
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn effective and versatile strategy for the synthesis of structurally diverse heteroarylsilanes via Ir(III)-catalyzed C–H silylation†
Zhi-Bo
Yan
a,
Meng
Peng
a,
Qi-Long
Chen
a,
Ka
Lu
a,
Yong-Qiang
Tu
 *ab,
Kun-Long
Dai
a,
Fu-Min
Zhang
a and
Xiao-Ming
Zhang
a
*ab,
Kun-Long
Dai
a,
Fu-Min
Zhang
a and
Xiao-Ming
Zhang
a
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: tuyq@lzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Frontiers Science Center for Transformative Molecules, Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, Shanghai Jiao Tong University, Shanghai 200240, P. R. China
First published on 8th June 2021
Abstract
A versatile silylation of heteroaryl C–H bonds is accomplished under the catalysis of a well-defined spirocyclic NHC Ir(III) complex (SNIr), generating a variety of heteroarylsilanes. A significant advantage of this catalytic system is that multiple types of intermolecular C–H silylation can be achieved using one catalytic system at α, β, γ, or δ positions of heteroatoms with excellent regioselectivities. Mechanistic experiments and DFT calculations indicate that the polycyclic ligand of SNIr can form an isolable cyclometalated intermediate, which leaves a phenyl dentate free and provides a hemi-open space for activating substrates. In general, favorable silylations occur at γ or δ positions of chelating heteroatoms, forming 5- or 6-membered C–Ir–N cyclic intermediates. If such an activation mode is prohibited sterically, silylations would take place at the α or β positions. The mechanistic studies would be helpful for further explaining the reactivity of the SNIr system.
Introduction
Organosilanes have emerged as an important class of compounds with diverse utilities,1 serving as versatile organic reagents to mediate many novel organic reactions,1a,2 functional materials,3 therapeutic pharmaceuticals, and bioactive chemicals (Scheme 1).4 Traditionally, silicon-containing molecules have been prepared through the reactions of equivalent organometallic species with electrophilic silicon reagents,5 which suffer from inferior atom-economy and low functional-group tolerance. For several decades, transition-metal-catalyzed intermolecular direct C–H silylation has been developed as a more efficient and attractive strategy.1c–e Among them, C(sp2)–H silylation can generally be promoted with a directing group, which could coordinate with the metal center to form cyclometalated species and thus improve the regioselectivity of reaction. In this field, a range of directing groups have been developed,6 which include strongly coordinating pyridines and various azoles, and more weakly coordinating imines, amides, esters, and ketones. In particular, Hou reported an alkoxyl-directed Sc-catalyzed silylation of various anisole derivatives.6a In 2009, a distinctive strategy of using easily-installed and -removed 2-pyrazol-5-ylaniline as a directing group for o-silylation of arylboronic acids has been developed by Suginome.6b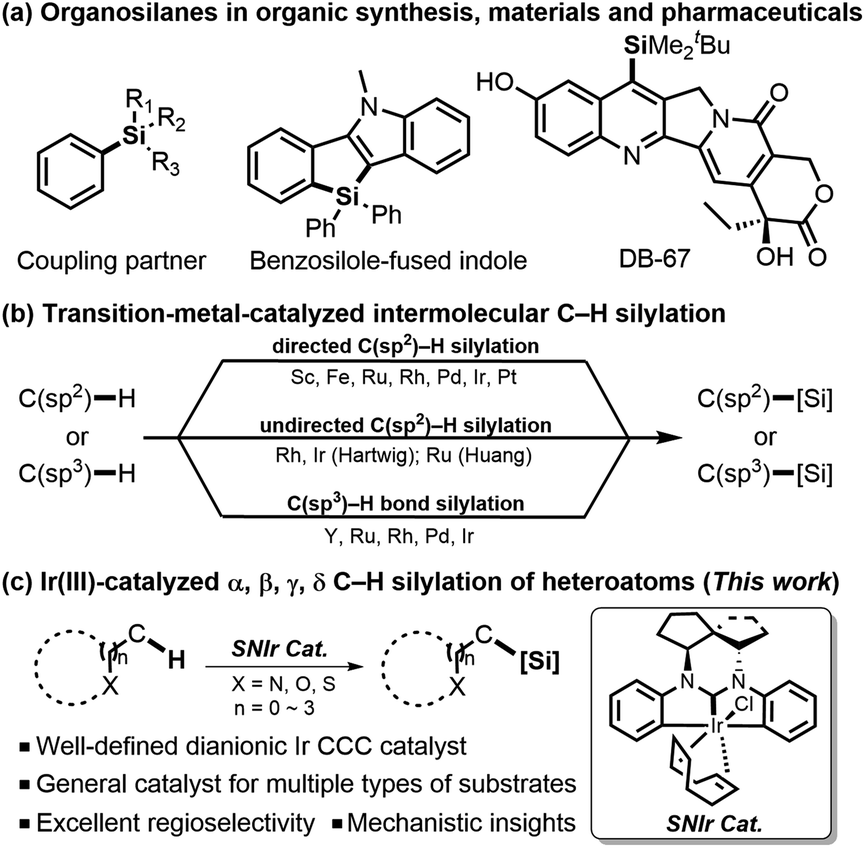 | ||
| Scheme 1 Representative organosilanes and intermolecular C–H silylation. | ||
In comparison, undirected C(sp2)–H silylation7 is more challenging due to the loss of interaction between the coordinating group and the catalyst. A breakthrough in undirected silylation was established by Hartwig,7a which takes advantage of steric effects in controlling regioselectivities. In addition, C(sp3)–H bond silylation at the benzylic position8 of the aromatic ring or next to the heteroatom such as nitrogen9 or sulfur10 was also reported, which has expanded the substrate scope and applicability of silylation reaction. Despite those precedent achievements on either directed or undirected C–H silylation reactions, a certain catalytic system could usually be used to activate a specific type of substrate. Therefore, development of a more general catalytic system for C–H silylation of multiple types of substrates with high regioselectivities for each type of reaction would be in high demand, considering the versatility of this strategy.
