DOI:
10.1039/D1SC01692J
(Edge Article)
Chem. Sci., 2021,
12, 7930-7936
Probe metal binding mode of imine covalent organic frameworks: cycloiridation for (photo)catalytic hydrogen evolution from formate†
Received
1st April 2021
, Accepted 17th April 2021
First published on 11th May 2021
Abstract
Metalation of covalent organic frameworks (COFs) is a critical strategy to functionalize COFs for advanced applications yet largely relies on the pre-installed specific metal docking sites in the network, such as porphyrin, salen, 2,2′-bipyridine, etc. We show in this study that the imine linkage of simple imine-based COFs, one of the most popular COFs, readily chelate transition metal (Ir in this work) via cyclometalation, which has not been explored before. The iridacycle decorated COF exhibited more than 10-fold efficiency enhancement in (photo)catalytic hydrogen evolution from aqueous formate solution than its molecular counterpart under mild conditions. This work will inspire more functional cyclometallated COFs to be explored beyond catalysis considering the large imine COF library and the rich metallacycle chemistry.
Introduction
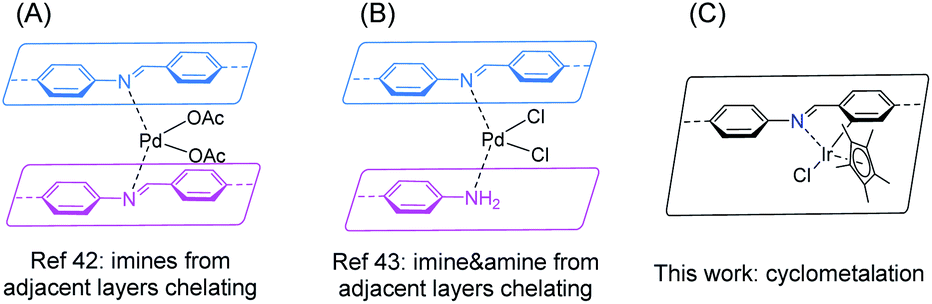
The designable ordered porous structure of covalent organic frameworks (COFs) has attracted broad research interest in the past decades.1–4 Although COFs are completely composed of light elements (typically H, B, C, N, O, Si), their advanced applications, particularly catalysis,5–8 have stressed the role of metal species anchored onto the network.8–10 To accommodate the target metal, a building unit with a suitable metal binding site is required to be incorporated into the framework, which usually meets complicated organic synthesis. Nevertheless, plenty of functional linkers have been successfully implemented into COFs, such as 2,2′-bipyridine,11–14 phenanthroline,15–17 porphyrin,18–22 phthalocyanine,23–25 salen,26–29 hydroxy groups,30–32 β-ketoenamine,33 phosphine,34,35 dehydrobenzoannulene,36,37 among others.9,10 As one of the most popular COFs featuring both high crystallinity and stability, Schiff base COFs possess uniformly distributed imine linkages,38 which have been well studied in coordination chemistry.39–41 If the imine linkages can be effectively exploited for binding metals, a huge COF catalyst library would be accessible considering the large number of imine COFs. While the first COF-based heterogeneous catalyst was constructed on an imine COF, in which palladium(II) was chelated by two imine moieties from adjacent COF layers in 2011 (Fig. 1A),42 not much progress has been achieved on using imines in COFs as intrinsic ligands for transition metals during the past decade. Only recently a new binding mode of stabilizing palladium(II) with imine and amine defect from adjacent layers of an imine COF was identified (Fig. 1B).43 Thus, it is of great interest to explore both the scope of metal species and binding modes of imine-based COFs.
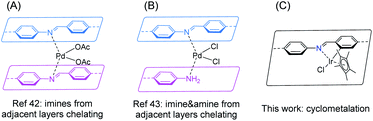 |
| | Fig. 1 Metal binding modes of imine-based COFs. | |
(Photo)catalytic hydrogen evolution reaction (HER) from water splitting is a promising clean energy production technique. COFs have been proved to be potential photocatalysts in promoting HER.44–49 The system is typically comprised of COF photocatalyst, Pt nanoparticle cocatalyst, and a sacrificial electron donor (e.g. ascorbic acid, triethanolamine) in an aqueous solution. Formic acid serves as an alternative high H2 storage density reservoir (4.3 wt%).50,51 The decomposition of HCOOH under basic conditions could release H2 with high purity. However, some iridium-based homogeneous HCOOH dehydrogenation catalysts displayed low stability and were prone to deactivate via nanoparticle formation.52,53 Immobilization of the catalyst on proper solid support is expected to extend the catalyst lifetime.54 Taking all of these considerations together, in this work, we show that cyclometalation at the imine site leads to an iridacycle functionalized imine COF for the first time (Fig. 1C), which exhibits fascinating performance in (photo)catalytic HER from aqueous formate solution.
Results and discussion
Synthesis and characterization
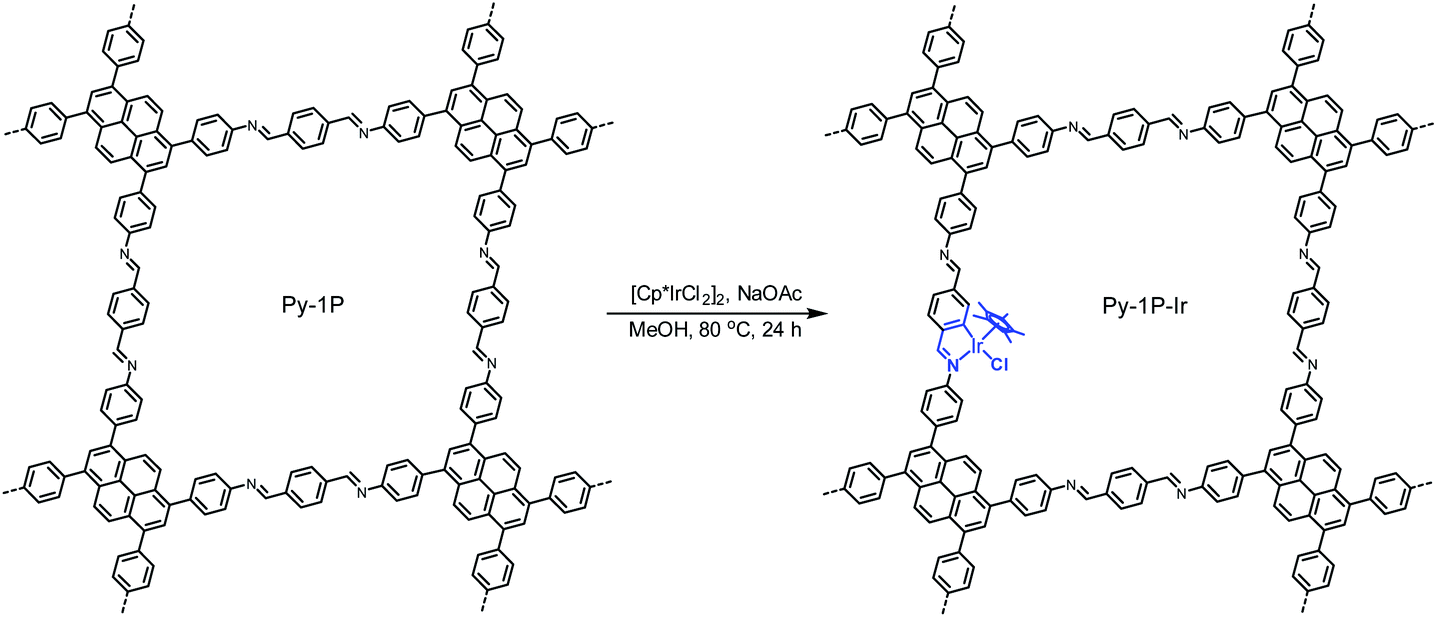
Metallacycles of Schiff base ligands have been well explored for transition metals in organometallic chemistry.55–57 Nevertheless, introducing such type of complexes into COFs is unprecedented. To explore this new binding mode, a pyrene-based imine COF (Py-1P) constructed from 1,3,6,8-tetrakis(4-aminophenyl)pyrene and terephthalaldehyde was chosen as a model system because of its high crystallinity58 and facile synthesis.59 Iridium became the metal choice because of the various catalytic applications of iridacycles.60,61 Metalation of Py-1P COF was carried out by refluxing with [Cp*IrCl2]2 (Cp*, pentamethylcyclopentadienyl) in methanol in the presence of NaOAc for 24 hours (Scheme 1, see ESI† for details).
