
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity of cyano- and isothiocyanatoborylenes: metal coordination, one-electron oxidation and boron-centred Brønsted basicity†
Stephan
Hagspiel
ab,
Dren
Elezi
ab,
Merle
Arrowsmith
 ab,
Felipe
Fantuzzi
ab,
Alfredo
Vargas
c,
Anna
Rempel
ab,
Marcel
Härterich
ab,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
ab,
Felipe
Fantuzzi
ab,
Alfredo
Vargas
c,
Anna
Rempel
ab,
Marcel
Härterich
ab,
Ivo
Krummenacher
ab and
Holger
Braunschweig
*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, Sussex, UK
First published on 4th May 2021
Abstract
Doubly base-stabilised cyano- and isothiocyanatoborylenes of the form LL′BY (L = CAAC = cyclic alkyl(amino)carbene; L′ = NHC = N-heterocyclic carbene; Y = CN, NCS) coordinate to group 6 carbonyl complexes via the terminal donor of the pseudohalide substituent and undergo facile and fully reversible one-electron oxidation to the corresponding boryl radical cations [LL′BY]˙+. Furthermore, calculations show that the borylenes have very similar proton affinities, both to each other and to NHC superbases. However, while the protonation of LL′B(CN) with PhSH yielding [LL′BH(CN)+][PhS−] is fully reversible, that of LL′B(NCS) is rendered irreversible by a subsequent B-to-CCAAC hydrogen shift and nucleophilic attack of PhS− at boron.
Introduction
With their lone pair at boron and two empty p orbitals monocoordinate borylenes (:BY, Y = anionic substituent), with boron in its +1 oxidation state, have only been observed spectroscopically by microwave and near-ultraviolet spectroscopy1 or in inert gas matrices at extremely low temperatures,2 and their transient generation has long been postulated by various trapping reactions.1–3 While at first the isolation of borylenes under ambient conditions was only achieved in the coordination sphere of electron-rich transition metals,4 the last decade has seen the emergence of metal-free borylenes stabilised by strong donor–acceptor ligands.5 The first metal-free borylene, designed by Bertrand and co-workers in 2011 (I, Fig. 1),6 owed its stability to the two cyclic (alkyl)(amino)carbene (CAAC) ligands,7 which stabilise the empty p orbitals of the borylene through σ donation and the lone pair