
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA therapeutic keypad lock decoded in drug resistant cancer cells†
Gulsen
Turkoglu
ab,
Gozde Kayadibi
Koygun
 c,
Mediha Nur
Zafer Yurt
b,
Seyda Nur
Pirencioglu
d and
Sundus
Erbas-Cakmak
*ab
c,
Mediha Nur
Zafer Yurt
b,
Seyda Nur
Pirencioglu
d and
Sundus
Erbas-Cakmak
*ab
aDepartment of Molecular Biology and Genetics, Konya Food and Agriculture University, Meram, Konya, Turkey. E-mail: sundus.erbascakmak@gidatarim.edu.tr
bResearch and Development Center for Diagnostic Kits (KIT-ARGEM), Konya Food and Agriculture University, Konya, Turkey
cDepartment of Nanotechnology and Advanced Materials, Selcuk University, Konya, Turkey
dDepartment of Molecular Biology and Genetics, Necmettin Erbakan University, Konya, Turkey
First published on 17th June 2021
Abstract
A molecular keypad lock that displays photodynamic activity when exposed to glutathione (GSH), esterase and light in the given order, is fabricated and its efficacy in drug resistant MCF7 cancer cells is investigated. The first two inputs are common drug resistant tumor markers. GSH reacts with the agent and shifts the absorption wavelength. Esterase separates the quencher from the structure, further activating the agent. After these sequential exposures, the molecular keypad lock is exposed to light and produces cytotoxic singlet oxygen. Among many possible combinations, only one ‘key’ can activate the agent, and initiate a photodynamic response. Paclitaxel resistant MCF7 cells are selectively killed. This work presents the first ever biological application of small molecular keypad locks.
The complex nature of diseases such as cancer necessitates smarter drugs that can discriminate each disease state or regulate drug efficacy spatially and/or temporally. With this intention, activatable drugs, drugs with on demand release properties are developed with promising selectivity.1–4 Information processing therapeutics which are based on molecular logic gate operations are another approach to solve this problem.5–7 Molecular logic gates are small compounds using Boolean logic operations to process inputs (i.e. the analyte concentration), and give an output as a result (fluorescence, and therapeutic activity etc.).8 Selective drug activation, release, multiple-analyte sensing and theranostic applications of these devices have been explored by us and others.5,9–19
Among the operations that can be carried out using small molecules, keypad locks provide an alternative application in information security.20 This logic operation can give a specific output when the inputs are given in the correct form and correct sequence. For the device, each input is considered as an AND logic operation where the history of the process is also considered. A pioneering example was reported by Margulies and Shanzer in 2007 where energy transfer is modulated by chelation of Fe3+ in a pH dependent manner.21 Later, various other devices were introduced with advanced properties such as more than 2 input responsiveness and error detection capability.22–24 All-photonic logic gates to address chemical waste production is extensively studied by Gust, Andréasson and Pischel.25,26 Beside small molecule keypad locks, enzymes, antibodies, and DNA hybrids are used to achieve the same goal.27–30 Although their potential use in molecular cryptology is highlighted, so far, there is no solid biological application of small molecule keypad locks.
In the research presented here, a molecular keypad lock is developed which displays a photodynamic therapeutic output when a molecule is exposed to analytes in the correct order and type (PS3, Fig. 1). Two inputs of the system are chosen to be the common markers of drug resistant tumours: glutathione (GSH) and esterase enzyme (E). Cancer cells develop resistance to traditional chemotherapy in time by changing the protein expression or metabolite content of the cell. This adaptation of cancer cells is an obstacle for their treatment and needs to be addressed. Glutathione is a tripeptide used in reductive biochemical synthesis and it is known to be present in elevated levels in rapidly dividing cells such as cancer cells.31 A high GSH level is reported to contribute to drug resistance, since GSH adducts of the drugs are exported out of the cell much more rapidly.32,33 Likewise, esterase enzyme activity is known to be associated with drug detoxification as this enzyme contributes to the chemical conversion of the drug.34,35 Glutathione and esterase enzyme are chosen to be the first two inputs of the molecular keypad lock, the first two digits of the password. In the research, light is used as the final input. Although trivial, light is essential for photodynamic activity and spatiotemporal control of irradiation, further improving selectivity of the therapy.
