
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclophane with eclipsed pyrene units enables construction of spin interfaces with chemical accuracy†
Marvin
Metzelaars
 a,
Sebastian
Schleicher
b,
Takuma
Hattori‡
b,
Bogdana
Borca
bc,
Frank
Matthes
b,
Sergio
Sanz
b,
Daniel E.
Bürgler
*b,
Jeff
Rawson§
*ab,
Claus M.
Schneider
b and
Paul
Kögerler
ab
a,
Sebastian
Schleicher
b,
Takuma
Hattori‡
b,
Bogdana
Borca
bc,
Frank
Matthes
b,
Sergio
Sanz
b,
Daniel E.
Bürgler
*b,
Jeff
Rawson§
*ab,
Claus M.
Schneider
b and
Paul
Kögerler
ab
aInstitute of Inorganic Chemistry, RWTH Aachen University, 52074 Aachen, Germany
bPeter Grünberg Institute (PGI-6), Forschungszentrum Jülich, 52428 Jülich, Germany. E-mail: d.buergler@fz-juelich.de
cNational Institute of Materials Physics, Atomistilor 405A, Magurele, 077125, Ilfov, Romania
First published on 18th May 2021
Abstract
Advanced functionality in molecular electronics and spintronics is orchestrated by exact molecular arrangements at metal surfaces, but the strategies for constructing such arrangements remain limited. Here, we report the synthesis and surface hybridization of a cyclophane that comprises two pyrene groups fastened together by two ferrocene pillars. Crystallographic structure analysis revealed pyrene planes separated by ∼352 pm and stacked in an eclipsed geometry that approximates the rare configuration of AA-stacked bilayer graphene. We deposited this cyclophane onto surfaces of Cu(111) and Co(111) at submonolayer coverage and studied the resulting hybrid entities with scanning tunnelling microscopy (STM). We found distinct characteristics of this cyclophane on each metal surface: on non-magnetic Cu(111), physisorption occurred and the two pyrene groups remained electronically coupled to each other; on ferromagnetic Co(111) nanoislands, chemisorption occurred and the two pyrene groups became electronically decoupled. Spin-polarized STM measurements revealed that the ferrocene groups had spin polarization opposite to that of the surrounding Co metal, while the pyrene stack had no spin polarization. Comparisons to the non-stacked analogue comprising only one pyrene group bolster our interpretation of the cyclophane's STM features. The design strategy presented herein can be extended to realize versatile, three-dimensional platforms in single-molecule electronics and spintronics.
Introduction
To design the electronic properties of nanoscale interfaces demands the precise arrangement of their structures. Magnetoresistance arises at interfaces between ferromagnetic electrodes and organic compounds if chemisorbed proximal molecules and subsequent layers are precisely aligned.1–9 The charge transport efficiencies of organic semiconductors are linked to the spatial overlap and orientation of adjacent π-orbitals.10–13 Superconductivity arises in a duplex of two graphene sheets if one is twisted by a “magic angle” (∼1.1°) with respect to the other.14,15 These uncommon electronic properties arise because atoms at interfaces have been arranged in precise configurations that are challenging to achieve.It is difficult to arrange two polyaromatic hydrocarbon (PAH) groups or continuous graphene sheets at their van der Waals (vdW) distance such that all carbon atoms of the upper sheet lie atop those of the bottom one. In such a fully eclipsed arrangement (AA-stacked), the frontier orbitals of the sheets overlap constructively and electronic repulsion is maximized (Fig. 1).10,11 The other limiting case is the half-eclipsed (AB-stacked) arrangement that is native to graphite.16 AA-stacked bilayer graphene has been a subject of theoretical interest,17–19 but it has been observed only in limited cases in which it could be characterized sparingly.17,20–26 We hypothesized that such an AA-packed graphene bilayer would exhibit interfacial magnetoresistance when placed in contact with a ferromagnetic electrode, as has been found at boundaries between zinc methyl phenalenyl molecules and cobalt.9 Chemisorption of the lower aromatic group would form a hybrid molecular magnet, while the upper ring would act as a spin analyser because it retains sharp states with spin-polarization induced by π–π interactions.3,9,27 Such spin interface effects have yet to be demonstrated at the level of a single molecule.
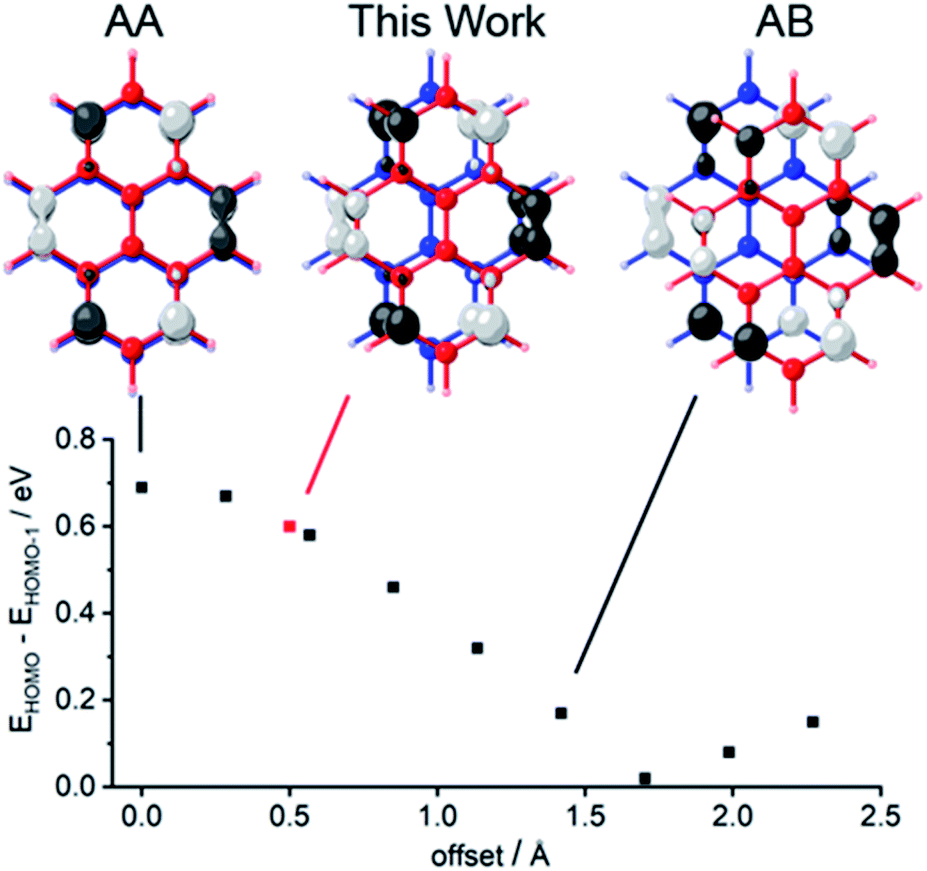 | ||
| Fig. 1 Dependence of the energy gap that separates the two highest occupied molecular orbitals (HOMO and HOMO−1) upon the lateral offset between pyrene molecules in a face-to-face dimer. Two optimized structures of pyrene were positioned in space with 352 pm vertical separation, and the lateral offset was varied from perfectly eclipsed (the configuration of AA graphene) along the coordinate through semi-eclipsed (the configuration of AB graphene) by increments of ∼28 pm. At each position we report the energy gap EHOMO − EHOMO−1, which is proportional to the electronic coupling between the pyrene groups,10 determined by single-point calculations using density functional theory, B3LYP functional, 6-311G(2d) basis set. We plotted ±0.04 (e bohr−3)1/2 isodensity surfaces of the HOMOs at the indicated offsets. | ||
