
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring benzylic gem-C(sp3)–boron–silicon and boron–tin centers as a synthetic platform†
Wei W.
Chen
 ab,
Nahiane Pipaon
Fernández
a,
Marta Díaz
Baranda
ab,
Anton
Cunillera
b,
Laura G.
Rodríguez
ab,
Alexandr
Shafir
*b and
Ana B.
Cuenca
*a
ab,
Nahiane Pipaon
Fernández
a,
Marta Díaz
Baranda
ab,
Anton
Cunillera
b,
Laura G.
Rodríguez
ab,
Alexandr
Shafir
*b and
Ana B.
Cuenca
*a
aDepartment of Organic and Pharmaceutical Chemistry, Institut Químic de Sarrià, URL, Vía Augusta 390, Barcelona 08017, Spain. E-mail: anabelen.cuenca@iqs.url.edu
bDepartment of Biological Chemistry, Institute of Advanced Chemistry of Catalonia, IQAC-CSIC, c/ Jordi Girona 20, Barcelona 08034, Spain. E-mail: alexandr.shafir@iqac.csic.es
First published on 2nd July 2021
Abstract
A stepwise build-up of multi-substituted Csp3 carbon centers is an attractive, conceptually simple, but often synthetically challenging type of disconnection. To this end, this report describes how gem-α,α-dimetalloid-substituted benzylic reagents bearing boron/silicon or boron/tin substituent sets are an excellent stepping stone towards diverse substitution patterns. These gem-dimetalloids were readily accessed, either by known carbenoid insertion into C–B bonds or by the newly developed scalable deprotonation/metallation approach. Highly chemoselective transformations of either the C–Si (or C–Sn) or the C–B bonds in the newly formed gem-Csp3 centers have been achieved through a set of approaches, with a particular focus on exploiting the synthetically versatile polarity reversal in organometalloids by λ3-aryliodanes. Of particular note is the metal-free arylation of the C–Si (or C–Sn) bonds in such gem-dimetalloids via the iodane-guided C–H coupling approach. DFT calculations show that this transfer of the (α-Bpin)benzyl group proceeds via unusual [5,5]-sigmatropic rearrangement and is driven by the high-energy iodine(III) center. As a complementary tool, the gem-dimetalloid C–B bond is shown to undergo a potent and chemoselective Suzuki–Miyaura arylation with diverse Ar–Cl, thanks to the development of the reactive gem-α,α-silyl/BF3K building blocks.
Background and motivation
In the context of diversity-oriented synthesis, reagents bearing geminally disposed bis-metalloid units represent an attractive class of building blocks. Expected to act as bis-nucleophilic components in a variety of reactivity patterns, such molecules may set the stage for densely 1,1-disubstituted carbon centers through sequential reactions with two electrophilic partners, including via catalytic cross-coupling. A potentially rewarding aim is the use of the Csp3-gem-dimetalloid group as a platform for straightforward access to multi-substituted methanes.1 This basic unit received a renewed spotlight as part of medicinal chemistry's “escape from the flatland” endeavor,2 and has been the subject of important recent methodological breakthroughs.3 In terms of the substitution pattern, compounds featuring a carbon center anchoring a gem-boron/boron pair have been particularly popular, whereby a variety of methods have been developed to form such Csp3-gem-diboronates and to selectively transform one or both of their C–B sites.4Along these lines, the prospect of selective introduction of two different substituents has also prompted interest in derivatives bearing mutually distinct metalloids, either as chemically differentiated boryl fragments5 or as a hetero-metalloid pair. Inherent reactivity differences between the two carbon–metalloid bonds can then be leveraged to gain chemo-control in modular C–C and/or C–heteroatom bond-forming sequences, with 1,1-silyl-boranes constituting an obvious paradigm for such a hetero-gem-dimetalloid platform.6 As part of our interest in this area, some of us recently showed that 1,1-SiMe3/B(OR)2 disubstituted alkenes could be obtained by a novel boron-Wittig-type olefination using the 1,1,1-B,B,Si-trimetalloid methane species,7 and that the newly formed olefinic Csp2-gem-Si,B group could be engaged in stereoselective access to tetra-substituted olefins. Encouraged by these earlier advances, we set our sight on the gem-B/Si-benzylic scaffold of type 1 (Scheme 1). Creative recent approaches to 1 include a Suzuki–Miyaura mono-arylation of the aforementioned 1,1,1-bis-(Bpin)-trialkylsilyl methane 2,8 including its enantioselective desymmetrization variant,8b or a homologation of an Ar–B bond using the diazo-reagent Me3SiCHN2, 3 (Scheme 1A and B).9 However, these advances in the preparation of 1 contrast with the scarcity of methods that employ such gem-dimetalloids in the modular build-up of poly-substituted Csp3 centers. For example, while the C–Bpin bond in such species was recently shown to undergo a Pd-catalyzed coupling with iodoarenes (Scheme 1C),9b the chemistry at the C–Si bond has been largely limited to the more established desilylative SN2 alkylations.9b In this report, we describe a series of approaches to expand the arsenal of chemoselective transformations of the gem-B/Si-benzylic scaffold 1. In particular, our prior experience in polarity reversal and rearrangement reactions of organosilanes induced by hypervalent iodine compounds has now been leveraged to achieve a metal-free selective C–Si and C–Sn arylation, along with a series of umpolung reactions (Scheme 1D). Among interesting gem-dimetalloid targets, we also show that the trifluoroborate analogue of 1 can be prepared, allowing for the Suzuki–Miyaura arylation (see Scheme 1D left) using bromo- and chloro-arenes.