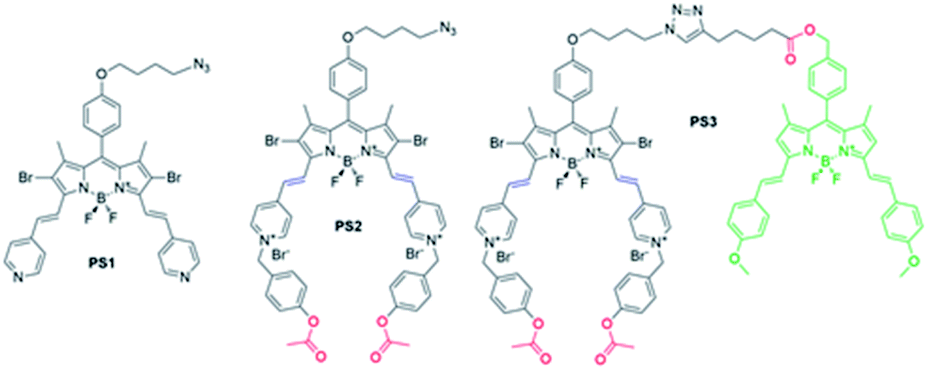 | ||
| Fig. 1 Chemical structures of model photosensitizers (PS1 and PS2) and a molecular keypad lock (PS3). Ester bonds (red) are prone to hydrolysis by the esterase enzyme. Distyryl sites of the photosensitizers (blue) can react with thiol nucleophile provided that it is bound to an electron deficient group (i.e. pyri-dinium). | ||
Keypad lock PS3 is a photodynamic therapy (PDT) agent. PDT is a non-invasive method used for the treatment of surface cancers and certain other diseases ranging from atherosclerosis to macular degeneration.36–39 In this therapy, a photosensitizer is excited with light, and produces cytotoxic singlet oxygen (1O2) thereby triggering apoptosis or necrosis of the cell, initiating an immune response and blocking microvasculature.40 In the research, a boradiazaindecene (BODIPY) photosensitizer is used to benefit from versatile chemistry and spectroscopic properties.41–45
Near-IR absorbing PS3 shown in Fig. 1 is the molecular keypad lock and it is synthesized in 13 steps (Scheme S1†). PS3 and model compound PS2 have heavy atoms on the structure to favour intersystem crossing required for transition to the triplet state and hence 1O2 generation occurs.43 Ester bonds on the structure of PS3 are prone to cleavage by esterase enzyme. Distryryl bonds on the PS3 (blue) tend to reduce or form an adduct with thiol nucleophiles when it is activated by the pyridinium electron withdrawing group.46 This property lies at the heart of sequential operation of esterase and GSH. When GSH reacts with electron poor double bonds, the extended conjugated structure is broken and PS3-a is generated (Fig. 2). This structure has absorption below 550 nm, like brominated core BODIPY molecules (compound 8, Scheme S1†), and therefore can be excited with a green light. A quencher (green) is attached to ensure that photodynamic activity is OFF until esterase cleaves the ester bond. This is because of the energy transfer from the photosensitizer to this module, until esterase separates the photosensitizer. Since PS3 lacks absorption around the 500–550 nm region, it is inactive until GSH reacts with the compound. However, the GSH reacted photosensitizer does show absorption in this region; so, in order to avoid full activation just by GSH, a quencher module is attached. Spectral overlap between the BODIPY core (see the structure of compound 8 in the ESI,† similar to that of PS3-a in terms of conjugation) and quencher (Q) can be seen from UV-Vis absorption and fluorescence spectra (Fig. 3 and S1†). By this way, the photosensitizer is chemically modulated by GSH to ensure excitation, and then esterase enzyme inhibits energy transfer by removing the quencher. Lastly a green light is used to excite the photosensitizer leading to generation of photodynamic action. Since light is necessary for the final excitation of the molecule, it should always be the last input. If the order of esterase and GSH changes, as shown in Fig. 2, activation is not expected to take place since cleavage of the ester bonds generates 4-hydroxybenzyl derivative on PS3, which spontaneously faces 1,4-elimination to generate pyridine (Fig. S2†).47 Pyridine on its own is not sufficiently electron withdrawing to favour nucleophilic attack of double bonds by GSH and to activate it as demonstrated below. Therefore, the photosensitizer preserves extended conjugation and essentially lacks absorption at the wavelength of excitation.