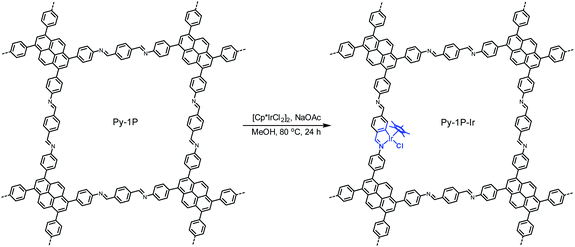 |
| | Scheme 1 Cyclometalation of Py-1P COF with iridium. | |
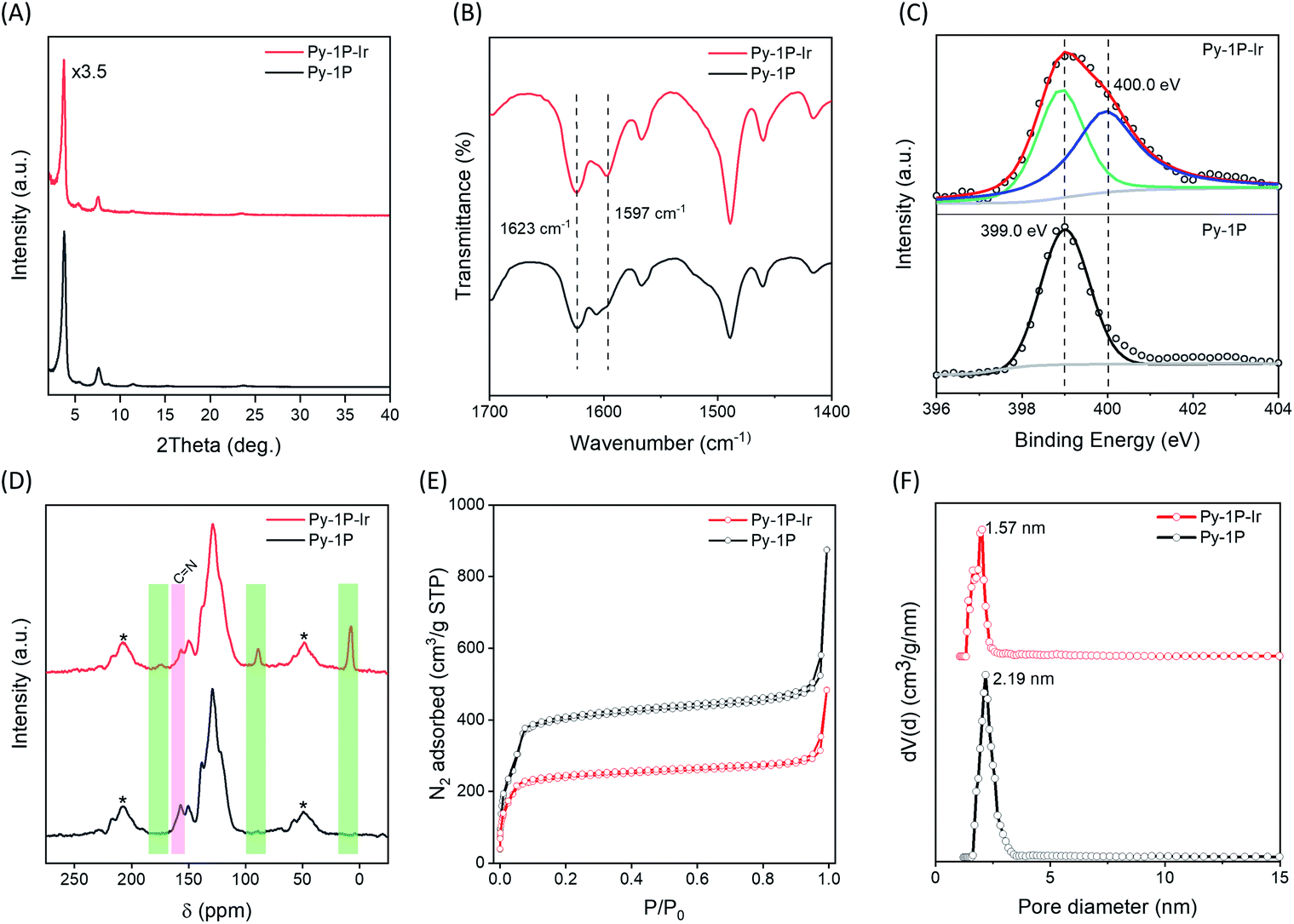
The successful formation of the iridacycle functionalized COF Py-1P–Ir was confirmed by FT-IR spectroscopy, X-ray photoelectron spectroscopy (XPS), and solid-state NMR characterization, and the retention of the framework crystallinity and porosity was examined by powder X-ray diffraction (PXRD) and N2 adsorption–desorption isotherm analysis, respectively. Py-1P–Ir COF showed strong diffraction peaks at 2θ = 3.74, 5.36, 7.56, 11.40, 23.52°, which are slightly shifted to low 2θ direction (0.04 to 0.18° difference) compared to the diffraction pattern of the parent Py-1P COF (Fig. 2A). This might result from the crystal cell expansion after introducing the bulky Ir organometallic unit. FT-IR analysis revealed a new C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration band at 1597 cm−1, corresponding to the metal coordinated imines, in addition to the free ones at 1623 cm−1 for Py-1P–Ir COF and (Fig. 2B). The shifting of CN vibration to lower wavenumber upon coordination to Ir was also observed for N-benzylideneaniline (L1) when forming the model iridacycle complex (L1–Ir, 1626 to 1582 cm−1, Fig. S1†).62 The coordination of imine N to iridium was further proved by XPS analysis (Fig. S2† and 2C). The N 1s XPS spectrum of Py-1P–Ir COF showed two subpeaks with binding energies of 399.0 and 400.0 eV, which were assigned to the free and coordinated imine nitrogen, respectively (Fig. 2C). The increase of N 1s binding energy upon coordination is consistent with that of the model complex L1–Ir (399.2 to 399.8 eV, Fig. S3†). Solid-state cross-polarization/magic angle spinning (CP/MAS) 13C NMR spectroscopy unambiguously confirmed the formation of the iridacycle in Py-1P–Ir COF. Three new signals at 173.7, 89.0, and 7.6 ppm appeared in the CP/MAS 13C NMR spectrum of Py-1P–Ir COF (Fig. 2D). The broad peak at 173.7 ppm is assigned to the iridium bonded carbon and the imine carbon of the iridacycle, while the peaks at 89.0 and 7.6 ppm originate from the aromatic and methyl carbons of Cp* ring, respectively, which matches well with the 13C NMR spectrum of the model complex L1–Ir (Fig. S4†). Meanwhile, the relative intensity of the free imine carbon peak at 157.0 ppm of Py-1P–Ir COF decreased accordingly compared to that of Py-1P COF. N2 adsorption experiment was carried out to investigate the porosity of Py-1P–Ir COF (Fig. 2E). The Brunauer–Emmett–Teller (BET) surface area of Py-1P–Ir COF is 972 m2 g−1, which is lower than 1960 m2 g−1 of Py-1P COF (Fig. S5 and S6†). The pore size is decreased to 1.57 nm for Py-1P–Ir COF, compared to 2.19 nm for Py-1P COF, which is expected for the pore wall metalation (Fig. 2F). The iridium loading was determined to be 11.8 wt% by inductively coupled plasma mass spectrometry (ICP-MS), corresponding to ca. 20% imine metalation. Scanning electron microscope analysis revealed that Py-1P–Ir COF was composed of aggregates of nanometer-sized particles, similar to that of Py-1P COF (Fig. S7†). Energy dispersive X-ray (EDX) analysis revealed the homogeneous distributions of Ir and Cl elements over the framework, and the Ir/Cl ratio is close to 1/1, as expected for the proposed structure (Fig. S8†). Besides, the formation of the iridacycle was also successfully performed on an azine linked COF (see ESI† for details), demonstrating the generality of the cyclometalation modification of imine-based COFs.