Results and discussion
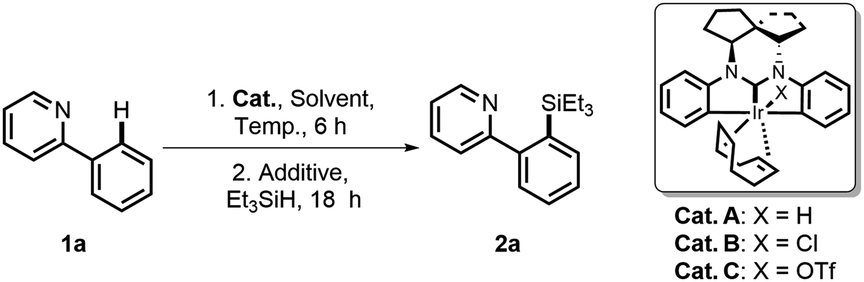
In our previous work, we have developed a well-defined dianionic Ir(III) CCC pincer catalyst (SNIr),11 which features unique double C(sp2)–H bond activation in a polycyclic ligand framework. This unexpected chelation mode reminds us that the central Ir may potentially enable C–H activation upon cleavage of the phenyl Ir–C bond to provide a hemi-open space for substrate activation under certain conditions. Base on this hypothesis, we have developed a versatile strategy for Ir(III)-catalyzed C–H silylation of diverse heteroarylsilanes. Herein we present our research results.We started our investigation with 2-phenylpyridine 1a as the model substrate and Et3SiH as the silane source to screen the catalysts A–C. A mixture of 1a and A–C (2.5–5 mol%) was first stirred at 100 °C for 6 h. Then a hydrogen acceptor (3 equiv.) and Et3SiH (2 equiv.) were added for further reaction. The results are summarized in Table 1. To our delight, the chloride catalyst B could give the highest yield of the desired silylation product 2a (entries 2 vs. 1 and 3), and no reaction was observed in the absence of Ir catalysts or hydrogen acceptors (entries 4 and 5). Further investigation found that tert-butylethylene (tbe) was the most effective hydrogen acceptor (entries 7 vs. 2 and 6). When an increased loading (5 mol%) of B was used in o-xylene solvent, the yield was improved to 85% (entries 9 vs. 7 and 8). Notably, when all reactants and catalysts were added to the reaction simultaneously, the system would become complicated and give a relatively low yield (entry 11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | Additive | Solvent | Temp. (°C) | Yieldb (%) |
| a Unless otherwise specified, reactions were conducted by pretreatment of a solution of 1a (0.5 mmol), cat. (2.5–5 mol%) and solvent (2 mL) at a given temperature for 6 h, and then the additive (3 equiv.) and Et3SiH (2 equiv.) were added for further reaction. b Determined by GC-MS (internal standard: dodecane). c Isolated yield in parentheses. d No pretreatment. nbe = 2-norbornene, tbe = tert-butylethylene. | |||||
| 1 | A (2.5) | Cyclohexene | Toluene | 100 | 22 |
| 2 | B (2.5) | Cyclohexene | Toluene | 100 | 46 |
| 3 | C (2.5) | Cyclohexene | Toluene | 100 | <5 |
| 4 | None | Cyclohexene | Toluene | 150 | 0 |
| 5 | B (2.5) | None | Toluene | 150 | 0 |
| 6 | B (2.5) | nbe | Toluene | 100 | 26 |