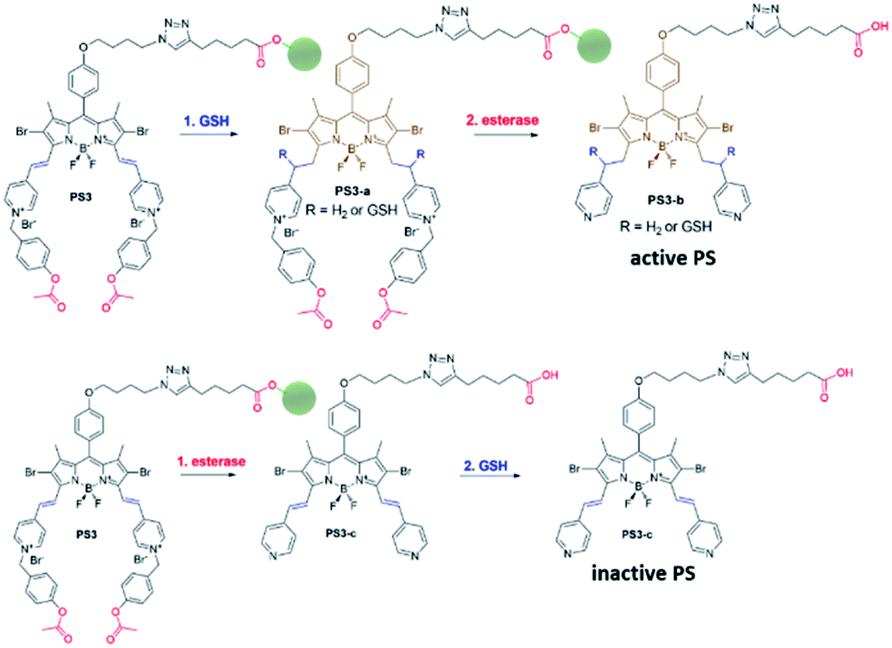 | ||
| Fig. 2 Sequential operation of GSH and esterase. GSH can only react with BODIPY distyryl units when the structure has electron withdrawing pyridinium, either reducing it or forming an adduct. Esterase enzyme cleaves ester bonds, liberating the photosensitizer from the quencher module (green). Initial esterase activity converts the pyridinium unit to pyridine, thereby decreasing the reactivity of double bonds with GSH. | ||
In order to understand the response of the PS3 to GSH, a molecule is incubated with 0.5 mM of GSH at 37 °C for 90 min. A new peak at 544 nm appears in UV-Vis absorption spectra consistent with the hypothesis (Fig. 3c, S1 and S9†). The formation of the GSH adduct (PS3-a) is demonstrated by Liquid Chromatography Mass spectrometry analysis (Fig. S3†). When control module PS1 is exposed to the same conditions, this new peak is not detected indicating that the pyridine bearing structure is neither activated enough for the nucleophilic substitution by GSH nor did it display PDT activity (Fig. S4 and S5†). On the other hand, GSH treated pyridinium bearing PS2 immediately displayed a colour change indicative of broken conjugation (Fig. S6†). When PS3 is incubated with esterase for 1 h, a small hypsochromic shift in the absorption peak is detected as a shoulder to the parent peak which is attributed to the conversion of pyridinium to pyridine (PS3-c, Fig. 3d). The control PS3 sample which is incubated under the same conditions but lacks esterase does not show an enhancement of this peak (Fig. 3d, black). High Resolution Mass Spectrometry analysis of the esterase treated PS2 samples confirm the hydrolysis of the ester and subsequent formation of the pyridine compound (Fig. S7†). Esterase treated samples display an increase in the emission intensity when excited at 620 nm (Fig. S8†). This is attributed to the initial quenching of the quencher module by the pyridinium photosensitizer. Analysis of the absorption and emission spectra suggest that the quencher module of PS3 can induce energy transfer to the pyridinium photosensitizer (Fig. 3). Once separated by esterase, fluorescence of the quencher module increases. In the case of GSH treated sample, a small enhancement in emission upon excitation at 500 nm is observed (Fig. S9†). Note that the GSH adduct (or PS3 with reduced double bonds) has higher absorption at this wavelength, which would be the reason for the increase in emission intensity. In the spectral analysis organic solvents with a low water content are used to monitor the formation of water-insoluble, neutral, pyridine-bearing intermediate species.
