
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolution-processable porous graphitic carbon from bottom-up synthesis and low-temperature graphitization†
Sai
Che
 ac,
Chenxuan
Li
a,
Chenxu
Wang
b,
Wasif
Zaheer
a,
Xiaozhou
Ji
a,
Bailey
Phillips
a,
Guvanch
Gurbandurdyyev
a,
Jessica
Glynn
a,
Zi-Hao
Guo
d,
Mohammed
Al-Hashimi
e,
Hong-Cai
Zhou
ab,
Sarbajit
Banerjee
*ab and
Lei
Fang
*ab
ac,
Chenxuan
Li
a,
Chenxu
Wang
b,
Wasif
Zaheer
a,
Xiaozhou
Ji
a,
Bailey
Phillips
a,
Guvanch
Gurbandurdyyev
a,
Jessica
Glynn
a,
Zi-Hao
Guo
d,
Mohammed
Al-Hashimi
e,
Hong-Cai
Zhou
ab,
Sarbajit
Banerjee
*ab and
Lei
Fang
*ab
aDepartment of Chemistry, Texas A&M University, College Station, Texas 77843, USA. E-mail: fang@chem.tamu.edu; banerjee@chem.tamu.edu
bDepartment of Materials Science & Engineering, Texas A&M University, College Station, Texas 77843, USA
cState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Changping, Beijing 102249, China
dSouth China Advanced Institute for Soft Matter Science and Technology, South China University of Technology, Guangzhou, Guangdong 510640, China
eDepartment of Chemistry, Texas A&M University at Qatar, P. O. Box 23874, Doha, Qatar
First published on 18th May 2021
Abstract
It is urgently desired yet challenging to synthesize porous graphitic carbon (PGC) in a bottom-up manner while circumventing the need for high-temperature pyrolysis. Here we present an effective and scalable strategy to synthesize PGC through acid-mediated aldol triple condensation followed by low-temperature graphitization. The deliberate structural design enables its graphitization in situ in solution and at low pyrolysis temperature. The resulting material features ultramicroporosity characterized by a sharp pore size distribution. In addition, the pristine homogeneous composition of the reaction mixture allows for solution-processability of the material for further characterization and applications. Thin films of this PGC exhibit several orders of magnitude higher electrical conductivity compared to analogous control materials that are carbonized at the same temperatures. The integration of low-temperature graphitization and solution-processability not only allows for an energy-efficient method for the production and fabrication of PGC, but also paves the way for its wider employment in applications such as electrocatalysis, sensing, and energy storage.
Introduction
Porous graphitic carbon (PGC) represents a new class of porous material with a regular distribution of micropores arrayed across graphitic basal planes.1–3 The continuous graphitic backbone enables high electrical and thermal conductivity,4 as well as extraordinary chemical and thermal stability.5 Meanwhile, the microporous structure imparts a number of unique properties and functions that are not available with nonporous graphene, such as permeability towards specific molecules;6 high surface area that allows for high capacity storage of ions and electrons;7 and abundant edge structures on pore peripheries as active catalytic sites.8 In certain cases, the porous structure opens up the energy band gap of originally metallic graphene to afford semiconducting characteristics.9 Combining the properties of micropores and graphitic backbone, PGC has been used in a wide range of applications, such as supercapacitors,10 lithium-ion batteries,11 electrocatalysis,8,12 field-effect transistors,13 water desalination,14 and DNA sequencing.15In terms of synthesis, PGC can be formed through a top-down process that involves chemical exfoliation or chemical/thermal etching.16 These approaches, however, often confront challenges when attempting to achieve micropores with narrow size distribution.17,18 In contrast, the strategy of bottom-up synthesis from specifically designed monomers has shown considerable promise. Most reported bottom-up syntheses involve template-mediated chemical vapor deposition.19 Several other examples have been reported to achieve microporous graphitic structures through on-surface interfacial synthesis,20,21 and metal–organic frameworks pyrolysis.22–25 With rationally targeted monomer design and temperature regulation, precise control of pore size and edge structure with regulated backbone constitution can be achieved.26 However, many materials synthesized through this strategy still contain large amounts of single-stranded bonds in the backbones, i.e., incomplete graphitization. In addition, high cost, poor scalability, and lack of processability have hindered the mass production and broader application of PGC synthesized from these methods.
Solution-phase bottom-up synthesis represents a promising approach to prepare PGC. Several milligram- and gram-scale examples have been reported.27–29 Typically, the synthesis starts with crosslinking of organic monomers to afford porous precursors, followed by graphitization. High temperature pyrolysis (≥700 °C) is usually required for such materials to achieve a reasonable level of graphitization.12,29–31 The demanding energy requirement is likely a result of the non-regulated precursor backbone that is not preorganized for ring-fusing reaction. We envision that if the precursor backbone is pre-organized so that ring-fusing is sterically favored—in a manner similar to carbonization of polyacrylonitrile32 or cyclodehydrogenative aromatization in the synthesis of graphene nanoribbons,33 high-quality PGC can be synthesized at a relatively low pyrolysis temperature, enabling integration of PGC in applications that involve temperature sensitive species, or wherein high thermal budget represents a substantial constraint. To verify this hypothesis, herein we report a highly promising bottom-up approach to synthesize PGC from a specifically designed monomer. This approach features feasible and scalable preparation, built-in solution processability, and an extraordinarily low graphitization temperature.
