
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn amplification-free ultra-sensitive electrochemical CRISPR/Cas biosensor for drug-resistant bacteria detection†
Akkapol
Suea-Ngam
 a,
Philip D.
Howes
*ab and
Andrew J.
deMello
*a
a,
Philip D.
Howes
*ab and
Andrew J.
deMello
*a
aInstitute for Chemical and Bioengineering, Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg1, 8093, Zürich, Switzerland. E-mail: andrew.demello@chem.ethz.ch
bDivision of Mechanical Engineering and Design, School of Engineering, London South Bank University, 103 Borough Road, London SE1 0AA, UK. E-mail: howesp@lsbu.ac.uk
First published on 3rd September 2021
Abstract
Continued development of high-performance and cost-effective in vitro diagnostic tools is vital for improving infectious disease treatment and transmission control. For nucleic acid diagnostics, moving beyond enzyme-mediated amplification assays will be critical in reducing the time and complexity of diagnostic technologies. Further, an emerging area of threat, in which in vitro diagnostics will play an increasingly important role, is antimicrobial resistance (AMR) in bacterial infections. Herein, we present an amplification-free electrochemical CRISPR/Cas biosensor utilizing silver metallization (termed E-Si-CRISPR) to detect methicillin-resistant Staphylococcus aureus (MRSA). Using a custom-designed guide RNA (gRNA) targeting the mecA gene of MRSA, the Cas12a enzyme allows highly sensitive and specific detection when employed with silver metallization and square wave voltammetry (SWV). Our biosensor exhibits excellent analytical performance, with detection and quantitation limits of 3.5 and 10 fM, respectively, and linearity over five orders of magnitude (from 10 fM to 0.1 nM). Importantly, we observe no degradation in performance when moving from buffer to human serum samples, and achieve excellent selectivity for MRSA in human serum in the presence of other common bacteria. The E-Si-CRISPR method shows significant promise as an ultrasensitive field-deployable device for nucleic acid-based diagnostics, without requiring nucleic acid amplification. Finally, adjustment to a different disease target can be achieved by simple modification of the gRNA protospacer.
1. Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-related technologies encompass a set of versatile and effective enzyme-based tools in the biological sciences.1–3 The rapid rise of CRISPR-based diagnostics has already had an immense impact on the in vitro diagnostics and biosensors fields, with approaches derived from these now proliferating and beginning to have real impact.4A key development CRISPR-based diagnostics was the demonstration of the CRISPR/Cas-based SHERLOCK technique by Zhang and co-workers in 2017.5,6 By leveraging the trans-cleavage activity of Cas13, probe-modified ssRNA is cleaved only after Cas13 has bound to a specific recombinase polymerase amplification (RPA) amplicon, allowing highly sensitive detection of Zika and Dengue viruses within a paper-based platform. At the same time, Doudna and co-workers developed the DETECTR system, using Cas12a to cleave the probe-modified ssDNA.7–9 Here, the trans-cleavage activity of Cas12a is activated by capturing loop-mediated isothermal amplification (LAMP) amplicons, allowing precise and ultrasensitive fluorometric detection of HPV16 or HPV18 fragments. Together, these discoveries have initiated the development of several other CRISPR/Cas biosensor platforms, including colorimetric, fluorometric and electrochemical approaches with outstanding analytical performance and selectivity down to single nucleotide level.5,10–12 In particular, the electrochemical (E-CRISPR) approach has proved adept in the highly sensitive detection of pathogens and toxins without the need for DNA amplification, either through use of ssDNA conjugated methylene blue (MB-ssDNA) or enzyme-assisted signal amplification.13,14 However, to date, E-CRISPR methods for gene-based analysis are associated with picomolar or higher limits of detection, leaving room for improvement toward ultrasensitive analysis.
Antimicrobial-resistance (AMR) in bacterial infections has become one of the most urgent problems in global healthcare. The number of antibiotic resistant strains is rapidly increasing, whilst there is a dearth of new antibiotic discoveries.15 Indeed, it is predicted that by 2050, AMR will be the cause of up to 150 million death per year.16 Methicillin-resistant Staphylococcus aureus (MRSA) is a prevalent AMR infection found in healthcare settings, and causes skin and bloodstream infections that lead to significant mortality and morbidity.17 Culture-based assays are the norm for AMR detection, and require from days to weeks determine if AMR is present.18 In recent years, nucleic acid amplification tests (NAATs) have begun to be employed for MRSA detection, and allow for swabbing-to-result times less than a day.19 The polymerase chain reaction (PCR) is the gold-standard in this regard,20 however it is compromised by its need for elaborate reaction optimization and precise temperature cycling. This is particularly problematic in resource-limited settings, but also in many point-of-care scenarios.21 Emerging isothermal NAATs (e.g. loop-mediated isothermal amplification, rolling circle amplification) have shown great promise in field-deployable diagnostics,19 however they are typically unable to differentiate different DNA/RNA targets down to the single nucleotide level which can be a crucial factor in gene-based detection (for example in the diagnosis of various cancers and hereditary diseases).22 However genotyping of single-nucleotide polymorphisms (SNPs) usually involves PCR and either fluorescence- or mass spectrometry-based detection, requiring extensive sample processing and expensive equipment which prevents field-deployability.23 Regarding MRSA, SNPs play a vital role in the dynamics of the disease and in tracking its epidemiology.24
In this work, we report a new sensing strategy that incorporates silver metallization into an electrochemical CRISPR-based biosensor (which we term E-Si-CRISPR) and achieves amplification-free gene-based detection down to the femtomolar level. Specifically, we target the detection of MRSA bacteria via the mecA gene, as it is common amongst the different strains and contains a protospacer adjacent motif (PAM) for the enzyme Cas12a. The developed E-Si-CRISPR platform achieves excellent analytical performance, with selectivity down to the single nucleotide level in both PAM and protospacer-binding regimes, and highly specific detection of MRSA in human serum against common bacteria. Accordingly, we believe this ultrasensitive gene-based detection platform has significant promise in field-deployable diagnostics.
2. Methodology
2.1 Materials and methods
DNA and RNA (sequences shown in Table S1†) were purchased from Microsynth (Balgach, Switzerland).25 AgNO3, NaBH4, HEPES, EDTA, Trizma® base, NaNO3, CaCl2, 6-mercapto-1-hexanol (MCH), K3[Fe(CN)6], K4[Fe(CN)6], PBS tablets, NaOH, H2SO4, NaCl, and KCl (all analytical grade) were purchased from Sigma Aldrich (Buchs, Switzerland). All other reagents, TCEP (Alfa Aesar, Karlsruhe, Germany), HCl (Fluka, Buchs, Switzerland), MgCl2 (Acros Organics, Geel, Belgium) were analytical reagent grade and used as received. Cas12a and its working buffer (50×) were purchased from New England Biolabs (Frankfurt am Main, Germany). All aqueous solutions were prepared with DNase and RNase free water (Invitrogen/Thermo Fisher, Reinach, Switzerland). Electrochemical measurements were performed with a PGSTAT204 AutoLab (Metrohm, Zofingen, Switzerland). Screen-printed gold electrodes (SPGE) were purchased from Dropsens (Metrohm, Zofingen, Switzerland). Cyclic voltammetry (CV) data for conductivity characterization were collected at 100 mV s−1 in 10 mM K3[Fe(CN)6], 1 M KCl, and 100 mM PBS (pH 7.42), unless otherwise indicated. Electrochemical impedance spectroscopy (EIS) was performed using an Impedance Analyzer SP-300 (Bio-Logic, Seyssinet-Pariset, France) in 0.1 M PBS containing 10 mM K3[Fe(CN)6]/K4 [Fe(CN)6] (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 1 M KCl as the supporting electrolyte. Impedance spectra were recorded within the frequency range of 10−2 to 105 Hz. The amplitude of the applied sine wave potential in each case was 5 mV. Curves were fitted in Origin2018 (64-bit) using the nonlinear curve fit function. Further, coulometric measurements were performed after silver metallization using 0.5 V applied potential for 60 seconds to evaluate the total amount of Ag deposited on the electrodes, by applying Q = nFNA (n is the number of electrons per mole of analyte, F is Faraday's constant (96487 C mol−1) and NA is the number of moles of analyte).