| 7 | B (2.5) | tbe | Toluene | 100 | 60 |
| 8 | B (5.0) | tbe | Toluene | 100 | 78 |
| 9 | B (5.0) | tbe | o-Xylene | 100 | 85 (80)c |
| 10 | B (5.0) | tbe | o-Xylene | 80 | 63 |
| 11d | B (5.0) | tbe | o-Xylene | 100 | 66 |
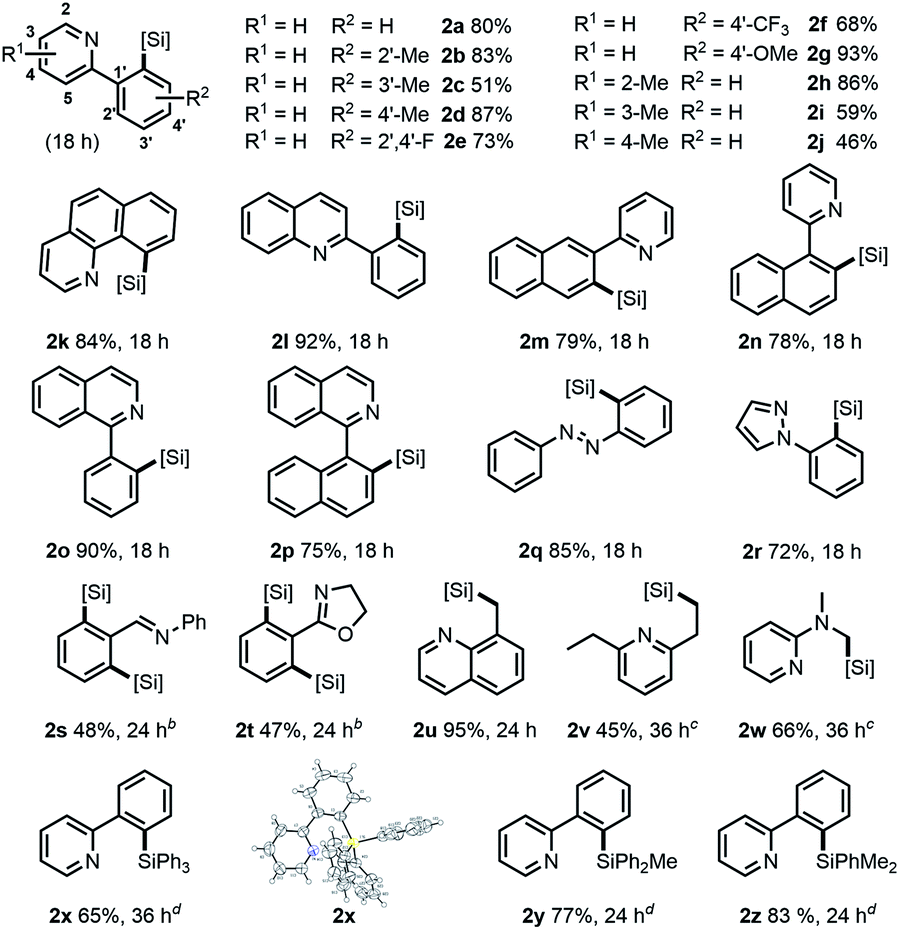
With the optimized conditions in hand,12 the substrate scope of γ silylations with a series of 2-phenylpyridine substrates was first explored. As shown in Table 2, high yields and regioselectivities were obtained in most cases, while the reaction efficiency could be influenced with the variation of the substitution pattern of substrates. Specifically, for substituted 2-phenylpyridine (1a–1j), the o- or p-methyl substitution on the benzene ring gave better product yields (83% for 2b, 87% for 2d) compared with the m-substitution (51% for 2c). The substrates with the p-EDG substituted phenyl group could give much higher yields than those with p-EWD substitution (2d and 2gvs.2e and 2f). A significant substituent effect was also observed on different positions of the pyridine ring. For example, 2-methyl substitution afforded a higher yield than 3,4-substitutions (2hvs.2i and 2j). The scope could be further extended to benzofused substrates (1k–1p), whose reactions could generally afford the desired products in good to high yields (75–92%). Notably, 2-phenylquinoline and 1-phenylisoquinoline could give excellent higher yields (92% for 2l, 90% for 2o). Moreover, for other N-heteroarenes, such as azo-, pyrazolyl-, and iminyl-arenes (1q–1t), they were also amenable in the reaction, affording the corresponding products with high efficiency. In particular, substrates 1s and 1t could mainly give disilylation products in moderate yields along with a trace amount of monosilylation product. Besides, our catalytic system was also well effective toward more inert γ C(sp3)–H bonds linked to heteroarenes. As a representative example, 8-methylquinoline could afford the γ-silylation product 2u in 95% yield. The reactions of 2,6-diethylpyridine (1v) and 2-dimethylaminopyridine (1w) were also feasible, giving products in moderate yields under conditions with elevated temperature. Remarkably, our catalytic system also accommodated the silylation of 1a with other hydrosilanes, such as Ph3SiH, Ph2MeSiH, or PhMe2SiH with good regioselectivities (2x–2z). It is worth nothing that in all cases we were not able to detect other α, β or δ silylation products.