Structure design and synthesis
We designed a bottom-up strategy for the synthesis of PGC involving aldol triple condensation (ATC) and subsequent ring-fusing pyrolysis. The ATC approach is feasible and cost-effective for the preparation of aromatic porous polymer networks,34–40 and more intriguingly allows for solution processing of such membranes (p-PPN in Fig. 1a) for organic solvent nanofiltration.41 Through this acid-mediated reaction, a diacetyl-functionalized molecule can be crosslinked to afford a network mainly composed of aromatic units.42,43 Here methanesulfonic acid (MSA) serves as both the catalyst and the solvent, rendering feasibility and low cost of the reaction procedure, meanwhile allowing for solution processing of the insoluble network into membranes and thin films through in situ crosslinking. Through this advantageous ATC method, PGC can be synthesized (Fig. 1b) from a diacetyl functionalized biphenyl monomer, namely, 2,2′-diacetylbiphenyl (DAB). Because the acetyl groups of DAB are on the 2-positions of the biphenyl units, the intermediate after crosslinking would have C–H groups of adjacent benzene units in proximity to each other. Such a favorable steric arrangement can facilitate cyclodehydrogenative C–C coupling with minimum conformational reorganization,20,44,45 therefore translating into feasible graphitization that requires low energy input. It is noteworthy that other possible monomers, such as 1,7-diacetylperylene and 1,9-diacetyldibenzo[g,p]chrysene, can also be potentially used to accomplish this synthetic goal, while DAB is by far the most easily accessible one.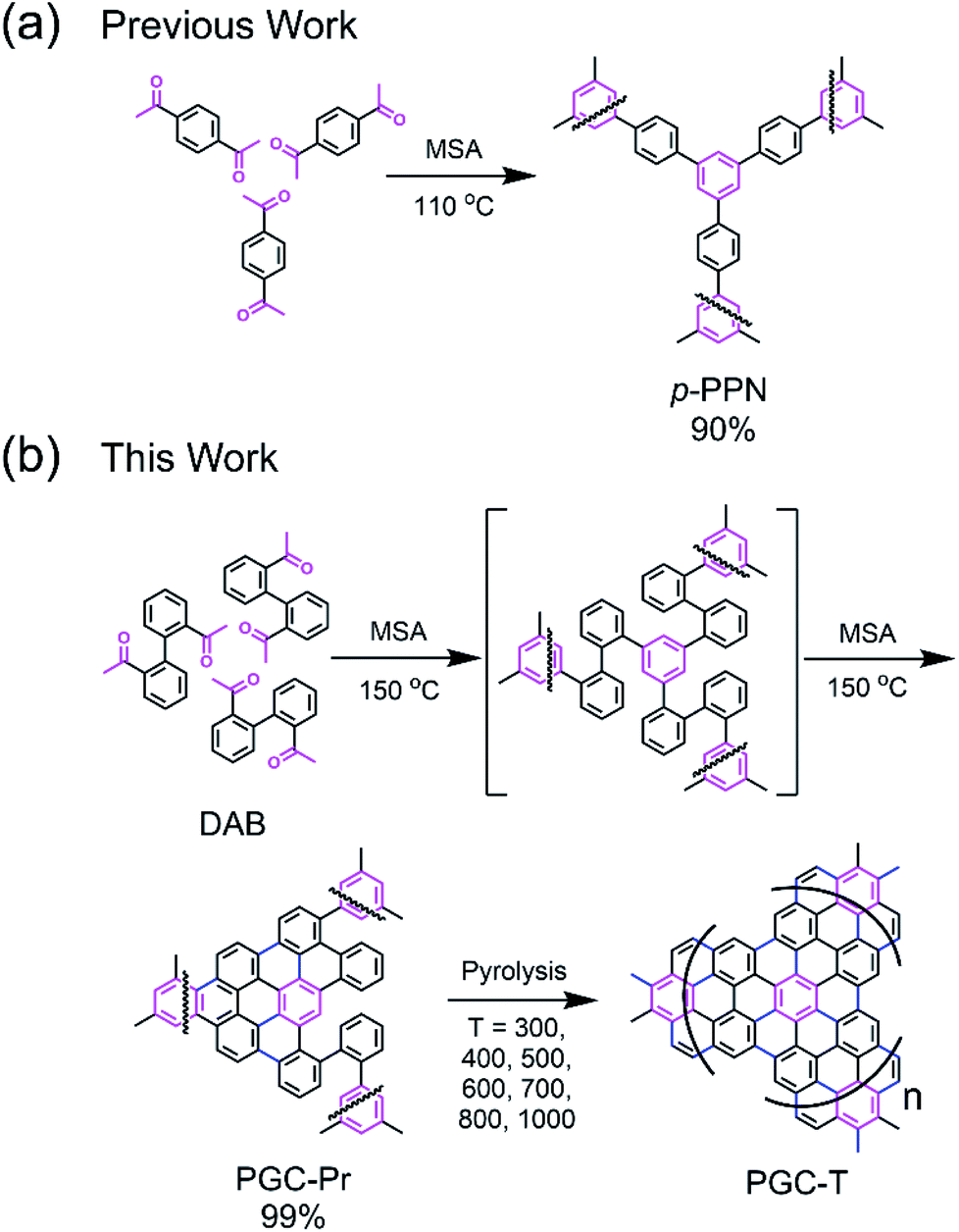 | ||
| Fig. 1 (a) Previously reported synthesis of non-graphitic p-PPN. (b) Solution-phase synthesis of the pro-graphitic precursor PGC-Pr from DAB monomer, and the subsequent pyrolysis to afford PGC materials PGC-T (T represents the pyrolysis temperature ranging from 300–1000 °C). | ||
The DAB monomer was synthesized at gram scale through a one-step Cu-catalyzed Ullman coupling of commercially available starting material 1-(2-iodophenyl)ethan-1-one (see ESI†). To conduct the ATC crosslinking, a homogeneous solution of DAB in MSA was first prepared by bath sonication. Upon heating the solution to 150 °C in an open vial set-up, the color of the mixture rapidly changed from orange to black, indicating the formation of extended π-conjugation through the reaction. After heating for 24 hours and subsequent thorough washing, insoluble black solid was isolated in nearly quantitative yield as the PGC precursor PGC-Pr. The reaction is tolerable to ambient oxygen and moisture so that no protective procedure was needed. No distinct sp3 peaks at ∼30 ppm was observed in the solid-state Cross-Polarization Magic Angle Spinning 13CNMR of PGC-Pr (Fig. S4†), confirming the high conversion of the acetyl groups. Interestingly, at this stage, the Raman spectrum of PGC-Pr (Fig. 2a) already showed characteristic D- and G-bands associated to graphitic structure, though PGC-Pr has not been subjected to any deliberate oxidation or thermal dehydrogenation. This observation suggests that the MSA-mediated condition is able to promote the cyclodehydrogenation aromatization to generate a partially graphitized structure, corroborating the hypothesis that the pre-organized pro-graphitic structure has a strong tendency to graphitize. To further confirm this point, a previously reported aromatic para-phenylene-derived porous network (p-PPN) (Fig. 1a) was synthesized and tested in the same condition as a control. Not surprisingly, p-PPN did not show any signals with respect to D- and G-bands after the ATC reaction (Fig. 2a), as a result of the lack of C–H groups in proximity for dehydrogenative C–C coupling. To probe the source of oxidant in this reaction, an air-free reaction in a sealed tube with degassed MSA was carried out and afforded similar PGC-Pr materials featuring the clear D- and G-bands in Raman spectra, suggesting that the oxidant is likely MSA itself, instead of oxygen in air. However, it is worth noting that PGC-Pr is only partially graphitized at this stage, indicated by the presence of both 1,2-disubstituted and 1,3,5-trisubstituted benzene rings in Fourier Transform Infrared Spectrum (Fig. S3†) and the relatively low carbon content observed in elemental analysis (Table S2†).
