CAS PubMed.
-
(a) J. Su and J. T. Groves, Inorg. Chem., 2010, 49, 6317–6329 CrossRef CAS PubMed;
(b) G. Ferrer-Sueta, N. Campolo, M. Trujillo, S. Bartesaghi, S. Carballal, N. Romero, B. Alvarez and R. Radi, Chem. Rev., 2018, 118, 1338–1408 CrossRef CAS PubMed.
-
(a) M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed;
(b) T. Matsui, M. Unno and M. Ikeda-Saito, Acc. Chem. Res., 2010, 43, 240–247 CrossRef CAS PubMed.
-
(a) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2278 CrossRef CAS PubMed;
(b) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed;
(c)
Handbook of Porphyrin Science, World Scientific Publishing Co., New Jersey, 2010 Search PubMed;
(d) P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed;
(e) S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949–1017 CrossRef CAS;
(f) H. M. Girvan and A. W. Munro, Curr. Opin. Chem. Biol., 2016, 31, 136–145 CrossRef CAS PubMed.
-
(a) D. Ghosh, J. Griswold, M. Erman and W. Pangborn, Nature, 2009, 457, 219–223 CrossRef CAS PubMed;
(b) K. R. Korzekwa, W. F. Trager, J. Mancewicz and Y. Osawa, J. Steroid Biochem. Mol. Biol., 1993, 44, 367–373 CrossRef CAS PubMed.
-
(a) Y. Zhu and R. B. Silverman, Biochemistry, 2008, 47, 2231–2243 CrossRef CAS PubMed;
(b) M. A. Marletta, J. Biol. Chem., 1993, 268, 12231–12234 CrossRef CAS PubMed;
(c) H. Huang, J.-M. Hah and R. B. Silverman, J. Am. Chem. Soc., 2001, 123, 2674–2676 CrossRef CAS PubMed.
- T. Doukov, H. Li, M. Soltis and T. L. Poulos, Biochemistry, 2009, 48, 10246–10254 CrossRef CAS PubMed.
-
(a) W. J. Wallace, R. A. Houtchens, J. C. Maxwell and W. S. Caughey, J. Biol. Chem., 1982, 257, 4966–4977 CrossRef CAS PubMed;
(b) K. Shikama, Chem. Rev., 1998, 98, 1357–1374 CrossRef CAS PubMed.
-
(a) M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Chem. Rev., 2018, 118, 2340–2391 CrossRef CAS PubMed;
(b) N. Mano and A. de Poulpiquet, Chem. Rev., 2018, 118, 2392–2468 CrossRef CAS PubMed;
(c) J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed;
(d) M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed;
(e) E. S. Dy, T. A. Roman, Y. Kubota, K. Miyamoto and H. Kasai, J. Phys.: Condens. Matter, 2007, 19, 445010 CrossRef;
(f) W. Zhang, W. Lai and R. Cao, Chem.
Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
-
(a) S. Hammes-Schiffer and A. A. Stuchebrukhov, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed;
(b) R. Tyburski, T. Liu, S. D. Glover and L. Hammarström, J. Am. Chem. Soc., 2021, 143, 560–576 CrossRef CAS PubMed;
(c) V. R. I. Kaila, M. I. Verkhovsky and M. Wikström, Chem. Rev., 2010, 110, 7062–7081 CrossRef CAS PubMed.
-
(a) J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed;
(b) J. W. Darcy, B. Koronkiewicz, G. A. Parada and J. M. Mayer, Acc. Chem. Res., 2018, 51, 2391–2399 CrossRef CAS PubMed;
(c) G. B. Wijeratne, B. Corzine, V. W. Day and T. A. Jackson, Inorg. Chem., 2014, 53, 7622–7634 CrossRef CAS PubMed;
(d) A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed;
(e) J. J. Warren and J. M. Mayer, Biochemistry, 2015, 54, 1863–1878 CrossRef CAS PubMed.