Here, we show how ferrocene (Fc) pillars can be used to construct a cyclophane whose undistorted planar aromatic groups are aligned at their vdW distance with a configuration very close to AA-stacked (red point in Fig. 1). Single-crystal X-ray crystallographic analysis of this compound provides the most precise measure to date of the vdW distance between two undistorted AA-stacked graphene fragments. Previously described bilayer molecules with shorter or contorted spacers have aromatic planes that are bent or rotated with respect to each other,28–34 and structures with longer spacers relax into AB-stacked arrangements that are typical for intermolecular contacts between PAHs.29,30,35–38
We deposited a cyclophane onto a ferromagnetic metal surface, nanoscale Co(111) islands, and onto non-magnetic Cu(111). We evaluated the electron transport across the hybrid (molecule–metal) entity by spin-polarized scanning tunnelling microscopy and spectroscopy (SP-STM/STS) under ultrahigh vacuum conditions at low temperatures (∼4 K). Our design provides an approach that can be generalized to craft well-defined AA-stacked graphene bilayers, a first step to building hybrid entities with precise arrangements of their atoms in three dimensions. Incidentally, we show that a pendant pyrene group can force face-on hybridization of Fc cyclopentadienyl rings onto a reactive Co surface, giving an entity with spin polarization that is inverted with respect to the bulk Co.
Results and discussion
Cyclophane design, synthesis, and structure
Our cyclophane design uses pyrene as an aromatic core that balances size, chemical processability, and vapor pressure.39–42 Pyrene chemisorbs onto metal surfaces with a face-on geometry,42 and its electronic states are influenced minimally by substituents at the 2- and 7-positions because the relevant frontier orbitals are bisected by a nodal plane (Fig. 1 and S13†).43,44Using the advances that have been made in Suzuki–Miyaura cross-couplings, we avoided the multistep approaches that have often been used to synthesize cyclophanes.30 2,7-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrene ([Bpin]2pyrene), readily produced via direct Ir-catalyzed borylation,45,46 was cross-coupled with 1,1′-diiodoferrocene47 utilizing [1,1′-bis(di-tert-butylphosphino)ferrocene]dichloropalladium(II) (PdCl2(dtbpf))48 as catalyst in N,N-dimethylformamide (DMF) (Scheme 1).
 | ||
| Scheme 1 Synthesis of 1 and 2via Suzuki–Miyaura cross-coupling. | ||
We obtained the target cyclophane 2 in <1% yield after purifying it by gradient sublimation (Fig. S1†). Several oligomers and larger macrocycles were also obtained. We synthesized 2,7-diferrocenylpyrene 1 under similar catalytic conditions with a yield of 65%, compared with the 2% yield obtained by Preuß et al. using Negishi coupling, illustrating the advantage of our optimized procedure.49 We confirmed the identities of 1 and 2 by matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS, Fig. S3 and S4†), elemental analysis, nuclear magnetic resonance (NMR, Fig. S2 (1)†) and Fourier-transform infrared (FT-IR) spectroscopy (Fig. S6†).
X-ray diffraction of a single crystal of 2 that we obtained from gradient sublimation confirms the resemblance of the pyrene groups to AA-stacked graphene bilayers (Fig. 2). The space group of cyclophane 2 is the same (P21/c) as reported for 1,49 with intermolecular C–H⋯π contacts that resemble the dimeric herringbone packing of pyrene (Fig. S7 and S8†).50 The intramolecular spacing between the consensus planes of the pyrene groups is 351.64(15) pm, and these planes have bending angles30 of just 3.8(4)°. The pyrene groups are offset by only ∼50 pm from perfectly eclipsed. The vdW contact distance between two AB-stacked graphene sheets is ∼335 pm;16,36 the spacing of the AA arrangement has not previously been measured with the accuracy of our X-ray diffraction, but a distance of 355 pm has been estimated.21 The distance of ∼352 pm between pyrene groups in 2 appears to be the closest that two PAHs can be forced with the AA arrangement without distortion. The distance between cyclopentadienyl centroids in 2 is 330.10(18) pm, only marginally larger than the 329.6 pm of ferrocene;51 the pyrene groups are neither stretching nor contracting these pillars and are thus at the vdW distance for the AA configuration.