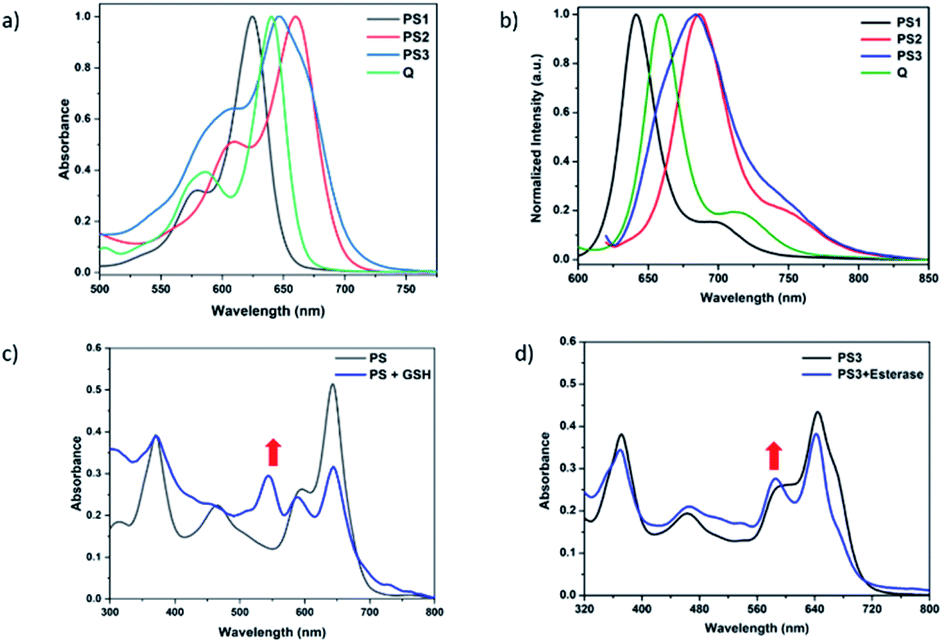 | ||
| Fig. 3 Normalized UV-Vis absorption and fluorescence spectra of PS1–3 in 2% water in THF (a and b). Samples are excited at 600 nm. Spectral changes of PS3 (10 μM) alone (black) or PS3 upon exposure to 0.5 mM GSH (c) and 10U esterase (d) for 90 min and 60 min at 37 °C, in 2% water in THF, respectively. A new peak at 544 nm appears upon incubation with GSH which is attributed to reduced PS3 and/orthe GSH-adduct. Esterase treatment increases the relative intensity of the shoulder peak around 600 nm. | ||
In the project, the molecular keypad lock is aimed to unlock in the presence of drug resistant tumour markers and get activated. Activation cannot take place when the input order differs. To demonstrate this, photodynamic action in the presence of all three inputs in a different order is investigated. 1O2 production can be followed by using trap molecule, 1,3-diphenylisobenzofuran (DPBF).48 This molecule reacts with 1O2 and loses its absorption at 418 nm. The effect of different input combinations on the PDT action are given in Fig. 4. In the first 15 min, all samples are kept in the dark. Under such conditions no 1O2 generation is detected, which indicates lack of dark activity. DPBF is exposed to light from a LED source (peak 505 nm) under the same experimental conditions and no decrease in the absorption is detected. This control experiment eliminates the photodegradation of DPBF in the absence of a photosensitizer. Upon irradiation before the activation of the photosensitizer by GSH and esterase, no 1O2 generation is observed as expected. The results show that 1O2 generation, and the subsequent decrease in DPBF absorption, are significantly more in the input order of glutathione, esterase enzyme and light, consistent with the proposed mode of activation.