:1) with 1 M KCl as the supporting electrolyte. Impedance spectra were recorded within the frequency range of 10−2 to 105 Hz. The amplitude of the applied sine wave potential in each case was 5 mV. Curves were fitted in Origin2018 (64-bit) using the nonlinear curve fit function. Further, coulometric measurements were performed after silver metallization using 0.5 V applied potential for 60 seconds to evaluate the total amount of Ag deposited on the electrodes, by applying Q = nFNA (n is the number of electrons per mole of analyte, F is Faraday's constant (96487 C mol−1) and NA is the number of moles of analyte).
2.2 MecA gene preparation
A synthetic mecA gene fragment of length of 28 bp was used in a fluorometric assay to test the CRISPR/Cas approach for this gene. Next, the full mecA gene (685 bp) was amplified from a lysed MRSA solution by PCR, as described previously.26 Briefly, 1 μL of lysed MRSA solution was added into the PCR master mix with the forward and backward primers. PCR was then performed by preheating the reaction mixture at 95 °C for 5 minutes, before thermocycling through 55 °C (30 seconds), 72 °C (1 minute) and 95 °C (30 seconds) for 30 repeats. After this, the reaction was held at 72 °C for 10 minutes for the post-PCR extension step to complete the amplification of incomplete amplicons, yielding a 2 μM mecA gene stock solution, which was stored at −20 °C before use. DNA reactions were analyzed using both a Quanstudio Real-time PCR machine (Thermo Fisher Scientific, Reinach, Switzerland) and gel electrophoresis (85 V applied potential for 75 minutes in TBE buffer) using a 1% agarose gel (Sigma-Aldrich, Buchs, Switzerland) prepared in TBE buffer.2.3 Electrochemical detection
2.4 Fluorometric assay
The fluorometric assay was performed according to the protocol of Zhang and co-workers.5 Briefly, Cas12a was incubated with the gRNA at an equimolar ratio, followed by a ssDNA reporter (1 μM) modified with a 5′ dye (FAM) and 3′ quencher (BHQ1). The mecA gene was introduced into this solution, mixed well, at room temperature and allowed to incubate for 30 minutes. During this period, the evolution of fluorescence was monitored using a real-time PCR machine.2.5 Mismatch analysis
For mismatch analysis, the mismatch ssDNA of the mecA gene was inserted into the target DNA strand. Mismatched nucleotide positions were assigned to the second position of the protospacer adjacent motif, and the first, fifth and tenth position on the upper DNA target strand. Electrochemical detection was then performed to assess Cas selectivity.2.6 Human sample and bacterial sample analyses
All lysed bacteria solutions were prepared by dissolving bacterial colonies in TE buffer (Fischer Scientific, Reinach, Switzerland), with incubation at 95 °C for 5 minutes, and storage at −20 °C before use. All bacteria were obtained as a gift from Professor Mathias Schmelcher, University of Zurich, including, MRSA strain (Staphylococcus aureus USA300 JE2; source: Annelies Zinkernagel, Division of Infectious Diseases and Hospital Epidemiology, University Hospital Zurich, University of Zurich, Zurich, Switzerland; properties: methicillin-resistant S. aureus), E. coli (Escherichia coli JM109; source: Stratagene, San Diego, CA, USA; properties: E. coli lab strain, non-pathogenic), E. faecalis (Enterococcus faecalis ATCC 19433; source: ATCC 19433; properties: type strain), L. monocytogenes (Listeria monocytogenes Scott A; source: Weihenstephan Listeria Collection; properties: clinical isolate, serovar 4b), S. epidermidis (Staphylococcus epidermidis MP04; source: Max Paape, ARS, USDA, Beltsville, MD, USA; properties: bovine mastitis isolate) and methicillin-sensitive S. aureus (MSSA) (Staphylococcus aureus Newman; source: NCTC 8178, Annelies Zinkernagel, Division of Infectious Diseases and Hospital Epidemiology, University Hospital Zurich, University of Zurich, Zurich, Switzerland).Human serum (Sigma Aldrich, Buchs, Switzerland) was employed to prepare a dilution series of the mecA gene from the stock amplicon solution. In order to construct a calibration curve against the clean buffer solution. Briefly, the PCR amplicon of mecA at 1 μM was prepared in human serum (1:10), yielding a 100 nM stock solution which was then used to prepare the mecA concentration series from 10−7 to 10 nM. For the bacterial sample analyses, each lysed bacterial solution was prepared by 1:1 dilution with the human serum before use. Here, 1 μL lysate was added into the Cas–gRNA complex solution on the electrode, incubated for 60 minutes, and followed by electrochemical analysis.
3. Results and discussion
3.1 Electrode characterization
As previously noted, we chose the mecA gene as the target for detection of MRSA as it is common across many MRSA strains.26 To detect mecA using a CRISPR/Cas biosensor, a protospacer adjacent motif (PAM) is required on a specific site of the mecA gene.27 To initially test our CRISPR/Cas biosensor, we used a synthetic DNA analog oligo (5′-TTT ACG ATA AAA AGC TCC AAC ATG AAG A-3′), containing the “TTTA” sequence, with its complement. To design the protospacer binding sequence, we searched for the segment “TTTA” in the mecA gene, and used the adjacent sequence to construct the full protospacer. Then, the protospacer sequence was analyzed using the internet-based basic local alignment search tool (BLAST, National Center for Biotechnology Information), which revealed that the full sequence is specific to the mecA gene. Thus, the sequence was validated for mecA detection.When the mecA gene is bound to Cas12a and the gRNA, cis-cleavage (where the Cas enzyme cleaves the target gene bound to the protospacer regime specifically) is initiated. This activates trans-cleavage activity (random cleavage of ssDNA), leading to degradation of the FAM-ssDNA-BQ1 oligos and evolution of a fluorometric signal (Scheme S1†). This method was used to test our system using the PCR-derived mecA gene amplicon (685 bp). We observed that the negative control (no mecA target) yielded no fluorescence, whilst the positive controls (50 pM and 1 nM mecA) both exhibited significant fluorescence (Fig. S1†). This result demonstrated that the designed sequences work in combination with the Cas12a for mecA gene detection.
We have previously demonstrated the utility of silver metallization in enhancing the sensitivity of enzyme-assisted aptamer-based electrochemical biosensors.28 In the current work, we coupled this technique with the CRISPR/Cas method to produce a new silver-enhanced E-CRISPR biosensor (E-Si-CRISPR) to detect MRSA. Scheme 1 illustrates the sensing mechanism. In the presence of mecA, the Cas12a–gRNA complex first cleaves the target gene via cis-cleavage. This initiates trans-cleavage activity, which sees the enzyme cleave ssDNA at random locations, thus degrading the electrode surface of its ssDNA surface layer. Conversely, when no mecA target is present, the trans-cleavage mechanism is not activated and the ssDNA remains intact. When the subsequent silver metallization step is performed, the degree of silver deposition will be proportional to the amount of ssDNA remaining, and thus proportional to the starting amount of mecA. The final electrochemical signal is simply read via square wave voltammetry.