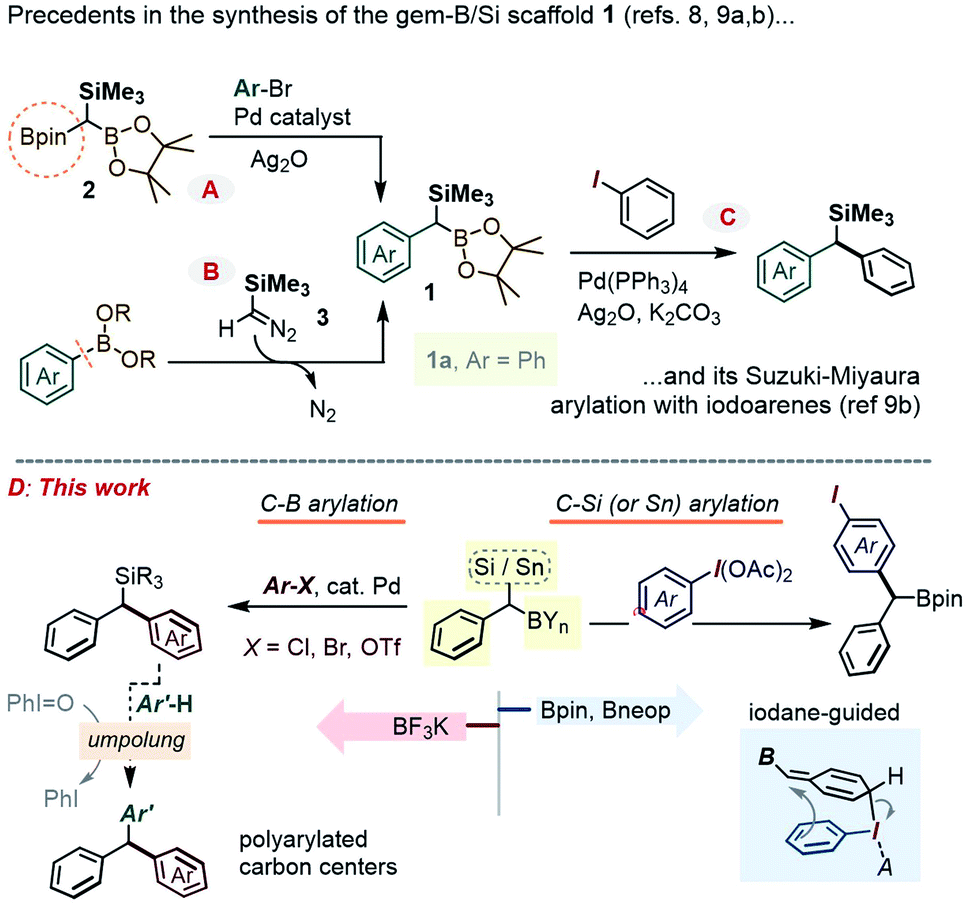 | ||
| Scheme 1 (A and B) Recent approaches to the gem-Si/B compound class 1, and (C) an example of Pd-catalyzed arylation of 1 with Ar–I; (D) the outline of the gem-dimetalloid diversification strategy described herein. | ||
Results and discussion
As was readily appreciated from the outset, the successful usage of the gem-Si/B molecular platform such as 1 would be contingent upon more ready access to this compound class. Our initial approach to 1a involved the insertion of the carbenoid-type fragment “SiMe3(H)C:” of the diazo species Me3SiCHN2, 3, into the B–C bond of arylboroxines, as reported recently by Ley and co-workers.9a Following this route (as in Scheme 1B), the phenyl derivative 1a was obtained in good yields by heating phenylboroxine, (PhBO)3, with 3 in toluene. Despite the success of this approach on small scales (1–2 mmol), we continued our search for a complementary method to access gram quantities of the reagent. After some trials, it was found that 1a could be conveniently prepared from commercial benzyl(trimethyl)silane by deprotonation at the benzylic position, followed by quenching the resulting benzyllithium species with an electrophilic B(OR)3 reagent (Scheme 2, approach A). In our hands, treating the α-B(OH)2 intermediate 4a with pinacol provided 1a in reproducibly good yields on scales of up to 10–15 mmol. Similarly, capping the Csp3–B unit with dimethylpropane-1,3-diol could be used to obtain the Bneop derivative 1a′ in 52%; the diminished yield in the latter case is likely due to the instability of the Bneop derivatives under column chromatography on SiO2. A second approach was also developed starting with a commercial benzylboronate core via deprotonation using a hindered lithium 2,2,6,6-tetramethylpiperidide base (Li-TMP), followed by the addition of a chlorosilane.10 This route was convenient for producing derivatives with silyl groups other than SiMe3, such as the gem-SiEt3 species 1b (84%, Scheme 2, approach B) or the Si(tBu)Me2 analogue 1c (60%). Finally, when a suitable B- or Si-benzylic precursor was not readily available, a Matteson-type homologation9c of several Ar–Bpin species with an in situ generated Me3Si(Cl)CH–Li reagent was found to proceed smoothly. Following this route, the meta-halo gem derivatives 1d and 1e were obtained in 82% and 85% yields, respectively (Scheme 2, approach C).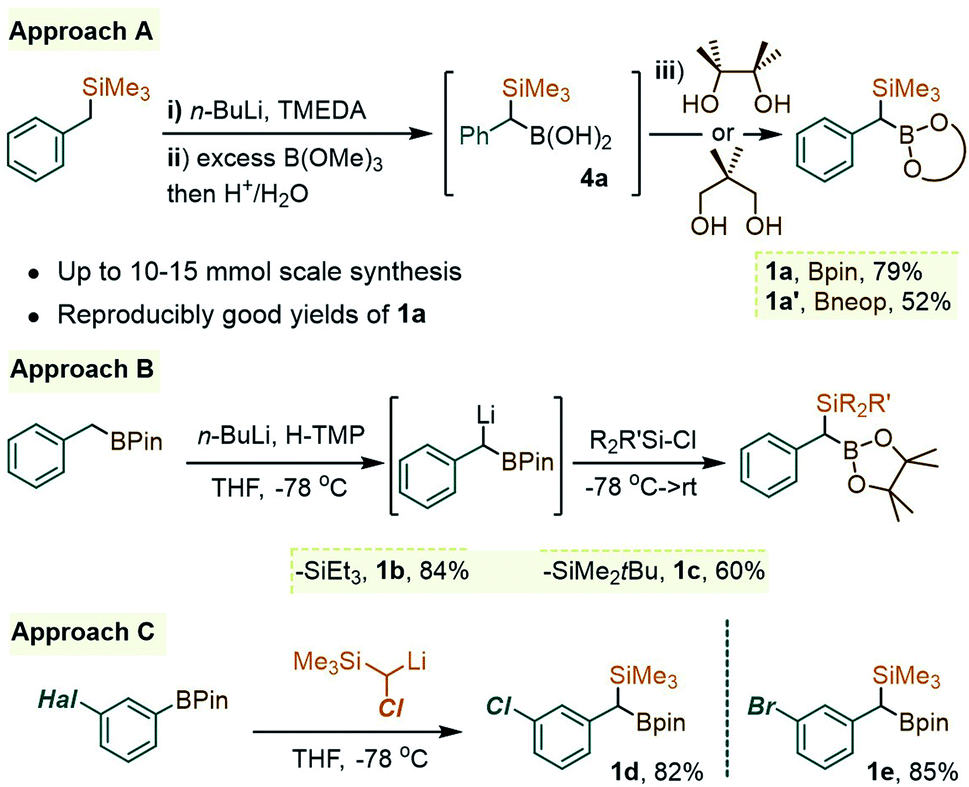 | ||
| Scheme 2 Complementary deprotonation/quench approaches to the benzylic gem-Si/B core 1a. Approach A: (i) in hexane using 1.0 equiv. of n-BuLi at rt; (ii): with 3 equiv. of B(OMe)3, −78 °C to rt. Approach B: Li-TMP generated at −40 °C prior to addition of BnBpin at −78 °C. Approach C: lithiation of Me3SiCH2Cl with sec-BuLi/TMEDA followed by addition of Ar–Bpin. | ||
With gram quantities of the model gem-hetero-dimetalloid 1a now accessible, we turned our attention to a selective transformation of its C–Si moiety. Although a Hiyama-type metal-catalyzed C–Si arylation coupling would be a valuable tool in this system, the applications of this reaction even to simple benzylsilanes are scarce, and generally require an activated silicon group.11 As an alternative, we sought to engage 1a in metal-free C–Si arylation with λ3-ArI(OAc)2 species. This possibility was based on our recent experience in the iodane-guided C–H benzylation of iodoarene cores, a process reported independently by the Hyatt and our laboratories.12 As depicted in Scheme 3A for a benzylsilane model, this unusual manifold would begin with the benzyl group transfer to the iodine(III) center. While related to the iodane-guided ortho-C–H coupling processes,13,14 the benzyl group in such case gave rise to a cationic head-to-tail π-stacked intermediate I, which evolves via the C–C bond formation at the ArI para-C–H position.12b We note that the Hyatt lab also showed the possibility of similarly engaging benzylic trifluoroborate nucleophiles.12a