|
|
|---|
| a Unless otherwise specified, reactions were conducted by pretreatment of a solution of 1 (0.5 mmol) and cat. B (5 mol%) in o-xylene (2 mL) at 100 °C for 6 h, and then tbe (3 equiv.) and Et3SiH (2 equiv.) were added for further reaction. b Trace amount (<5%) of monosilylation product could be detected by GC-MS. c At 140 °C, starting materials recycled. d Ph3SiH, Ph2MeSiH, or PhMe2SiH was used instead of Et3SiH. |
|
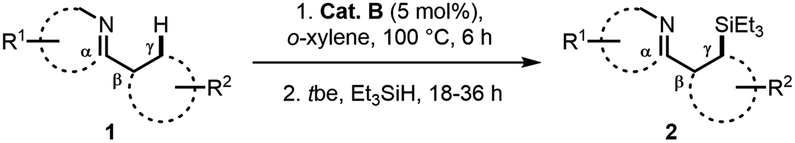
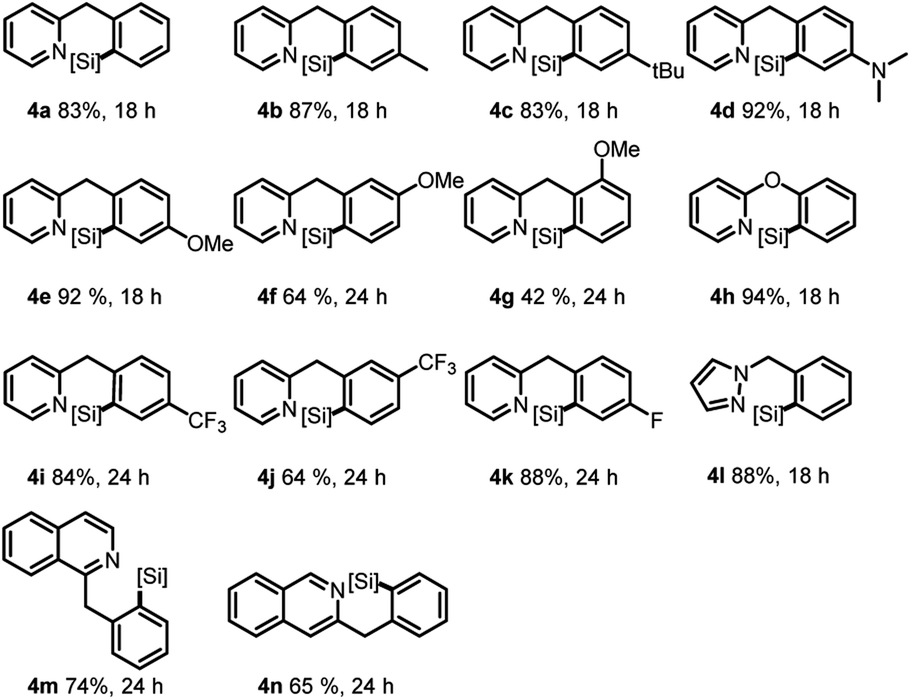
Subsequent investigation was carried out toward the δ-silylation of 2-benzylpyridine 3,13 and the desired products could be generated in good to high yields in most cases (Table 3). Generally, a higher reaction temperature (120 °C) was required than the corresponding γ C–H silylation, possibly due to a higher activation energy for the formation of the 6-membered cyclometalated intermediates. Similarly, both electron and steric effects of the benzyl group showed significant influence on the reaction outcome. For example, p-EDG substituted substrates gave higher yields than the p-EWG substituted ones in general sense (4d and 4evs.4i and 4k). However, for the o- or m-substitutions, both reactions were sluggish and gave poor to moderate yields regardless of either EDG or EWG substituents (4f, 4g and 4j). Delightedly, 2-phenoxypyridine afforded the best result (94% for 4h), probably because of the double activation of the same C–H bond (N to δ-C and O to β-C) and electro-donating effect of the ether group. Compared with γ C–H silylations of benzo-phenyl pyridines (92% for 2l, 90% for 2o, Table 2), a slow reaction rate and decreased product yields were observed for these δ-silylations (74% for 4m, 65% for 4n).