N vibration band at 1597 cm−1, corresponding to the metal coordinated imines, in addition to the free ones at 1623 cm−1 for Py-1P–Ir COF and (Fig. 2B). The shifting of CN vibration to lower wavenumber upon coordination to Ir was also observed for N-benzylideneaniline (L1) when forming the model iridacycle complex (L1–Ir, 1626 to 1582 cm−1, Fig. S1†).62 The coordination of imine N to iridium was further proved by XPS analysis (Fig. S2† and 2C). The N 1s XPS spectrum of Py-1P–Ir COF showed two subpeaks with binding energies of 399.0 and 400.0 eV, which were assigned to the free and coordinated imine nitrogen, respectively (Fig. 2C). The increase of N 1s binding energy upon coordination is consistent with that of the model complex L1–Ir (399.2 to 399.8 eV, Fig. S3†). Solid-state cross-polarization/magic angle spinning (CP/MAS) 13C NMR spectroscopy unambiguously confirmed the formation of the iridacycle in Py-1P–Ir COF. Three new signals at 173.7, 89.0, and 7.6 ppm appeared in the CP/MAS 13C NMR spectrum of Py-1P–Ir COF (Fig. 2D). The broad peak at 173.7 ppm is assigned to the iridium bonded carbon and the imine carbon of the iridacycle, while the peaks at 89.0 and 7.6 ppm originate from the aromatic and methyl carbons of Cp* ring, respectively, which matches well with the 13C NMR spectrum of the model complex L1–Ir (Fig. S4†). Meanwhile, the relative intensity of the free imine carbon peak at 157.0 ppm of Py-1P–Ir COF decreased accordingly compared to that of Py-1P COF. N2 adsorption experiment was carried out to investigate the porosity of Py-1P–Ir COF (Fig. 2E). The Brunauer–Emmett–Teller (BET) surface area of Py-1P–Ir COF is 972 m2 g−1, which is lower than 1960 m2 g−1 of Py-1P COF (Fig. S5 and S6†). The pore size is decreased to 1.57 nm for Py-1P–Ir COF, compared to 2.19 nm for Py-1P COF, which is expected for the pore wall metalation (Fig. 2F). The iridium loading was determined to be 11.8 wt% by inductively coupled plasma mass spectrometry (ICP-MS), corresponding to ca. 20% imine metalation. Scanning electron microscope analysis revealed that Py-1P–Ir COF was composed of aggregates of nanometer-sized particles, similar to that of Py-1P COF (Fig. S7†). Energy dispersive X-ray (EDX) analysis revealed the homogeneous distributions of Ir and Cl elements over the framework, and the Ir/Cl ratio is close to 1/1, as expected for the proposed structure (Fig. S8†). Besides, the formation of the iridacycle was also successfully performed on an azine linked COF (see ESI† for details), demonstrating the generality of the cyclometalation modification of imine-based COFs.