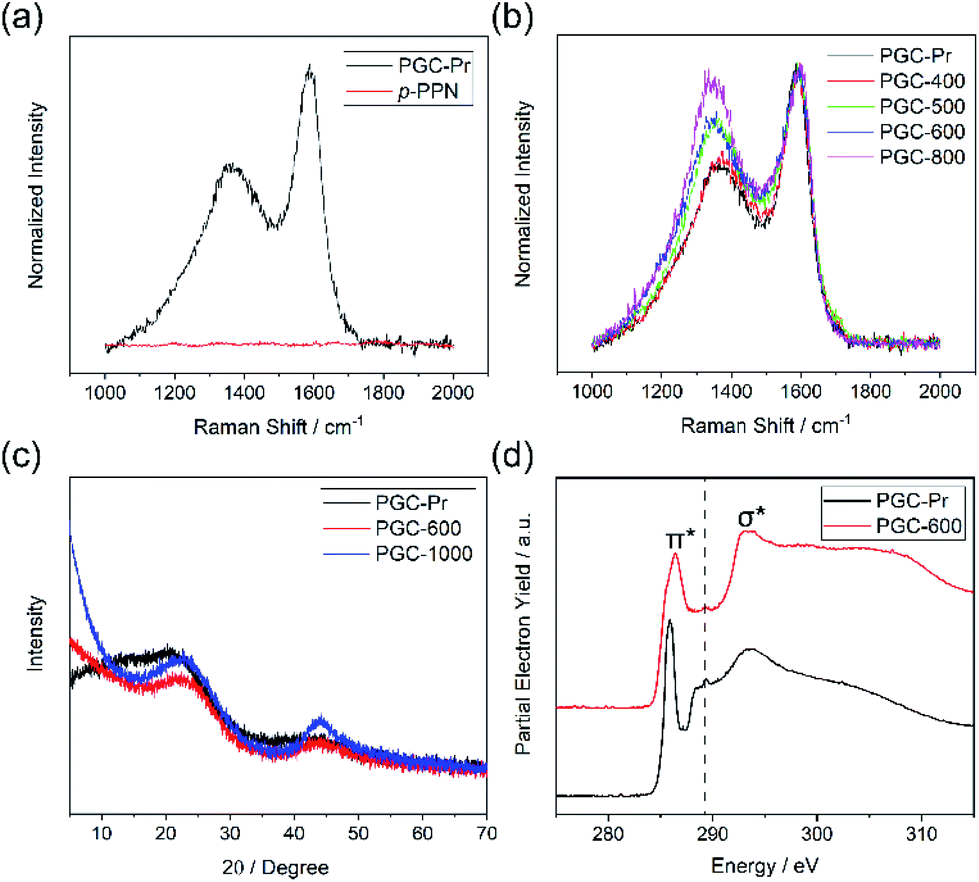 | ||
| Fig. 2 (a) Comparison of Raman spectra of PGC-Pr and p-PPN. (b) Raman spectra of PGC after pyrolysis at varied temperatures. (c) PXRD data of PGC-Pr, PGC-600, and PGC-1000. (d) XANES data at the C K-edge of PGC-Pr and PGC-600. | ||
To further graphitize PGC-Pr, the material was subsequently pyrolyzed in N2 atmosphere at different temperatures to afford the products PGC-T (PGC-300, -400, -500, -600, -700, -800, and -1000, respectively), where the number corresponds to the pyrolysis temperature. The Raman spectra of the pyrolyzed samples exhibited a more pronounced D-band with a higher pyrolysis temperature (Fig. 2b), which can be attributed to the construction of more edge structures. The control material p-PPN was also pyrolyzed at same temperatures, showing no D-band or G-band signals until the temperature was raised above 500 °C. The sharp contrast between pyrolyzed PGC-T and p-PPN materials again demonstrated that the rational structural design of DAB monomer and PGC-Pr was a key to enable the efficient graphitization at a relatively low temperature. It is worth noting that the term “graphitic” here is used here to describe the local graphitic nature of PGC-T backbone structure. Crystallinity of these materials is in between fully amorphous glassy carbon and crystalline graphite.