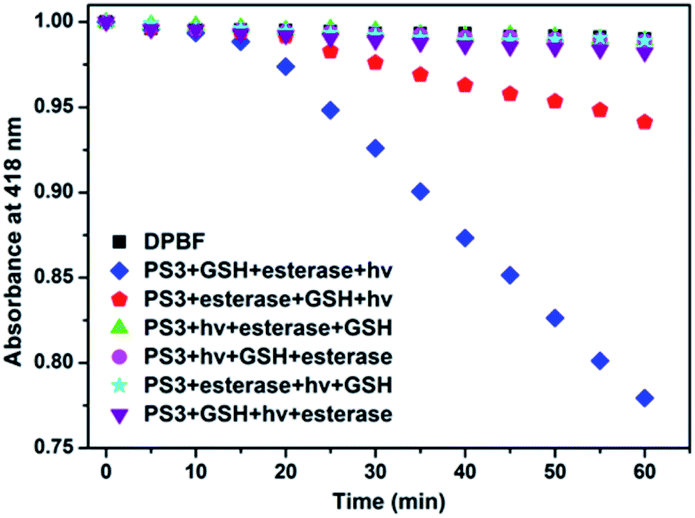 | ||
| Fig. 4 1O2 generation ability of PS3 (0.1 μM) when three inputs are given in a different order. All samples contain 50 μM of 1O2 trap molecule DPBF. In the first 15 minutes samples are kept in the dark. GSH is added in 0.5 mM concentration and incubated for 90 min at 37 °C. Samples are incubated with 10U esterase for 1 h at 37 °C. An LED light is irradiated from a 30 cm distance for 45 min. | ||
To analyse the effect of PDT action in the cell, a drug resistant cell line is generated. MCF7 cells are exposed to an increased dose of traditional cancer therapeutic agent paclitaxel as described in the literature.49 When the spindle-shaped morphology is obtained following maximum drug dose application, cells are reported to have drug resistance. At this stage, PS3 is applied to both normal and drug resistant cells. When cell viabilities at various concentrations are analysed, it has been found that the light toxicity of PS3 is significantly enhanced in drug resistant cells (Fig. 5). The IC50 values of irradiated samples are calculated to be 124.8 μM for MCF7 cells. This value is reduced to 52.5 μM in paclitaxel resistant MCF7 (Pac-MCF7) indicating improved cytotoxicity in these cells. Efficient induction of apoptosis is also proved by Annexin V and PI staining (Fig. 6). Under dark conditions, cells do not have significant loss of viability. Upon irradiation, resistant cells are more prone to apoptosis by the photosensitizer. Relative singlet oxygen generation abilities and results of cell culture experiments altogether confirm selective activation in drug resistant cells.
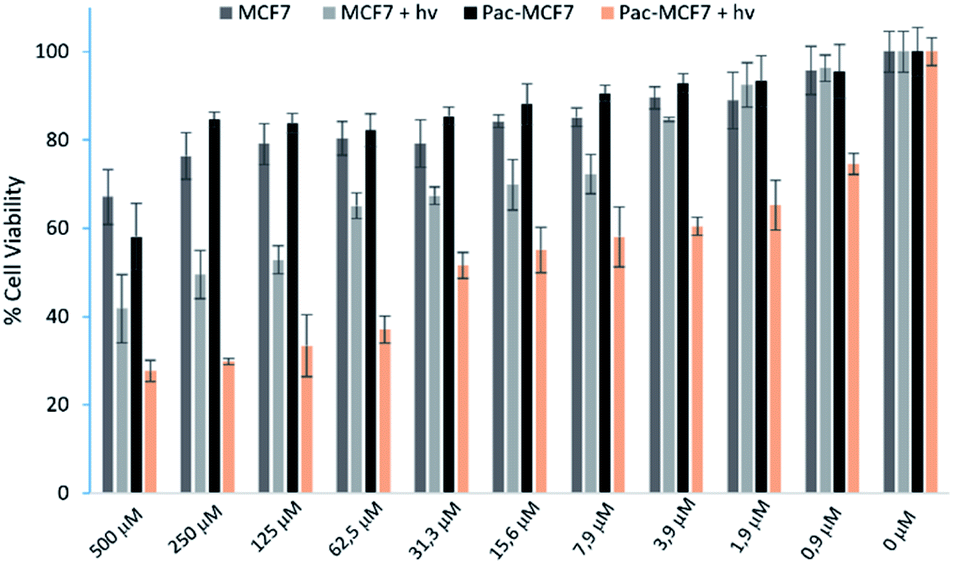 | ||
| Fig. 5 Change in the cell viability of normal and paclitaxel resistant MCF7 cells (Pac-MCF7) in the presence of PS3 at various concentrations. For each group, cell viability is analysed both after incubation in the dark or after irradiation with a 505 nm LED light source from a distance of 10 cm. Average values of three independent experiments are used. | ||
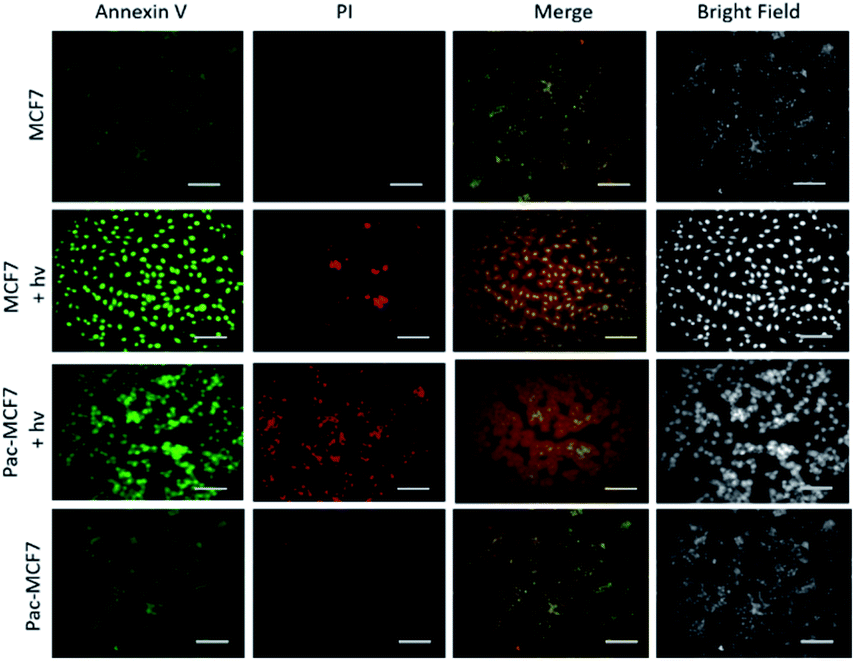 | ||
| Fig. 6 Apoptosis induction by PS3 (25 μM) in normal and paclitaxel resistant MCF7 cancer cells under dark conditions and upon irradiation with a 505 nm LED light from 10 cm distance. Scale bars: 50 μm. | ||
Conclusions
As a conclusion, in this research a molecular keypad lock device with selective therapeutic activity in drug resistant cells is demonstrated. The order of inputs is shown to determine singlet oxygen generation ability of the photosensitizer. Irradiation dependent apoptosis induction is enhanced in drug resistant cells and viability analysis indicates a significantly lowered IC50 value in irradiated resistant cells. In recent years, fluorescent probes, therapeutic or theranostic agents are being used