at boron through π backdonation to the π-acidic carbene centres. Since then CAAC ligands have been successfully employed to access the first stable dicoordinate aminoborylene,8 as well as a wide range of tricoordinate borylenes of the form (CAAC)LBY, where L is a neutral donor ligand (e.g. N-heterocyclic carbene (NHC), phosphine, CO, N2, etc.) and Y is an anionic substituent (e.g. aryl,9 amino,8 boryl,10 H,10a,11 F,12 Cl,13 CN,14 NCS,15etc.).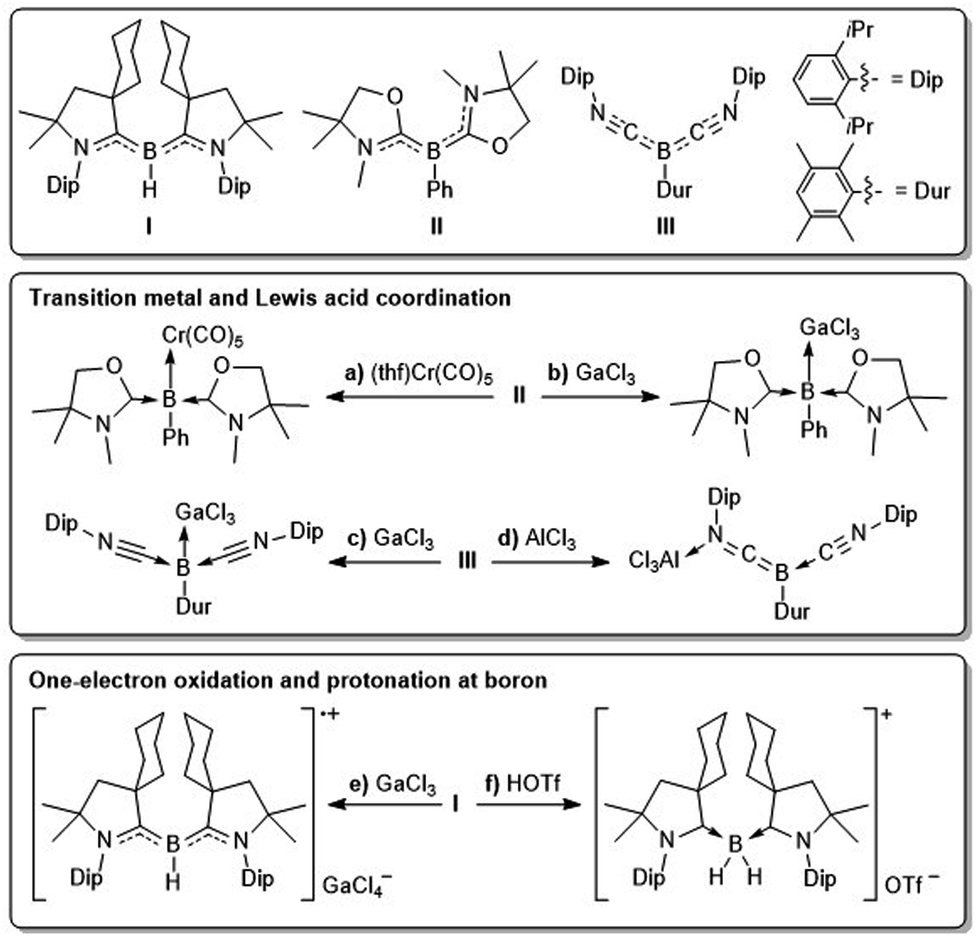 | ||
| Fig. 1 Selected examples of reactivity of tricoordinate borylenes. | ||
While much less reactive than mono- or dicoordinate borylenes, tricoordinate borylenes display typical boron-centred borylene reactivity, including, but not limited to, boron coordination to Lewis acids and transition metals, one-electron oxidation and Brønsted basicity.
Kinjo showed, for example, that bis(oxazol-2-ylidene)-stabilised phenylborylene II can coordinate as a neutral σ donor to chromium(0) and group 9 precursors, as well as GaCl3 (Fig. 1a,b).16 Similarly, our group has reported Lewis adduct formation between the bis(isonitrile)-stabilised arylborylene III and GaCl3 (Fig. 1c), whereas the hard Lewis acid AlCl3 coordinates at the isonitrile nitrogen (Fig. 1d).17 Furthermore, borylene I undergoes facile one-electron oxidation to the corresponding boryl radical cation,6 in which boron adopts a formal +2 oxidation state and spin density is mainly delocalised over the B–CCAAC π bonds (Fig. 1e). Finally, while dicoordinate neutral and cationic borylenes are known to activate H2,8,18 and transient dicoordinate borylenes have been observed to insert into intramolecular C–H and C–C σ bonds,3b,19 examples of σ bond activation by tricoordinate borylenes remain limited to the protonation at boron by strong Brønsted acids. Thus borylenes I and II can be protonated with triflic acid, generating the corresponding hydroboronium triflates (Fig. 1f).6,16b
In this study, we explore the reactivity of tricoordinate pseudohaloborylenes of the form (CAAC)(NHC)BY (Y = CN, NCS), as neutral donors to group 6 transition metals, in one-electron oxidation reactions, and as boron-centred Brønsted bases in reversible and irreversible protonations with thiophenol.