In other PAH-containing cyclophanes,30 the conflict between shorter bridge groups and repulsive interactions bends the aromatic planes dramatically (e.g. by ∼37.9° in ethyl28,30 or ∼16.9° in propyl29,30,35 bridged cyclophanes). For discrete aromatic bilayers linked by longer bridges (e.g. butylene29 or –CH2CH2OCH2CH2–36) lesser bending and large lateral offset values were reported that are similar to the values observed in natural graphite (AB-stacking). The two Fc bridges in 2 gently force the two pyrene planes into an eclipsed arrangement at a threshold distance that prevents both bending and relaxation into an AB-stacked arrangement.
 | ||
| Fig. 2 Crystal structure of cyclophane 2 represented with thermal ellipsoids (50% probability); important structural features are indicated in grey with standard uncertainties given in parenthesis. | ||
STM measurements of cyclophane 2 and reference compound 1
We performed STM measurements at ∼4 K of 1 and 2 that were deposited at submonolayer coverages atop nanoscale Co(111) islands on a Cu(111) surface. Triangle-shaped, double-layered Co(111) islands (5–25 nm across) on Cu(111) have out-of-plane magnetization,52–59 and the Cu(111) regions are nonmagnetic. The interactions of aromatic adsorbates with transition metal surfaces have been the subject of extensive theoretical and experimental investigations.1–9 Co has 3d bands that are narrow, spin-split, close to the Fermi level, and partly filled, leading to strong chemisorption. This may be understood to result from Co d-orbitals that protrude at the positions of the Co surface atoms normal to the surface and interact with π-orbitals of pyrene. The broad 3d bands of Cu are fully occupied with energies well below the Fermi level, thus fostering weaker molecule–metal interactions than Co, i.e. physisorption.60A topographic constant-current STM plot (Fig. 3a) imaged intact molecules of 2 as protrusions of length and symmetry consistent with our crystallographic measurements on both Cu (dashed red ellipse) and Co (dashed green ellipse). We noted other fragments (dashed blue ellipse) that appeared after surface deposition. In pristine areas of Co(111), cyclophane 2 adsorbed with the longitudinal axis perpendicular to one of the island edges, which run along the crystallographic [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01], [10] or [01] axis. Wang et al. previously showed that pyrene adsorbed to Cu(111) with its four ring centroids occupying bridge sites upon the metal,61 since the spacings of these centroids (∼244 pm) are only slightly mismatched from the Cu lattice constant (aCu = 256 pm). A similar geometry of adsorption for 2 on Co(111) (aCo = 251 pm)62 is consistent with our data; we propose such an arrangement in Fig. 4. This excellent match of the benzenoid repeat units with the Co(111) surface lattice favours strong hybridization of out-of-plane d-orbitals to molecular π-orbitals.
01], [10] or [01] axis. Wang et al. previously showed that pyrene adsorbed to Cu(111) with its four ring centroids occupying bridge sites upon the metal,61 since the spacings of these centroids (∼244 pm) are only slightly mismatched from the Cu lattice constant (aCu = 256 pm). A similar geometry of adsorption for 2 on Co(111) (aCo = 251 pm)62 is consistent with our data; we propose such an arrangement in Fig. 4. This excellent match of the benzenoid repeat units with the Co(111) surface lattice favours strong hybridization of out-of-plane d-orbitals to molecular π-orbitals.
 | ||
| Fig. 3 Comparison of topographic constant-current STM images showing (a) cyclophane molecules 2 (Vb = 500 mV, It = 0.5 nA) and (b) non-stacked molecules 1 (Vb = 100 mV, It = 0.1 nA) on Cu(111) substrates (e.g. red ellipses), and intact symmetric (e.g. green ellipses) as well as modified asymmetric (e.g. black ellipses) molecules on Co(111) nanoislands. The white ellipse highlights aggregated molecules 2 on Cu(111) and the blue ellipse molecular fragments. (c and d) Show representative cross-sections of apparent height surfaces along the longitudinal molecular axes of several molecules of (c) cyclophane 2 and (d) non-stacked molecule 1 on Cu (red) and Co (green) as well as for modified molecules on Co (black). Depths of the dips measured for 1 and 2 and peak height differences Δh of the double-peak profiles measured for modified molecules are indicated. | ||
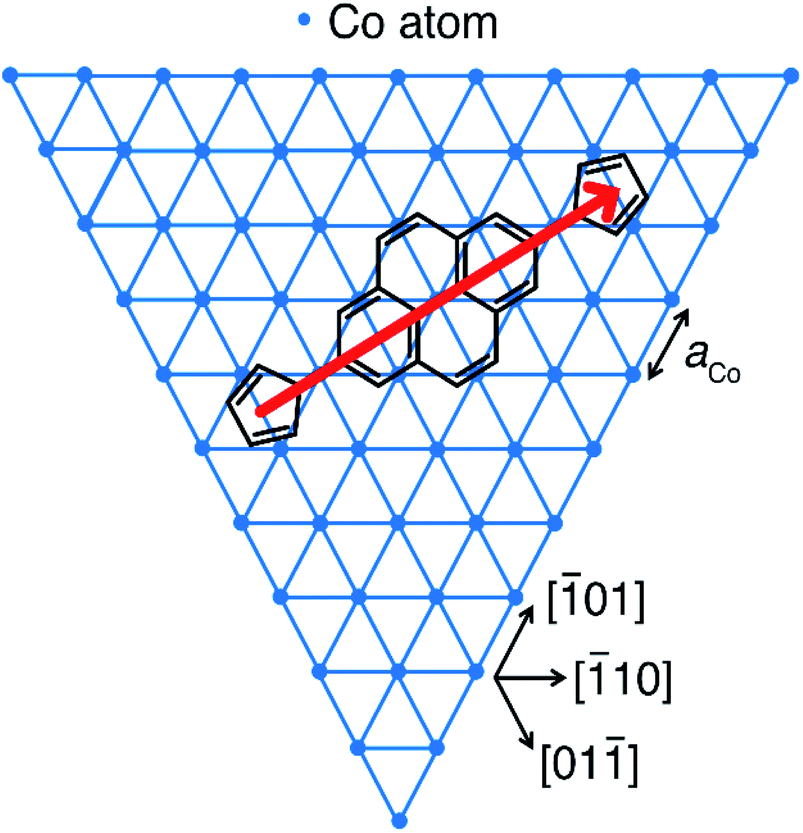 | ||
Fig. 4 Proposed adsorption geometry for 1 and 2 on the topmost atomic layer of Co nanoislands on Cu(111). The border of the hexagonal Co lattice with the lattice constant aCo = 251 pm (ref. 62) indicates the directions of the energetically most favourable islands edges. The longitudinal axis of the molecule (red arrow) points along the [![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 11] direction. Rotation by ±60° yields symmetry-equivalent, energetically degenerate adsorption geometries for which the longitudinal axes of the molecule point in [11] and [2] directions. 11] direction. Rotation by ±60° yields symmetry-equivalent, energetically degenerate adsorption geometries for which the longitudinal axes of the molecule point in [11] and [2] directions. | ||
Cyclophane 2 adsorbed to Co(111) more strongly than to Cu(111). On Cu(111), the profile of the apparent height traced along the main axis of 2 (red plot in Fig. 3c) is flat and featureless. The corresponding profile of 2 on Co(111) (green plot in Fig. 3c) clearly differs, with a pronounced dip at the centre. The ∼300 pm maxima of both of these apparent height profiles are similar to previous reports of Fc adsorbed with a face-on configuration,63 and the symmetries and lengths of the profiles agree with the adsorption scheme in Fig. 4. Cyclophane 2 tended to form aggregates on Cu(111) (white dashed ellipse in Fig. 3a), and was often found at the edges of islands, indications that its interactions with this metal were weak and physisorptive in character. On the Co islands, we found many isolated molecules of 2, suggesting that lateral diffusion was restricted and that 2 chemisorbs to Co(111).