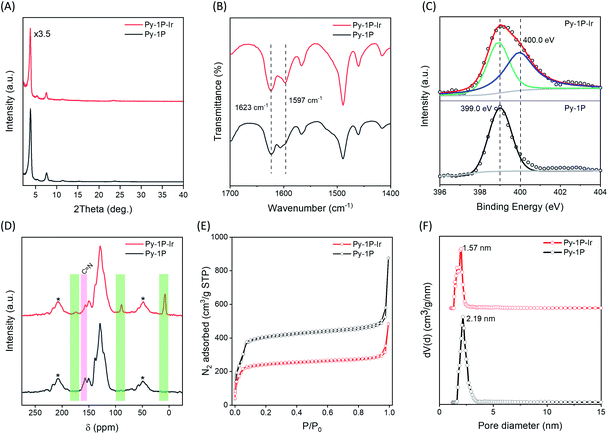 |
| | Fig. 2 Comparison of the (A) PXRD patterns, (B) FT-IR spectra, (C) N 1s XPS spectra, (D) CP/MAS 13C NMR spectra, (E) N2 adsorption–desorption isotherms, and (F) pore size distributions of Py-1P and Py-1P–Ir COFs. Pore size distribution was analysed using the nonlocal density functional theory equilibrium model. The NMR spinning sidebands in (D) are marked with asterisks (*). | |
Catalytic HER from formate
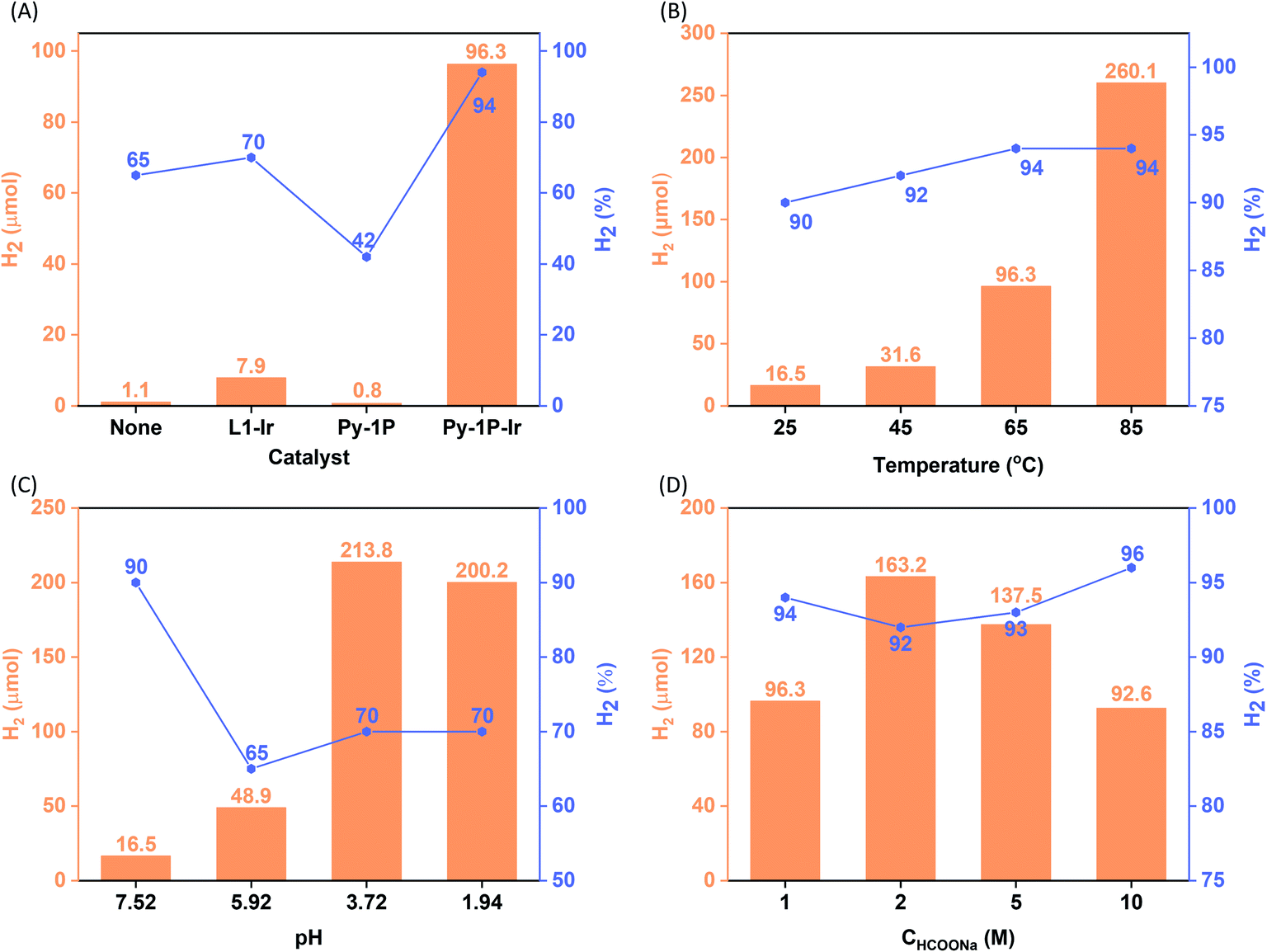
Iridium complexes have demonstrated their potential to be homogeneous catalysts for hydrogen production from formate.50–53,63 The homogeneous distribution of the single-site iridacycle over the Py-1P–Ir COF with high surface area prompted us to test its capability in promoting formate decomposition to release H2. To our delight, Py-1P–Ir COF produced 96.3 μmol H2 with a purity of 94% from 10 mL of 1.0 M HCOONa solution at 65 °C in 6 hours (6.35 μmol catalyst based on Ir, Fig. 3A). In contrast, L1–Ir gave only 7.9 μmol H2 with 70% purity under otherwise identical conditions. Py-1P COF was catalytically inactive, showing even lower H2 production (0.8 μmol, 42%) than the control reaction (2.3 μmol, 65%). These results highlight the importance of the unique structure of Py-1P–Ir COF. Then, we set to investigate the influence of reaction parameters on the hydrogen production efficiency catalyzed by Py-1P–Ir COF, including temperature, pH, and formate concentration.