Results and discussion
Synthesis of tricoordinate pseudohaloborylenes
The tricoordinate cyanoborylene (CAAC)(IiPr)B(CN) (2CN, CAAC = 1-(2,6-diisopropylphenyl)-3,3,5,5-tetramethylpyrrolidin-2-ylidene; IiPr = 1,3-diisopropylimidazol-2-ylidene) and isothiocyanatoborylene (CAAC)(IiPr)B(NCS) (2NCS)15 were synthesised by the reduction of the corresponding dibromoborane precursors, (CAAC)BBr2(CN) (1CN)14 and (CAAC)BBr2(NCS) (1NCS),15 respectively, with 2.2 equiv. KC8 in benzene in the presence of 2.2 equiv. IiPr (Scheme 1).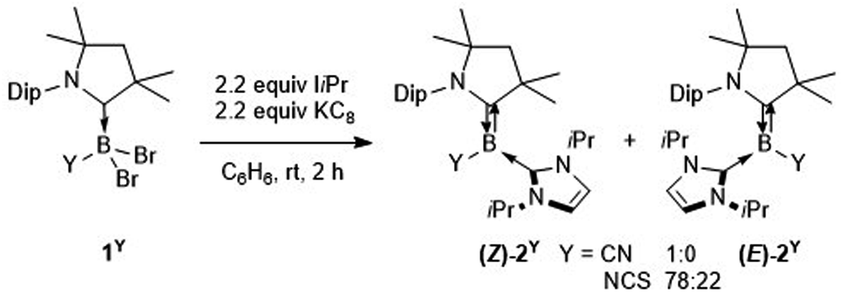 | ||
| Scheme 1 Synthesis of cyano- and isothiocyanatoborylenes 2CN and 2NCS. | ||
The cyanoborylene 2CN was isolated as a yellow crystalline solid and showed an 11B NMR resonance at −12.1 ppm, similar to the related cyanoborylene (CAAC)(IMe)B(CN) (δ11B = −11.3 ppm; IMe = 1,3,4,5-tetramethylimidazol-2-ylidene).14 This contrasts with 2NCS, which is formed as a 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :78 mixture of the (E)- and (Z)-isomer with regards to the arrangement of the NCS unit and CAAC nitrogen atom across the B
:78 mixture of the (E)- and (Z)-isomer with regards to the arrangement of the NCS unit and CAAC nitrogen atom across the B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCAAC double bond, each isomer showing a distinct 11B NMR resonance ((E)-2NCS: δ11B = 3.8 ppm; (Z)-2NCS: δ11B = −2.6 ppm).15 The solid-state structure of 2CN (Fig. 2a) shows the Z-isomer. Both (Z)-2CN and (Z)-2NCS display structural parameters typical for CAAC–NHC–stabilised tricoordinate borylenes,9–11,13,14 with trigonal planar boron centres and short B–CCAAC bonds (ca. 1.45 Å) with double bond character, indicating strong π backbonding from the borylene centre to the π-acidic CAAC ligand. In both compounds the NHC rings are rotated almost perpendicularly to the borylene plane (torsion angle C1–B1–C22–N3 ca. 85–98°) and coordinating as pure σ donors (B1–C22 ca. 1.59 Å).
CCAAC double bond, each isomer showing a distinct 11B NMR resonance ((E)-2NCS: δ11B = 3.8 ppm; (Z)-2NCS: δ11B = −2.6 ppm).15 The solid-state structure of 2CN (Fig. 2a) shows the Z-isomer. Both (Z)-2CN and (Z)-2NCS display structural parameters typical for CAAC–NHC–stabilised tricoordinate borylenes,9–11,13,14 with trigonal planar boron centres and short B–CCAAC bonds (ca. 1.45 Å) with double bond character, indicating strong π backbonding from the borylene centre to the π-acidic CAAC ligand. In both compounds the NHC rings are rotated almost perpendicularly to the borylene plane (torsion angle C1–B1–C22–N3 ca. 85–98°) and coordinating as pure σ donors (B1–C22 ca. 1.59 Å).