Reference compound 1 preferentially adsorbed on both metals with a face-on geometry, and bound to Cu(111) even more weakly than did 2 (Fig. 3b). We observed no aggregates, and few isolated molecules of 1 that were static on Cu(111). In fact, measuring the profile of the apparent height for 1 atop Cu(111) proved difficult, and the only plots we obtained (red in Fig. 3d) have lower maxima than we expect for intact molecules. We do not know the reason for this discrepancy but suspect that the only molecules of 1 that remained sufficiently static for this measurement were pinned at defect sites of the substrate or had been chemically modified in a way analogous to that which we induced using tip manipulation (vide infra). There are no indications that 1 diffused across Co(111), which corroborates its chemisorption to this metal, and its Fc groups gave apparent heights of the expected ∼300 pm.63
Hybridization of cyclophane 2 to Co(111) suppresses conductance through the pyrene stack
We investigated the role of the upper pyrene group on the electronic structure of cyclophane 2 by comparing the profile of its apparent height with that of 1 under similar imaging conditions. The apparent height at the centre of the profile of 2 is ∼245 pm, while for 1 it is ∼205 pm (Fig. 3). We might have expected the difference between these values to be larger based upon the crystallographic structures of 1 and 2, but the apparent height measured by STM in the constant-current mode convolutes the local topography with the local conductance between the surface and the tip. The lack of infinite sharpness of the STM tip hindered our plumbing of the depth of the crevice between Fc groups in 1, but the apparent height at the centre may be inflated by the strong chemisorption of pyrene to Co(111), providing a channel of conducting states between the tip and the substrate.The apparent dip of ∼55 pm at the centre of cyclophane 2 reflects the lesser tunnelling conductance of the stacked pyrene groups when compared with the Fc groups, a signal that the upper pyrene group of 2 is electronically decoupled from the lower one after it has chemisorbed to Co(111). This dip is unlikely to reflect a topographic depression because the upper pyrene group is suspended at a level that is even with the upper cyclopentadienyl rings. In contrast, on Cu(111) the upper pyrene of cyclophane 2 has an apparent height equal to those of the Fc groups.
Our observations suggest that the electronic coupling between the pyrene groups of 2 is stronger when the cyclophane is physisorbed to Cu(111) than when it is chemisorbed to Co(111). The coupling between group orbitals is proportional to their spatial overlap and inversely proportional to the difference in their energies when the groups are isolated. The reduced coupling between pyrene groups in 2 after chemisorption on Co(111) is likely to be caused by a reduced π–π overlap or a shift of the energies of the lower pyrene orbitals induced by hybridization with the metal.27,64–71 On Cu(111), hybridization of the lower pyrene group to the metal is weaker, this energetic shift as well as the impact on the orbital overlap are smaller, and the pyrene groups remain strongly coupled. Due to the wide HOMO–LUMO gap of pyrene (Fig. S14†), the isolated pyrene group of 2 contributes little to the density of states between this bias voltage of 500 mV and the Fermi level. Similar observations were made for naphthalenediimide (NDI) cyclophanes with large interplanar distances adsorbed to Au(111) or Ag(111). The NDIs are electronically decoupled, and the photophysical properties of the upper NDIs are partially preserved and could be exploited in photonics.67,68,72
Our efforts to directly probe the states of the upper pyrene group of cyclophane 2 by STM were hindered because the molecules were not stable to measurements at larger bias voltages. This instability manifests itself in the occurrence of molecules with asymmetric appearance (black ellipse in Fig. 3a). Molecules of 2 were readily modified by placing the STM tip over a pristine area of a Co island, applying a bias voltage Vb = 1.5 V at a setpoint current It = 50 pA, and moving the tip over a molecule (Fig. S9†). The tip retracted by ∼350 pm, the apparent height of 2 at Vb = 1.5 V, and then irreversibly advanced by more than 100 pm after ∼3 s. We subsequently collected topographic maps over the area in constant-current mode to find that the Fc portion of the molecule subjected to this treatment had been truncated (Fig. S9b†). This is consistent with other reports of the partial decomposition of Fc groups into cyclopentadienyl (Cp) and FeCp fragments on Au,73 Pt74,75 and Cu76,77 surfaces, and with the reported modification by an STM tip of a tetradecylene-bridged di-ferrocene molecule on Cu(110).76 We also applied this modification procedure to the reference molecule 1, with results that were generally similar (Fig. S10†). For a more detailed discussion see ESI.†
Spin-polarized STM of cyclophane 2 on Co(111) islands
We mapped the spin-polarized conductance of cyclophane 2 on Co(111) islands using an antiferromagnetic Cr tip at a low bias voltage of Vb = −0.5 V (It = 1.0 nA, ±1 T). Under these conditions, the molecules were sufficiently stable for us to measure SP-STM conductance maps with acquisition times of up to 6 h. Spin asymmetry maps were obtained from the difference of conductance maps measured at the same bias voltage Vb but for parallel and antiparallel alignment of tip and sample magnetizations. Spin asymmetry maps reflect spatial variations of the spin polarization of the sample's density of states at the energy |e|Vb.78,79 Further details on spin asymmetry and our acquisition methods can be found in the ESI.†We found positive spin asymmetry over the pristine Co(111) and inverted spin asymmetry over the Fc groups, while the spin asymmetry returned to zero as the tip passed over the pyrene stack (see traces along the lengths of two molecules of cyclophane 2 in Fig. 5). Theoretical calculations9,27 indicate that the upper plane of a discrete aromatic bilayer structure that is chemisorbed to a ferromagnetic metal retains sharp but spin-polarized electronic states. This polarization is induced by coupling between aromatic layers and may lead to resistive spin filter functionality. We did not observe spin polarization at the pyrene site of 2 after chemisorption to Co. One possible explanation is that the bias voltage we applied may have been too low to observe spin asymmetry at this site, since the upper pyrene group contributed so little to the density of states in this energy range. Another possibility is that weak coupling between the pyrene groups of 2 is responsible for the lack of spin asymmetry. These two explanations could both be supported with our measurements of the profiles of apparent height, which suggested that the coupling between pyrene groups was weak when cyclophane 2 was chemisorbed onto Co(111). Our data here do not allow us to distinguish between these possibilities but point to several possible extensions that may be explored in future work.