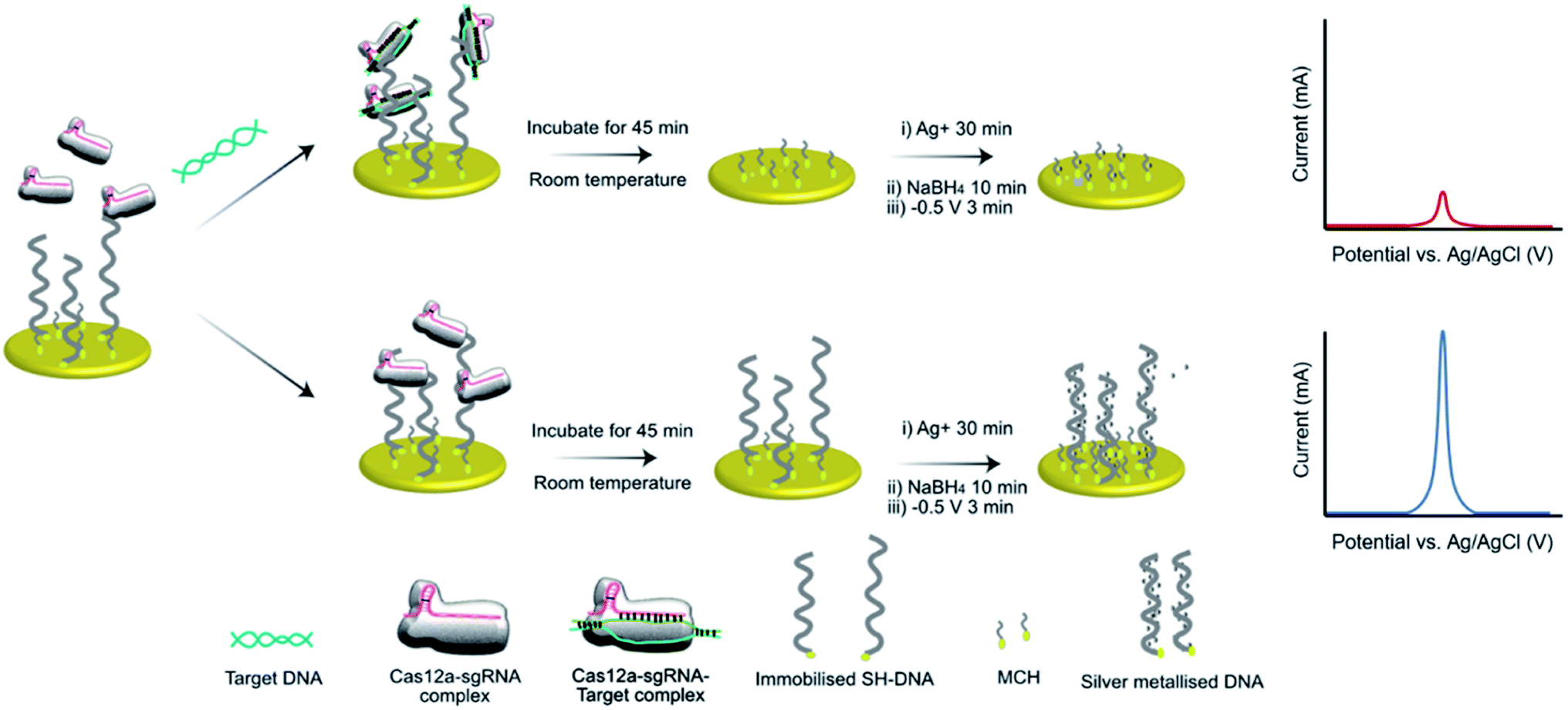 | ||
| Scheme 1 Schematic of the E-Si-CRISPR technique. ssDNA immobilized on the electrode are removed by the triplex Cas, only in the presence of a target gene. Subsequent addition of Ag+ and NaBH4 seeds the silver metallization, and an applied potential of −0.5 V initiates ‘double metallization’, yielding a minimized SWV signal (i.e. target positive result). In the absence of the target gene, ssDNA cannot be removed, yielding a higher electrochemical signal (i.e. target negative result). | ||
The assay was characterized using cyclic voltammetry, square wave voltammetry, electrochemical impedance spectroscopy and coulometry. To analyze the DNA metallization, CV was used to compare the redox signal before and after DNA metallization on the electrode (Fig. 1a). The oxidation and reduction peaks of silver on 30 nt ssDNA were observed at 0.16 V and −0.06 V, respectively. Signal reversibility of a 10 mM K3[Fe(CN)6]/K4[Fe(CN)6] solution was observed for both the ssDNA-SPGE and silver-metallized ssDNA-SPGE, suggesting an increase in the electroactive surface area.29 CV analysis indicated an increased electroactive area in the case of the silver-metalized electrode due to the reversible system relationship of the cyclic voltammogram, inferred from the Randle–Sevcik equation (ip = 2.69 × 105ACn3/2D1/2ν1/2, where ip is the peak current; A is the electrode surface area; C is the bulk concentration of analyte; D is the diffusion coefficient of the analyte; ν is the scan rate).30 With all parameters fixed, the increased peak current confirms an increased electroactive area. Next, the cleaved and non-cleaved ssDNA signals on the electrode were measured by SWV (Fig. 1b). A larger current is observed for the non-cleaved ssDNA on the SPGE (i.e. target negative), while the cleaved ssDNA showed only 30% of the original signal (i.e. target positive). This provides for greater sensitivity than previous methylene blue-based methods,13 as silver is a more sensitive electroactive species and highly resilient once deposited on ssDNA. Importantly, these results were in good agreement with previous reports,13,14 and explained by the strong electrostatic attraction between the negatively-charged ssDNA and positively-charged Ag+.31
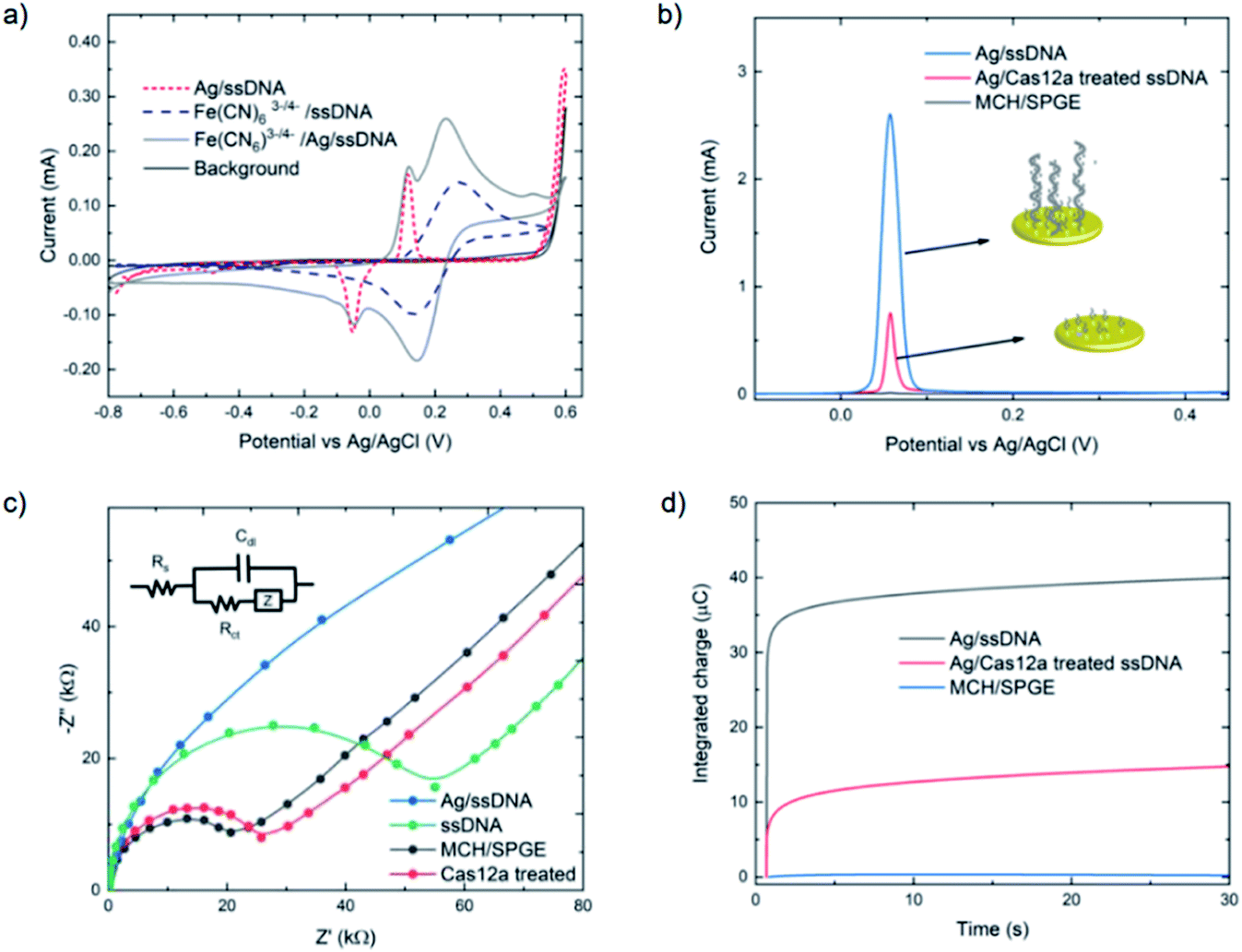 | ||
| Fig. 1 Comparison of MCH-modified, ssDNA-modified and Cas-treated electrodes (with 10 nM target gene) using (a) cyclic voltammetry (CV), (b) square wave voltammetry (SWV), (c) electrochemical impedance spectroscopy (EIS) and (d) controlled potential coulometry. For cyclic voltammograms and Nyquist plots, 10 mM [Fe(CN)6]3−/4− in 0.1 M PBS was used as the working solution, and 0.1 M PBS was used as the background electrolyte. Square wave voltammograms of silver-metallized electrodes were carried out using a step potential of 0.01 V, an amplitude of 60 mV, and a frequency of 200 Hz. The reversible cyclic voltammograms were carried out using a scan rate of 100 mV s−1, while Nyquist plots were obtained by applying a 5 mV sine wave potential within a frequency range of 10−2 to 105 Hz. For coulometry, silver was deposited using a similar protocol as SWV, then applying 0.5 V potential for 30 seconds to collect the silver metallization amount on the electrode. | ||
Electrochemical impedance spectroscopy was used to study the DNA cleavage/Cas activity and silver metallization on the electrode surface. EIS is highly effective for evaluating the interfacial electron transfer efficiency at different stages of biosensor preparation, as each step can be readily evaluated by monitoring the change in electron transfer resistance (Rct, whose value equals the radius of the semicircular response measured on the Z′ axis), allowing electrode activities to be monitored.32 As shown in Fig. 1c, the MCH/SPGE exhibited smaller Rct, indicating that the electroactive ions of [Fe(CN)6]3−/4− are