|
|
|---|
| a Unless otherwise specified, reactions were conducted by pretreatment of a solution of 3 (0.5 mmol) and cat. B (5 mol%) in o-xylene (0.5 mL) at 120 °C for 6 h, and then tbe (1.5 equiv.) and Et3SiH (3 equiv.) were added for further reaction. |
|
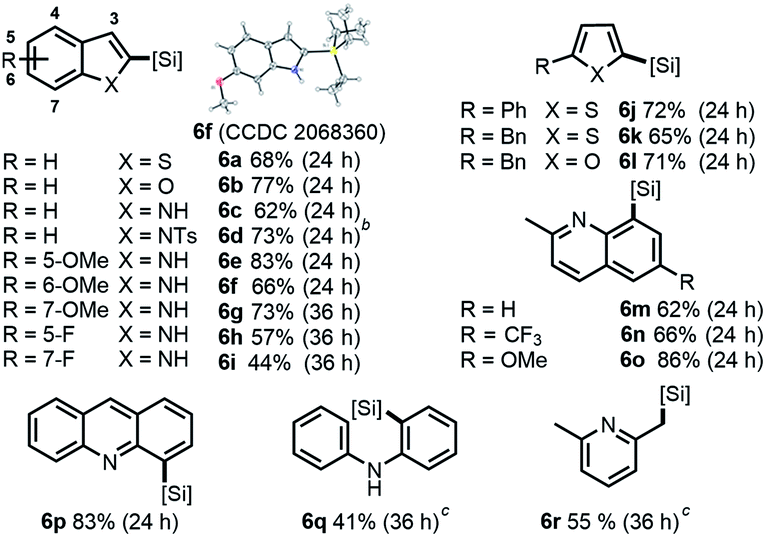
Finally, we investigated more universal and practically useful heteroarenes (Table 4), and these silylation reactions showed extremely good regioselectivities and broad substrate scope. For thiophene (5a, 5j and 5k) and furan (5b and 5l) derivatives, silylations generally took place at α positions with good yields, which complemented the normal electrophilic Friedel–Crafts silylation reactions.1e,14 Further investigation was focused on the derivatives of indole 5c–5i as they have practical utilities in the fields of natural products and drug discovery.15 In general, silylation always occurred at C-2 positions of indoles except for N-tosyl substituted indole, which directed the silylation to an unusual β-position (6d).7d The results of α-silylations of indoles indicated that EDG substitutions would give better outcomes than the EWG substitutions (6e–6gvs.6h and 6i). As for the substituted 2-methyl quinolines and benzo(b)quinoline, β-silylation would occur to afford 6m–6p in moderate to good yields. Moreover, this catalytic mode was also well effective toward the β C–H silylation of arene directed by sp3-N (5q) or the more inert β C(sp3)–H bond (5r).
|
|
|---|
| a Unless otherwise specified, reactions were conducted by pretreatment of a solution of 5 (0.5 mmol) and cat. B (5 mol%) in o-xylene (0.5 mL) at 120 °C for 6 h, and then tbe (3 equiv.) and Et3SiH (2 equiv.) were added for further reaction. b β-silylation was observed. c At 140 °C, starting materials recycled. |
|
Computational studies were next conducted to explore the mechanism using 3a (2-BnPy) as a model (Fig. 1).6f,16,17 Initially, the cod ligand of cat. B was dissociated from Ir before coordination of the N atom of 2-BnPy. Next, σ-metathesis between the phenyl C–Ir and δ C–H bonds of the 2-BnPy moiety easily took place through TS1 to form a stable 6-membered intermediate IM1, which then reacted with Et3SiH to give the Ir–H species IM3 with the formation of Et3SiCl. As the catalytic cycle begins, the hydride of IM3 was captured by the hydrogen acceptor (tbe) in the system to give Ir-alkyl species IM4. Then the o-C–H activation of the side-phenyl occurred through TS3 with the release of neohexane and formation of the Ir–Si intermediate IM5 in the presence of Et3SiH. Thereafter, Ir(I) species IM6 was generated by reductive elimination of IM5 through TS4. Finally, exchange of the silylation products 4a to 2-BnPy followed by oxidative addition of Ir to the δ C–H bond of intramolecular 2-BnPy regenerated the active Ir–H species IM3 and completed the catalytic cycle.