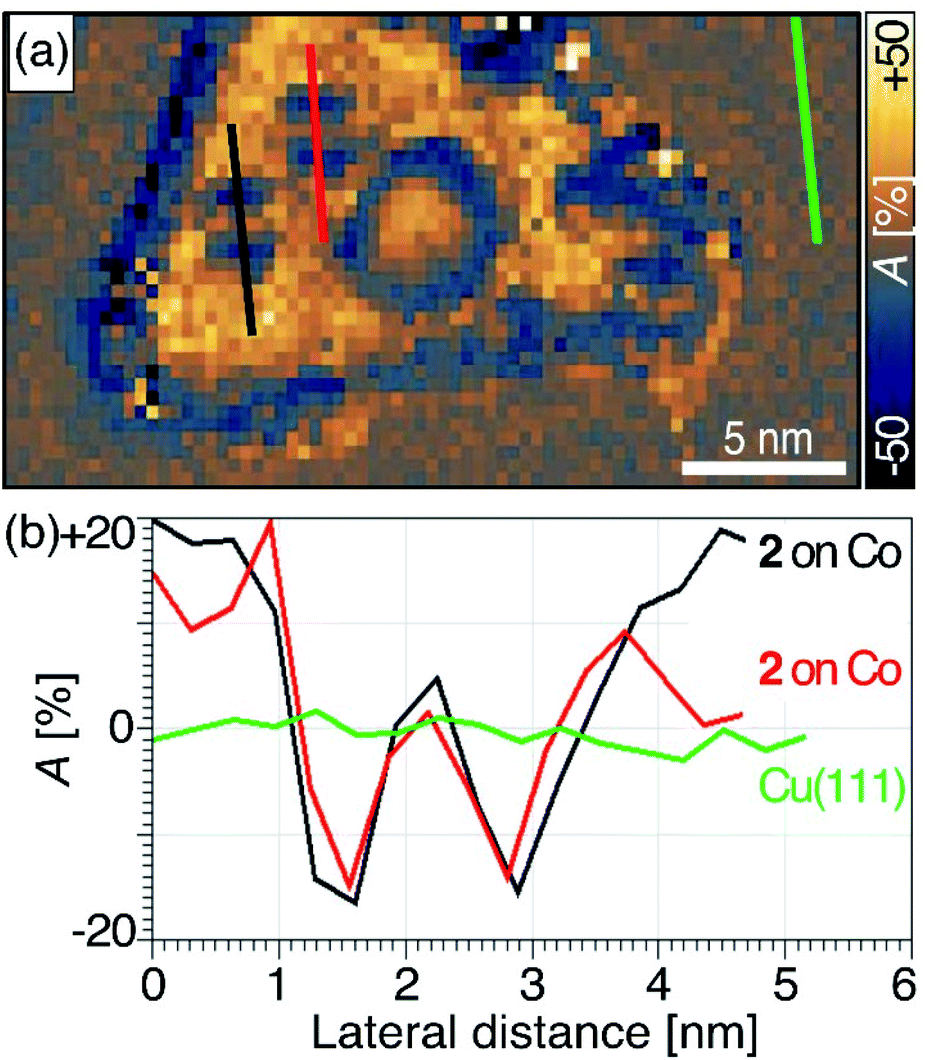 | ||
| Fig. 5 (a) Spin asymmetry map of cyclophane 2 chemisorbed to a Co nanoisland, generated from two spin-polarized conductance maps (Fig. S11†) recorded at Vb = −0.5 V and It = 1.0 nA while applying an external magnetic field of ±1 T, respectively, perpendicular to the surface. The definition and sign convention for the spin asymmetry A is given in eqn (S1) of the ESI.† (b) Spin asymmetry profiles along the longitudinal molecular axes of two molecules 2 (red and black) and for comparison on the bare Cu(111) surface (green). According to the discussion of eqn (S2) in the ESI,† the profiles show no spin polarization on Cu(111), inverted hybridization-induced spin polarization over the Fc sites nearly equal in magnitude to that on Co(111), and no significant spin polarization over the upper pyrene sites. | ||
Chemisorption of 2 to Co(111) leads to inverted spin asymmetry at the Fc sites. Ormaza et al. reported depositing a single Fc molecule atop a Co adatom on Cu(111) to give a magnetic entity, with ferromagnetic coupling between individual Fe and Co atoms.80 In their case, magnetic polarization of Fc was accompanied by zero-bias conductance, a manifestation of the Kondo effect. In our case, there is no reactive adatom bonded to Fc, and the negative spin asymmetry at the Fc site of 2 on Co(111) results from spin-dependent hybridization of the ferromagnetic metal surface with chemisorbed molecules, an effect that has been encountered in films (e.g. aluminium-tris(8-hydroxyquinolin) (Alq3)81) or individual molecules (e.g. phthalocyanine82 or 2,4,6-triphenyl-1,3,5-triazine (TPT)70), and theoretically explained in detail.9,65,66,81,82
Conclusions
We designed and synthesized a cyclophane in which ferrocene pillars hold two pyrene groups at their vdW distance in an eclipsed arrangement. The aromatic planes are virtually undistorted with a bending angle of just ∼3.8° and are displaced laterally from a fully-eclipsed alignment by only ∼50 pm. X-ray diffraction showed that the pyrene groups of this cyclophane are separated by 351.64(15) pm, the most precise measure to date of the vdW distance in AA-stacked graphene bilayers.The organization of molecules at metal surfaces determines the characteristics of devices in molecular electronics and spintronics. The synthesis of 1 and cyclophane 2, compounds amenable to deposition by evaporation, allowed us to investigate and compare spin interfaces at single- and double-layered hybrid entities with chemical accuracy. Using STM, we determined that cyclophane 2 physisorbs onto nonmagnetic Cu(111) with a face-on geometry, low barrier to lateral migration, and a maximum of the apparent height profile of ∼300 pm. On Co(111), 2 chemisorbs with a face-on geometry, no evidence of lateral migration, and an apparent height of ∼245 pm over the pyrene stack. We conclude that hybridization of the lower pyrene group to Co realigns the energy levels of the π-orbitals and reduces their overlap to weaken the electronic coupling between pyrene groups. This interlayer decoupling is consistent with our spin-polarized STM measurements that showed no spin polarization above the chemisorbed pyrene stacks. Further investigations at higher bias voltages were hindered by partial fragmentation of 2 that we confirmed with controlled manipulations using the STM tip.