Results and discussion
Powder X-ray diffraction (PXRD) patterns of PGC-Pr, PGC-600, and PGC-1000 all demonstrate characteristic reflections at 2θ = 22.7° and 2θ = 43.4°, which are consistent with that observed on reduced graphene oxide.46–49 The first reflection represents the interlayer separation between graphitic layers of 0.39 nm (Fig. 2c), confirming the successful formation of graphitic structure in PGC-600 and -1000. These peaks become narrower and increase in intensity with higher pyrolysis temperature, indicating that a higher pyrolysis temperature yielded better defined graphitic domains with extended X-ray coherence lengths despite the fact that PGC-Pr is already partially graphitic. In addition, the lack of other reflections suggested an amorphous nature of the porous backbone. X-ray photoelectron spectroscopy (XPS) further confirmed the increased content of sp2 carbon and thereby higher degree of graphitization with higher pyrolysis temperature (Fig. S8†).The structural composition of PGC-Pr and subsequent changes upon thermal annealing were further studied with X-ray absorption near-edge structure (XANES) measurements (Fig. 2d), in which the data collected at the C K-edge for PGC-Pr and PGC-600 are plotted. Two distinct features can be observed for both samples: the low energy feature at 285.9 eV corresponds to the electronic transition from C 1s core levels into the unoccupied C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π* states above the Fermi level. The broad feature centered at 293.5 eV can be attributed to transitions of C 1s core electrons to the states with σ* symmetry in the conduction band.50 The emergence of the double-peaked resonance is characteristic of excitonic origins of the electronic transition in well-crystallized local graphitic domains.51 The spectrum of PGC-Pr also exhibits a small peak centered at ∼289.2 eV, which is likely the result of electronic excitation from C 1s to π* states of carbonyl or carboxylic acid CO bonds.50 This feature is attributed to the presence of small amount of unreacted acetyl groups in PGC-Pr after the ATC reaction. The absence of such feature in the spectrum of PGC-600 indicates that these residual groups are consumed in the subsequent ring-fusing pyrolysis at 600 °C. The broad and slightly blue-shifted CC (1s–π*) feature in the spectrum of PGC-600 further suggests that the original acetyl groups in PGC-Pr likely contribute to oxygen doping in the conjugated structure during pyrolysis. Such oxygen doping process leads to broadened and shifted 1s–π* feature as a result of distinctive electronic transitions from C 1s to conjugated domains with and without oxygen incorporation. The presence and transformation of oxygen moieties in these samples is further verified by the XANES data at the O K-edge (Fig. S5†). The π* feature in the O K-edge data represents the transition of O 1s electrons to π* CO states derived from the acetyl groups. The decrease in the intensity of this feature for PGC-600 relative to PGC-Pr points to a loss of CO moieties.50
C π* states above the Fermi level. The broad feature centered at 293.5 eV can be attributed to transitions of C 1s core electrons to the states with σ* symmetry in the conduction band.50 The emergence of the double-peaked resonance is characteristic of excitonic origins of the electronic transition in well-crystallized local graphitic domains.51 The spectrum of PGC-Pr also exhibits a small peak centered at ∼289.2 eV, which is likely the result of electronic excitation from C 1s to π* states of carbonyl or carboxylic acid CO bonds.50 This feature is attributed to the presence of small amount of unreacted acetyl groups in PGC-Pr after the ATC reaction. The absence of such feature in the spectrum of PGC-600 indicates that these residual groups are consumed in the subsequent ring-fusing pyrolysis at 600 °C. The broad and slightly blue-shifted CC (1s–π*) feature in the spectrum of PGC-600 further suggests that the original acetyl groups in PGC-Pr likely contribute to oxygen doping in the conjugated structure during pyrolysis. Such oxygen doping process leads to broadened and shifted 1s–π* feature as a result of distinctive electronic transitions from C 1s to conjugated domains with and without oxygen incorporation. The presence and transformation of oxygen moieties in these samples is further verified by the XANES data at the O K-edge (Fig. S5†). The π* feature in the O K-edge data represents the transition of O 1s electrons to π* CO states derived from the acetyl groups. The decrease in the intensity of this feature for PGC-600 relative to PGC-Pr points to a loss of CO moieties.50
Thermogravimetric analysis (TGA) of PGC-Pr demonstrated its excellent thermal stability with over 80% weight remaining at 1000 °C in an oxygen-free measurement, in agreement with its partially graphitized, sp2 carbon-rich nature (Fig. 3a). N2 adsorption–desorption isotherm measurements at 77 K and Brunauer–Emmet–Teller (BET) surface area analysis was conducted to evaluate porosity. No distinct N2 adsorption was observed for PGC-Pr (Fig. 3c), which was likely due to the entrapped solvent and/or impurity in the pores similar to many other reported microporous materials.52 The PGC-T samples started to show porosity with >400 °C pyrolysis. The BET surface area increased with higher graphitization temperatures and reached 830 m2 g−1 for PGC-800 (Fig. 3b). Nitrogen adsorption of PGC-800 gave a typical type I isotherm, indicating the presence of dominant micropores. Pore size distribution revealed the major pore size of 0.6 nm with a narrow distribution (Fig. 3d), which matches well with the model structure. The high BET surface area and regulated pore size distribution also confirmed the retained microporous structure of PGC-800 after pyrolysis. The small amount of larger pores was attributed to defects generated during crosslinking and pyrolysis, which was in consistent with the observation of residual carbonyl groups in PGC-Pr and the amorphous nature of these materials. The stability of the microporous structure of PGC-800 was tested against various harsh chemical conditions, including strong acid, base, reductant, and oxidant (see ESI†). The BET surface areas showed only marginal changes after 7 days of treatments in these conditions (Fig. S2†), demonstrating the anticipated high chemical stability of these PGC materials.