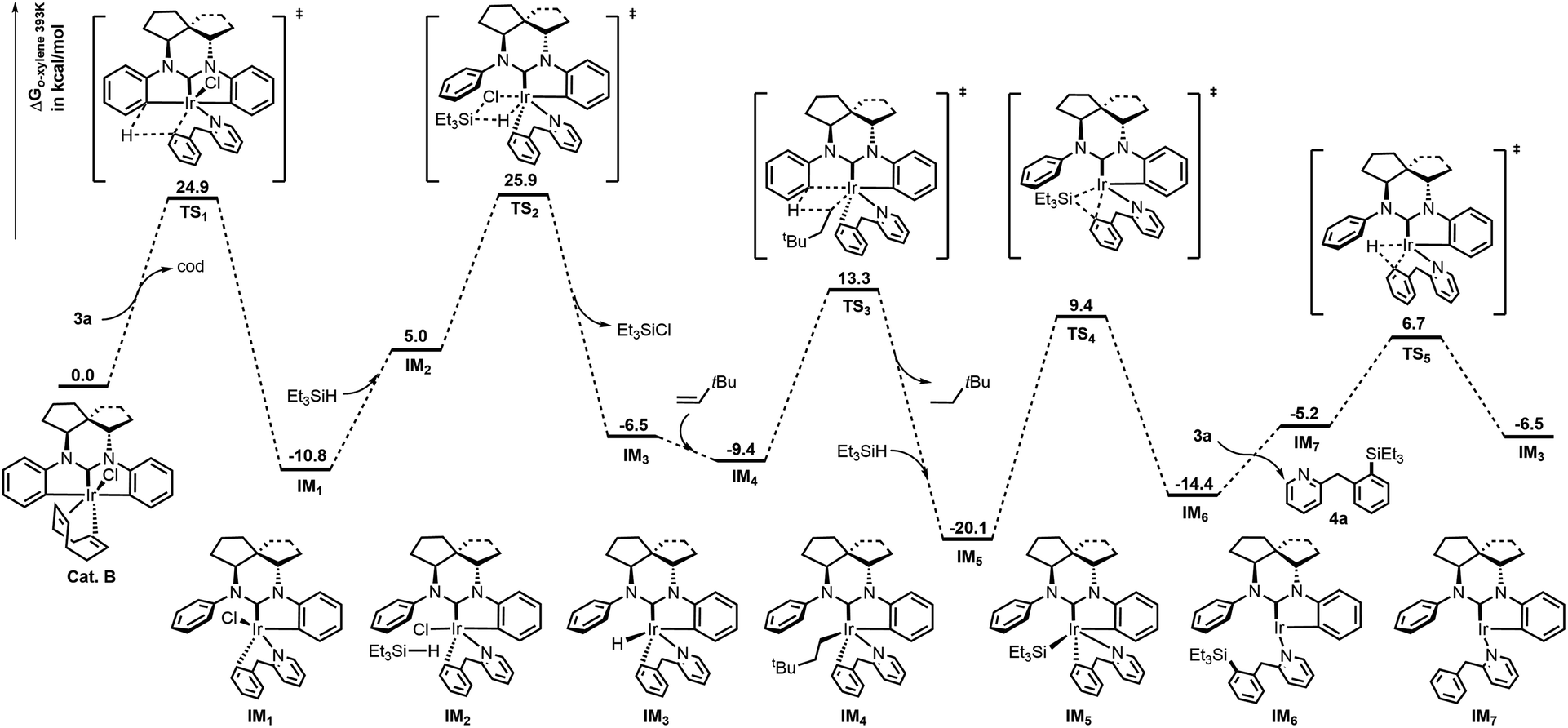 | ||
| Fig. 1 Energy profiles of the Ir-catalyzed silylation of 3a at 120 °C. | ||
To further probe the mechanism, several control experiments were conducted (Fig. 2). First, reactions of 1b or 3d and catalyst B without Et3SiH at 120 °C for 6 h could generate two brown complexes 1bB and 3dB in 88% and 92% yields, respectively. 1H NMR, high resolution mass spectroscopy (HRMS) and X-ray analysis confirmed that these complexes contained either a 5- or 6-membered C–Ir–N ring formed from the substrates and catalyst, and both intermediates had a free phenyl group dissociated with Ir.18 Furthermore, the silylation products 2b and 4d could be generated in 65% and 77% yields, respectively, when 5 mol% 1bB or 3dB was directly used as a catalyst under standard conditions. These results suggested that the iridacycle intermediates might serve as the pre-catalysts during the reaction process. Next, the H/D exchange experiment indicated that the C–H bond activation step might be irreversible (Fig. 2b). The kinetic isotope effect experiment showed a value of 3.1 from two parallel reactions and a KIE of 2.4 from intermolecular competition, which indicated that the C–H bond cleavage process was likely involved in the rate-determining step (Fig. 2c).