rapidly transported to the electrode interface. After the thiolated ssDNA was self-assembled onto the electrode surface, the Rct increased significantly as the oligonucleotides are non-electroactive, and yield a strong resistance to electron transfer. After digestion of the ssDNA by Cas12a, the electrode showed an Rct value only slightly different from the MCH-modified electrode, indicating successful trans-cleavage of the ssDNA, leaving MCH and remnant ssDNA on the electrode surface. Importantly, we observed that the semicircular domain was absent after silver metallization of the ssDNA-SPGE, suggesting full silver metallization of the ssDNA structure. These EIS results confirm that ssDNA modification, ssDNA silver metallization, and trans-cleavage of triplex Cas12a on the SPGE were successful.
Next, coulometry was employed to analyze the amount of Ag0 on the electrode surface (using Faraday's law, Q = nFNA, where n is the number of electrons per mole of analyte, F is Faraday's constant = 96487 C mol−1, and NA is the number of moles of analyte). Silver metallization of the ssDNA-SPGEs (with and without Cas12a treatment) were investigated using controlled potential coulometry, and compared to the MCH/SPGE. The data, as shown in Fig. 1d, indicates high integrated charge of the un-treated ssDNA-SPGE (∼40 ng of Ag0) and much lower integrated charge for the Cas12a-treated ssDNA-SPGE (∼10 ng of Ag0), suggesting that the Cas12a removes ∼70% of the oligonucleotides on the electrode surface.
3.2 Method optimization
SWV is especially useful in the current work as it allows background signal subtraction and gives well-defined redox peaks, yielding excellent sensitivity.33 To optimize the SWV protocol used in our study, we considered three key output variables, namely the peak full-width half maximum (FWHM), peak position (oxidative potential), and peak height (current), changing as a function of pulse amplitude, pulse frequency and step potential. According to SWV theory, the ideal parameters for an Ag+/Ag0 system (having only one electroactive species, an electron) would be a 90.5 mV FWHM, 0 mV oxidative potential, and maximum peak current.34 Following the recommendation of Osteryoung and O'Dea,35 we applied a 0.01 V step potential to obtain an approximately 90.5 mV FWHM. We then studied an amplitude range of 10–100 mV, and a frequency range of 100–500 Hz, to find the optimal values. Fig. 2a–c presents heat maps describing the parameter space, with FWHM, peak current and peak position varying as a function of amplitude and frequency. The regions defining optimal parameter combinations are indicated with a mesh. From this analysis, we estimate the optimal parameter values to be a 10 mV step potential, 200 Hz frequency, and 60 mV amplitude (noting that although a higher amplitude could provide a higher current, an amplitude of 60 mV yields a result closer the FWHM target value of 90.5 mV).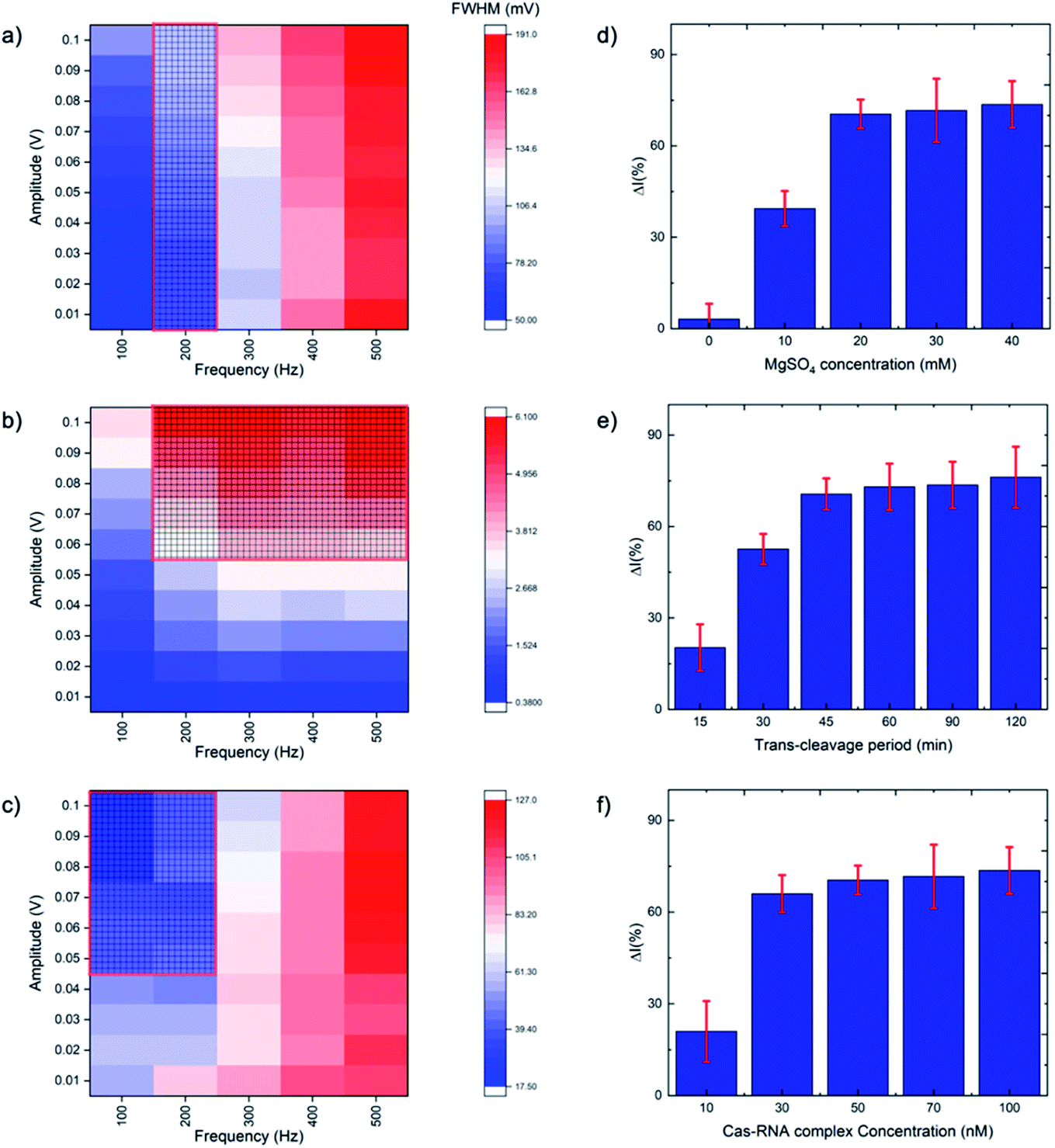 | ||
| Fig. 2 Heat maps of SWV optimization for (a) full width half maximum (FWHM), (b) peak current and (c) peak position, with meshed regions showing the preferred value ranges. SWV optimization was carried out by varying frequency from 100 to 500 Hz and amplitude from 0.01 to 0.1 V. The CRISPR/Cas system was optimized for the percentage difference between the treated and untreated electrode current as a function of (d) MgSO4 concentration, (e) trans-cleavage period, and (f) Cas–gRNA complex concentration (N = 3). | ||