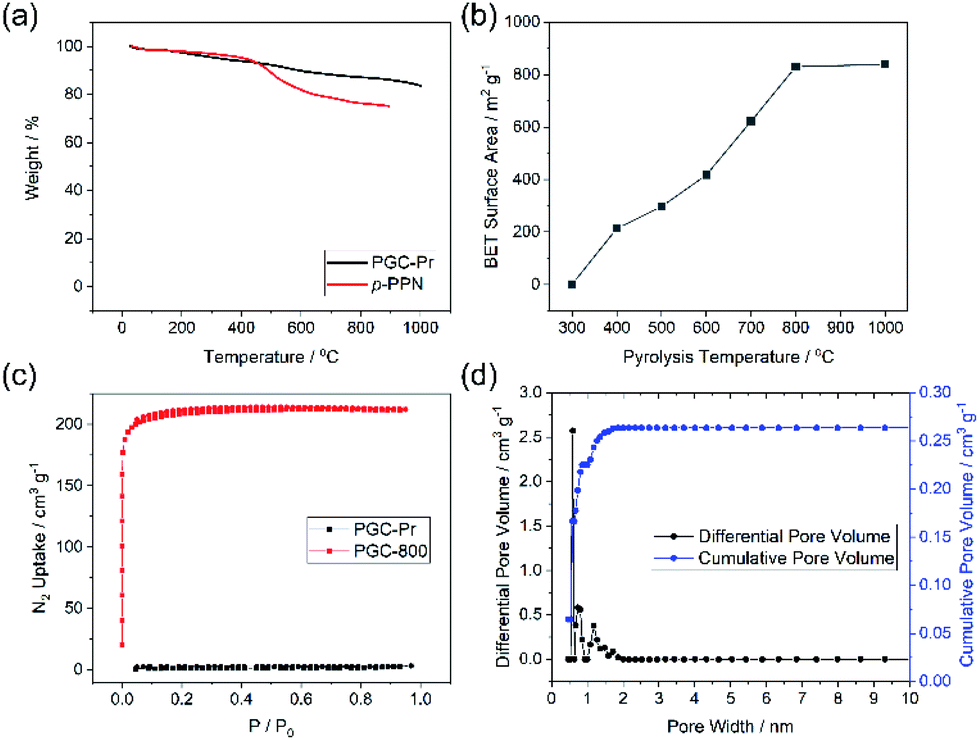 | ||
| Fig. 3 (a) TGA curves of PGC-Pr and p-PPN. (b) Relationship between BET surface areas of PGC and pyrolysis temperatures. (c) N2 adsorption isotherms (77 K) of PGC-Pr and PGC-800. (d) Pore size distribution of PGC-800. | ||
The crosslinked nature of PGC renders the material insoluble and unmeltable, hence extremely difficult to process. In this work, however, the pristine monomer solution of DAB in MSA allows for fabricating the PGC thin film through an approach of “casting followed by in situ crosslinking”. To demonstrate this, a solution of DAB in MSA was drop-cast onto a glass substrate and then sandwiched by another piece of cover glass with several pieces of small micrometer-thick glass in between as the spacers (Fig. S6a†). The set-up was pre-heated at 90 °C for 1 hour, 110 °C for 1 hour, and 150 °C for 72 hours to obtain the PGC-Pr films. These films were further pyrolyzed at varied temperatures similar to the synthesis of the bulk samples. SEM images of these pyrolyzed films revealed a continuous morphology (Fig. S6b–d†). For samples prepared with 300 μm-thick spacers, the thicknesses of the PGC-Pr, PGC-600, and PGC-1000 films were measured to be 151 ± 15, 90 ± 12, and 95 ± 10 μm, respectively (Fig. S6e–g†). The thin film processability, integrated with the aforementioned thermal and chemical stability, allows PGC to survive harsh processing and/or fabrication conditions, hence applicable in extreme environments.
Electrical conductivity is an important parameter reflecting the degree of carbonization and graphitization. The built-in processability of this method enables fabrication of continuous PGC thin films for the measurement of conductivity before and after thermal pyrolysis. Four-probe conductivity measurements were conducted on film samples of PGC-Pr, PGC-T, and p-PPN (Fig. S7†). Before pyrolysis, no sample exhibited measurable conductivity despite clear evidence of localized graphitic domains, presumably due to the presence of backbone defects that break the charge transport percolation. After pyrolysis at the temperature as low as 400 °C, PGC film started to exhibit measurable conductivity, suggesting that the graphitization process enabled a long-range percolated electrical conducting pathway in the sample. In contrast, p-PPN film remained insulating after 400 °C treatment (Fig. 4c). With 500 and 600 °C pyrolysis, PGC-500 and -600 films exhibited electrical conductivity of 2.1 × 10−4, and 0.031 S cm−1, respectively. These values are four orders and two orders of magnitude higher compared to the corresponding values measured on analogous p-PPN films pyrolyzed at the same temperatures (Fig. 4a and b). With the pyrolysis temperature over 700 °C, both materials exhibit comparable high conductivity, indicating that deeply graphitized backbones can be achieved in both materials at such high temperature. This comparison confirms that the designed structural constitution of PGC-Pr is favorable to facilitate graphitization at a relatively low temperature. So far, without metal catalyst, most reported amorphous carbon materials have to be pyrolyzed at high temperatures to obtain good degree of graphitization and electrical conductivity.53 For instance, the conductivity of PGC-400 film (∼10−6 S cm−1) is comparable with that of porous carbon pyrolyzed at 900 °C,54 while PGC-700 and -800 films exhibit much higher conductivities than most other porous carbon materials pyrolyzed at similar temperatures (Table S1†).53–61 The significantly higher conductivity of PGC pyrolyzed at low temperature is critically important to achieve a general low-energy consumption production of functional graphitic carbon materials, and to enable the employment of PGC in temperature-sensitive fabrications and applications.