-
(a) M. L. Pegis, D. J. Martin, C. F. Wise, A. C. Brezny, S. I. Johnson, L. E. Johnson, N. Kumar, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 8315–8326 CAS;
(b) M. L. Pegis, B. A. McKeown, N. Kumar, K. Lang, D. J. Wasylenko, X. P. Zhang, S. Raugei and J. M. Mayer, ACS Cent. Sci., 2016, 2, 850–856 CrossRef CAS PubMed;
(c) M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000–11003 CrossRef CAS PubMed.
- N. Kostopoulos, C. Achaibou, J.-M. Noël, F. Kanoufi, M. Robert, C. Fave and E. Anxolabéhère-Mallart, Inorg. Chem., 2020, 59, 11577–11583 CrossRef CAS PubMed.
-
(a) I. P. Gerothanassis, M. Momenteau and B. Loock, J. Am. Chem. Soc., 1989, 111, 7006–7012 CrossRef CAS;
(b) M. Momenteau and C. A. Reed, Chem. Rev., 1994, 94, 659–698 CrossRef CAS.
-
(a) A. C. Brezny, S. I. Johnson, S. Raugei and J. M. Mayer, J. Am. Chem. Soc., 2020, 142, 4108–4113 CrossRef CAS PubMed;
(b) A. C. Brezny, H. S. Nedzbala and J. M. Mayer, Chem. Commun., 2021, 57, 1202–1205 RSC;
(c) S. Bhunia, A. Rana, P. Roy, D. J. Martin, M. L. Pegis, B. Roy and A. Dey, J. Am. Chem. Soc., 2018, 140, 9444–9457 CrossRef CAS PubMed;
(d) S. Chatterjee, K. Sengupta, B. Mondal, S. Dey and A. Dey, Acc. Chem. Res., 2017, 50, 1744–1753 CrossRef CAS PubMed.
-
(a) K. Sengupta, S. Chatterjee, S. Samanta and A. Dey, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8431 CrossRef CAS PubMed;
(b) S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed;
(c) S. Samanta, K. Mittra, K. Sengupta, S. Chatterjee and A. Dey, Inorg. Chem., 2013, 52, 1443–1453 CrossRef CAS PubMed;
(d) S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 0098 CrossRef CAS.
- P. Mondal and G. B. Wijeratne, J. Am. Chem. Soc., 2020, 142, 1846–1856 CrossRef CAS PubMed.
-
(a) K. Tajima, M. Shigematsu, J. Jinno, K. Ishizu and H. Ohya-Nishiguchi, J. Chem. Soc., Chem. Commun., 1990, 144–145 RSC;
(b) E. E. Chufán and K. D. Karlin, J. Am. Chem. Soc., 2003, 125, 16160–16161 CrossRef PubMed;
(c) J.-G. Liu, Y. Shimizu, T. Ohta and Y. Naruta, J. Am. Chem. Soc., 2010, 132, 3672–3673 CrossRef CAS PubMed.
-
(a) P. Nagaraju, T. Ohta, J.-G. Liu, T. Ogura and Y. Naruta, Chem. Commun., 2016, 52, 7213–7216 RSC;
(b) J.-G. Liu, T. Ohta, S. Yamaguchi, T. Ogura, S. Sakamoto, Y. Maeda and Y. Naruta, Angew. Chem., Int. Ed., 2009, 48, 9262–9267 CrossRef CAS PubMed;
(c) T. Ohta, P. Nagaraju, J.-G. Liu, T. Ogura and Y. Naruta, J. Biol. Inorg Chem., 2016, 21, 745–755 CrossRef CAS PubMed.
-
(a) A. Singha, A. Mondal, A. Nayek, S. G. Dey and A. Dey, J. Am. Chem. Soc., 2020, 142, 21810–21828 CrossRef CAS PubMed;
(b) A. Singha and A. Dey, Chem. Commun., 2019, 55, 5591–5594 RSC.
-
(a) H. Kim, P. J. Rogler, S. K. Sharma, A. W. Schaefer, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2020, 142, 3104–3116 CrossRef CAS PubMed;
(b) H. Kim, P. J. Rogler, S. K. Sharma, A. W. Schaefer, E. I. Solomon and K. D. Karlin, Angew. Chem., Int. Ed., 2021, 60, 5907–5912 CrossRef CAS PubMed.