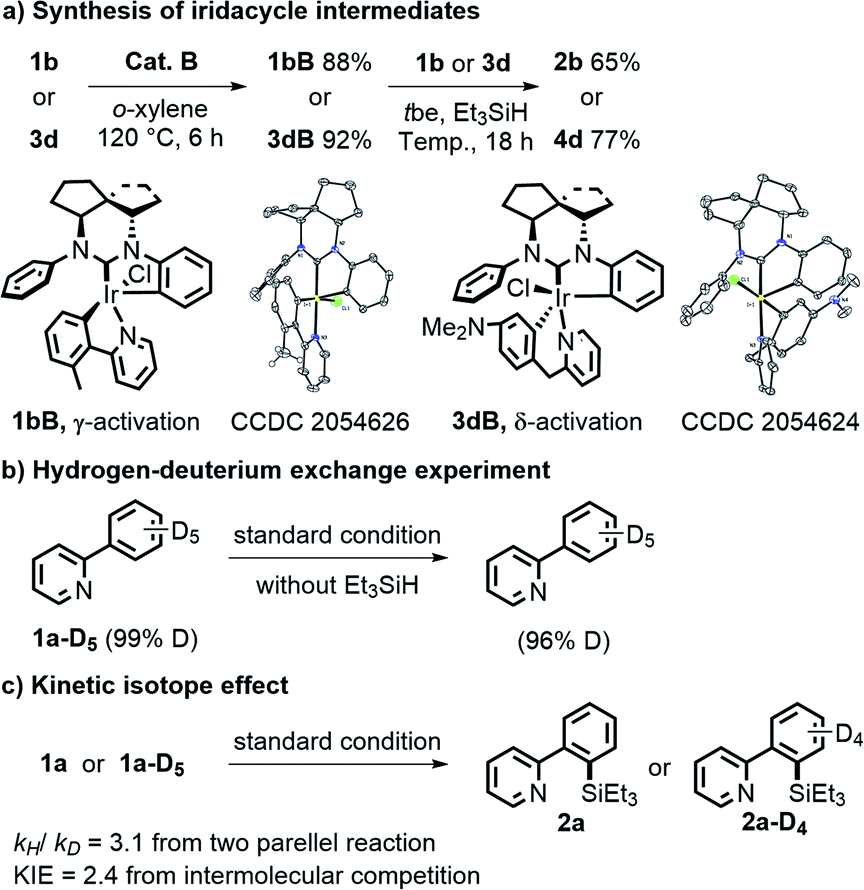 | ||
| Fig. 2 Mechanistic experiments. | ||
Conclusions
In summary, we have developed a general catalyst system based on SNIr for intermolecular C–H silylation of a wide range of substrate types with excellent regioselectivities and good to high yields. In all examples, single silylation products can be obtained in high regioselectivities. Mechanistic experiments and DFT calculations indicated the intermediate species and an Ir(III)/Ir(I) mechanism in the catalytic cycle. This methodology we established here would probably shed some new light on dianionic pincer complexes both in academic and applied research.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
Z.-B. Yan performed all of the experiments and prepared the ESI;† M. Peng prepared materials and catalysts for silylation reactions; K. Lu performed the computational studies; Y.-Q. Tu and Z.-B. Yan wrote the manuscript; all authors provided input on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the NSFC (No. 21871117, 21702136, 21502080, and 21772071), the “111” Program of MOE, the Major project (2018ZX09711001-005-002) of MOST, and the STCSM (19JC1430100) for the financial support. This work was supported by the High-Performance Computing Center (HPCC) of Shanghai Jiao Tong University. Also, we thank Zi-Hao Li from the Shanghai Jiao Tong University for his helpful discussion.Notes and references
- (a) C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946 CrossRef CAS PubMed; (b) R. Sharma, R. Kumar, I. Kumar, B. Singh and U. Sharma, Synthesis, 2015, 47, 2347 CrossRef CAS; (c) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631 CrossRef CAS; (d) H. T. Huang, T. Li, J. Z. Wang, G. P. Qin and T. B. Xiao, Chin. J. Org. Chem., 2019, 39, 1511 CrossRef CAS; (e) S. C. Richter and M. Oestreich, Trends Chem., 2020, 2, 13 CrossRef CAS.
- (a) S. E. Denmark and R. F. Sweis, Metal-Catalyzed Cross-Coupling Reactions, ed. A. De Meijere and F. Diederich, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1, pp. 163–216 Search PubMed; (b) B. Yang, W. Yang, Y. Guo, L. You and C. He, Angew. Chem., Int. Ed., 2020, 59, 22217 CrossRef CAS; (c) H. Chen, Y. Chen, X. Tang, S. Liu, R. Wang, T. Hu, L. Gao and Z. Song, Angew. Chem., Int. Ed., 2019, 58, 4695 CrossRef CAS PubMed; (d) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644 CrossRef CAS PubMed.
- (a) T. Kumagai and S. Itsuno, Macromolecules, 2002, 35, 5323 CrossRef CAS; (b) P. M. Zelisko, Bio-Inspired Silicon-Based Materials, Springer, Dordrecht, The Netherlands, 2014 Search PubMed.
- (a) A. K. Franz, Curr. Opin. Drug Discovery Dev., 2007, 10, 654 CAS; (b) M. Mortensen, R. Husmann, E. Veri and C. Bolm, Chem. Soc. Rev., 2009, 38, 1002 RSC; (c) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed; (d) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed.
- (a) S. E. Denmark and L. Neuville, Org. Lett., 2000, 2, 3221 CrossRef CAS PubMed; (b) M. Nakano, S. Shinamura, R. Sugimoto, I. Osaka, E. Miyazaki and K. Takimiya, Org. Lett., 2012, 14, 5448 CrossRef CAS PubMed.