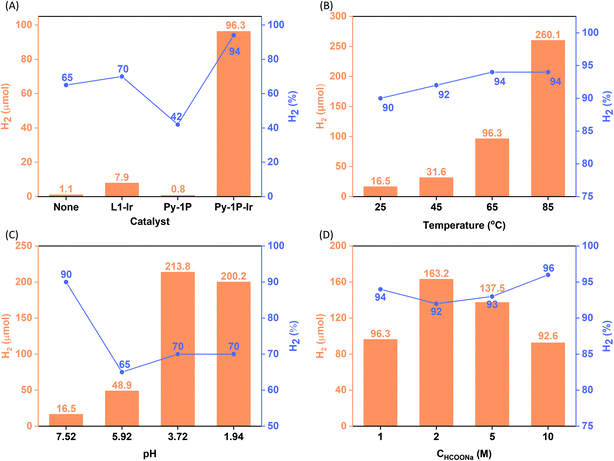 |
| | Fig. 3 Catalytic hydrogen evolution from aqueous formate solution. The influence of catalyst (A), temperature (B), pH (C), and formate concentration (D) on the reaction outcome (H2 quantity and purity are presented in orange column and grey line/square symbol form, respectively. Purity refers to the H2 composition in the generated H2/CO2 mixture). Standard reaction condition: the reactions were carried out by heating a 10 mL HCOONa solution (1.0 M, pH = 7.52) containing 6.35 μmol catalyst (based on Ir) at 65 °C for 6 h. The pH effect was studied at room temperature. The concentration effect was studied at 65 °C and the pHs of HCOONa solutions were not adjusted. See ESI† for details. | |
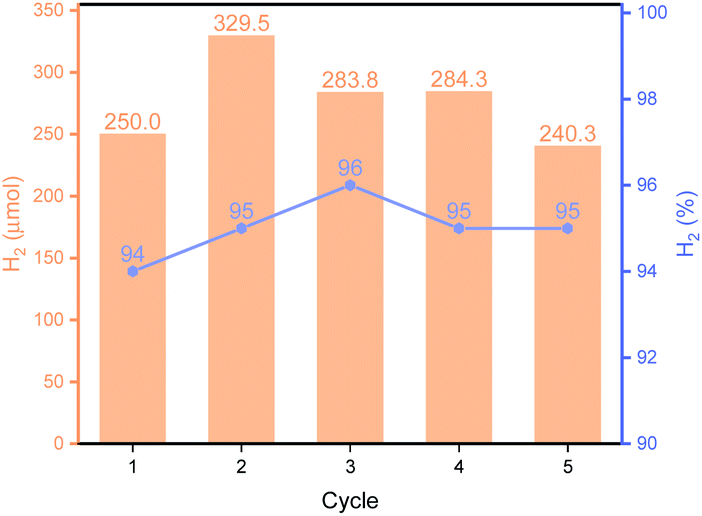
As shown in Fig. 3B, Py-1P–Ir COF displayed higher efficiency at higher temperature. An amount of 250.0 μmol H2 was obtained at 85 °C, which is ca. 2.6 times of that obtained at 65 °C. The high reactivity of Py-1P–Ir COF was manifested that 16.5 μmol H2 was formed even at 25 °C. In all the tested temperatures, the H2 purity was no less than 90%. Lowering the pH was found to favor the reaction that 48.9 and 213.8 μmol H2 were afforded at pH = 5.92, 3.72 respectively, while further lowering the pH to 1.94 did not lead to higher H2 production (200.2 μmol, Fig. 3C). The increased H2 production came with compromised H2 purity to ca. 70% under acidic conditions. The concentration of HCOONa on the HER showed a volcano-type effect (Fig. 3D). The H2 yield increased to 163.2 μmol from the reaction of 2 M HCOONa but started to fall back to 137.5 μmol for 5 M HCOONa and 92.6 μmol for 5 M HCOONa respectively thereafter. The H2 purities were all higher than 90% from the tested four HCOONa concentrations. The decreased reaction efficiency might be due to the decreased concentration of water, which is the proton source for H2 production. Py-1P–Ir COF exhibited excellent stability under all the tested conditions. All the recovered COF samples preserved the crystallinity as shown by PXRD analysis (Fig. S20–S22†). Interestingly, FT-IR and XPS indicate that the imine linkage was partially reduced (Fig. S20–S23†). The imine bond reduction is likely mediated by an iridium hydride intermediate via an outer-sphere process.64–66 The involvement of Ir in the imine reduction is supported by the fact that no imine reduction was observed in Py-1P COF during HER catalysis (Fig. S24†) and the catalytic reduction of N-benzylideneaniline as an exogenous substrate in the presence of Py-1P–Ir COF was observed (Fig. S25†). ICP-MS showed that the Ir concentrations in the reaction filtrates were all below 60 ppb (Table S2†), demonstrating the heterogeneous catalysis nature of the reaction. The high stability of Py-1P–Ir COF allowed it to be recycled for at least another four runs (Fig. 4). Interestingly, an initial performance improvement was observed in the second cycle. This improved reactivity of Py-1P–Ir COF is attributed to the presence of more reactive Ir species in recovered material, in which the Cl ligand has been replaced (see mechanism discussion below). This is supported by the fact that no Cl element was detected by EDX in Py-1P–Ir COF after one HER cycle (Fig. S26†). The average H2 production rate during the five cycles was calculated to be 4626 μmol g−1 h−1, corresponding to a TOF of 7.3 h−1. The recovered Py-1P–Ir COF after five cycles was still crystalline (Fig. S28†). No metal nanoparticle formation was observed by XPS (Fig. S29†) and TEM (Fig. S30†) analysis.
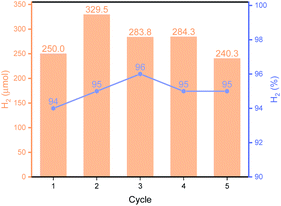 |
| | Fig. 4 Recyclability of Py-1P–Ir COF. The reactions were carried out in 1 M HCOONa at 85 °C for 6 hours. | |
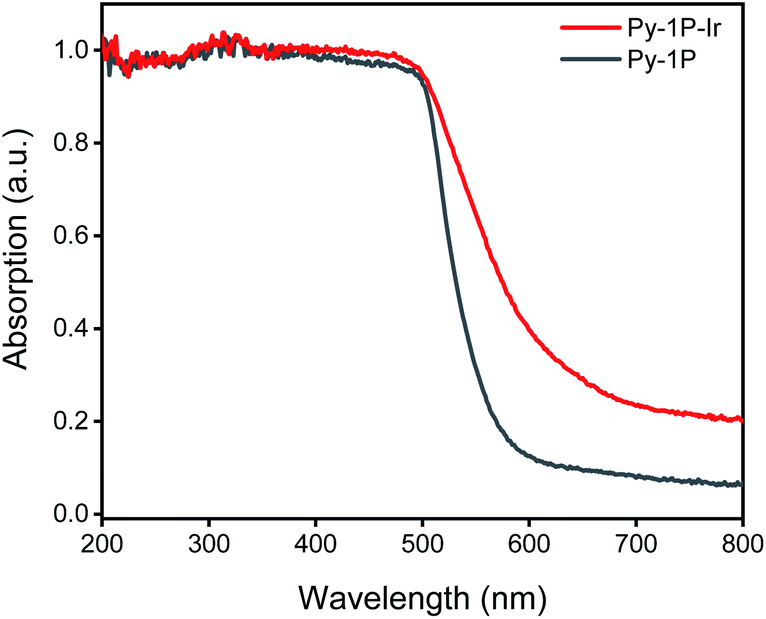
Pyrene-based COFs have exhibited excellent photophysical properties which benefit photocatalysis.67–70 Diffuse reflectance UV-vis absorption spectrum of Py-1P–Ir COF displayed prominent absorption in 200–500 nm and a long tail to near-IR range (500–800 nm, Fig. 5). The incorporation of iridium complex into Py-1P COF did not change the absorption in 200–500 nm band but strongly improved the absorption ability in the 500–800 nm region. Encouraged by the excellent light absorption ability across the whole UV-visible region of Py-1P–Ir COF, we further tested its photocatalytic HER performance from formate decomposition. The reactions were carried out in 1 M formate solution in the presence of 6.35 μmol catalyst (based on Ir) under 460 nm light irradiation. As shown in Table 1, only 0.7 μmol H2 was detected in the absence of catalyst (entry 1). Py-1P–Ir COF catalyzed the generation of 84.5 μmol H2 in high purity (93%) in 6 hours (entry 3), which is ca. 34 times higher than the molecular counterpart L1–Ir did (entry 2). The corresponding H2 generation rate of 1358 μmol g−1 h−1 (TOF 2.2 h−1) is comparable to the typical COF photocatalyst/Pt cocatalyst/sacrificial electron donor system.71Py-1P COF failed to exert any catalytic effect showing the critical role of iridium for the reaction (entry 5, 0.6 μmol H2). Under the dark condition, Py-1P–Ir COF produced 31.6 μmol H2 in 92% purity, proving it to be a photocatalytic process (entry 4). A physical mixture of Py-1P COF and L1–Ir displayed lower H2 production (19.5 μmol) and poorer selectivity (71% purity), highlighting the importance of covalent hybridization of Ir catalytic centers within the COF (entry 6).72 The recovered Py-1P–Ir COF almost retained its crystalline structure as evidenced by PXRD analysis (Fig. S31†). Similarly, reduction of the imine bond was observed (Fig. S31†). ICP-MS analysis of the Py-1P–Ir COF catalyzed reaction solution revealed a low Ir concentration of 0.99 ppm, corresponding to 0.8% of the total Ir in Py-1P–Ir COF. Besides, the reaction filtrate only produced 2.0 μmol H2 under the photocatalysis conditions (entry 7), demonstrating the heterogeneous catalytic process by Py-1P–Ir COF.