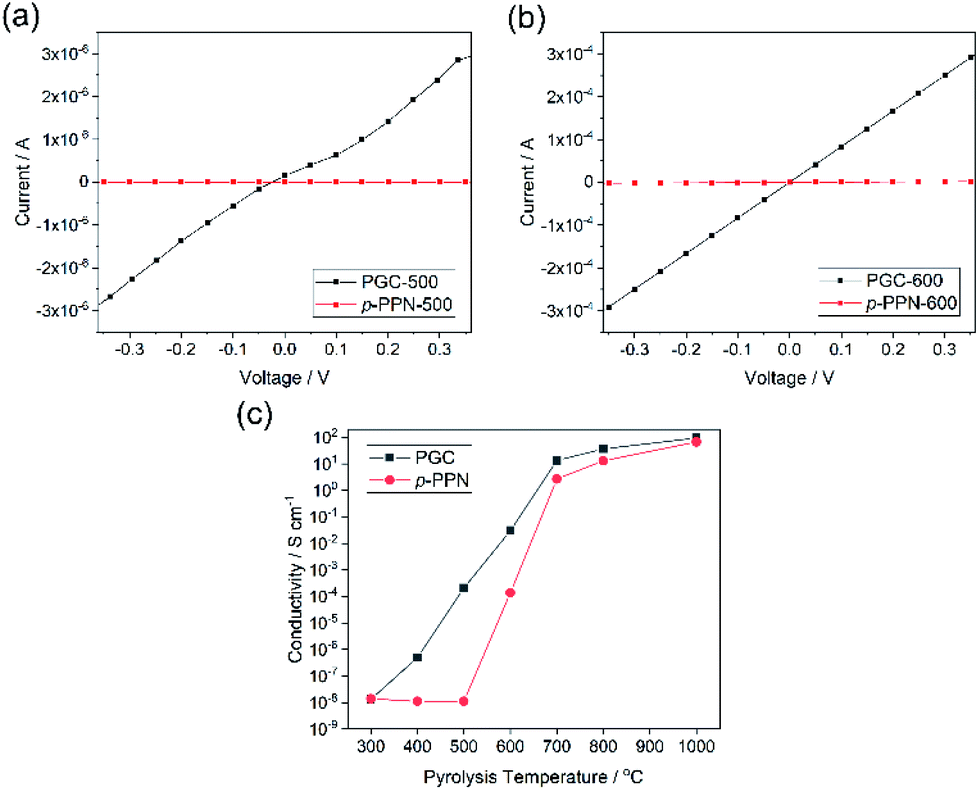 | ||
| Fig. 4 Four-point probe I–V plot of (a) PGC-500 and p-PPN-500 films (thickness ∼ 100 μm) and (b) PGC-600 and p-PPN-600 films (thickness ∼ 90 μm). (c) Comparison of conductivity of PGC films and p-PPN films with different temperature pyrolysis. | ||
Conclusions
In conclusion, the bottom-up synthesis and thin film fabrication of porous graphitic carbon is achieved through solution-crosslinking and thermal graphitization. Rational design of the monomer leads to the construction of pre-organized PGC precursor through aldol triple condensation reaction, in which in situ partial graphitization already takes place. The pre-organized precursor constitution allows for its favorable graphitization at relatively low pyrolysis temperatures. The resulting PGC materials possess ultramicropores with regulated pore size and ample edge structures. This scalable method features feasible starting materials preparation, inexpensive reagents, and simple reaction procedure. Meanwhile, the pristine homogeneous reaction allows for solution processing of this intrinsically insoluble graphitic materials into thin films, essential for a wide range of practical applications. This low-temperature method opens up the possibility to achieve diverse element-doped PGC materials by involving functionalized monomers into the synthesis. It enables the applications of PGC in situations that high temperature treatment cannot be tolerated, such as carbon-based catalysts functionalized with temperature-sensitive groups, supercapacitor electrode materials composited with redox-active species that cannot survive high heat, or PGC-derived device on heat-vulnerable substrates. In addition, the facile film construction paves the way for the direct fabrication of electrocatalyst device for a variety of essential reactions, such as hydrogen evolution reaction, oxygen evolution reaction, oxygen reduction reaction, etc.Author contributions
Sai Che: methodology, validation, formal analysis, investigation, resources, data curation, writing-original draft, writing-review & editing. Chenxuan Li: methodology, investigation, resources, data curation. Chenxu Wang: methodology, investigation, resources. Wasif Zaheer: investigation, resources, data curation. Xiaozhou Ji: investigation, resources. Bailey Phillips: investigation, resources. Guvanch Gurbandurdyyev: investigation, resources. Jessica Glynn: investigation, resources. Zi-Hao Guo: methodology, investigation, resources. Mohammed Al-Hashimi: supervision, funding acquisition. Hong-Cai Zhou: supervision, funding acquisition. Sarbajit Banerjee: writing-review & editing, supervision, funding acquisition. Lei Fang: conceptualization, writing-review & editing, supervision, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Welch Foundation (A-1898), the President's Excellence Fund of Texas A&M University (X-Grants), and the Qatar National Research Fund (NPRP10-0111-170152, NPRP11S-1204-170062, and NPRP11S-0116-180320). J. G. acknowledges support from the National Science Foundation REU Program (CHE 1851936). The gas sorption studies were supported by the Center for Gas Separations, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-SC0001015). This research used resources of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.Notes and references
- C. West, C. Elfakir and M. Lafosse, J. Chromatogr. A, 2010, 1217, 3201 CrossRef CAS PubMed.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS PubMed.
- X. Liu, M. He, D. Calvani, H. Qi, K. Gupta, H. J. M. de Groot, G. J. A. Sevink, F. Buda, U. Kaiser and G. F. Schneider, Nat. Nanotechnol., 2020, 15, 307 CrossRef CAS PubMed.