for many advanced bio-applications.50–53 Previous studies pave the way for the use of molecular logic gates such as AND, OR, and DEMUX for therapeutic applications.5 This study expands the list further with one other advanced information processing device. Motivation behind the research is to develop an agent which is decoded by the cellular parameters on its way as it diffuses towards the drug resistant cell. GSH is known to be present in elevated levels and effluxes in drug resistant cancer cells.53–56 Exported GSH would be the first input of the photosensitizer. Following the intake of the agent, further activation by esterase enzyme is aimed. Light is selected as a final input since more efficient excitation of the activated photosensitizer can be achieved in the end and local application of light would enable further selectivity. Future studies would shed light on the exact cellular mechanism of action.Cellular studies presented here indicate the potential use of PS3 as a treatment modality, yet, long term exposure of the compound to solvents such as DMSO and THF leads to de-composition and a cellular stability of the compound requires additional research. Thiol of GSH may lead to cleavage of the ester bond via the formation of thioester, which also requires further attention. Sequential activation demonstrated here eliminates this latest possibility but in a cellular environment it should be investigated. In the work, a relatively higher dose of the photosensitizer (25 μM) is used in cell studies. This value is chosen to have more selectivity towards resistant cells, since as shown in Fig. 5, around these concentrations, the difference in cell viability is more pronounced. This concentration is still much lower than the IC50 value of MCF7 cells with or without light. By further optimizations (i.e. the distance of the light source), lower, hence safer, doses can be used.
Based on this discussion, once structurally and functionally optimised, information processing devices such as PS3 can find applications in diagnosis or personalized treatment of complex diseases such as cancer. In that respect, this study may inspire researchers to develop such devices.
Author contributions
The manuscript was written through contributions of all authors. G. T., M. N. Z. Y. and S. N. P. synthesized the compounds, G. T. performed spectroscopic analysis, G. K. K. performed the cell culture experiments, and S. E. C. proposed, coordinated the research, analysed the results, and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Cooperation in Science and Technology (COST), action number 17104, and the Scientific and Technological Research Council of Turkey, grant number 118Z668.Notes and references
- I. Giang, E. L. Boland and G. M. K. Poon, AAPS J., 2014, 16, 899–913 CrossRef CAS PubMed.