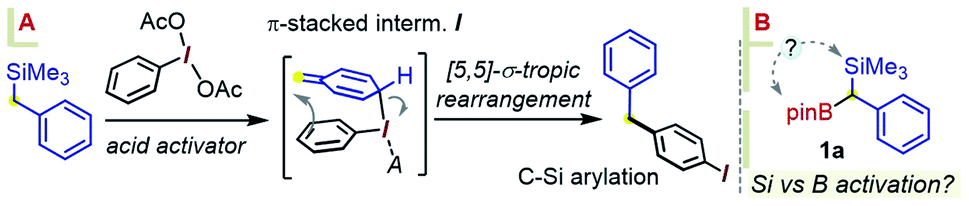 | ||
| Scheme 3 (A) A precedent in the iodane-guided C–Si arylation of benzylsilanes with λ3-iodanes (Hyatt et al. and Shafir et al.), 2018–19. (B) Potential selectivity dilemma in the analogous arylation of 1a. | ||
Considering an application of such a manifold to 1a, an interesting question arises as to which of the substrate's two metalloid groups, i.e. Si or B, would be retained in the product (Scheme 3B). Thus, the reactivity of 1a with the iodoanisole-based λ3-ArI(OAc)2 reagent 5 was probed at −78 °C. While no reaction took place between 1a and 5 in a CH3CN/CH2Cl2 mixture without an acid additive, enhancing the reactivity of the λ3-iodane with an acidic activator, either BF3·Et2O or TMSOTf,15 led to the new benzylated species 6 in 55% yield, with the new C–C created para to the iodine (Scheme 4). The product retains the Bpin group, suggesting a more reactive nature of the C–Si bond during the putative transmetallation step, a feature that contrasts with C–B-specific Pd-catalyzed coupling reactions (as in Scheme 1C). A DFT analysis was used to understand how the process could play out for a gem-B/Si-dimetalloid substrate. Calculations on the –B-glycolate analogue of 1a (–Bgly, Scheme 4B) point to a cationic π-stacked intermediate II with a head-to-tail alignment of the two aromatic fragments. This species is analogous to the adduct I, previously identified in a para-CH–benzylation (Scheme 3), and similarly evolves via a low-lying (∼0.5 kcal mol−1) transition state to form a new carbon–carbon bond para to iodine (Scheme 4B). The rearrangement is expected to initially provide the ring protonated intermediate III. Although the final aromatization may then take place through proton transfer to an external base, the presence of the boronate group in III may also enable an interesting low-barrier internal proton transfer from the acidic C–H site to one of the boronate O atoms (see TS2 in Scheme 4B).
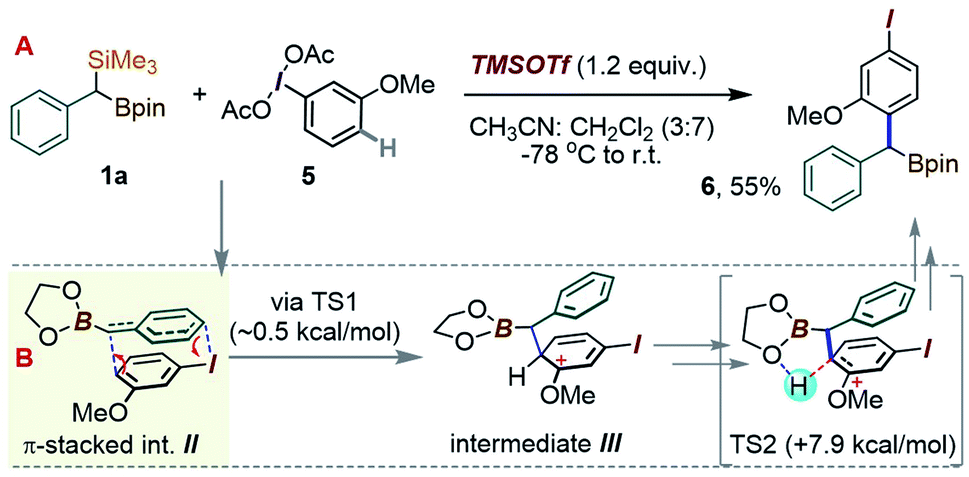 | ||
| Scheme 4 (A) Para-selective C–H-(α-borylbenzylation) of the iodoanisole core. (B) DFT proposal for the C–C bond forming step. | ||
A brief scope survey showed that the coupling efficiency may benefit from the presumably less encumbered Bneop substituent, as seen in the formation of the Bneop analogue 6′ in 65% yield starting from 1a′ (Table 1). A similar moderate improvement was also seen for the C–H benzylation at the 4-position of λ3-1-(diacetoxyiodo)-naphthalene, with a 43% yield obtained for the α-Bpin derivative 7 and a 57% yield (NMR) for the Bneop analogue 7′. The reaction was also successful in providing the para-(α-boryl)benzylated derivative of the protected 3-iodoaniline (prod. 8, 67%), the meta-halo derivatives 9 and 10 (63% and 57%), and the iodoxylene coupling product 11 (85%). The success of these reaction may partly be due to the presence of electron-releasing substituents in most of the iodoarene cores.16 Indeed, the use of the non-activated simple PhI(OAc)2 initially led to the target arylated species 12 in yields of ∼20%. Finally, as we previously observed for λ3-iodoanes blocked at the para position,12b exposing 1a to the λ3-iodane derived from iodomesitylene led to an efficient formation of the meta-CH–benzylated species 13.17 The scope of this meta-substitution was then extended to the m-xylyl derivative 14 and the bromo-target 15. The method was also tolerant of a gem-Si/B reagent bearing a second BPin substituent on the aromatic ring (Table 1, prod. 16).

In view of the initially low yields in the coupling between 1a and PhI(OAc)2, due to the formation of benzaldehyde and other benzylic oxidation side products, we considered replacing the SiMe3 fragment with a potentially more reactive SnBu3 group. With that in mind, two new gem-Sn/B-dimetalloids 17 and 17′ were synthesized from the benzylboronate 18via benzylic deprotonation followed by quenching with R3SnCl (Scheme 5).