Rather than using direct electrochemical measurements, the percentage difference in the electrochemical signal between the metallized ssDNA and triplex Cas12a-treated ssDNA, 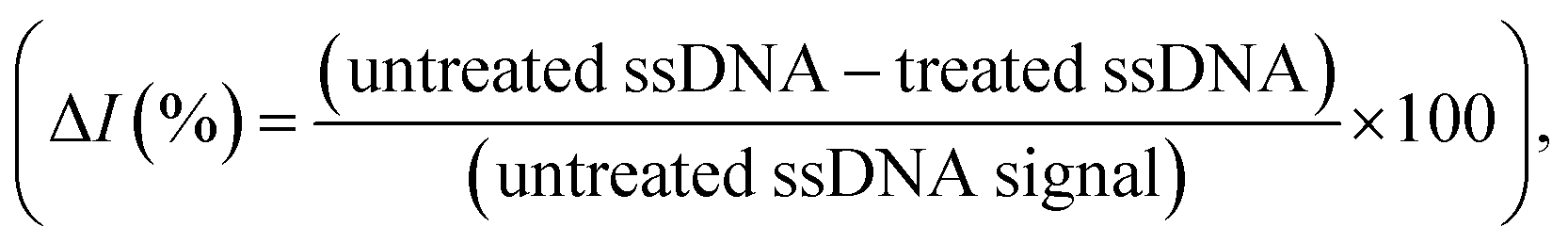 was used for quantitative analysis. This approach is preferable as it negates possible inconsistencies in equipment and consumables, e.g. variations in SPGE performance. Next, the silver metallization process was investigated. As described previously,28 combining electrochemical and chemical reduction of silver on ssDNA can provide a two-fold increase in oxidative signal. Here, electrostatic attraction between the Ag+ and the negatively-charged DNA allows the Ag+ to bind to the DNA, whilst the application of the reducing potential allows the formation of solid Ag0. It has previously been reported, however, that it is not only the electrostatics at play, but also an affinity bond between the Ag+ and the DNA bases.36 Here, a study of the reducing potential and electrochemical deposition time revealed signal plateaus at −0.5 V (Fig. S2a†) and 3 minutes (Fig. S2b†), respectively. The observed plateau points represent maximum values beyond which further silver deposition is negligible, thus we carried forward these values into the subsequent system development.
was used for quantitative analysis. This approach is preferable as it negates possible inconsistencies in equipment and consumables, e.g. variations in SPGE performance. Next, the silver metallization process was investigated. As described previously,28 combining electrochemical and chemical reduction of silver on ssDNA can provide a two-fold increase in oxidative signal. Here, electrostatic attraction between the Ag+ and the negatively-charged DNA allows the Ag+ to bind to the DNA, whilst the application of the reducing potential allows the formation of solid Ag0. It has previously been reported, however, that it is not only the electrostatics at play, but also an affinity bond between the Ag+ and the DNA bases.36 Here, a study of the reducing potential and electrochemical deposition time revealed signal plateaus at −0.5 V (Fig. S2a†) and 3 minutes (Fig. S2b†), respectively. The observed plateau points represent maximum values beyond which further silver deposition is negligible, thus we carried forward these values into the subsequent system development.
The CRISPR/Cas system was optimized by considering five parameters: MgSO4 concentration, triplex Cas12a concentration, trans-cleavage incubation time, ssDNA length and ssDNA concentration. The Mg2+ divalent cation concentration is important as the Cas12a RuvC domain (the Cas12a active pocket) cleaves ssDNA through a two metal ion mechanism, in which the Mg2+ ions induce conformational coordination of the RuvC domain and the ssDNA by shifting the spatial distribution of ssDNA around the RuvC active cutting center.37 Thus, we studied the Mg2+ concentration with a view to promoting the trans-cleavage activity of Cas12a, and observed that Cas12a trans-cleavage is only activated in the presence of the Mg2+, and that the signal plateaus at an MgSO4 concentration of 20 mM (Fig. 2d). For the Cas12a incubation time, we observed a signal plateau at 45 minutes (Fig. 2e). Finally, by varying the Cas12a–RNA complex concentration, we observed a signal plateau at 50 nM (Fig. 2f). Therefore, we carried forward values of 20 mM Mg2+, 45 minutes incubation time and 50 nM enzyme concentration.
Given the role of the Cas12a in degrading electrode surface-bound ssDNA, it was important to understand the effect of oligonucleotide length on the ΔI (%). From the perspective of silver metallization, a longer DNA length will yield a greater signal. However, this must be balanced against how much ssDNA remains after Cas12a trans-cleavage, given that we want to minimize remnant DNA to maximize ΔI (%). We studied ssDNA lengths of 10, 20, 30 and 40 bases (Fig. S3†). The ΔI (%) was lowest for the 10 nt ssDNA, followed by the 20 nt. The 30 and 40 nt oligos yielded very similar ΔI (%) values, however the error was larger for the 40 nt DNA. Therefore, we chose the 30 nt length to carry forward. Finally, we studied the effect ssDNA concentration (at incubation and before MCH addition) on ΔI (%), in the range of 0.01 to 50 μM, and observed that 1 μM yielded the highest signal (Fig. S4†).