We expect that the cyclophane design presented in this study can be extended to craft well-defined structures with variable aromatic stacking patterns and sizes. Over the Fc units of 2 on Co(111), we measured spin polarization of comparable strength, but opposite direction, to that of the pristine Co surface. This shows the efficiency with which Fc transmits spin polarization by the spin-dependent hybridization mechanism, and that covalent extension of Fc is an approach to enforce face-on hybridization of Fc groups to ferromagnetic surfaces. This opens a path to metallocene nanopatterning and the construction of more complicated systems. The chemisorption of such structures to ferromagnetic metal surfaces is an important step toward engineering spin interfaces by precise orientation of their constituents in three dimensions.
Author contributions
D. E. B., C. M. S., and P. K. resources, funding acquisition, and project administration. D. E. B., J. R., and P. K. conceptualization and supervision of the study. S. S. (Schleicher), T. H., B. B. F. M., and D. E. B. STM investigation. M. M. (lead), S. S. (Sanz), J. R., and P. K. synthesis and chemical investigation. M. M., F. M., D. E. B., J. R., and P. K. writing – original manuscript. M. M., S. S., T. K., B. B., F. M., S. S. (Sanz), D. E. B., J. R., C.M.S., and P. K. formal analysis and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. and P. K. gratefully acknowledge financial support from the Deutsche Forschungsgemeinschaft (DFG, project 380626961). We thank Dr Nicolae Atodiresei (Research Center Jülich) for helpful discussions and Dr Natalya Izarova for crystallographic measurements (RWTH Aachen University). We acknowledge NMR, MS and EA measurements performed by the Central Institute for Engineering, Electronics and Analytics (ZEA-3) at Research Centre Jülich.Notes and references
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- P. Lazić, V. Caciuc, N. Atodiresei, M. Callsen and S. Blügel, J. Phys.: Condens. Matter, 2014, 26, 263001 CrossRef PubMed.
- K. V. Raman, Appl. Phys. Rev., 2014, 1, 031101 Search PubMed.
- M. Cinchetti, V. A. Dediu and L. E. Hueso, Nat. Mater., 2017, 16, 507–515 CrossRef CAS PubMed.
- S. Delprat, M. Galbiati, S. Tatay, B. Quinard, C. Barraud, F. Petroff, P. Seneor and R. Mattana, J. Phys. D: Appl. Phys., 2018, 51, 473001 CrossRef.
- M. Sun and W. Mi, J. Mater. Chem. C, 2018, 6, 6619–6636 RSC.
- L. Guo, X. Gu, X. Zhu and X. Sun, Adv. Mater., 2019, 31, 1805355 CrossRef CAS PubMed.
- I. Bergenti and V. Dediu, Nano Mater. Sci., 2019, 1, 149–155 CrossRef.
- K. V. Raman, A. M. Kamerbeek, A. Mukherjee, N. Atodiresei, T. K. Sen, P. Lazić, V. Caciuc, R. Michel, D. Stalke, S. K. Mandal, S. Blügel, M. Münzenberg and J. S. Moodera, Nature, 2013, 493, 509–513 CrossRef CAS PubMed.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.-L. Brédas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- A. Batra, G. Kladnik, H. Vázquez, J. S. Meisner, L. Floreano, C. Nuckolls, D. Cvetko, A. Morgante and L. Venkataraman, Nat. Commun., 2012, 3, 1086 CrossRef PubMed.
- M. Carini, M. P. Ruiz, I. Usabiaga, J. A. Fernández, E. J. Cocinero, M. Melle-Franco, I. Diez-Perez and A. Mateo-Alonso, Nat. Commun., 2017, 8, 15195 CrossRef CAS PubMed.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS PubMed.
- A. Nimbalkar and H. Kim, Nano-Micro Lett., 2020, 12, 126 CrossRef CAS PubMed.
- J. D. Bernal and W. L. Bragg, Proc. R. Soc. London, Ser. A, 1924, 106, 749–773 CAS.
- A. V. Rozhkov, A. O. Sboychakov, A. L. Rakhmanov and F. Nori, Phys. Rep., 2016, 648, 1–104 CrossRef CAS.
- H. M. Abdullah, M. A. Ezzi and H. Bahlouli, J. Appl. Phys., 2018, 124, 204303 CrossRef.
- H. S. Qasem, H. M. Abdullah, M. A. Shukri, H. Bahlouli and U. Schwingenschlögl, Phys. Rev. B, 2020, 102, 075429 CrossRef CAS.
- H. V. Roy, C. Kallinger and K. Sattler, Surf. Sci., 1998, 407, 1–6 CrossRef CAS.
- J.-K. Lee, S.-C. Lee, J.-P. Ahn, S.-C. Kim, J. I. B. Wilson and P. John, J. Chem. Phys., 2008, 129, 234709 CrossRef PubMed.