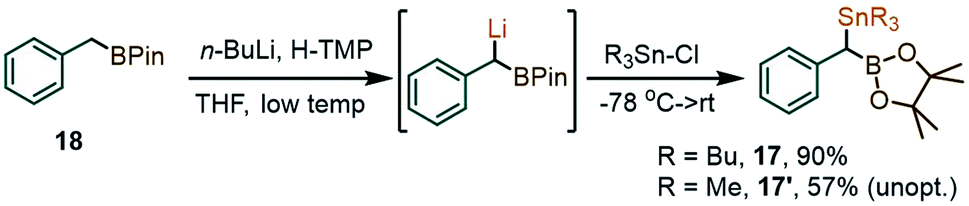 | ||
| Scheme 5 Formation of the gem-stannyl-boryl precursors 17 and 17′. | ||
To our delight, a reaction between 17 and the λ3-iodane PhI(OAc)2 led to the para-benzylated target 12 in 60% yield (65% by NMR), i.e. is ∼3 times higher than for the silyl analogue 1a (Scheme 6A). This improvement may be tentatively explained by the enhanced ability of organo-stannanes to undergo transmetallation in iodane-guided C–H coupling reactions, a feature illustrated in the 1990's by the Ochiai group,13 and recently leveraged by Peng and co-workers in ortho-directed C–H cyanoalkylation.18 Similarly, while barely a trace amount of coupling took place between 1a and the 2-iodothiophene-derived λ3-iodane 19, switching to 17 led to the thienyl product 20 in 50% yield (Scheme 6B). We note that although the 2-iodothiophene core does not possess a para position per se with respect to I, the substrate's C(5)–H unit does correspond to a C–C coupling site predicted for a putative [5,5]-sigmatropic rearrangement path (see ESI†). Indeed, a DFT optimization of the expected π-stacked intermediate IV (analogous to III in Scheme 4) converges to a conformation with the benzylic CH(BPin) group placed directly above the thiophene C(5)–H site (Scheme 6 bottom). Finally, the use of the Bu3Sn species 17 allowed for the synthesis of the naphthyl-derived 7 to be improved from the 43% reported in Table 1 using 1a to 65%. The use of 17′ also allowed for the C–H coupling of an o-Br, m-OMe iodane precursor to give 21 in 51% yield.
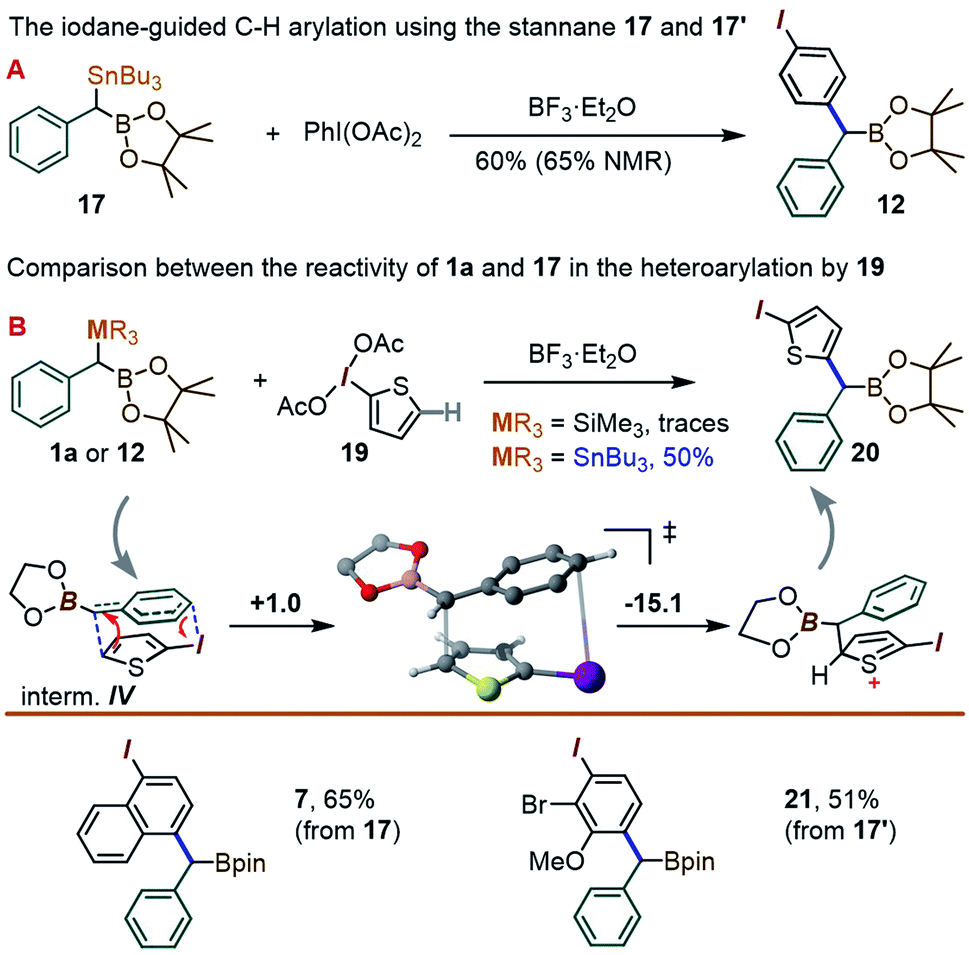 | ||
| Scheme 6 The C–H coupling reaction of the new gem-Sn–B benzylic derivatives 17 and 17′. The free energies in the mechanistic scheme are in kcal mol−1. | ||
Along with the transfer of an intact boryl substituent, an important feature of the iodane-guided arylation reactions is the retention of the synthetically versatile iodine handle. A variety of downstream diversification routes could thus be assayed, including Pd-catalyzed cross-coupling reactions, e.g. Sonogashira alkynylation, Stille coupling or stanylation reactions (Scheme 7, prods. 22–24).
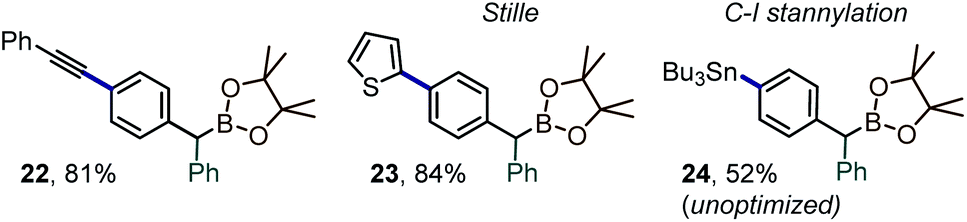 | ||
| Scheme 7 Examples of downstream C–I diversification of 12. | ||
While the pronounced para-C–H selectivity in the arylation of 1a and 17 is consistent with the iodane-guided rearrangement mechanism outlined in Scheme 4, the outcome could in principle be rationalized through a particularly selective Friedel–Crafts type benzylation mechanism. Indeed, organosilanes can undergo the umpolung of the C–Si unit in the presence of simple λ3-iodane oxidants. In the case of the gem-dimetalloids 1 or 17, such a process might involve the benzyl iodonium intermediate such as II, which would evolve by a dissociation into the parent iodoarene and the α-boryl benzyl cation V. In such a sequence, the product would arise through an electrophilic attack at the ArI para position (see Scheme 8A). To test for the presence of free carbocationic species V, 17 was allowed to react with PhI(OAc)2 in the presence of 1 equiv. of mesitylene, 25. A concerted iodane-guided path would still favor the iodobenzene-derived 12, while any free V would be expected to cause the functionalization of mesitylene. Here, the highly nucleophilic mesitylene was chosen so as to stack the odds in favor of such a hypothetical Friedel–Crafts process. Although the mesitylene-derived diarylmethane 26 was observed, this compound was present as a minor component, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio with 12 (Scheme 8B, also see the ESI for Fig. S2†). This result indicates that while the electrophilic aromatic substitution is possible, such a route does not represent the major path in the formation of 12.