- B. Wang, B. V. Cunning, N. Y. Kim, F. Kargar, S. Y. Park, Z. Li, S. R. Joshi, L. Peng, V. Modepalli, X. Chen, Y. Shen, W. K. Seong, Y. Kwon, J. Jang, H. Shi, C. Gao, G. H. Kim, T. J. Shin, K. Kim, J. Y. Kim, A. A. Balandin, Z. Lee and R. S. Ruoff, Adv. Mater., 2019, 31, 1903039 CrossRef PubMed.
- K. H. Thebo, X. Qian, Q. Zhang, L. Chen, H. M. Cheng and W. Ren, Nat. Commun., 2018, 9, 1486 CrossRef PubMed.
- M. Shibukawa, A. Unno, T. Miura, A. Nagoya and K. Oguma, Anal. Chem., 2003, 75, 2775 CrossRef CAS PubMed.
- L. Sun, C. Tian, L. Wang, J. Zou, G. Mu and H. Fu, J. Mater. Chem., 2011, 21, 7232 RSC.
- R. Paul, L. Zhu, H. Chen, J. Qu and L. Dai, Adv. Mater., 2019, 31, 1806403 CrossRef PubMed.
- A. Sinitskii and J. M. Tour, J. Am. Chem. Soc., 2010, 132, 14730 CrossRef CAS PubMed.
- V. Strauss, K. Marsh, M. D. Kowal, M. El-Kady and R. B. Kaner, Adv. Mater., 2018, 30, 1704449 CrossRef PubMed.
- R. Wang, J. Yang, X. Chen, Y. Zhao, W. Zhao, G. Qian, S. Li, Y. Xiao, H. Chen, Y. Ye, G. Zhou and F. Pan, Adv. Energy Mater., 2020, 10, 1903550 CrossRef CAS.
- K. Yuan, D. Lutzenkirchen-Hecht, L. Li, L. Shuai, Y. Li, R. Cao, M. Qiu, X. Zhuang, M. K. H. Leung, Y. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404 CrossRef CAS PubMed.
- P. Hu, M. Yan, X. Wang, C. Han, L. He, X. Wei, C. Niu, K. Zhao, X. Tian, Q. Wei, Z. Li and L. Mai, Nano Lett., 2016, 16, 1523 CrossRef CAS PubMed.
- K. Celebi, J. Buchheim, R. M. Wyss, A. Droudian, P. Gasser, I. Shorubalko, J. I. Kye, C. Lee and H. G. Park, Science, 2014, 344, 289 CrossRef CAS PubMed.
- G. F. Schneider and C. Dekker, Nat. Biotechnol., 2012, 30, 326 CrossRef CAS PubMed.
- W. Tian, H. Zhang, X. Duan, H. Sun, G. Shao and S. Wang, Adv. Funct. Mater., 2020, 30, 1909265 CrossRef CAS.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806 CrossRef CAS PubMed.
- J. Bai, X. Zhong, S. Jiang, Y. Huang and X. Duan, Nat. Nanotechnol., 2010, 5, 190 CrossRef CAS PubMed.
- L. Zhang and M. Jaroniec, Chem. Soc. Rev., 2020, 49, 6039 RSC.
- C. Moreno, M. Vilas-Varela, B. Kretz, A. Garcia-Lekue, M. V. Costache, M. Paradinas, M. Panighel, G. Ceballos, S. O. Valenzuela, D. Pena and A. Mugarza, Science, 2018, 360, 199 CrossRef CAS PubMed.
- K. Xu, J. I. Urgel, K. Eimre, M. D. Giovannantonio, A. Keerthi, H. Komber, S. Wang, A. Narita, R. Berger, P. Ruffieux, C. A. Pignedoli, J. Liu, K. Mullen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 7726 CrossRef CAS PubMed.
- C. Wang, J. Kim, J. Tang, J. Na, Y. M. Kang, M. Kim, H. Lim, Y. Bando, J. Li and Y. Yamauchi, Angew. Chem., Int. Ed., 2020, 59, 2066 CrossRef CAS PubMed.
- C. Wang, J. Kim, J. Tang, M. Kim, H. Lim, V. Malgras, J. You, Q. Xu, J. Li and Y. Yamauchi, Chem, 2020, 6, 19 CAS.
- Y. V. Kaneti, J. Zhang, Y.-B. He, Z. Wang, S. Tanaka, M. S. A. Hossain, Z.-Z. Pan, B. Xiang, Q.-H. Yang and Y. Yamauchi, J. Mater. Chem. A, 2017, 5, 15356 RSC.
- J. Tang, R. R. Salunkhe, H. Zhang, V. Malgras, T. Ahamad, S. M. Alshehri, N. Kobayashi, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Sci. Rep., 2016, 6, 30295 CrossRef CAS PubMed.
- A. Azhar, Y. Li, Z. Cai, M. B. Zakaria, M. K. Masud, M. S. A. Hossain, J. Kim, W. Zhang, J. Na, Y. Yamauchi and M. Hu, Bull. Chem. Soc. Jpn., 2019, 92, 875 CrossRef CAS.
- Y. Wang, J. Chen, G. Wang, Y. Li and Z. Wen, Angew. Chem., Int. Ed., 2018, 57, 13120 CrossRef CAS PubMed.
- H. Lu, R. Chen, Y. Hu, X. Wang, Y. Wang, L. Ma, G. Zhu, T. Chen, Z. Tie, Z. Jin and J. Liu, Nanoscale, 2017, 9, 1972 RSC.
- K. Yuan, X. Zhuang, H. Fu, G. Brunklaus, M. Forster, Y. Chen, X. Feng and U. Scherf, Angew. Chem., Int. Ed., 2016, 55, 6858 CrossRef CAS PubMed.