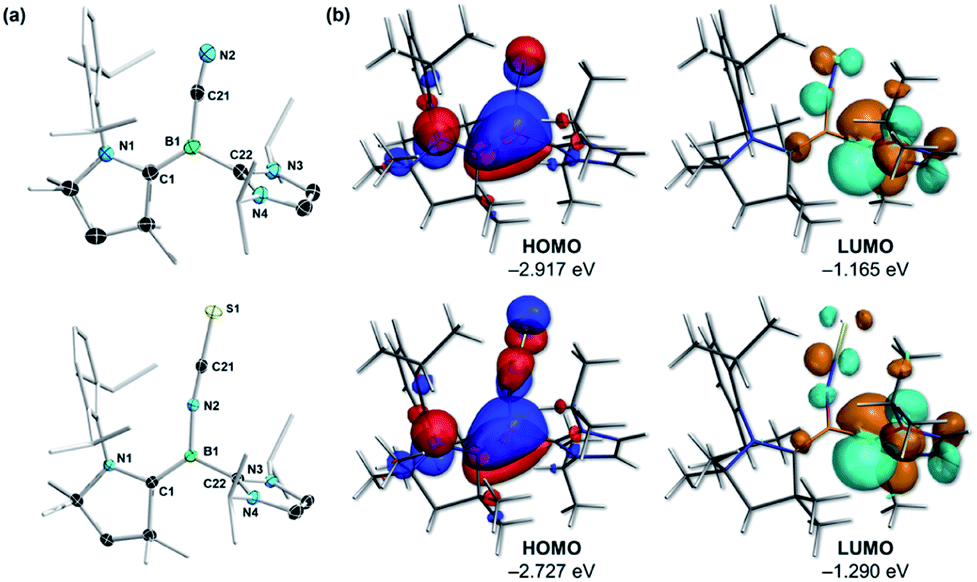 | ||
| Fig. 2 (a) Crystallographically-derived molecular structures of (Z)-2CN and (Z)-2NCS (only one of the two molecules of (Z)-2CN and (Z)-2NCS present in the asymmetric unit is represented). Thermal ellipsoids drawn at the 50% probability level. Ellipsoids on the CAAC ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) for (Z)-2CN: N1–C1 1.4076(14), C1–B1 1.4630(17), B1–C21 1.5490(18), C21–N2 1.1618(16), B1–C22 1.5918(16). For (Z)-2NCS: N1–C1 1.4080(14), C1–B1 1.4591(16), B1–N2 1.4836(15), N2–C21 1.1706(15), C21–S1 1.6195(12). (b) Canonical Kohn–Sham molecular orbitals of (Z)-2CN (top) and (Z)-2NCS (bottom) at the OLYP/TZ2P level of theory. Selected Hirshfeld atomic charges for (Z)-2CN: N1: −0.055; C1: −0.050; B1: −0.064; C21: −0.032; N2: −0.282; C22: +0.076; for (Z)-2NCS: N1: −0.061; C1: −0.068; B1: −0.012; N2: −0.142; C21: +0.035; S1: −0.210; C22: +0.072. | ||
Accordingly, their HOMOs are composed of strong π contributions in the B–CCAAC bonding region, with negligible involvement of the CNHC atom, while their LUMOs feature the vacant CNHC p orbitals (Fig. 2b). The more negative Hirshfeld charge at boron for (Z)-2CN (−0.064) than for (Z)-2NCS (−0.012) indicates that the former is more electron-rich than the latter, in agreement with the relative 11B NMR shifts of the two compounds.
Coordination to group 6 transition metal carbonyls
The addition of (Z)-2CN or (Z)-2NCS to irradiated THF solutions of group 6 carbonyl precursors, [M(CO)6] (M = Cr, Mo, W), resulted in an instant intensification of the solution colour. After removal of the solvent, complexes 2CN-M (M = Cr, Mo, W) and 2NCS-M (M = Cr, W) were isolated as yellow-brown and red-brown solids, respectively (Scheme 2).‡ The 11B NMR resonances of these compounds were only shifted ca. 1 ppm upfield from those of the corresponding borylene precursors (2CN-M: δ11B = −13.2 to −13.8 ppm; 2NCS-M: δ11B = −3.8 to −3.9 ppm), thus indicating that the boron centres remain tricoordinate. Significant shifts in the 1H NMR resonances of the CAAC and IiPr ligands, however, suggested the formation of adducts by metal coordination to the terminal nitrogen and sulphur donors of 2CN and 2NCS, respectively (see Fig. S30 in the ESI†). The solid-state IR spectra of 2CN-M and 2NCS-M each showed four CO stretching bands in the 1850–2070 cm−1 and 1880–2060 cm−1 regions, respectively, confirming that one CO ligand had indeed been replaced. While the 11B NMR spectrum of 2CN-Cr showed only the (Z)-isomer, 