:5 ratio with 12 (Scheme 8B, also see the ESI for Fig. S2†). This result indicates that while the electrophilic aromatic substitution is possible, such a route does not represent the major path in the formation of 12.
 | ||
| Scheme 8 Mechanistic assessment of the umpolung manifold in the oxidative coupling of 17 promoted by λ3-iodanes. | ||
While these studies were underway, a parallel effort was directed towards securing a complementary method for the arylation of the C–B bond in gem-Si/B precursors, which we thought to be possible through a metal-catalyzed coupling approach. In fact, at the time when our early efforts were getting underway (late 2018), there existed no literature reports on the coupling of α-silylated benzylboranes such as 1a. More recently, however, Wang and co-workers described the arylation of 1 with iodoarenes using a Pd-catalyzed Ag-promoted coupling.9b,19 In our case, the study centered on solving a more difficult coupling of gem-silylborylated cores with bromo- and chloro-arenes, which we hoped could be achieved using a series of newer Pd catalysts. Initial attempts to engage the Bpin precursor 1a in the coupling with chloroarenes under a variety of catalytic conditions led to unsatisfactory results. Reasoning that a more reactive boryl substituent might prove beneficial, we sought to access the trifluoroborate derivative of 1a. Borrowing a page from the Molander group's synthesis of the methylene reagent Me3Si–CH2BF3K,20 a treatment of 1a with KHF2 in an Et2O/H2O mixture led to the precipitation of a white solid identified as the gem-silyl potassium trifluoroborate 27a (Scheme 9, top path). The NMR spectra of this species presented the benzylic 1H CH signal at 1.26 ppm (broadened quartet, JH–F = 7.1 Hz), and showed a displacement of the 11B-NMR resonance from ∼33 ppm for the –Bpin group in 1a to ∼1 ppm for 27a. Noteworthily, no fluoro-desilylation of 27a was observed at this stage. It was subsequently found that the lithiation/borylation approach, used earlier for the synthesis of 1a (i.e. as in Scheme 2), may be redirected toward 27a by intercepting the crude intermediate 4a (identified by GC-MS as two diastereomeric boroxine trimers, m/z = 570, see the ESI†), with KHF2 (Scheme 9). This method circumvents the need for the isolation of 1a and allows for the ready synthesis of 27a on a gram scale. A small family of additional trifluoroborate salts were also prepared, including 27-CF3 and 27-anis, as well the species 27b and 27c, the latter with variations at the silyl group (Scheme 9), either from the corresponding –Bpin precursor or via deprotonation/silylation sequences. All were isolated as white or off-white powders.
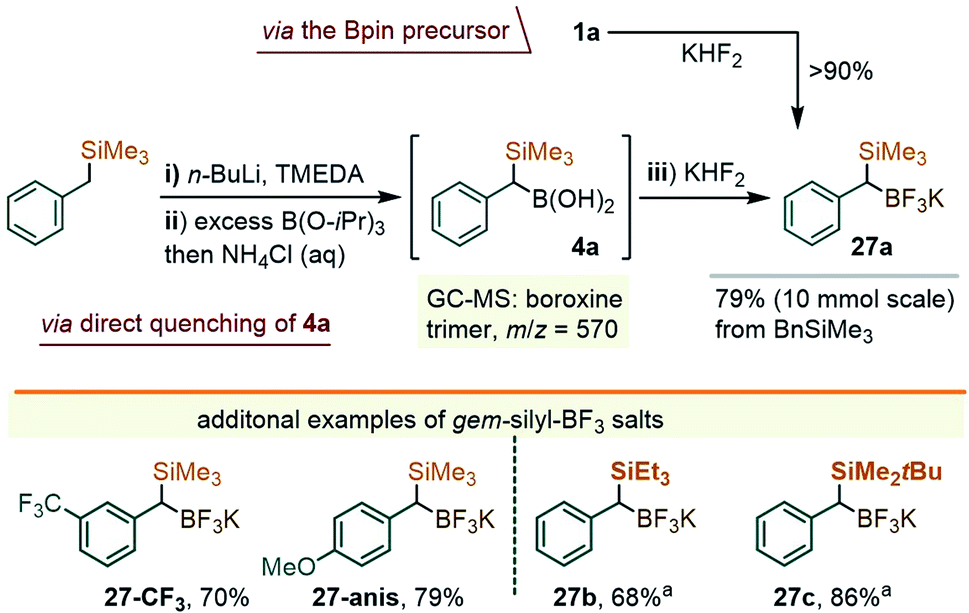 | ||
| Scheme 9 A telescope synthesis of 27a from benzyl(trimethyl)silane. (i) In hexane using 1.0 equiv. of n-BuLi at rt; (ii) with 3 equiv. of B(O-iPr)3, −78 °C to rt, and overnight; (iii) in Et2O/H2O 4:1. aPrepared from the corresponding –Bpin precursors. | ||
As we hoped for, a switch from a Bpin to a BF3K gem-dimetalloid proved highly beneficial for the Suzuki–Miyaura coupling with chloroarenes. While in our hands 1a failed to undergo a C–B arylation with the p-Cl–methylbenzoate 28, the use of 27a led to very promising yields of the target diarylmethane 29 using a number of modern catalytic systems. In particular, virtually quantitative conversions were achieved by using the RuPhos/Pd(OAc)2 combination,21 or by applying the 3rd generation tBu3P–Pd palladacycle precatalyst (tBu3P–Pd G3), the latter as used by Biscoe and co-workers for the aliphatic secondary trifluoroborate species (Scheme 10, gray inset).22 The system was also suitable for bromoarenes, but gave unsatisfactory results with iodoarenes. This coupling efficiency is remarkable, given that the challenging Ar–Cl electrophile is joined with the bulky α-SiMe3-substituted C(sp3) nucleophile without the loss of the potentially sensitive α-silyl substituent. While the two catalyst systems showed similar performances, the scope of the arylation of 27a (Scheme 10) is mainly illustrated using the Pd-G3 metallacycle. Hence, steric hindrance at the Ar–X partner was found to be well tolerated, with nearly quantitative yields obtained with the ortho-bromotoluene (prod. 30, 98%), and a somewhat diminished yield for o,o-dimethyl bromoarene (31, 65%). Efficient coupling was also observed for a series of substituted haloarenes to give diarylmethanes 32–35 in generally excellent yields. As a limitation, the coupling of the strongly π-deficient chloroquinoline was inefficient due to rampant product desilylation (prod. 36, <20%, observed by GC-MS). Nevertheless, the C–C coupling was also well-suited for naphthalene and thiophene-based haloarenes (see prods. 37–39), with a somewhat lower yield in the isolation of 38 possibly due to mechanical losses during purification. The method is also suited for structure variations on the gem-Si-BF3K component, including the successful formation of trifluorotolyl and anisyl derivatives 40 and 41. Additionally, the method allowed for the ready formation of SiEt3 and tBuMe2Si analogues 42 and 43, albeit with a somewhat lower yield for the latter. Our initial venture into bio-active core functionalization involved the coupling of the estrone moiety, which was possible starting with the corresponding 3-Br derivative. Interestingly, the RuPhos-based catalyst (but not the Pd-G3 system) also allowed for the direct coupling of the more readily available 3-OTf form of the precursor (prod. 44, 94%). The method was also applied to the C–Cl moiety of the antihistaminic drug Loratadine, a polycyclic N-containing bioactive chloroarene, providing the α-silyl-benzylated core 45 in an excellent 94% yield. Likewise, the reaction proved almost quantitative for the PPARα activator Fenofibrate as the Ar–Cl partner. Incidentally, the latter hyperlipidemic prodrug was recently considered for repurposing as a SARS-CoV-2 infection inhibitor.23 Only mono-arylation was observed in all cases, confirming the expected resistance of the Csp3–SiMe3 group towards conventional cross-coupling. Interestingly, although the two pre-catalysts were largely interchangeable, reactions conducted with the tBu3P–Pd G3 system frequently showed a minor GC-MS peak with the same m/z value as the target product (typically in a ∼25–30:1 area ratio). For the –CN derivative 33, this minor isomer, 33inv, was isolated and shown by NMR to correspond to the arylation at the para C–H position of the benzylic precursor (Scheme 10C). We note that while related Pd-catalyzed para-selective coupling in benzylic systems is known, it typically relies on the use of benzylic electrophiles (e.g. benzyl chlorides), and not nucleophiles, as is the case in our system.24 While a number of mechanistic manifolds can be envisioned leading to such side-products, one possibility is illustrated in the beige inset in Scheme 10C. Assuming a canonical oxidative addition/transmetallation path, species 33inv could arise from the isomerization of the Pd(II)-benzyl fragment to one of its para-palladated forms prior to reductive elimination. The bulky α-SiMe3 group appears to influence the process, as no such product was observed in the control coupling using the simple benzyltrifluoroborate precursor (see the ESI†). In a few cases, e.g. for the Loratadine-derived 46, the para-coupled species were formed in yields of up to 10%. Gratifyingly, this side reaction is fully suppressed by switching to the RuPhos-based catalyst system (i.e. the system used to obtain the 94% yield of 46 as shown).