- F. G. Bordwell, J. P. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229–1231 CrossRef CAS.
- R. A. Ghiladi, R. M. Kretzer, I. Guzei, A. L. Rheingold, Y.-M. Neuhold, K. R. Hatwell, A. D. Zuberbühler and K. D. Karlin, Inorg. Chem., 2001, 40, 5754–5767 CrossRef CAS PubMed.
- D. A. Quist, M. A. Ehudin, A. W. Schaefer, G. L. Schneider, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2019, 141, 12682–12696 CrossRef CAS PubMed.
- J. J. Warren and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 7546–7547 CrossRef CAS PubMed.
-
(a) D. M. Corsi, N. N. Murthy, V. G. Young and K. D. Karlin, Inorg. Chem., 1999, 38, 848–858 CrossRef CAS PubMed;
(b) S. K. Sharma, A. W. Schaefer, H. Lim, H. Matsumura, P. Moënne-Loccoz, B. Hedman, K. O. Hodgson, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2017, 139, 17421–17430 CrossRef CAS PubMed;
(c) M. Nakamura, T. Ikeue, S. Neya, N. Funasaki and N. Nakamura, Inorg. Chem., 1996, 35, 3731–3732 CrossRef CAS PubMed.
-
(a) K. Tajima, S. Oka, T. Edo, S. Miyake, H. Mano, K. Mukai, H. Sakurai and K. Ishizu, J. Chem. Soc., Chem. Commun., 1995, 1507–1508 RSC;
(b) R. Oliveira, W. Zouari, C. Herrero, F. Banse, B. Schöllhorn, C. Fave and E. Anxolabéhère-Mallart, Inorg. Chem., 2016, 55, 12204–12210 CrossRef CAS PubMed.
- The (O–O) feature for [(F20TPP)FeIII(OOH)] was observed at 954 (Δ18O2 = −30) cm−1 (Fig. 3), which is at much higher energy compared other similar systems. Whether this higher energy O–O feature is due to some unexpected interaction in solution is unclear. We note that detailed computational analyses may be needed to unambiguously assign the O–O vibration of [(F20TPP)FeIII(OOH)].
-
(a) D. L. Wertz, M. F. Sisemore, M. Selke, J. Driscoll and J. S. Valentine, J. Am. Chem. Soc., 1998, 120, 5331–5332 CrossRef CAS;
(b) M. Selke, M. F. Sisemore and J. S. Valentine, J. Am. Chem. Soc., 1996, 118, 2008–2012 CrossRef CAS.
-
(a) J. J. D. Sacramento and D. P. Goldberg, Chem. Commun., 2019, 55, 913–916 RSC;
(b) J. B. Gordon, A. C. Vilbert, M. A. Siegler, K. M. Lancaster, P. Moënne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2019, 141, 3641–3653 CrossRef CAS;
(c) J. Y. Lee, R. L. Peterson, K. Ohkubo, I. Garcia-Bosch, R. A. Himes, J. Woertink, C. D. Moore, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 9925–9937 CrossRef CAS PubMed;
(d) Y.-M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668–10670 CrossRef CAS PubMed;
(e) C.-W. Chiang, S. T. Kleespies, H. D. Stout, K. K. Meier, P.-Y. Li, E. L. Bominaar, L. Que, E. Münck and W.-Z. Lee, J. Am. Chem. Soc., 2014, 136, 10846–10849 CrossRef CAS PubMed;
(f) R. L. Peterson, R. A. Himes, H. Kotani, T. Suenobu, L. Tian, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2011, 133, 1702–1705 CrossRef CAS PubMed;
(g) M. N. Blakely, M. A. Dedushko, P. C. Y. Poon, G. Villar-Acevedo and J. A. Kovacs, J. Am. Chem. Soc., 2019, 141, 1867–1870 CrossRef CAS PubMed.
-
(a) N. Kindermann, C.-J. Günes, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2017, 139, 9831–9834 CrossRef CAS PubMed;
(b) The difference in pKa values among the TEMPOH/TEMPOD couple is likely not adequate to result in the large KIE’s observed here, see K. M. Erickson,
et al.