3.3 Analytical performance
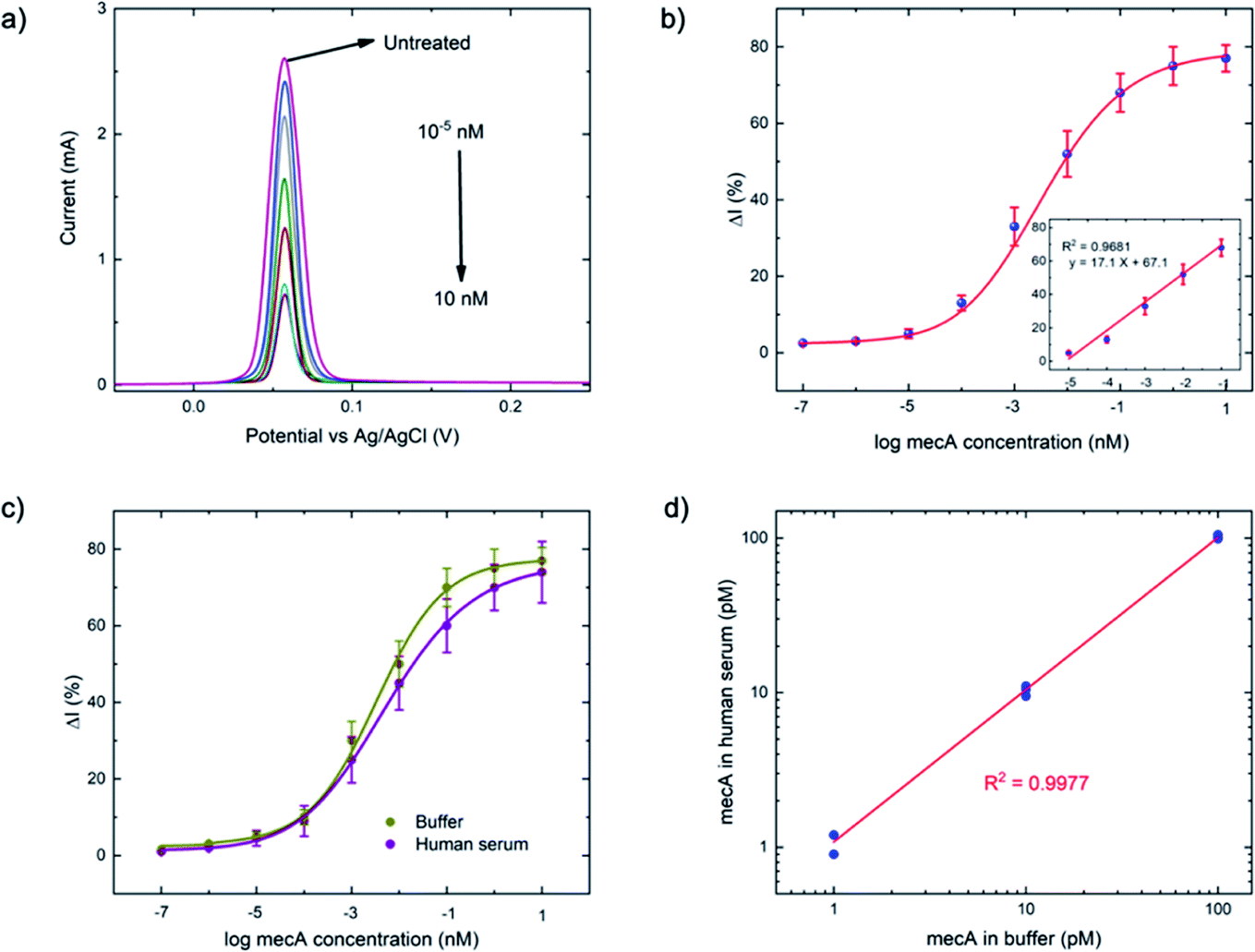 | ||
| Fig. 3 (a) Square wave voltammograms of a series of mecA concentrations (10−7 to 10 nM) using previously optimized conditions: ssDNA concentration = 1 μM, ssDNA length = 30 nt, applied potential = −0.5 V, electrochemical deposition time = 3 minutes, Cas–gRNA complex concentration = 50 nM, a trans-cleavage period = 45 min, MgSO4 concentration = 20 mM, step potential = 0.01 V, amplitude = 0.06 V, and frequency = 200 Hz. (b) Calibration curve of anodic current (N = 3) obtained from (a), versus mecA concentration (the linear range between 10 fM and 100 pM is shown in the inset). (c) Comparison of the calibration curves for buffer- and human serum-based tests, using the concentration series of mecA (as in (b)) and (d) detection of the mecA gene in both media, at 1, 10 and 100 pM (N = 3). | ||
As a comparison, the gold-standard PCR approach, which requires ca. 2–4 hours total operation time, requires sophisticated instrumentation to perform thermocycling, and often cannot achieve single-nucleotide mismatch discrimination. In contrast, the E-Si-CRISPR technique achieves its excellent analytical performance in less time (90 minutes), without nucleic acid amplification, and uses portable instrumentation, whilst achieving excellent mismatch discrimination. These attributes would facilitate its use as a field-deployable diagnostic tool. Further, as can be seen in Table S2†, the 90 minute assay time is equal to or faster than comparable E-CRISPR assays published to date.
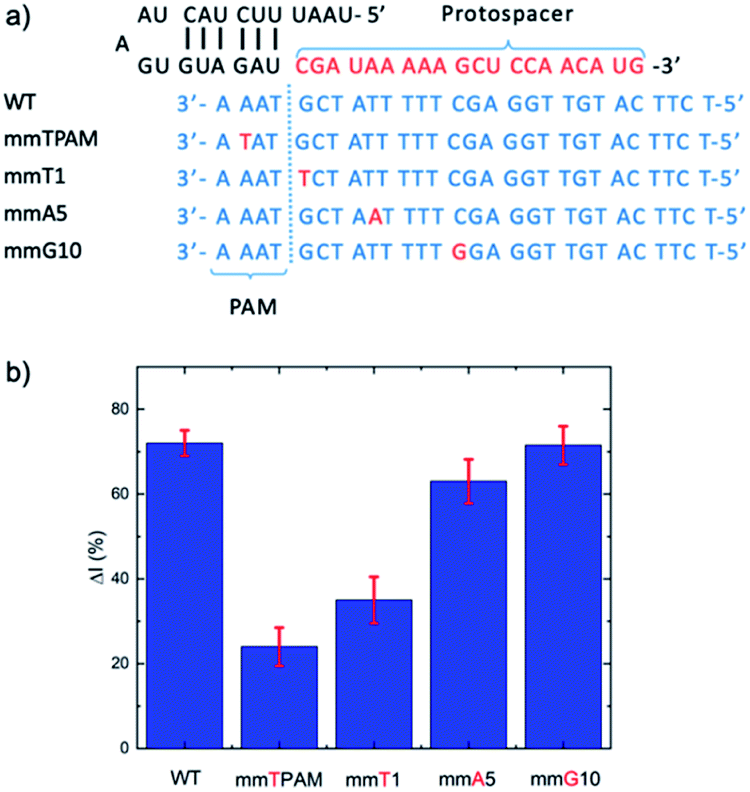 | ||
| Fig. 4 (a) Target strands with mismatches at different positions, including in the PAM region and crRNA complement at different positions (1, 5, and 10). (b) Evaluation of the influence of mismatches at different positions on the E-CRISPR signal. A target concentration of 1 pM was applied for all the targets (wild type (WT) and mismatched targets). | ||
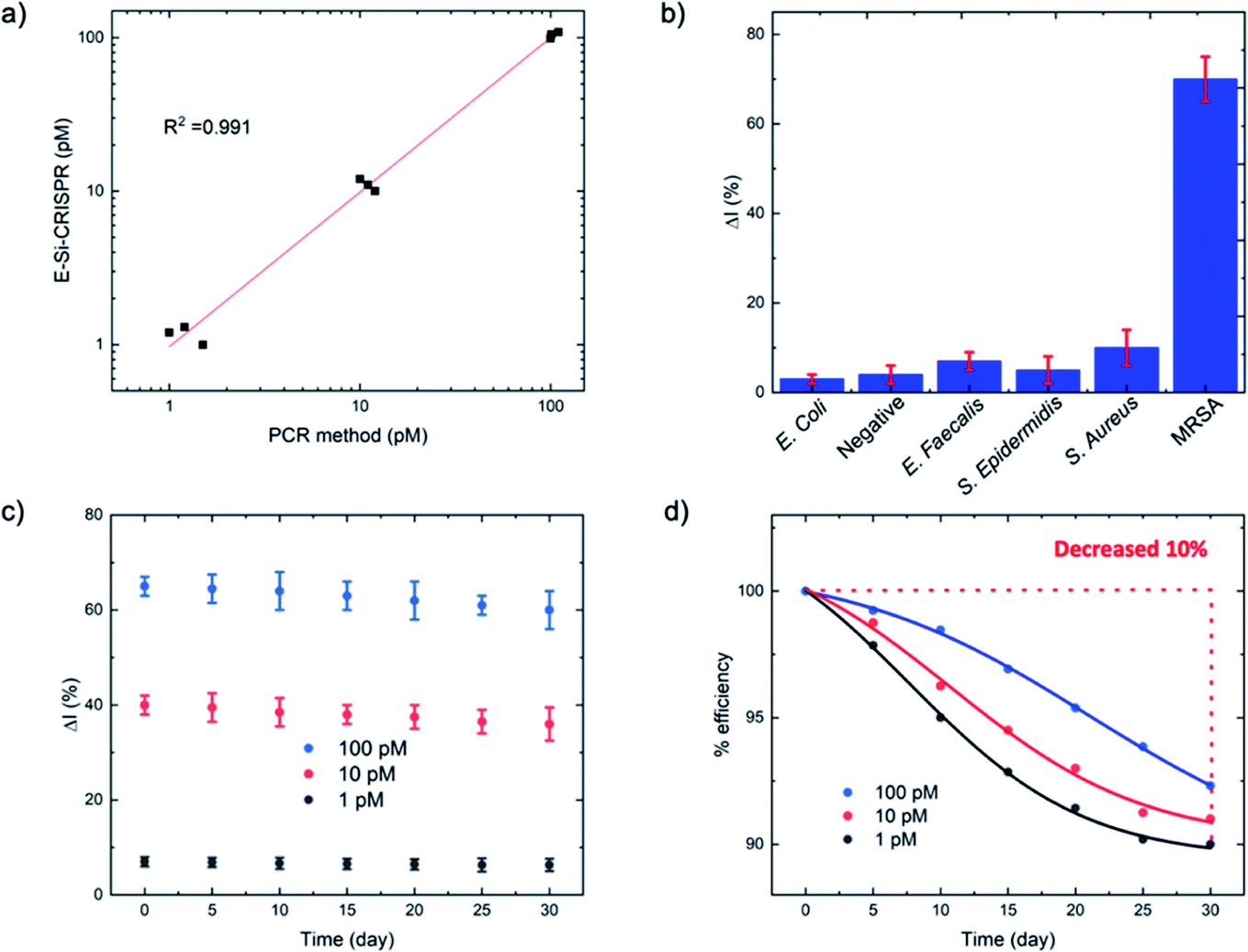 | ||
| Fig. 5 (a) Comparison of mecA concentrations between the E-Si-CRISPR and PCR at 1, 10 and 100 pM. (b) Specificity test for MRSA versus other bacteria for the E-Si-CRISPR technique, revealing excellent specificity (N = 3). (c and d) Stability of the ssDNA-immobilized electrode for detecting mecA (1, 10 and 100 pM) over 30 days (N = 3). | ||
:1 volume dilution in human serum before application to the E-Si-CRISPR electrode.