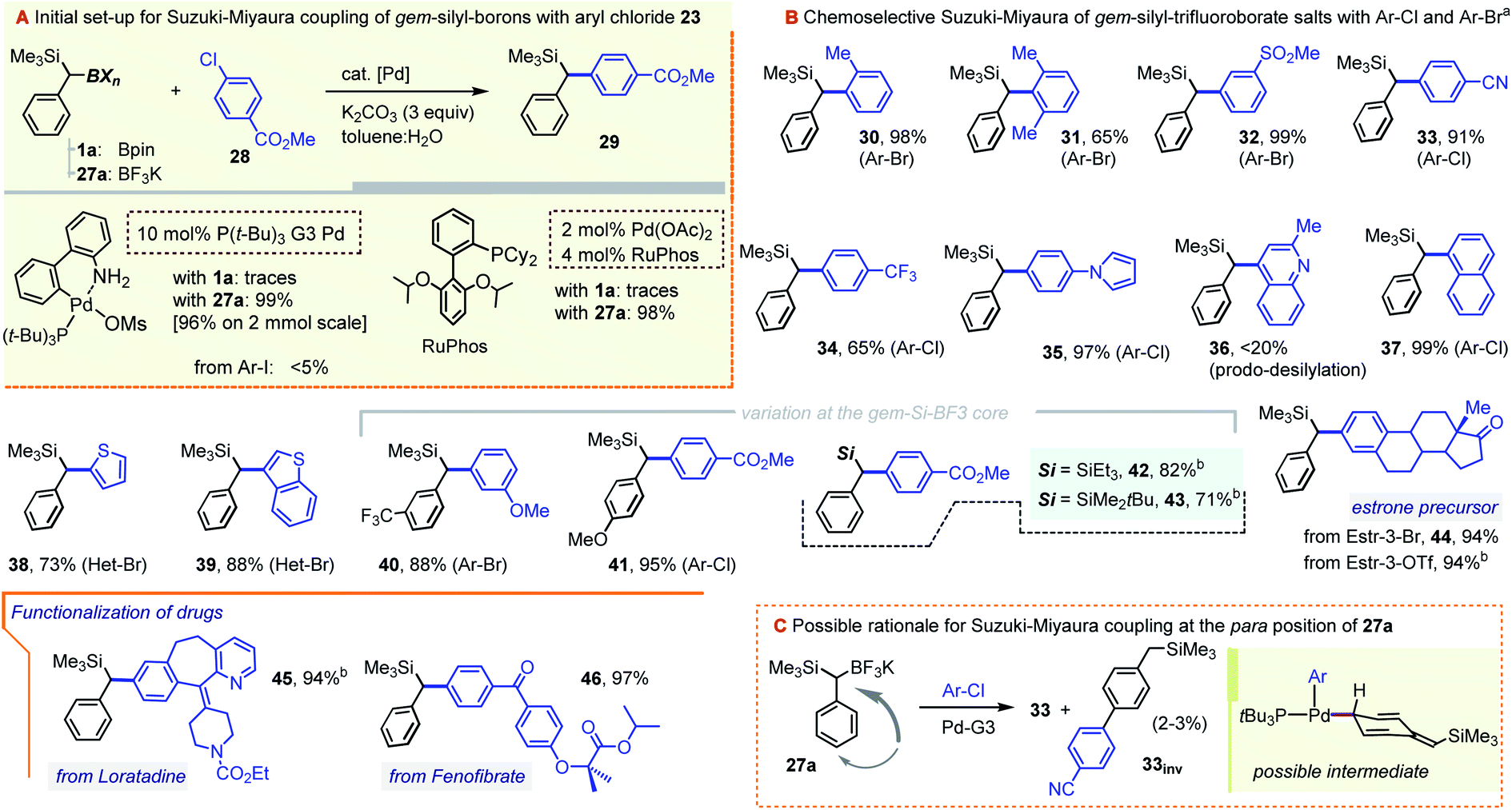 | ||
| Scheme 10 Pd-catalyzed Suzuki–Miyaura coupling between benzyl-gem-silyl-trifluoroborates and chloro- or bromo-arenes. aMost examples were carried out using the tBu3P–Pd G3 catalyst system: aryl halide (1 equiv.), gem-Si-BF3K precursor (2.0 equiv.), 2–10 mol% of Pd-G3 catalyst, and K2CO3 (3.0 equiv.), in a toluene:H2O mixture; see the ESI† for details in each case. bEmploying a system composed of Pd(OAc)2 (2–3%) and RuPhos (4–6%) and using the gem-Si-BF3K precursor (1.1–1.3 equiv.) and K2CO3 (3.0 equiv.). | ||
As a final note, we envision that the retention of the C–Si bond under Pd catalysis opens the door for the introduction of a 3rd substituent at the central carbon. While this task was not extensively pursued, some preliminary results shown below attest to its viability. Specifically, our experience with hypervalent iodine chemistry14a,b led us to seek the introduction of a 3rd substituent via an umpolung transformation. It was found that exposing the silylated diarylmethane 29 to PhI(OAc)2 and BF3·Et2O in the standard CH2Cl2/CH3CN medium led to the acetamide 47 as the main product (93%); in the absence of acetonitrile, the O-acetate 48 was obtained instead (84%, Scheme 11A). Seeking to suppress this C–O/N bond formation, further tests were conducted in CH2Cl2 using iodosobenzene, PhI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, as an “acetate-free” λ3-iodane oxidant. Under such conditions, a reaction between 29 and mesitylene (10 equiv.) afforded the differentially substituted triarylmethane 49 in an 82% yield, presumably via the intermediacy of the corresponding doubly benzylic carbocation VI (Scheme 11B). In a similar manner, a family of π-excessive arenes were also engaged, including anisole, 2-iodothiophene and N-tosyl-5-iodoindole (see triarylmethanes 50–52, Scheme 11B, bottom).