, J. Phys. Chem. B, 2011, 115, 3038–3051 CrossRef CAS PubMed.
- W. Lai and S. Shaik, J. Am. Chem. Soc., 2011, 133, 5444–5452 CrossRef CAS PubMed.
- G. B. Wijeratne, V. W. Day and T. A. Jackson, Dalton Trans., 2015, 44, 3295–3306 RSC.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
-
(a) J.-M. Noel, N. Kostopoulos, C. Achaibou, C. Fave, E. Anxolabéhère-Mallart and F. Kanoufi, Angew. Chem., Int. Ed., 2020, 59, 16376–16380 CrossRef CAS PubMed;
(b) All reduction potentials in the manuscript are referenced against the Fc+/0 redox couple, and hence some have been converted from the originally reported values.
- I. Kaljurand, J. Saame, T. Rodima, I. Koppel, I. A. Koppel, J. F. Kogel, J. Sundermeyer, U. Kohn, M. P. Coles and I. Leito, J. Phys.
Chem. A, 2016, 120, 2591–2604 CrossRef CAS.
- Addition of stoichiometric or excess amounts of weak bases (such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or Et3N) did not result in the complete conversion of [(Por)FeIII(OOH)] to [(Por)FeIII(O22−)]−. All pKa values of acids and bases used in this study have been previously reported (i.e., either measured in or converted to THF solvent) in the literature.
- E. P. Cappellani, S. D. Drouin, G. Jia, P. A. Maltby, R. H. Morris and C. T. Schweitzer, J. Am. Chem. Soc., 1994, 116, 3375–3388 CrossRef CAS.
- L. W. Chung, X. Li, H. Hirao and K. Morokuma, J. Am. Chem. Soc., 2011, 133, 20076–20079 CrossRef CAS PubMed.
- S. Shaik, D. Kumar and S. P. de Visser, J. Am. Chem. Soc., 2008, 130, 10128–10140 CrossRef CAS PubMed.
-
(a) D. C. Cummins, J. G. Alvarado, J. P. T. Zaragoza, M. Q. Effendy Mubarak, Y.-T. Lin, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2020, 59, 16053–16064 CrossRef CAS PubMed;
(b) F. G. Cantú Reinhard, A. S. Faponle and S. P. de Visser, J. Phys. Chem. A, 2016, 120, 9805–9814 CrossRef PubMed.
- R. Latifi, M. Bagherzadeh and S. P. de Visser, Chem.–Eur. J., 2009, 15, 6651–6662 CrossRef CAS PubMed.
- H. S. Ali, R. H. Henchman and S. P. de Visser, Chem.–Eur. J., 2021, 27, 1795–1809 CrossRef CAS PubMed.
- S. P. de Visser, J. Am. Chem. Soc., 2010, 132, 1087–1097 CrossRef CAS PubMed.
-
E. P. Hunter and S. G. Lias, in NIST Chemistry Webbook, ed. P. J. Linstrom and W. G. Mallard, NIST Standard Reference Database, Number 69, National Institute of Standards and Technology, Gaithersburg, MD, 20899, http://webbook.nist.gov Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text, figures and tables depicting synthetic procedures, detailed experimental procedures, characterisation data including UV-vis, EPR, resonance Raman, and computational data including Cartesian coordinates of all structures. See DOI: 10.1039/d1sc01952j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Izumi
Ishigami
b,
Emilie F.
Gérard
c,
Chaeeun
Lim
a,
Syun-Ru
Yeh
b,
Sam P.
de Visser
a,
Izumi
Ishigami
b,
Emilie F.