2CN-Mo and 2CN-W were formed as a mixture of the (Z)- and (E)-isomers in a 95:5 and 90:10 ratio, respectively. Furthermore, while (Z)-2CN-Cr and (Z)-2CN-Mo did not isomerise at higher temperatures, prolonged heating of a C6D6 solution of the isolated 90:10 (Z)/(E) mixture of 2CN-W at 60 °C afforded a 2:3 (Z)/(E) mixture, showing that the (Z)-isomer is the kinetic and the (E)-isomer the thermodynamic product (see Fig. S31 and S32 in the ESI†).§
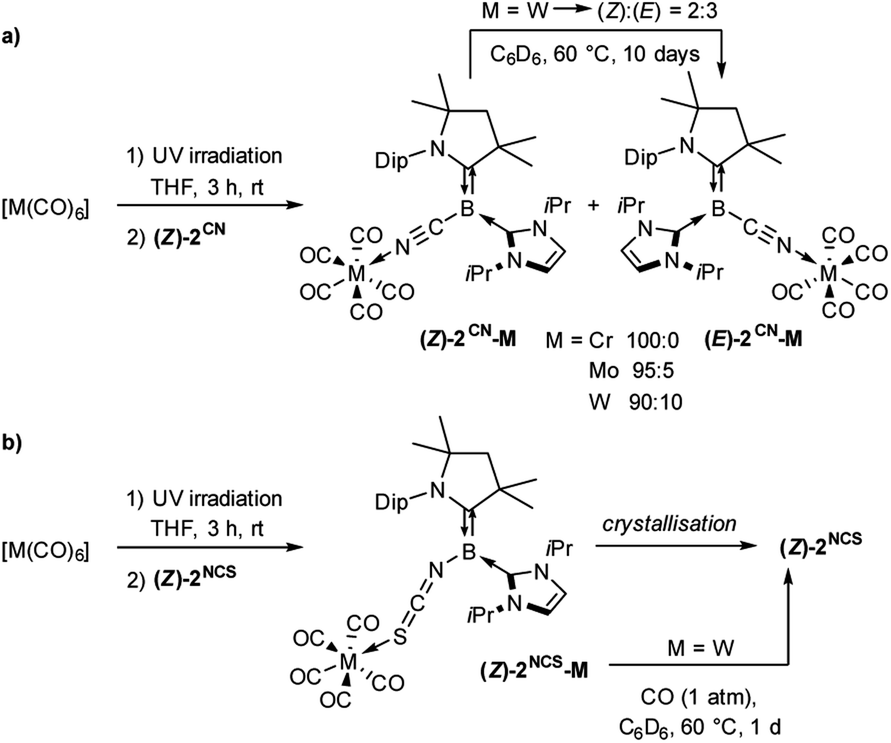 | ||
| Scheme 2 Adduct formation between (Z)-2Y and group 6 hexacarbonyls. | ||
X-ray crystallographic analyses of 2CN-M (Fig. 3) confirmed that adduct formation occurred through the cyano nitrogen atom rather than the electron-rich boron centre, and furthermore confirmed the configuration of the BCCAAC bonds in (Z)-2CN-Cr, (Z)-2CN-Mo and (E)-2CN-W (Fig. 2a). The bond lengths and angles in the borylene moiety of these complexes do not differ significantly from those in (Z)-2CN. The N2–M bond lengths (N2–Cr 2.0614(17), N2–Mo 2.200(3), N2–W 2.163(3) Å) are typical for nitrile–carbonyl complexes of the chromium triade.20 Although 2NCS-M could be isolated as powders by removal of the reaction solvent in vacuo, all attempts to obtain single crystals of 2NCS-M resulted in quantitative recovery of the starting borylene (Z)-2NCS, indicating a very weak S→M interaction. Similarly, heating a C6D6 solution of 2NCS-W under a CO atmosphere led to full recovery of (Z)-2NCS and [W(CO)6] within one day. A literature search showed that there have been no other reports of group 6 M0 complexes with RNCS donor ligands. At first sight the preferred coordination of M(CO)5 at the pendant CN and NCS ligands of 2CN and 2NCS, respectively, rather than at the borylene centres, seems mostly due to the excessive steric congestion of the latter by the CAAC and IiPr ligands. Indeed, a comparison with the 11B NMR shift of borylene II (δ11B = −1.1 ppm, Fig. 1), which coordinates to transition metals as a borylene donor,16 suggests that the borylene centre in (Z)-2NCS (δ11B = −2.6 ppm) should be similarly nucleophilic to II, while that of (Z)-2CN (δ11B = −12.1 ppm) should be even more nucleophilic. However, the calculated Hirshfeld charges of the terminal CN nitrogen (−0.282) and NCS sulphur atoms (−0.210) in (Z)-2CN and (Z)-2NCS, respectively, are significantly more negative than those of the boron atoms ((Z)-2CN: −0.064; (Z)-2NCS: −0.012, see legend of Fig. 2b), which further favours coordination at the CN and NCS ligands.