- Selected examples of directed C(sp2)–H silylation: (a) J. Oyamada, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2011, 50, 10720 CrossRef CAS PubMed; (b) H. Ihara and M. Suginome, J. Am. Chem. Soc., 2009, 131, 7502 CrossRef CAS PubMed; (c) T. Mita, K. Michigami and Y. Sato, Org. Lett., 2012, 14, 3462 CrossRef CAS PubMed; (d) N. A. Williams, Y. Uchimaru and M. Tanaka, J. Chem. Soc., Chem. Commun., 1995, 1129 RSC; (e) G. Choi, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2013, 135, 13149 CrossRef CAS PubMed; (f) L. Rubio-Pérez, M. Iglesias, J. Munárriz, V. Polo, V. Passarelli, J. J. Pérez-Torrente and L. A. Oro, Chem. Sci., 2017, 8, 4811 RSC; (g) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 14903 CrossRef CAS PubMed; (h) E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092 CrossRef CAS PubMed; (i) T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534 CrossRef CAS PubMed; (j) J.-L. Pan, C. Chen, Z.-G. Ma, J. Zhou, L.-R. Wang and S.-Y. Zhang, Org. Lett., 2017, 19, 5216 CrossRef CAS PubMed; (k) W. Li, W. Chen, B. Zhou, Y. Xu, G. Deng, Y. Liang and Y. Yang, Org. Lett., 2019, 21, 2718 CrossRef CAS PubMed.
- Selected examples of undirected C(sp2)–H silylation: (a) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed; (b) H. Fang, L. Guo, Y. Zhang, W. Yao and Z. Huang, Org. Lett., 2016, 18, 5624 CrossRef CAS PubMed; (c) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592 CrossRef CAS; (d) B. Lu and J. R. Falck, Angew. Chem., Int. Ed., 2008, 47, 7508 CrossRef CAS PubMed; (e) A. A. Toutov, W.-B. Liu, K. N. Betz, A. Fedorov, B. M. Stoltz and R. H. Grubbs, Nature, 2015, 518, 80 CrossRef CAS PubMed; (f) A. A. Toutov, W.-B. Liu, K. N. Betz, B. M. Stoltz and R. H. Grubbs, Nat. Protoc., 2015, 10, 1897 CrossRef CAS PubMed; (g) Q. Yin, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2016, 55, 3204 CrossRef CAS; (h) Y. Ma, B. Wang, L. Zhang and Z. Hou, J. Am. Chem. Soc., 2016, 138, 3663 CrossRef CAS; (i) M. Murai, N. Nishinaka and K. Takai, Angew. Chem., Int. Ed., 2018, 57, 5843 CrossRef CAS PubMed; (j) L. Zhang, K. An, Y. Wang, Y.-D. Wu, X. Zhang, Z.-X. Yu and W. He, J. Am. Chem. Soc., 2021, 143, 3571 CrossRef CAS.
- (a) F. Kakiuchi, K. Tsuchiya, M. Matsumoto, E. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792 CrossRef CAS PubMed; (b) Y. Kuninobu, T. Nakahara, H. Takeshima and K. Takai, Org. Lett., 2013, 15, 426 CrossRef CAS PubMed.
- (a) T. Mita, K. Michigami and Y. Sato, Chem.–Asian J., 2013, 8, 2970 CrossRef CAS; (b) H. Fang, W. Hou, G. Liu and Z. Huang, J. Am. Chem. Soc., 2017, 139, 11601 CrossRef CAS.
- T. Luo, H.-L. Teng, C. Xue, M. Nishiura and Z. Hou, ACS Catal., 2018, 8, 8027 CrossRef.
- Z.-B. Yan, K.-L. Dai, B.-M. Yang, Z.-H. Li, Y.-Q. Tu, F.-M. Zhang, X.-M. Zhang, M. Peng, Q.-L. Chen and Z.-R. Jing, Sci. China: Chem., 2020, 63, 1761 CrossRef CAS.
- For detailed condition screening of γ C–H silylations, see ESI, Section 3.1.†.
- For detailed condition screening of δ C–H silylations, see ESI, Section 4.1.†.
- (a) S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52 CrossRef PubMed; (b) Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868 CrossRef CAS PubMed.
- H.-J. Borschberg, Curr. Org. Chem., 2005, 9, 1465 CrossRef CAS.
- For computational details, see ESI, Section 7.†.
- (a) T. Sperger, I. A. Sanhueza, I. Kalve and F. Schoenebeck, Chem. Rev., 2015, 115, 9532 CrossRef CAS PubMed; (b) C. Karmel and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 10494 CrossRef CAS PubMed.
- For details of 1H NMR, mass spectroscopy, infrared spectrum and X-ray data of complexes 1bB and 3dB, see ESI, Sections 6.1 and 8.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2068360, 2054626 and 2054624. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02344f |
| This journal is © The Royal Society of Chemistry 2021 |