O, as an “acetate-free” λ3-iodane oxidant. Under such conditions, a reaction between 29 and mesitylene (10 equiv.) afforded the differentially substituted triarylmethane 49 in an 82% yield, presumably via the intermediacy of the corresponding doubly benzylic carbocation VI (Scheme 11B). In a similar manner, a family of π-excessive arenes were also engaged, including anisole, 2-iodothiophene and N-tosyl-5-iodoindole (see triarylmethanes 50–52, Scheme 11B, bottom).
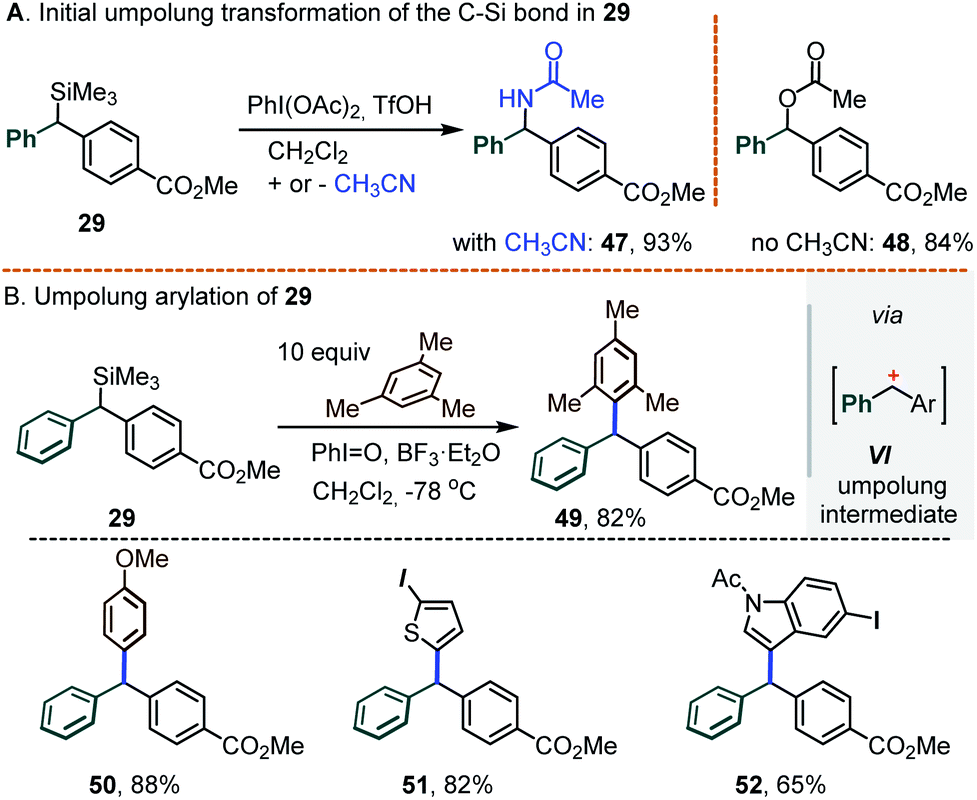 | ||
| Scheme 11 Iodane-promoted umpolung transformations of the gem-diaryl-α-silyl substrate 29. | ||
Conclusions
In summary, this work expands the synthetic landscape of versatile benzylic gem-boron–silicon and gem-boron–tin molecules, going from the generation of these species to their C–M reactivity applications. A new approach has been developed, which complements prior routes and by which gram quantities of benzylic gem-boron–silicon and gem-boron–tin reagents are obtained via the straightforward electrophilic α-borylation of the benzylsilane core. The products were isolated either as –Bpin or novel gem-silylated trifluoroborate salt derivatives. The C–Si or C–Sn sites in these species undergo metal-free oxidative arylation employing λ3-bis(acetoxy)iodoarenes. The “iodane-guided” mechanism leads to selective coupling at the Ar–I para C–H site, with the molecule thus retaining both the C–I and the C–B functional groups. In addition, a protocol was identified for the Pd-catalyzed C–B arylation of hindered gem-SiMe3–BF3K substrates with chloro- and bromo-arenes. We envisage that these advances provide a strong foothold for further method development directed towards the modular iterative construction of multi-substituted carbon centers, a task of great importance in modern synthetic chemistry.Data availability
Details of the experimental procedures, spectroscopic data and the results of the DFT calculations are available in the ESI file.Author contributions
W. W. C., N. P. F., M. D. B., A. C. and L. G. R. contributed to the experimental work; A. B. C. and A. S. contributed to ideation and writing of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by MINECO (CTQ2017-86936-P), MICINN (PID2020-113661GB-I00) and AGAUR (2017 SGR 01051 and 2017 SGR 00294), as well as financial support from URL (2019-URL_Proj-034), IQS-Obra Social La Caixa (2017-URL-Intermac-010) and an entry-level grant from CSIC. IQS is also acknowledged for a doctoral scholarship to W. W. C., and CSIC for the JAE Intro scholarship for L. R. G.Notes and references
- For a specific review on Csp I. Marek and J. F. Normant, Chem. Rev., 1996, 96, 3241–3267 CrossRef CAS PubMed -gem-dimetallated species, see:.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (c) T. T. Talele, J. Med. Chem., 2020, 63, 13291–13315 CrossRef CAS PubMed.
- For the selected recent reviews on advances in triarylmethane synthesis, see: (a) M. Nambo and C. M. Crudden, ACS Catal., 2015, 5, 4734–4742 CrossRef CAS; (b) X. Liu, X. Wu, L. Zhang, X. Lin and D. Huang, Synthesis, 2020, 52, 2311–2329 CrossRef CAS.
- For reviews on the chemistry of gem-diborylalkanes, see: (a) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050–1064 RSC; (b) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306–1327 CrossRef CAS; (c) C. Wu and J. Wang, Tetrahedron Lett., 2018, 59, 2128–2140 CrossRef CAS. For Pd-catalyzed mono-arylation of gem-diboryl species, including enantioselective variants, see (d) H.-Y. Sun, K. Kubota and D. Hall, Chem.–Eur. J., 2015, 21, 19186–19194 CrossRef CAS PubMed; (e) C. Sun, B. Potter and J. Morken, J. Am. Chem. Soc., 2014, 136, 6534–6537 CrossRef CAS PubMed; (f) S. H. Cho and J. F. Hartwig, Chem. Sci., 2014, 5, 694–698 RSC; (g) K. Endo, T. Ohkubo, M. Hirokami and T. Shibata, J. Am. Chem. Soc., 2010, 132, 11033–11035 CrossRef CAS PubMed. For additional selective functionalization of Csp3-gem-diboryls, see: (h) C. E. Iacono, T. C. Stephens, T. S. Rajan and G. Pattison, J. Am. Chem. Soc., 2018, 140, 2036–2040 CrossRef CAS PubMed; (i) S. A. Murray, J. C. Green, S. B. Tailor and S. J. Meek, Angew. Chem., Int. Ed., 2016, 55, 9065–9069 CrossRef CAS PubMed.