- X. Zhang, X. Li, Q. You and X. Zhang, Eur. J. Med. Chem., 2017, 139, 542–563 CrossRef CAS PubMed.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- X. Li, S. Kolemen, J. Yoon and E. U. Akkaya, Adv. Funct. Mater., 2017, 27, 1604053 CrossRef.
- S. Erbas-Cakmak, S. Kolemen, A. C. Sedgwick, T. Gunnlaugsson, T. D. James, J. Yoon and E. U. Akkaya, Chem. Soc. Rev., 2018, 47, 2228–2248 RSC.
- U. Pischel, J. Andréasson, D. Gust and V. F. Pais, ChemPhysChem, 2013, 14, 28–46 CrossRef CAS PubMed.
- C. Luo, L. He, F. Chen, T. Fu, P. Zhang, Z. Xiao, Y. Liu and W. Tan, Cell Rep., 2021, 2, 100350 Search PubMed.
- A. P. de Silva and S. Uchiyama, Nat. Nanotechnol., 2007, 2, 399–410 CrossRef CAS PubMed.
- S. Erbas-Cakmak and E. U. Akkaya, Angew. Chem., Int. Ed., 2013, 52, 11364–11368 CrossRef CAS PubMed.
- R. J. Amir, M. Popkov, R. A. Lerner, C. F. III Barbas and D. Shabat, Angew. Chem., Int. Ed., 2005, 44, 4378–4381 CrossRef CAS PubMed.
- S. Ozlem and E. U. Akkaya, J. Am. Chem. Soc., 2009, 131(1), 48–49 CrossRef CAS PubMed.
- S. Erbas-Cakmak, O. A. Bozdemir, Y. Cakmak and E. U. Akkaya, Chem. Sci., 2013, 4, 858–862 RSC.
- S. Erbas-Cakmak, F. P. Cakmak, S. D. Topel, T. B. Uyar and E. U. Akkaya, Chem. Commun., 2015, 51, 12258–12261 RSC.
- P. Zhang, J. Li, B. Li, J. Xu, F. Zeng, J. Lv and S. Wu, Chem. Commun., 2015, 51, 4414–4416 RSC.
- I. S. Turan, G. Gunaydin, S. Ayan and E. U. Akkaya, Nat. Commun., 2018, 9, 805 CrossRef PubMed.
- K. S. Hettie, J. L. Klockow and T. E. Glass, ACS Chem. Neurosci., 2013, 4, 1334–1338 CrossRef PubMed.
- D. P. Murale, H. Liew, Y.-H. Suh and D. G. Churchill, Anal. Methods, 2013, 5, 2650–2652 RSC.
- G. Upendar Reddy, J. Axthelm, P. Hoffmann, N. Taye, S. Glaser, H. Görls, S. L. Hopkins, W. Plass, U. Neugebauer, S. Bonnet and A. Schiller, J. Am. Chem. Soc., 2017, 139, 4991–4994 CrossRef PubMed.
- L. Unger-Angel, L. Motiei and D. Margulies, Front. Chem., 2019, 7, 243 CrossRef CAS PubMed.
- J. Andréasson and U. Pischel, Chem. Soc. Rev., 2018, 47, 2266–2279 RSC.
- D. Margulies, C. E. Felder, G. Melman and A. Shanzer, J. Am. Chem. Soc., 2007, 129, 347–354 CrossRef CAS PubMed.
- X.-J. Jiang and D. K. P. Ng, Angew. Chem., Int. Ed., 2014, 53, 10481–10484 CrossRef CAS PubMed.
- H. L. Li, W. Hong, S. J. Dong, Y. Q. Liu and E. K. Wang, ACS Nano, 2014, 8, 2796–2803 CrossRef CAS PubMed.
- B. Rout, P. Milko, M. A. Iron, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2013, 135, 15330–15333 CrossRef CAS PubMed.
- J. Andréasson, U. Pischel, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, J. Am. Chem. Soc., 2011, 133, 11641–11648 CrossRef PubMed.
- J. Andréasson, S. D. Straight, T. A. Moore, A. L. Moore and D. Gust, Chem.–Eur. J., 2009, 15, 3936–3939 CrossRef PubMed.
- G. Strack, M. Ornatska, M. Pita and E. Katz, J. Am. Chem. Soc., 2008, 130, 4234–4235 CrossRef CAS PubMed.