- P. Lauffer, K. V. Emtsev, R. Graupner, T. Seyller, L. Ley, S. A. Reshanov and H. B. Weber, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155426 CrossRef.
- Z. Liu, K. Suenaga, P. J. F. Harris and S. Iijima, Phys. Rev. Lett., 2009, 102, 015501 CrossRef.
- J. Borysiuk, J. Sołtys and J. Piechota, J. Appl. Phys., 2011, 109, 093523 CrossRef.
- I. Brihuega, P. Mallet, H. González-Herrero, G. Trambly de Laissardière, M. M. Ugeda, L. Magaud, J. M. Gómez-Rodríguez, F. Ynduráin and J. Y. Veuillen, Phys. Rev. Lett., 2012, 109, 196802 CrossRef CAS PubMed.
- G. Jeong, B. Choi, D.-S. Kim, S. Ahn, B. Park, J. H. Kang, H. Min, B. H. Hong and Z. H. Kim, Nanoscale, 2017, 9, 4191–4195 RSC.
- M. Callsen, V. Caciuc, N. Kiselev, N. Atodiresei and S. Blügel, Phys. Rev. Lett., 2013, 111, 106805 CrossRef PubMed.
- H. Irngartinger, R. G. H. Kirrstetter, C. Krieger, H. Rodewald and H. A. Staab, Tetrahedron Lett., 1977, 18, 1425–1428 CrossRef.
- H. A. Staab, N. Riegler, F. Diederich, C. Krieger and D. Schweitzer, Chem. Ber., 1984, 117, 246–259 CrossRef CAS.
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494–6518 RSC.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476–9481 CrossRef CAS PubMed.
- P. J. Evans, J. Ouyang, L. Favereau, J. Crassous, I. Fernández, J. Perles and N. Martín, Angew. Chem., Int. Ed., 2018, 57, 6774–6779 CrossRef CAS PubMed.
- X.-J. Zhao, H. Hou, X.-T. Fan, Y. Wang, Y.-M. Liu, C. Tang, S.-H. Liu, P.-P. Ding, J. Cheng, D.-H. Lin, C. Wang, Y. Yang and Y.-Z. Tan, Nat. Commun., 2019, 10, 3057 CrossRef PubMed.
- X.-J. Zhao, H. Hou, P.-P. Ding, Z.-Y. Deng, Y.-Y. Ju, S.-H. Liu, Y.-M. Liu, C. Tang, L.-B. Feng and Y.-Z. Tan, Sci. Adv., 2020, 6, eaay8541 CrossRef CAS PubMed.
- H. A. Staab, D. Q. Zhang and C. Krieger, Liebigs Ann./Recl., 1997, 1997, 1551–1556 CrossRef.
- S. Akine, T. Onuma and T. Nabeshima, New J. Chem., 2018, 42, 9369–9372 RSC.
- F. Jäckel, M. D. Watson, K. Müllen and J. P. Rabe, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045423 CrossRef.
- M. D. Watson, F. Jäckel, N. Severin, J. P. Rabe and K. Müllen, J. Am. Chem. Soc., 2004, 126, 1402–1407 CrossRef CAS.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260–7314 CrossRef CAS.
- T. A. Pham, F. Song, M.-T. Nguyen and M. Stöhr, Chem. Commun., 2014, 50, 14089–14092 RSC.
- T. Kaposi, S. Joshi, T. Hoh, A. Wiengarten, K. Seufert, M. Paszkiewicz, F. Klappenberger, D. Ecija, L. Đorđević, T. Marangoni, D. Bonifazi, J. V. Barth and W. Auwärter, ACS Nano, 2016, 10, 7665–7674 CrossRef CAS PubMed.
- S. Schleicher, B. Borca, J. Rawson, F. Matthes, D. E. Bürgler, P. Kögerler and C. M. Schneider, Phys. Status Solidi B, 2018, 255, 1800235 CrossRef.
- A. G. Crawford, A. D. Dwyer, Z. Liu, A. Steffen, A. Beeby, L.-O. Pålsson, D. J. Tozer and T. B. Marder, J. Am. Chem. Soc., 2011, 133, 13349–13362 CrossRef CAS PubMed.
- S. Sangtarash, C. Huang, H. Sadeghi, G. Sorohhov, J. Hauser, T. Wandlowski, W. Hong, S. Decurtins, S.-X. Liu and C. J. Lambert, J. Am. Chem. Soc., 2015, 137, 11425–11431 CrossRef CAS.
- D. N. Coventry, A. S. Batsanov, A. E. Goeta, J. A. K. Howard, T. B. Marder and R. N. Perutz, Chem. Commun., 2005, 2172–2174 RSC.
- A. G. Crawford, Z. Liu, I. A. I. Mkhalid, M. H. Thibault, N. Schwarz, G. Alcaraz, A. Steffen, J. C. Collings, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Chem.–Eur. J., 2012, 18, 5022–5035 CrossRef CAS PubMed.
- M. Roemer and C. A. Nijhuis, Dalton Trans., 2014, 43, 11815–11818 RSC.
- T. J. Colacot and H. A. Shea, Org. Lett., 2004, 6, 3731–3734 CrossRef CAS PubMed.
- A. Preuß, S. Notz, E. Kovalski, M. Korb, T. Blaudeck, X. Hu, J. Schuster, D. Miesel, T. Rüffer, A. Hildebrandt, K. Schreiter, S. Spange, S. E. Schulz and H. Lang, Chem.–Eur. J., 2020, 26, 2635–2652 CrossRef.
- Y. Kai, F. Hama, N. Yasuoka and N. Kasai, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1263–1270 CrossRef.
- P. Seiler and J. D. Dunitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 2020–2032 CrossRef.
- L. Diekhöner, M. A. Schneider, A. N. Baranov, V. S. Stepanyuk, P. Bruno and K. Kern, Phys. Rev. Lett., 2003, 90, 236801 CrossRef PubMed.
- O. Pietzsch, A. Kubetzka, M. Bode and R. Wiesendanger, Phys. Rev. Lett., 2004, 92, 057202 CrossRef CAS PubMed.