- (a) K. N. Babu, F. Massarwe, R. R. Reddy, N. Eghbarieh, M. Jakob and A. Masarwa, Molecules, 2020, 25, 959–989 CrossRef CAS PubMed; (b) A. B. Cuenca, J. Cid, D. García-López, J. J. Carbó and E. Fernández, Org. Biomol. Chem., 2015, 13, 9659–9664 RSC.
- W. Sun, Y. Hu, C. Xia and C. Liu, New J. Chem., 2021 10.1039/d0nj01344g.
- (a) E. La Cascia, A. B. Cuenca and E. Fernández, Chem.–Eur. J., 2016, 22, 18737–18741 CrossRef CAS PubMed. For a recent tutorial review on boron-Wittig olefination with gem-bis(boryl)alkanes, see: (b) A. B. Cuenca and E. Fernández, Chem. Soc. Rev., 2021, 50, 72–86 RSC.
- (a) K. Endo, F. Kurosawa and Y. Ukaji, Chem. Lett., 2013, 42, 1363–1365 CrossRef CAS; (b) J. Kim and S. H. Cho, ACS Catal., 2019, 9, 230–235 CrossRef CAS.
- (a) C. Bomio, M. A. Kabeshov, A. R. Lit, S. H. Lau, J. Ehlert, C. Battilocchio and S. V. Ley, Chem. Sci., 2017, 8, 6071–6075 RSC; (b) C. Wu, Z. Bao, X. Xua and J. Wang, Org. Biomol. Chem., 2019, 17, 5714–5724 RSC. For the original Matteson C–boron homologation: (c) D. J. S. Tsai and D. S. Matteson, Organometallics, 1983, 2, 236–241 CrossRef CAS. For an interesting gem-silylborylation approach, see (d) L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257–5264 CrossRef CAS PubMed.
- While this manuscript was in preparation, the formation of analogous α-boryl carbanions was reported by Lee and Chirik (in a different methodological context) via the mono-deborylation of gem-bis-boryl precursors: B. Lee and P. J. Chirik, J. Am. Chem. Soc., 2020, 142, 2429–2437 CrossRef CAS PubMed.
- For a notable example of a Hiyama-type benzylation, see: K. Itami, M. Mineno, T. Kamei and J. I. Yoshida, Org. Lett., 2002, 4, 3635–3638 CrossRef CAS PubMed.
- (a) C. Mowdawalla, F. Ahmed, T. Li, K. Pham, L. Dave, G. Kim and I. F. D. Hyatt, Beilstein J. Org. Chem., 2018, 14, 1039–1045 CrossRef CAS PubMed; (b) Y. Wu, S. Bouvet, S. Izquierdo and A. Shafir, Angew. Chem., Int. Ed., 2019, 58, 2617–2621 CrossRef CAS PubMed.
- For one of the earliest forays into iodane-guided C–H coupling, see M. Ochiai, T. Ito, Y. Takaoka and Y. Masaki, J. Am. Chem. Soc., 1991, 113, 1319–1323 CrossRef CAS.
- (a) For a recent overview on iodane-guided C–H coupling, including a discussion on the unproductive Umpolung phenomenon, see W. W. Chen, A. B. Cuenca and A. Shafir, Angew. Chem., Int. Ed., 2020, 59, 16294–16309 CrossRef CAS PubMed. For related review articles, see: (b) A. Bauer and N. Maulide, Chem. Sci., 2021, 12, 853–864 RSC; (c) I. F. D. Hyatt, L. Dave, N. David, K. Kaur, M. Medard and C. Mowdawalla, Org. Biomol. Chem., 2019, 17, 7822–7848 RSC.
- For the activation of ArI(OAc)2 reagents by acids such as HOTf and BF3·Et2O, see: S. Izquierdo, S. Essafi, I. del Rosal, P. Vidossich, R. Pleixats, A. Vallribera, G. Ujaque, A. Lledós and A. Shafir, J. Am. Chem. Soc., 2016, 138, 12747–12750 CrossRef CAS PubMed.
- For earlier examples of improved performance of electron-rich λ3-iodanes in related processes, see: (a) M. Ochiai, M. Kida and T. Okuyama, Tetrahedron Lett., 1998, 39, 6207–6210 CrossRef CAS; (b) H. R. Khatri and J. Zhu, Chem.–Eur. J., 2012, 18, 12232–12236 CrossRef CAS PubMed.
- The meta selectivity arises from the blocked aromatization for the initial cationic intermediate which forms immediately after the rearrangement step (species similar to III). In such a case, the benzyl group in III may undergo a [1,2] shift to the ring meta position prior to final aromatization.
- J. Tian, F. Luo, C. Zhang, X. Huang, Y. Zhang, L. Zhang, L. Kong, X. Hu, Z. X. Wang and B. Peng, Angew. Chem., Int. Ed., 2018, 57, 9078–9082 CrossRef CAS PubMed.
- For the closely related precedent on the use of Ag2O, see: (a) D. Imao, B. W. Glasspoole, V. S. Laberge and C. M. Crudden, J. Am. Chem. Soc., 2009, 131, 5024–5025 CrossRef CAS PubMed. For the origin of the acceleration of certain Suzuki–Miyaura coupling by Ag2O, see: (b) J. Uenishi, J. M. Beau, R. W. Armstrong and Y. Kishi, J. Am. Chem. Soc., 1987, 109, 4756–4758 CrossRef CAS.
- G. A. Molander, C. S. Yun, M. Ribagorda and B. Biolatto, J. Org. Chem., 2003, 68, 5534–5539 CrossRef CAS PubMed.
- S. D. Dreher, S.-E. Lim, D. L. Sandrock and G. A. Molander, J. Org. Chem., 2009, 74, 3626–3631 CrossRef CAS PubMed.
- (a) L. Li, S. Zhao, A. Joshi-Pangu, M. Diane and M. R. Biscoe, J. Am. Chem. Soc., 2014, 136, 14027–14030 CrossRef CAS PubMed; (b) N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- S. P. Davies, C. J. Mycroft-West, I. Pagani, H. J. Hill, Y.-H. Chen, R. Karlsson, I. Bagdonaite, S. E. Guimond, Z. Stamataki, M. Andrade De Lima, J. E. Turnbull, Z. Yang, E. Vicenzi, M. A. Skidmore, F. Khanim and A. Richardson, bioRxiv, 2021 DOI:10.1101/2021.01.10.426114.
- For a recent example of a related para C–H coupling in a benzylic system, see: (a) F. de Azambuja, M.-H. Yang, T. Feoktistova, M. Selvaraju, A. C. Brueckner, M. A. Grove, S. Koley, P. H.-Y. Cheong and R. A. Altman, Nat. Chem., 2020, 12, 489–496 CrossRef CAS PubMed. For a review on the reactivity of Pd-benzyl intermediates, see: (b) S. Zhang, Y. Yamamoto and M. Bao, Adv. Synth. Catal., 2021, 363, 587–601 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01741a |
| This journal is © The Royal Society of Chemistry 2021 |