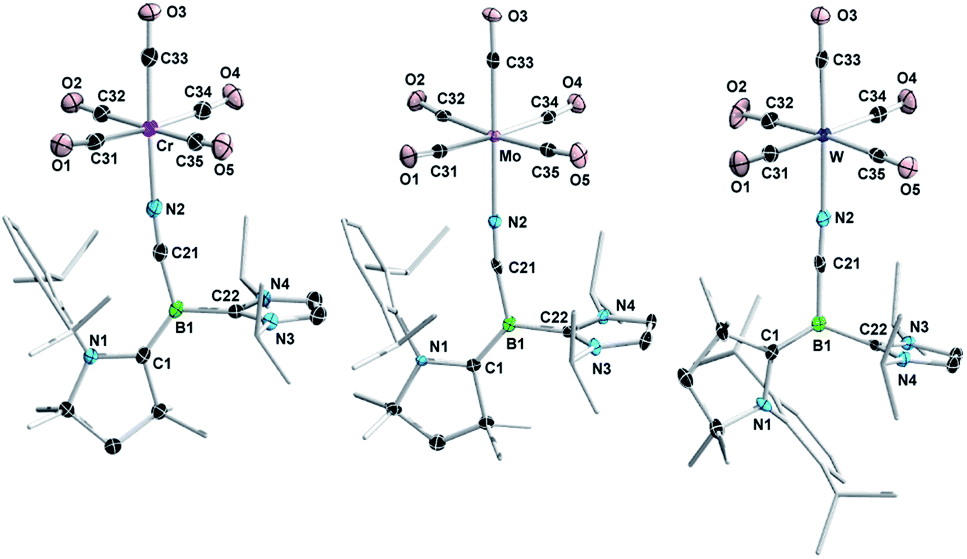 | ||
| Fig. 3 Crystallographically-derived molecular structures of (Z)-2CN-Cr, (Z)-2CN-Mo and (E)-2CN-W (from left to right). Thermal ellipsoids drawn at 50% probability level. Ellipsoids on the CAAC ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) for (Z)-2CN-Cr: N1–C1 1.399(2), C1–B1 1.456(3), B1–C21 1.542(3), C21–N2 1.158(2), B1–C22 1.588(3), N2–Cr 2.0614(17), Cr–C33 1.842(2), C33–O3 1.154(2). For (Z)-2CN-Mo: N1–C1 1.382(5), C1–B1 1.468(5), B1–C21 1.544(5), C21–N2 1.157(4), B1–C22 1.573(6), N2–Mo 2.200(3), Mo–C33 1.961(3), C33–O3 1.166(4). For (E)-2CN-W: N1–C1 1.408(5), C1–B1 1.436(6), B1–C21 1.541(6), C21–N2 1.156(5), B1–C22 1.596(5), N2–W 2.163(3), W–C33 1.978(4), C33–O3 1.154(5). | ||
One-electron oxidation
The cyclovoltammograms of (Z)-2Y in THF showed a fully reversible redox event at E1/2 = −1.06 V (Y = NCS) and a partially reversible redox event at E1/2 = −0.89 V (Y = CN), respectively (vs. the ferrocene standard Fc/Fc+), suggesting the possibility of selective chemical one-electron oxidation (see Fig. S51 and S52 in the ESI†). Indeed, the room temperature reactions of (Z)-2Y with exactly one equivalent of silver triflate (AgOTf) in benzene resulted in a colour change from red to yellow (Y = NCS) or intensification of the yellow colour (Y = CN), respectively, accompanied by the formation of a black silver precipitate. After workup, the boryl radical cations [(Z)-2Y˙+][OTf−] were isolated as yellow crystals in 81% (Y = NCS) and 85% (Y = CN) yield (Scheme 3a). Conversely, the reduction of isolated [(Z)-2Y˙+][OTf−] with KC8 in benzene afforded borylenes (Z)-2Y in quantitative yield (Scheme 3b). The EPR spectrum of [(Z)-2NCS˙+][OTf−] (Fig. 4) showed a broad resonance at giso = 2.003. In contrast, that of [(Z)-2CN˙+][OTf−] displayed a multiplet at giso = 2.0025 with hyperfine coupling parameters to the boron (a10,11B = 9.0 MHz), as well as the CAAC- and cyano-nitrogen nuclei (a14N = 21.7, 18.0 MHz). X-ray crystallographic analyses confirmed that the boryl radical cations [(Z)-2Y˙+][OTf−] retain the (Z)-configuration of the starting borylenes (Fig. 4). The boron centres remain trigonal planar (∑(∠B) = 360°), with a slight elongation of the C1–B1 bonds to a partial B–C double bond (ca. 1.51 Å), concomitant with shortening of the N1–C1 (ca. 1.34 Å) bonds, as expected upon one-electron oxidation of CAAC-stabilised borylenes.6,12 Calculations show that the spin density in both radical cations is delocalised over the [(N–C)CAAC–B–Y] π framework (Fig. 4), with the highest contribution at boron (0.388 for Y = CN; 0.295 for Y = NCS). The lower spin density at boron compared to Bertrand's CAAC-stabilized hydroboryl radical cations (0.50)6,11 is owed to the additional delocalization of the spin density over the CN and NCS fragments, respectively.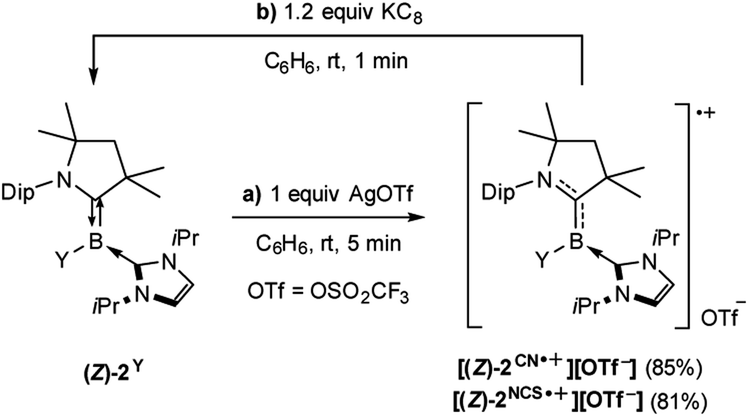 | ||
| Scheme 3 Reversible chemical one-electron oxidation of (Z)-2Y to [(Z)-2Y˙+][OTf−], Y = CN, NCS. | ||
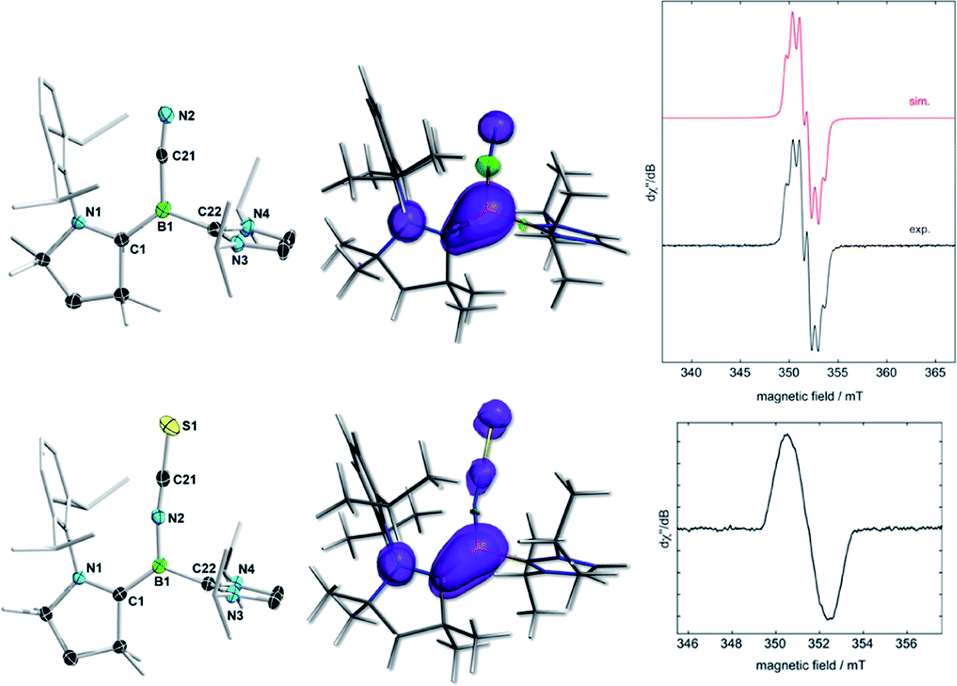 | ||
| Fig. 4 Left: crystallographically-derived molecular structures of [(Z)-2CN˙+][OTf−] (top) and [(Z)-2NCS˙+][OTf−] (bottom). Thermal ellipsoids drawn at the 50% probability level. Triflate anion, ellipsoids on the CAAC ligand periphery and hydrogen atoms omitted for clarity. Selected bond lengths (Å) for [(Z)-2CN˙+][OTf−]: N1–C1 1.335(6), C1–B1 1.510(7), B1–C21 1.548(7), C21–N2 1.146(6), B1–C22 1.590(7); for [(Z)-2NCS˙+][OTf−]: N1–C1 1.342(2), C1–B1 1.510(2), B1–N2 1.454(2), N2–C21 1.170(2), C21–S1 1.5835(18), B1–C22 1.590(2). Centre: plots of spin density of [(Z)-2CN˙+] (top) and [(Z)-2NCS˙+] (bottom) obtained from the multipole-derived charges up to quadruple expansion (MDC-q) at the OLYP/TZ2P level of theory. Selected spin densities for [(Z)-2CN˙+]: N1: 