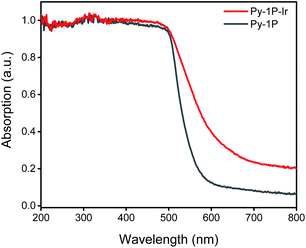 |
| | Fig. 5 Diffuse reflectance UV-vis spectra of Py-1P and Py-1P–Ir COF. | |
Table 1 Photocatalytic HER from aqueous formate solutiona
| Entry |
Catalyst |
H2 (μmol) |
Purity (%) |
|
Reaction conditions: 10 mL formate solution (1.0 M) containing 6.35 μmol catalyst was irradiated with 460 nm LED light for 6 h.
Without light at 45 °C (the photothermal effect raised the reaction temperature to ca. 42 °C).
Physical mixture of Py-1P COF and L1–Ir with the same Ir and total mass loading as Py-1P–Ir COF.
Reaction filtrate of entry 3.
|
| 1 |
— |
0.7 |
10 |
| 2 |
L1–Ir
|
2.5 |
82 |
| 3 |
Py-1P–Ir
|
84.5 |
93 |
| 4b |
Py-1P–Ir
|
31.6 |
92 |
| 5 |
Py-1P
|
0.6 |
68 |
| 6c |
Py-1P/L1–Ir |
19.5 |
71 |
| 7d |
— |
2.0 |
58 |
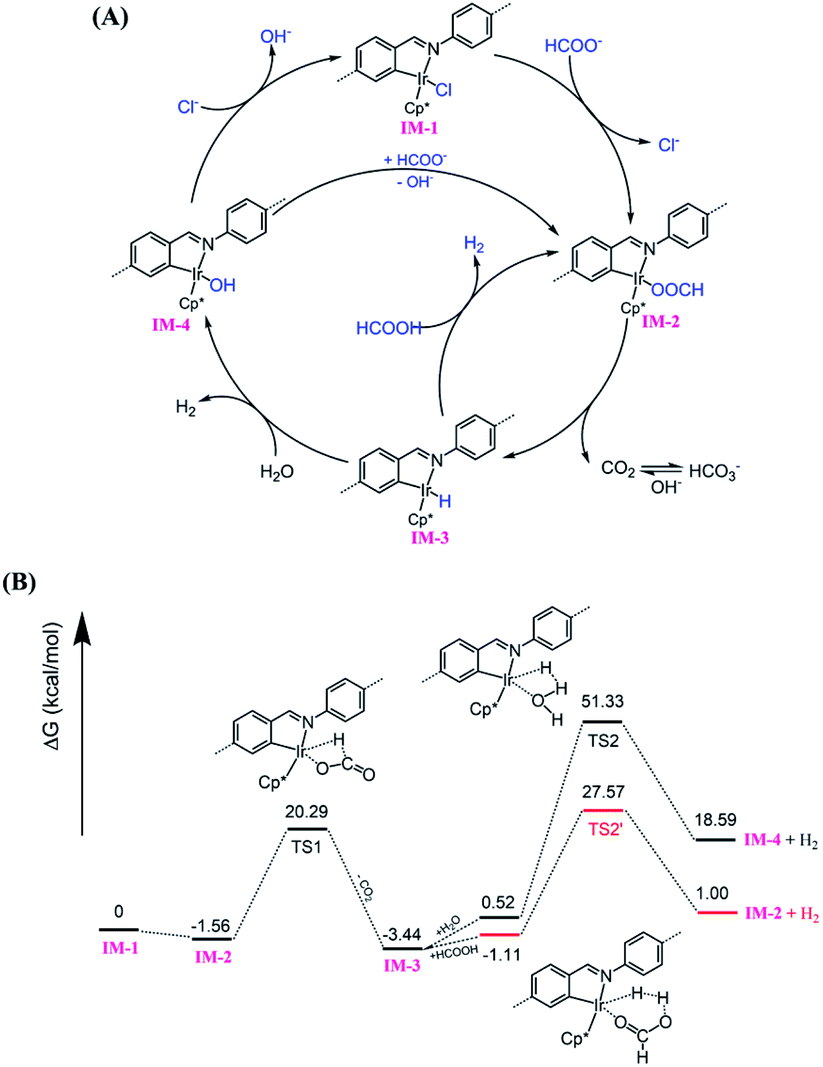
Based on literature reports,52,53 a proposed mechanism is shown in Fig. 6A. The catalytic cycle is initiated by Cl ligand replacement of IM-1 with formate to give HCOO− coordinated intermediate IM-2. CO2 extrusion from IM-2 affords the key iridium hydride species IM-3. Then two pathways account for hydrogen production depending on the reaction conditions (pH). Under neutral or basic conditions, IM-3 reacts with one molecular H2O to generate H2 and OH− bonded Ir intermediate IM-4, which could be reconverted to IM-1 or IM-2via ligand exchange to close the catalytic cycle. While under acidic conditions, IM-3 reacts with HCOOH to release H2 and regenerate IM-2. The involvement of iridium hydride intermediate was supported by NMR studies on L1–Ir in solution. Mixing L1–Ir and excess HCOONa in CD3CN/D2O (v/v, 4/1) generated an iridium hydride complex showing a hydride signal at −12.45 ppm in the 1H NMR spectrum (Fig. S32†).52,73 To further support the proposed mechanism, we carried out DFT calculations. The computed Gibbs free energy profile of the prosed reaction pathway is shown in Fig. 6B. IM-2 is slightly lower (−1.56 kcal mol−1) in energy than IM-1, suggesting the first step ligand exchange is feasible. The transformation of IM-2 to IM-3via CO2 extrusion is thermodynamically favored (ΔG = 1.88 kcal mol−1) with a small energy barrier of 21.85 kcal mol−1 (TS1). This is consistent with our experimental findings that the iridium hydride formed immediately at room temperature. The reaction of IM-3 with H2O to give IM-4 and H2 is an energy demanding process (ΔG = 18.07 kcal mol−1) with an energy barrier of 50.81 kcal mol−1 (TS2). In contrast, the reaction of IM-3 with HCOOH to give IM-2 and H2 is energetically less demanding (ΔG = 2.11 kcal mol−1) and the activation energy is also significantly reduced to 28.68 kcal mol−1 (TS2′). These results match well with the observed higher reactivity of the catalyst in lower pH conditions.