- W. Ye, S. Chen, Y. Lin, L. Yang, S. Chen, X. Zheng, Z. Qi, C. Wang, R. Long, M. Chen, J. Zhu, P. Gao, L. Song, J. Jiang and Y. Xiong, Chem, 2019, 5, 2865 CAS.
- J. M. Lee, M. E. Briggs, T. Hasell and A. I. Cooper, Adv. Mater., 2016, 28, 9804 CrossRef CAS PubMed.
- X. Zhang, T. Kitao, D. Piga, R. Hongu, S. Bracco, A. Comotti, P. Sozzani and T. Uemura, Chem. Sci., 2020, 11, 10844 RSC.
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed.
- Z. H. Guo, C. X. Wang, Q. Zhang, S. Che, H. C. Zhou and L. Fang, Mater. Chem. Front., 2018, 2, 396 RSC.
- M. Rose, N. Klein, I. Senkovska, C. Schrage, P. Wollmann, W. Böhlmann, B. Böhringer, S. Fichtner and S. Kaskel, J. Mater. Chem., 2011, 21, 711 RSC.
- Y.-C. Zhao, D. Zhou, Q. Chen, X.-J. Zhang, N. Bian, A.-D. Qi and B.-H. Han, Macromolecules, 2011, 44, 6382 CrossRef CAS.
- F. M. Wisser, K. Eckhardt, D. Wisser, W. Böhlmann, J. Grothe, E. Brunner and S. Kaskel, Macromolecules, 2014, 47, 4210 CrossRef CAS.
- S. K. Samanta, E. Preis, C. W. Lehmann, R. Goddard, S. Bag, P. K. Maiti, G. Brunklaus and U. Scherf, Chem. Commun., 2015, 51, 9046 RSC.
- B. Yang, J. Bjork, H. Lin, X. Zhang, H. Zhang, Y. Li, J. Fan, Q. Li and L. Chi, J. Am. Chem. Soc., 2015, 137, 4904 CrossRef CAS PubMed.
- S. M. Jung, J. Park, D. Shin, H. Y. Jeong, D. Lee, I. Y. Jeon, H. Cho, N. Park, J. W. Yoo and J. B. Baek, Angew. Chem., Int. Ed., 2019, 58, 11670 CrossRef CAS.
- C. Wang, C. Li, E. R. C. Rutledge, S. Che, J. Lee, A. J. Kalin, C. Zhang, H.-C. Zhou, Z.-H. Guo and L. Fang, J. Mater. Chem. A, 2020, 8, 15891 RSC.
- S. S. Elmorsy, A. Pelter and K. Smith, Tetrahedron Lett., 1991, 32, 4175 CrossRef CAS.
- R. S. Sprick, A. Thomas and U. Scherf, Polym. Chem., 2010, 1, 283 RSC.
- M. Kolmer, R. Zuzak, A. K. Steiner, L. Zajac, M. Engelund, S. Godlewski, M. Szymonski and K. Amsharov, Science, 2019, 363, 57 CrossRef CAS PubMed.
- Y. Byun and A. Coskun, Chem. Mater., 2015, 27, 2576 CrossRef CAS.
- K. Govind Raj and P. A. Joy, J. Am. Ceram. Soc., 2017, 100, 5151 CrossRef CAS.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019 CrossRef CAS.
- X. Jiao, Y. Qiu, L. Zhang and X. Zhang, RSC Adv., 2017, 7, 52337 RSC.
- B. Gupta, N. Kumar, K. Panda, V. Kanan, S. Joshi and I. Visoly-Fisher, Sci. Rep., 2017, 7, 45030 CrossRef CAS PubMed.
- V. Lee, R. V. Dennis, C. Jaye, X. Wang, D. A. Fischer, A. N. Cartwright and S. Banerjee, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2012, 30, 061206 Search PubMed.
- V. Lee, R. V. Dennis, B. J. Schultz, C. Jaye, D. A. Fischer and S. Banerjee, J. Phys. Chem. C, 2012, 116, 20591 CrossRef CAS.
- J. Mahmood, E. K. Lee, M. Jung, D. Shin, I. Y. Jeon, S. M. Jung, H. J. Choi, J. M. Seo, S. Y. Bae, S. D. Sohn, N. Park, J. H. Oh, H. J. Shin and J. B. Baek, Nat. Commun., 2015, 6, 6486 CrossRef CAS PubMed.
- W. Yang, L. Hou, X. Xu, Z. Li, X. Ma, F. Yang and Y. Li, Carbon, 2018, 130, 325 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2006, 44, 468 CrossRef CAS.
- K. S. Yang and B.-H. Kim, Electrochim. Acta, 2015, 186, 337 CrossRef CAS.
- S. I. Yun, H. J. Lee and B. H. Kim, J. Electroanal. Chem., 2020, 858, 113815 CrossRef CAS.
- N. K. Chaudhari, M. Y. Song and J. S. Yu, Sci. Rep., 2014, 4, 5221 CrossRef CAS PubMed.
- F. Destyorini, Y. Irmawati, A. Hardiansyah, H. Widodo, I. N. D. Yahya, N. Indayaningsih, R. Yudianti, Y.-I. Hsu and H. Uyama, Eng. Sci. Technol. an Int. J., 2021, 24, 514 CrossRef.
- S. Shi, X. Zhou, W. Chen, M. Chen, T. Nguyen, X. Wang and W. Zhang, RSC Adv., 2017, 7, 44632 RSC.
- A. Gupta, S. R. Dhakate, P. Pal, A. Dey, P. K. Iyer and D. K. Singh, Diamond Relat. Mater., 2017, 78, 31 CrossRef CAS.
- C. Chen, K. Sun, A. Wang, S. Q. Wang and J. C. Jiang, Bioresources, 2018, 13, 3165 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and Fig. S1–S8. See DOI: 10.1039/d1sc01902c |
| This journal is © The Royal Society of Chemistry 2021 |