- O. Pietzsch, S. Okatov, A. Kubetzka, M. Bode, S. Heinze, A. Lichtenstein and R. Wiesendanger, Phys. Rev. Lett., 2006, 96, 237203 CrossRef CAS PubMed.
- M. V. Rastei, B. Heinrich, L. Limot, P. A. Ignatiev, V. S. Stepanyuk, P. Bruno and J. P. Bucher, Phys. Rev. Lett., 2007, 99, 246102 CrossRef CAS.
- G. Rodary, S. Wedekind, D. Sander and J. Kirschner, Jpn. J. Appl. Phys., 2008, 47, 9013–9015 CrossRef CAS.
- H. Oka, P. A. Ignatiev, S. Wedekind, G. Rodary, L. Niebergall, V. S. Stepanyuk, D. Sander and J. Kirschner, Science, 2010, 327, 843–846 CrossRef CAS PubMed.
- S. Wedekind, G. Rodary, J. Borme, S. Ouazi, Y. Nahas, M. Corbetta, H. Oka, D. Sander and J. Kirschner, IEEE Trans. Magn., 2011, 47, 3351–3354 CAS.
- S. Ouazi, S. Wedekind, G. Rodary, H. Oka, D. Sander and J. Kirschner, Phys. Rev. Lett., 2012, 108, 107206 CrossRef CAS PubMed.
- X. Jia and W. An, J. Phys. Chem. C, 2018, 122, 21897–21909 CrossRef CAS.
- D. Wang, L.-J. Wan, Q.-M. Xu, C. Wang and C.-L. Bai, Surf. Sci., 2001, 478, L320–L326 CrossRef CAS.
- A. Hahlin, J. H. Dunn, O. Karis, P. Poulopoulos, R. Nünthel, J. Lindner and D. Arvanitis, J. Phys.: Condens. Matter, 2003, 15, S573–S586 CrossRef CAS.
- M. Ormaza, P. Abufager, N. Bachellier, R. Robles, M. Verot, T. Le Bahers, M.-L. Bocquet, N. Lorente and L. Limot, J. Phys. Chem. Lett., 2015, 6, 395–400 CrossRef CAS.
- S. Gabutti, M. Knutzen, M. Neuburger, G. Schull, R. Berndt and M. Mayor, Chem. Commun., 2008, 2370–2372 RSC.
- N. Atodiresei, J. Brede, P. Lazić, V. Caciuc, G. Hoffmann, R. Wiesendanger and S. Blügel, Phys. Rev. Lett., 2010, 105, 066601 CrossRef PubMed.
- N. Atodiresei, V. Caciuc, P. Lazić and S. Blügel, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 172402 CrossRef.
- F. Matino, G. Schull, F. Köhler, S. Gabutti, M. Mayor and R. Berndt, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 961–964 CrossRef CAS.
- N. L. Schneider, F. Matino, G. Schull, S. Gabutti, M. Mayor and R. Berndt, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 153403 CrossRef.
- J. Brede, N. Atodiresei, V. Caciuc, M. Bazarnik, A. Al-Zubi, S. Blügel and R. Wiesendanger, Nat. Nanotechnol., 2014, 9, 1018–1023 CrossRef CAS PubMed.
- V. Heß, R. Friedrich, F. Matthes, V. Caciuc, N. Atodiresei, D. E. Bürgler, S. Blügel and C. M. Schneider, New J. Phys., 2017, 19, 053016 CrossRef.
- J. Kröger, N. Néel, R. Berndt, Y. F. Wang and T. G. Gopakumar, in Encyclopedia of Interfacial Chemistry, ed. K. Wandelt, Elsevier, Oxford, 2018, pp. 81–98 Search PubMed.
- M. Valášek and M. Mayor, Chem.–Eur. J., 2017, 23, 13538–13548 CrossRef PubMed.
- K. F. Braun, V. Iancu, N. Pertaya, K. H. Rieder and S. W. Hla, Phys. Rev. Lett., 2006, 96, 246102 CrossRef PubMed.
- R. Paul, R. G. Reifenberger, T. S. Fisher and D. Y. Zemlyanov, Chem. Mater., 2015, 27, 5915–5924 CrossRef CAS.
- J. R. Reimers, Y. Wang and D. S. Kosov, J. Phys. Chem. C, 2019, 123, 15569–15574 CrossRef CAS.
- D. Y. Zhong, J. Franke, T. Blömker, G. Erker, L. F. Chi and H. Fuchs, Nano Lett., 2009, 9, 132–136 CrossRef CAS PubMed.
- B. W. Heinrich, L. Limot, M. V. Rastei, C. Iacovita, J. P. Bucher, D. M. Djimbi, C. Massobrio and M. Boero, Phys. Rev. Lett., 2011, 107, 216801 CrossRef CAS PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. Lett., 1983, 50, 1998–2001 CrossRef CAS.
- D. Wortmann, S. Heinze, P. Kurz, G. Bihlmayer and S. Blügel, Phys. Rev. Lett., 2001, 86, 4132–4135 CrossRef CAS PubMed.
- M. Ormaza, R. Robles, N. Bachellier, P. Abufager, N. Lorente and L. Limot, Nano Lett., 2016, 16, 588–593 CrossRef CAS PubMed.
- C. Barraud, P. Seneor, R. Mattana, S. Fusil, K. Bouzehouane, C. Deranlot, P. Graziosi, L. Hueso, I. Bergenti, V. Dediu, F. Petroff and A. Fert, Nat. Phys., 2010, 6, 615–620 Search PubMed.
- J. Brede, N. Atodiresei, S. Kuck, P. Lazić, V. Caciuc, Y. Morikawa, G. Hoffmann, S. Blügel and R. Wiesendanger, Phys. Rev. Lett., 2010, 105, 047204 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and synthetic details; NMR, MALDI-MS, UV-Vis, and FT-IR spectra; crystallographic information; additional STM and SP-STM data; computational methods. CCDC 2056848 and 2056849. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01036k |
| ‡ Present address: Institute for Solid State Physics, The University of Tokyo, Kashiwa, Chiba 277-8581, Japan. |
| § Present address: Department of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. Email: E-mail: jrawson@gmwgroup.harvard.edu |
| This journal is © The Royal Society of Chemistry 2021 |