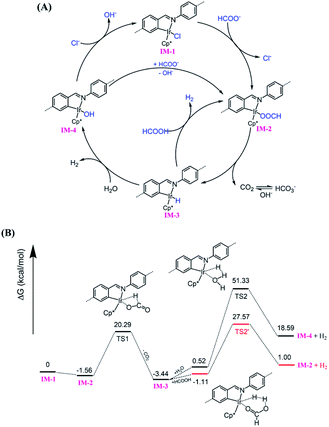 |
| | Fig. 6 (A) Proposed mechanism for Py-1P–Ir COF catalyzed HER from formate; (B) calculated Gibbs free energy profile. | |
Conclusions
In summary, we identified a new metal binding mode of imine COFs where cyclometalation with iridium readily occurred. The iridium functionalized COF exhibited much enhanced catalytic efficiency in HER from an aqueous formate solution than the parent COF and the molecular counterpart in both thermal- and photo-triggered reactions. The easily available imine-based COFs, as well as various transition metals capable of forming metallacycles will largely advance surface organometallic chemistry in COFs beyond catalysis.
Author contributions
J. Hu and H. Beyzavi conceived the work. J. Hu performed all experimental studies and data analysis. H. Mehrabi and R. H. Coridan performed GC analysis. Y.-S. Meng performed TGA and N2-sorption tests. M. Taylor was involved in linkers synthesis. J.-H. Zhan performed the DFT calculations. Q. Yan and M. Benamara performed XPS and TEM studies. J. Hu and H. Beyzavi wrote the manuscript with valuable inputs from all the authors. All authors reviewed and agreed with the content of the paper. H. Beyzavi led the project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
H. B. gratefully acknowledges the financial support through the startup funds from the University of Arkansas. J.-H. Zhan is grateful for the financial support from the National Natural Science Foundation of China (No. 21875255). Work by R. H. C was supported by the U.S. Department of Energy (D.O.E.), Office of Science, Office of Basic Energy Sciences under Award Number DE-SC-0020301. We thank Dr Barry J. Shaulis (University of Arkansas, Department of Geosciences) for the ICP tests.
Notes and references
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- H. Vardhan, A. Nafady, A. M. Al-Enizi and S. Ma, Nanoscale, 2019, 11, 21679–21708 RSC.
- K. Geng, T. He, R. Liu, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- J. Hu, S. K. Gupta, J. Ozdemir and M. H. Beyzavi, ACS Appl. Nano Mater., 2020, 3, 6239–6269 CrossRef CAS.
- W. Tu, Y. Xu, S. Yin and R. Xu, Adv. Mater., 2018, 30, 1707582 CrossRef PubMed.
- J. Liu, N. Wang and L. Ma, Chem.–Asian J., 2020, 15, 338–351 CrossRef CAS PubMed.
- R. K. Sharma, P. Yadav, M. Yadav, R. Gupta, P. Rana, A. Srivastava, R. Zbořil, R. S. Varma, M. Antonietti and M. B. Gawande, Mater. Horiz., 2020, 7, 411–454 RSC.
- J. Guo and D. Jiang, ACS Cent. Sci., 2020, 6, 869–879 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. M. Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- Q. Sun, B. Aguila, J. Perman, N. Nguyen and S. Ma, J. Am. Chem. Soc., 2016, 138, 15790–15796 CrossRef CAS PubMed.
- W. Leng, Y. Peng, J. Zhang, H. Lu, X. Feng, R. Ge, B. Dong, B. Wang, X. Hu and Y. Gao, Chem.–Eur. J., 2016, 22, 9087–9091 CrossRef CAS PubMed.
- H. B. Aiyappa, J. Thote, D. B. Shinde, R. Banerjee and S. Kurungot, Chem. Mater., 2016, 28, 4375–4379 CrossRef CAS.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed.
- Y. Liu, Y. Ma, J. Yang, C. S. Diercks, N. Tamura, F. Jin and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 16015–16019 CrossRef CAS PubMed.
- H.-S. Xu, Y. Luo, X. Li, P. Z. See, Z. Chen, T. Ma, L. Liang, K. Leng, I. Abdelwahab, L. Wang, R. Li, X. Shi, Y. Zhou, X. F. Lu, X. Zhao, C. Liu, J. Sun and K. P. Loh, Nat. Commun., 2020, 11, 1434 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS PubMed.
- H. Wang, H. Ding, X. Meng and C. Wang, Chin. Chem. Lett., 2016, 27, 1376–1382 CrossRef CAS.
- G. Lin, H. Ding, R. Chen, Z. Peng, B. Wang and C. Wang, J. Am. Chem. Soc., 2017, 139, 8705–8709 CrossRef CAS PubMed.
- Y. Meng, Y. Luo, J. L. Shi, H. Ding, X. Lang, W. Chen, A. Zheng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 3624–3629 CrossRef CAS PubMed.
- X. Ding, J. Guo, X. Feng, Y. Honsho, J. Guo, S. Seki, P. Maitarad, A. Saeki, S. Nagase and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 1289–1293 CrossRef CAS PubMed.
- S. Jin, X. Ding, X. Feng, M. Supur, K. Furukawa, S. Takahashi, M. Addicoat, M. E. El-Khouly, T. Nakamura and S. Irle, Angew. Chem., Int. Ed., 2013, 52, 2017–2021 CrossRef CAS PubMed.
- X. Feng, X. Ding, L. Chen, Y. Wu, L. Liu, M. Addicoat, S. Irle, Y. Dong and D. Jiang, Sci. Rep., 2016, 6, 32944 CrossRef CAS PubMed.
- L.-H. Li, X.-L. Feng, X.-H. Cui, Y.-X. Ma, S.-Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed.
- X. Han, Q. Xia, J. Huang, Y. Liu, C. Tan and Y. Cui, J. Am. Chem. Soc., 2017, 139, 8693–8697 CrossRef CAS PubMed.