For the MRSA sample, we observed a drop in the ΔI (%) of ca. 10% (with a 45 minute incubation) versus our previous testing with the mecA amplicon. The mecA gene amplicon is 685 bp, whereas the real genomic material is far longer. Thus, the Cas12a takes longer to ‘find’ its specific binding site. Jeon and coworkers reported that Cas enzymes use a ‘walking’ mechanism, where dsDNA is randomly captured before the enzyme then moves along to find the target gene.40 Therefore, we expect that the longer the target sequence, the longer the required incubation time will be. Accordingly, we decided to study ΔI (%) signal evolution over time as a function of the Cas12a incubation time for the lysate/serum sample (Fig. S5†). We observed that, compared to operation in clean buffer (Fig. 2e), the signal plateaus slightly later, at approximately 60 minutes (compared to 45 minutes). Thus, for the following selectivity study, we carried forward an incubation time of 60 minutes.
For the full selectivity test, we tested all lysate/serum samples using the optimized E-Si-CRISPR technique. The results shown in Fig. 5b clearly demonstrate excellent selectivity for MRSA in the current configuration, proving that the chosen protospacer binding with the PAM sequence is highly selective for MRSA, and that the cleavage activity of the chosen Cas system is still excellent, even in a complex matrix. Further, in order to study whether the E-Si-CRISPR technique could feasibly detect MRSA in a real sample at a concentration similar to that obtainable with PCR, we tested a spiked serum sample containing ca. 102 cells per μL, which is representative of the amount in a clinical sample of human serum,41 and compared it to the results obtained with 103 cells. Detection at 102 copies of MRSA shows that the assay signal is still ca. 30 ΔI (%) (compared to ca. 65 ΔI (%) for 103 copies), whilst all of the non-MRSA targets in Fig. 5b exhibited signals below 15 ΔI (%). This result demonstrates the feasibility for detecting MRSA at clinically relevant levels using the E-Si-CRISPR technique.
4. Conclusions
We have presented a novel ultra-sensitive electrochemical CRISPR/Cas biosensor using silver metallization to enable high-sensitivity detection of drug-resistant bacteria, without the need for DNA amplification. The E-Si-CRISPR technique exhibited excellent specificity for the mecA gene, activating the cis- and trans-cleavage activities of Cas12a. Removal of immobilized ssDNA was performed on the electrode surface after trans-cleavage activation in the presence of the target gene. The addition of Ag+ followed by a double reduction metallized the remaining ssDNA, yielding a significant signal enhancement in square wave voltammetry-based measurements. CRISPR/Cas and square wave voltammetry were optimized carefully to achieve an excellent analytical performance for the E-Si-CRISPR technique, with an LoD of 3.5 fM, an LoQ of 10 fM, and linearity between 10 fM to 100 pM (five orders of magnitude) against the mecA amplicon (685 bp). E-Si-CRISPR was compared to the gold-standard PCR analysis, and showed excellent agreement in terms of accuracy. Significantly, E-Si-CRISPR analytical performance was maintained even in human serum. Furthermore, mismatch analysis was performed to study the Cas12a selectivity, providing excellent selectivity down to the single nucleotide level. The selectivity of the E-Si-CRISPR technique was demonstrated using a panel of common bacteria, yielding excellent selectivity in the detection of specific genes. It should be noted that the use of an enzyme in such a sensor could pose potential complications for field-deployable applications, however we expect that careful freeze-drying of enzymes will be useful in extending the system shelf life.42 To conclude, the developed E-Si-CRISPR technique allows the detection of MRSA with high sensitivity, selectivity, accuracy, precision, and robustness even in lysed bacteria solution, and should be further developed for real samples in the near future.Author contributions
Akkapol Suea-Ngam: conceptualization, methodology, validation, formal analysis, investigation, writing - original draft. Philip Howes: methodology, supervision, project administration, visualization, writing - review & editing. Andrew deMello: methodology, supervision, project administration, review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge ETH Zurich for partial financial support. A. S. N. would like to thank the State Secretariat for Education, Research and Innovation (SERI) for a Swiss Government Excellence Scholarship (ESKAS No. 2016.0728).References
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang and L. A. Marraffini, Multiplex genome engineering using CRISPR/Cas systems, Science, 2013, 339(6121), 819–823 CrossRef CAS PubMed.
- Y. Li, S. Li, J. Wang and G. Liu, CRISPR/Cas systems towards next-generation biosensing, Trends Biotechnol., 2019, 37(7), 730–743 CrossRef CAS PubMed.
- G. Amitai and R. Sorek, CRISPR–Cas adaptation: insights into the mechanism of action, Nat. Rev. Microbiol., 2016, 14(2), 67–76 CrossRef CAS PubMed.
- Portable CRISPR-based diagnostics, Nat. Biotechnol., 2019, 37(8), 832 Search PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, SHERLOCK: nucleic acid detection with CRISPR nucleases, Nat. Biotechnol., 2019, 14(10), 2986–3012 CAS.
- J. Engreitz, O. Abudayyeh, J. Gootenberg and F. Zhang, CRISPR tools for systematic studies of RNA regulation, Cold Spring Harbor Perspect. Biol., 2019, 11(8), a035386 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity, Science, 2018, 360(6387), 436–439 CrossRef CAS PubMed.
- G. J. Knott and J. A. Doudna, CRISPR-Cas guides the future of genetic engineering, Science, 2018, 361(6405), 866–869 CrossRef CAS PubMed.
- C.-H. Huang, K.-C. Lee and J. A. Doudna, Applications of CRISPR-Cas enzymes in cancer therapeutics and detection, Trends Cancer, 2018, 4(7), 499–512 CrossRef CAS PubMed.
- P. Qin, M. Park, K. J. Alfson, M. Tamhankar, R. Carrion, J. L. Patterson, A. Griffiths, Q. He, A. Yildiz and R. Mathies, Rapid and fully microfluidic Ebola virus detection with CRISPR-Cas13a, ACS Sens., 2019, 4(4), 1048–1054 CrossRef CAS PubMed.
- Y. Dai, Y. Wu, G. Liu and J. J. Gooding, CRISPR Mediated Biosensing Toward Understanding Cellular Biology and Point-of-Care Diagnosis, Angew. Chem., Int. Ed., 2020, 59, 20754–20766 CrossRef CAS PubMed.
- K. Pardee, A. A. Green, M. K. Takahashi, D. Braff, G. Lambert, J. W. Lee, T. Ferrante, D. Ma, N. Donghia and M. Fan, Rapid, low-cost detection of Zika virus using programmable biomolecular components, Cell, 2016, 165(5), 1255–1266 CrossRef CAS PubMed.