Gérard
c,
Chaeeun
Lim
a,
Syun-Ru
Yeh
b,
Sam P.
de Visser

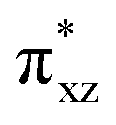 and
and  orbitals. The former orbital is a dominant 3dxz atomic orbital on iron in the plane of the FeOO group, while the latter is the antibonding interaction along the O–O bond that also interacts with the 3dyz orbital on iron. The triplet spin state has the same electron configuration but ferromagnetically coupled and its energy with zero-point energy correction is ΔE + ZPE = 5.6 kcal mol−1 higher in energy. Our calculated ground state; therefore, matches experimental assignments (see above) that the system is EPR silent. Subsequently, we calculated the hydrogen atom abstraction transition states (TSHA) for the reaction of 1,3[(TPP)FeIII(O2−˙)] from TEMPO–H and the optimized geometries are shown in Fig. 7. The reactions are concerted with a single hydrogen atom transfer leading to an iron(III)–hydroperoxo product. The transition states are early with short TEMPO–H distances of 1.075 and 1.057 Å in the singlet and triplet spin states, respectively. At the same time, the accepting O–H distance is long: 1.419 Å in 1TSHA and 1.473 Å in 3TSHA. Generally, early transition states correspond with low-energy hydrogen atom abstraction reactions, while later transition states have much higher energy barriers.45
orbitals. The former orbital is a dominant 3dxz atomic orbital on iron in the plane of the FeOO group, while the latter is the antibonding interaction along the O–O bond that also interacts with the 3dyz orbital on iron. The triplet spin state has the same electron configuration but ferromagnetically coupled and its energy with zero-point energy correction is ΔE + ZPE = 5.6 kcal mol−1 higher in energy. Our calculated ground state; therefore, matches experimental assignments (see above) that the system is EPR silent. Subsequently, we calculated the hydrogen atom abstraction transition states (TSHA) for the reaction of 1,3[(TPP)FeIII(O2−˙)] from TEMPO–H and the optimized geometries are shown in Fig. 7. The reactions are concerted with a single hydrogen atom transfer leading to an iron(III)–hydroperoxo product. The transition states are early with short TEMPO–H distances of 1.075 and 1.057 Å in the singlet and triplet spin states, respectively. At the same time, the accepting O–H distance is long: 1.419 Å in 1TSHA and 1.473 Å in 3TSHA. Generally, early transition states correspond with low-energy hydrogen atom abstraction reactions, while later transition states have much higher energy barriers.45
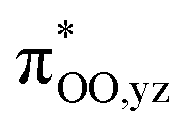 orbital and generates a doublet spin [(TPP)FeIII(OOH)] coupled to a TEMPO radical. This electron transfer is confirmed by the group spin densities that show an increase of spin on the TEMPO group to ρSub = 0.42, while at the same time spin is lost on the dioxygen moiety ρOO = 0.69. The hydrogen atom abstraction barrier is ΔE‡ + ZPE = 3.2 kcal mol−1 in the singlet spin state and 9.9 kcal mol−1 for the triplet spin state barrier. The low-spin barrier matches the experimentally determined ΔH‡ = 2.9 kcal mol−1 excellently. The experimentally determined entropy contribution is relatively large, probably due to a solvent cage surrounding the molecular complex. As shown by previous work of ours, reactions in solution often have a solvent cage around the active complex that affect entropies and particularly reduces vibrational contributions.46 Because of the fact that the solvent cage was not included in the model, the computational free energy of activation is relatively low and much lower than that observed in experiment as the gas-phase entropy is overestimated.
orbital and generates a doublet spin [(TPP)FeIII(OOH)] coupled to a TEMPO radical. This electron transfer is confirmed by the group spin densities that show an increase of spin on the TEMPO group to ρSub = 0.42, while at the same time spin is lost on the dioxygen moiety ρOO = 0.69. The hydrogen atom abstraction barrier is ΔE‡ + ZPE = 3.2 kcal mol−1 in the singlet spin state and 9.9 kcal mol−1 for the triplet spin state barrier. The low-spin barrier matches the experimentally determined ΔH‡ = 2.9 kcal mol−1 excellently. The experimentally determined entropy contribution is relatively large, probably due to a solvent cage surrounding the molecular complex. As shown by previous work of ours, reactions in solution often have a solvent cage around the active complex that affect entropies and particularly reduces vibrational contributions.46 Because of the fact that the solvent cage was not included in the model, the computational free energy of activation is relatively low and much lower than that observed in experiment as the gas-phase entropy is overestimated.