- G. Yuan, H. Jiang, L. Zhang, Y. Liu and Y. Cui, Coord. Chem. Rev., 2019, 378, 483–499 CrossRef CAS.
- S. Yan, X. Guan, H. Li, D. Li, M. Xue, Y. Yan, V. Valtchev, S. Qiu and Q. Fang, J. Am. Chem. Soc., 2019, 141, 2920–2924 CrossRef CAS PubMed.
- X. Chen, N. Huang, J. Gao, H. Xu, F. Xu and D. Jiang, Chem. Commun., 2014, 50, 6161–6163 RSC.
- W. Zhang, P. Jiang, Y. Wang, J. Zhang, Y. Gao and P. Zhang, RSC Adv., 2014, 4, 51544–51547 RSC.
- H. Vardhan, G. Verma, S. Ramani, A. Nafady, A. M. Al-Enizi, Y. Pan, Z. Yang, H. Yang and S. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 3070–3079 CrossRef CAS PubMed.
- X. Han, J. Zhang, J. Huang, X. Wu, D. Yuan, Y. Liu and Y. Cui, Nat. Commun., 2018, 9, 1294 CrossRef PubMed.
- R. Tao, X. Shen, Y. Hu, K. Kang, Y. Zheng, S. Luo, S. Yang, W. Li, S. Lu and Y. Jin, Small, 2020, 16, 1906005 CrossRef CAS PubMed.
- Y. Liu, A. Dikhtiarenko, N. Xu, J. Sun, J. Tang, K. Wang, B. Xu, Q. Tong, H. J. Heeres, S. He, J. Gascon and Y. Fan, Chem.–Eur. J., 2020, 26, 12134–12139 CrossRef CAS PubMed.
- L. A. Baldwin, J. W. Crowe, D. A. Pyles and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 15134–15137 CrossRef CAS PubMed.
- W. K. Haug, E. R. Wolfson, B. T. Morman, C. M. Thomas and P. L. McGrier, J. Am. Chem. Soc., 2020, 142, 5521–5525 CrossRef CAS.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
-
R. H. Holm, G. W. Everett Jr and A. Chakravorty, in Prog. Inorg. Chem., ed. F. A. Cotton, 1966, pp. 83–214 Search PubMed.
- E. Sinn and C. M. Harris, Coord. Chem. Rev., 1969, 4, 391–422 CrossRef CAS.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Angew. Chem., Int. Ed., 2020, 59, 13013–13020 CrossRef PubMed.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 1–9 RSC.
- T. Banerjee, F. Haase, G. k. Savasci, K. Gottschling, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2017, 139, 16228–16234 CrossRef CAS PubMed.
- X. Wang, L. Chen, S. Y. Chong, M. A. Little, Y. Wu, W.-H. Zhu, R. Clowes, Y. Yan, M. A. Zwijnenburg and R. S. Sprick, Nat. Chem., 2018, 10, 1180–1189 CrossRef CAS PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomäcker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- T. Banerjee, K. Gottschling, G. Savasci, C. Ochsenfeld and B. V. Lotsch, ACS Energy Lett., 2018, 3, 400–409 CrossRef CAS PubMed.
- D. Mellmann, P. Sponholz, H. Junge and M. Beller, Chem. Soc. Rev., 2016, 45, 3954–3988 RSC.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- J. H. Barnard, C. Wang, N. G. Berry and J. Xiao, Chem. Sci., 2013, 4, 1234–1244 RSC.
- S. M. Barrett, S. A. Slattery and A. J. Miller, ACS Catal., 2015, 5, 6320–6327 CrossRef CAS.
- S. Zhang, M. Li, Q. Wu, H. Yang, J. Han, H. Wang and X. Liu, Appl. Catal., B, 2018, 236, 466–474 CrossRef CAS.
-
I. Omae, Cyclometalation reactions, Springer, 2016 Search PubMed.
- Y.-F. Han and G.-X. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS.
- F. Auras, L. Ascherl, A. H. Hakimioun, J. T. Margraf, F. C. Hanusch, S. Reuter, D. Bessinger, M. Döblinger, C. Hettstedt, K. Karaghiosoff, S. Herbert, P. Knochel, T. Clark and T. Bein, J. Am. Chem. Soc., 2016, 138, 16703–16710 CrossRef CAS PubMed.
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Adv. Mater., 2019, 1905776 Search PubMed.
- C. Michon, K. MacIntyre, Y. Corre and F. Agbossou-Niedercorn, ChemCatChem, 2016, 8, 1755–1762 CrossRef CAS.
- C. Wang and J. Xiao, Chem. Commun., 2017, 53, 3399–3411 RSC.
- D. L. Davies, O. Al-Duaij, J. Fawcett, M. Giardiello, S. T. Hilton and D. R. Russell, Dalton Trans., 2003, 4132–4138 RSC.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- B. Villa-Marcos, W. Tang, X. Wu and J. Xiao, Org. Biomol. Chem., 2013, 11, 6934–6939 RSC.
- H. Y. T. Chen, C. Wang, X. Wu, X. Jiang, C. R. A. Catlow and J. Xiao, Chem.–Eur. J., 2015, 21, 16564–16577 CrossRef CAS PubMed.
- Y.-N. Gong, W. Zhong, Y. Li, Y. Qiu, L. Zheng, J. Jiang and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 16723–16731 CrossRef CAS PubMed.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Nat. Commun., 2018, 9, 3802 CrossRef PubMed.
- L. Stegbauer, S. Zech, G. Savasci, T. Banerjee, F. Podjaski, K. Schwinghammer, C. Ochsenfeld and B. V. Lotsch, Adv. Energy Mater., 2018, 8, 1703278 CrossRef.
- E. Jin, Z. Lan, Q. Jiang, K. Geng, G. Li, X. Wang and D. Jiang, Chem, 2019, 5, 1632–1647 CAS.
- K. Wang, Z. Jia, Y. Bai, X. Wang, S. E. Hodgkiss, L. Chen, S. Y. Chong, X. Wang, H. Yang, Y. Xu, F. Feng, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2020, 142, 11131–11138 CrossRef CAS.
- G.-B. Wang, S. Li, C.-X. Yan, F.-C. Zhu, Q.-Q. Lin, K.-H. Xie, Y. Geng and Y.-B. Dong, J. Mater. Chem. A, 2020, 8, 6957–6983 RSC.
- K. Gottschling, G. Savasci, H. Vignolo-González, S. Schmidt, P. Mauker, T. Banerjee, P. Rovó, C. Ochsenfeld and B. V. Lotsch, J. Am. Chem. Soc., 2020, 142, 12146–12156 CrossRef CAS PubMed.
- C. Wang, H.-Y. T. Chen, J. Bacsa, C. R. A. Catlow and J. Xiao, Dalton Trans., 2013, 42, 935–940 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01692j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Hamed
Mehrabi
a,
Hamed
Mehrabi