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Exploring the Trans-Cleavage Activity of CRISPR-Cas12a (cpf1) for the Development of a Universal Electrochemical Biosensor, Angew. Chem., Int. Ed., 2019, 58(48), 17399–17405 CrossRef CAS PubMed.
- R. Bruch, J. Baaske, C. Chatelle, M. Meirich, S. Madlener, W. Weber, C. Dincer and G. A. Urban, CRISPR/Cas13a-powered electrochemical microfluidic biosensor for nucleic acid amplification-free miRNA diagnostics, Adv. Mater., 2019, 31(51), 1905311 CrossRef CAS PubMed.
- World Health Organization, Technical consultation on in vitro diagnostics for AMR, 27–28 March 2019, WHO Headquarters, Geneva: meeting report, World Health Organization, 2019 Search PubMed.
- J. O'Neill, Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations, 2014, p. 14 Search PubMed.
- M. D. Bartels, P. Worning, L. P. Andersen, M. Bès, H. Enger, C. G. Ås, T. A. Hansen, B. J. Holzknecht, K. W. Larssen and F. Laurent, Repeated introduction and spread of the MRSA clone t304/ST6 in Northern Europe, Clin. Microbiol. Infect., 2020, 27(2), 284 Search PubMed.
- I. Choopara, A. Suea-Ngam, Y. Teethaisong, P. D. Howes, M. Schmelcher, A. Leelahavanichkul, S. Thunyaharn, D. Wongsawaeng, A. J. deMello and D. Dean, Fluorometric Paper-Based, Loop-Mediated Isothermal Amplification Devices for Quantitative Point-of-Care Detection of Methicillin-Resistant Staphylococcus aureus (MRSA), ACS Sens., 2021, 6(3), 742–751 CrossRef CAS PubMed.
- A. Suea-Ngam, L. Bezinge, B. Mateescu, P. D. Howes, A. J. deMello and D. Richards, Enzyme-assisted nucleic acid detection for infectious disease diagnostics: moving towards the point-of-care, ACS Sens., 2020, 5(9), 2701–2723 CrossRef CAS PubMed.
- R. Cunningham, P. Jenks, J. Northwood, M. Wallis, S. Ferguson and S. Hunt, Effect on MRSA transmission of rapid PCR testing of patients admitted to critical care, J. Hosp. Infect., 2007, 65(1), 24–28 CrossRef CAS PubMed.
- C. A. Holland and F. L. Kiechle, Point-of-care molecular diagnostic systems—past, present and future, Curr. Opin. Microbiol., 2005, 8(5), 504–509 CrossRef PubMed.
- H.-B. Wang, L.-H. Ma, T. Zhang, K.-C. Huang, Y.-D. Zhao and T.-C. Liu, Simple and accurate visual detection of single nucleotide polymorphism based on colloidal gold nucleic acid strip biosensor and primer-specific PCR, Anal. Chim. Acta, 2020, 1093, 106–114 CrossRef CAS PubMed.
- S. Kim and A. Misra, SNP genotyping: technologies and biomedical applications, Annu. Rev. Biomed. Eng., 2007, 9, 289–320 CrossRef CAS PubMed.
- M. McDonald, A. Dougall, D. Holt, F. Huygens, F. Oppedisano, P. M. Giffard, J. Inman-Bamber, A. J. Stephens, R. Towers and J. R. Carapetis, Use of a single-nucleotide polymorphism genotyping system to demonstrate the unique epidemiology of methicillin-resistant Staphylococcus aureus in remote aboriginal communities, J. Clin. Microbiol., 2006, 44(10), 3720–3727 CrossRef CAS PubMed.
- L. Lv, D. Li, R. Liu, C. Cui and Z. Guo, Label-free aptasensor for ochratoxin A detection using SYBR Gold as a probe, Sens. Actuators, B, 2017, 246, 647–652 CrossRef CAS.
- A. Suea-Ngam, I. Choopara, S. Li, M. Schmelcher, N. Somboonna, P. D. Howes and A. J. deMello, In Situ Nucleic Acid Amplification and Ultrasensitive Colorimetric Readout in a Paper-Based Analytical Device Using Silver Nanoplates, Adv. Healthcare Mater., 2020, 2001755 Search PubMed.
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. Van Der Oost and A. Regev, Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system, Cell, 2015, 163(3), 759–771 CrossRef CAS PubMed.
- A. Suea-Ngam, P. D. Howes, C. E. Stanley and A. J. deMello, An Exonuclease I-Assisted Silver-Metallized Electrochemical Aptasensor for Ochratoxin A Detection, ACS Sens., 2019, 4(6), 1560–1568 CrossRef CAS PubMed.
- A. Suea-Ngam, P. Rattanarat, O. Chailapakul and M. Srisa-Art, Electrochemical droplet-based microfluidics using chip-based carbon paste electrodes for high-throughput analysis in pharmaceutical applications, Anal. Chim. Acta, 2015, 883, 45–54 CrossRef CAS PubMed.
- A. Suea-Ngam, P. Rattanarat, K. Wongravee, O. Chailapakul and M. Srisa-Art, Droplet-based glucosamine sensor using gold nanoparticles and polyaniline-modified electrode, Talanta, 2016, 158, 134–141 CrossRef CAS PubMed.
- L. Wu, J. Wang, J. Ren and X. Qu, Ultrasensitive telomerase activity detection in circulating tumor cells based on DNA metallization and sharp solid-state electrochemical techniques, Adv. Funct. Mater., 2014, 24(18), 2727–2733 CrossRef CAS.
- M. E. Orazem and B. Tribollet, Electrochemical impedance spectroscopy, New Jersey, 2008, pp. 383–389 Search PubMed.
- J. G. Osteryoung and R. A. Osteryoung, Square wave voltammetry, Anal. Chem., 1985, 57(1), 101–110 CrossRef.
- L. Ramaley and M. S. Krause, Theory of square wave voltammetry, Anal. Chem., 1969, 41(11), 1362–1365 CrossRef CAS.
- J. O'Dea and J. Osteryoung, Square wave voltammetry, Electroanal. Chem., 1986, 14, 209–308 Search PubMed.
- J. Kondo, Y. Tada, T. Dairaku, Y. Hattori, H. Saneyoshi, A. Ono and Y. Tanaka, A metallo-DNA nanowire with uninterrupted one-dimensional silver array, Nat. Chem., 2017, 9(10), 956–960 CrossRef CAS PubMed.
- R. Sundaresan, H. P. Parameshwaran, S. Yogesha, M. W. Keilbarth and R. Rajan, RNA-independent DNA cleavage activities of Cas9 and Cas12a, Cell Rep., 2017, 21(13), 3728–3739 CrossRef CAS PubMed.
- P. Puthongkham and B. J. Venton, Nanodiamond coating improves the sensitivity and antifouling properties of carbon fiber microelectrodes, ACS Sens., 2019, 4(9), 2403–2411 CrossRef CAS PubMed.
- A. Suea-Ngam, L.-T. Deck, P. D. Howes and A. J. deMello, An ultrasensitive non-noble metal colorimetric assay using starch–iodide complexation for Ochratoxin A detection, Anal. Chim. Acta, 2020, 1135, 29–37 CrossRef CAS PubMed.
- Y. Jeon, Y. H. Choi, Y. Jang, J. Yu, J. Goo, G. Lee, Y. K. Jeong, S. H. Lee, I.-S. Kim and J.-S. Kim, Direct observation of DNA target searching and cleavage by CRISPR-Cas12a, Nat. Commun., 2018, 9(1), 1–11 CrossRef PubMed.
- V. K. Rajendran, P. Bakthavathsalam, P. L. Bergquist and A. Sunna, Sens. Actuators, B, 2019, 298, 126849 CrossRef CAS.
- J. S. Gootenberg, O. O. Abudayyeh, J. W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer and C. A. Freije, Nucleic acid detection with CRISPR-Cas13a/C2c2, Science, 2017, 356(6336), 438–442 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02197d |
| This journal is © The Royal Society of Chemistry 2021 |