- J. Halámek, T. K. Tam, G. Strack, V. Bocharova, M. Pita and E. Katz, Chem. Commun., 2010, 46, 2405–2407 RSC.
- J. Halámek, T. K. Tam, S. Chinnapareddy, V. Bocharova and E. Katz, J. Phys. Chem. Lett., 2010, 1, 973–977 CrossRef.
- J. B. Zhu, X. Yang, L. B. Zhang, L. L. Zhang, B. H. Lou, S. J. Dong and E. K. Wang, Chem. Commun., 2013, 49, 5459–5461 RSC.
- N. Traverso, R. Ricciarelli, M. Nitti, B. Marengo, A. L. Furfaro, M. A. Pronzato, U. M. Marinari and C. Domenicotti, Oxid. Med. Cell. Longevity, 2013, 2013, 972913 Search PubMed.
- K. Zhang, P. Mack and K. P. Wong, Int. J. Oncol., 1998, 12, 871–953 CAS.
- E. Karwicka, Med. J. Cell Biol., 2010, 2, 105–124 Search PubMed.
- T. Satoh and M. Hosokawa, Annu. Rev. Pharmacol. Toxicol., 1998, 38, 257–288 CrossRef CAS.
- M. J. Hatfield, R. A. Umans, J. L. Hyatt, C. C. Edwards, M. Wierdl, L. Tsurkan, M. R. Taylor and P. M. Potter, Chem.-Biol. Interact., 2016, 259, 327–331 CrossRef CAS PubMed.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS.
- R. Wormald, J. Evans, L. Smeeth and K. Henshaw, Chem.-Biol. Interact., 2005, 4, CD002030 Search PubMed.
- M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47(16), 3897–3915 CrossRef CAS PubMed.
- A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535–545 CrossRef CAS.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107(11), 4891–4932 CrossRef CAS PubMed.
- O. Büyükçakır, O. A. Bozdemir, S. Kölemen, S. Erbas and E. U. Akkaya, Org. Lett., 2009, 11, 4644–4647 CrossRef PubMed.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169–11183 RSC.
- G. Turkoglu, G. K. Koygun, M. N. Z. Yurt, N. Demirok and S. Erbas-Cakmak, Org. Biomol. Chem., 2020, 18, 9433–9437 RSC.
- M. N. Z. Yurt, Y. Cakmak, G. Tekin, S. Karakurt and S. Erbas-Cakmak, ACS Omega, 2019, 4, 12293–12299 CrossRef CAS PubMed.
- S. Kolemen, M. Isik, G. M. Kim, D. Kim, H. Geng, M. Buyuktemiz, T. Karatas, X.-F. Zhang, Y. Dede, J. Yoon and E. U. Akkaya, Angew. Chem., Int. Ed., 2015, 54, 5340–5344 CrossRef CAS PubMed.
- Y. Wang, L. Yang, X.-R. Wei, R. Sun, Y.-J. Xu and J.-F. Ge, Anal. Methods, 2018, 10, 5291–5296 RSC.
- J. A. Howard and G. D. Mendenhall, Can. J. Chem., 1975, 53, 2199–2201 CrossRef CAS.
- M. D. Kars, O. D. Iseri, U. Gunduz and J. Molnar, Chemotherapy, 2008, 54, 194–200 CrossRef CAS PubMed.
- S. He, Y. Jiang, J. Li and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 10633–10638 CrossRef CAS PubMed.
- J. Huang and K. Pu, Chem. Sci., 2021, 12, 3379–3392 RSC.
- J. Huang, Y. Jiang, J. Li, S. He, J. Huang and K. Pu, Angew. Chem., Int. Ed., 2020, 59, 4415–4420 CrossRef CAS.
- S. P. Cole and R. G. Deeley, Trends Pharmacol. Sci., 2006, 27, 438–446 CrossRef CAS PubMed.
- L. Homolva, A. Varadi and B. Sarkadi, Biofactors, 2003, 17, 103–114 CrossRef PubMed.
- R. Marchand, C. L. Hammond and N. Ballatori, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2413–2420 CrossRef PubMed.
- D. Lorendeau, R. Nasr, A. Bounebdiel, E. Teodori, M. Gutschow, P. Falson, A. Di Pietro and H. Baubichon-Cortay, Curr. Med. Chem., 2017, 24, 1186–1213 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, experimental procedures, and spectral data. See DOI: 10.1039/d1sc02521j |
| This journal is © The Royal Society of Chemistry 2021 |