0.219; C1: 0.125; B1: 0.388; C21: −0.009; N2: 0.134; for [(Z)-2NCS˙+]: N1: 0.220; C1: 0.179; B1: 0.295; N2: 0.013; C21: 0.058; S1: 0.103. Right: Experimental (black line) and simulated (red line) continuous-wave X-band EPR spectra of [(Z)-2CN˙+][OTf−] (top) and [(Z)-2NCS˙+][OTf−] (bottom) in benzene at room temperature. Best-fit simulation parameters for [(Z)-2CN˙+]: giso = 2.0025, a(10,11B) = 9.0 MHz, a(14N) = 21.7 MHz, and a(14N) = 18.0 MHz. | ||
Boron-centred Brønsted basicity
Given that tricoordinate borylenes can be protonated by the strong Brønsted acid HOTf (pKa(DMSO) = −14.3),6,16b,21 we sought to determine the relative Brønsted basicity of 2CN and 2NCS using the much weaker acid thiophenol (pKa(DMSO) = 10.3).22 The reaction of (Z)-2NCS with 1.2 equiv. PhSH at room temperature in benzene resulted in a 3:2 mixture of the thiolatoborane 3NCS (δ11B = −6.0 ppm) and the hydroboronium species [2NCS-H+][PhS−] (δ11B = −18.9 ppm). The 1H{11B} NMR spectrum of the product mixture showed a broad BH resonance at 3.76 ppm for [2NCS-H+] and a 1H singlet at 3.70 ppm for 3NCS, characteristic of a C1-protonated CAAC ligand.23 Over the course of three days at room temperature in solution [2NCS-H+][PhS−] converted entirely to 3NCS, which was identified by X-ray crystallographic analysis as the product of the formal 1,2-addition of the S–H bond to the BCCAAC double bond (Scheme 4b, Fig. S33–S36†). The conversion of [2NCS-H+][PhS−] to 3NCS proceeds via a B-to-CCAAC hydride shift typical for CAAC-stabilised hydroboranes and hydroboronium species,23 followed by nucleophilic attack of the PhS– anion at the three-coordinate cationic boron centre. Both the reactions of [2NCS-H+][PhS−] and (Z)-2NCS with an excess of thiophenol at 60 °C cleanly yielded the tricoordinate dithiolatoborane 4 (δ11B = 42.6 ppm), alongside the imidazolium salt ([IiPr-H][NCS]) as a by-product (Scheme 4d and e).
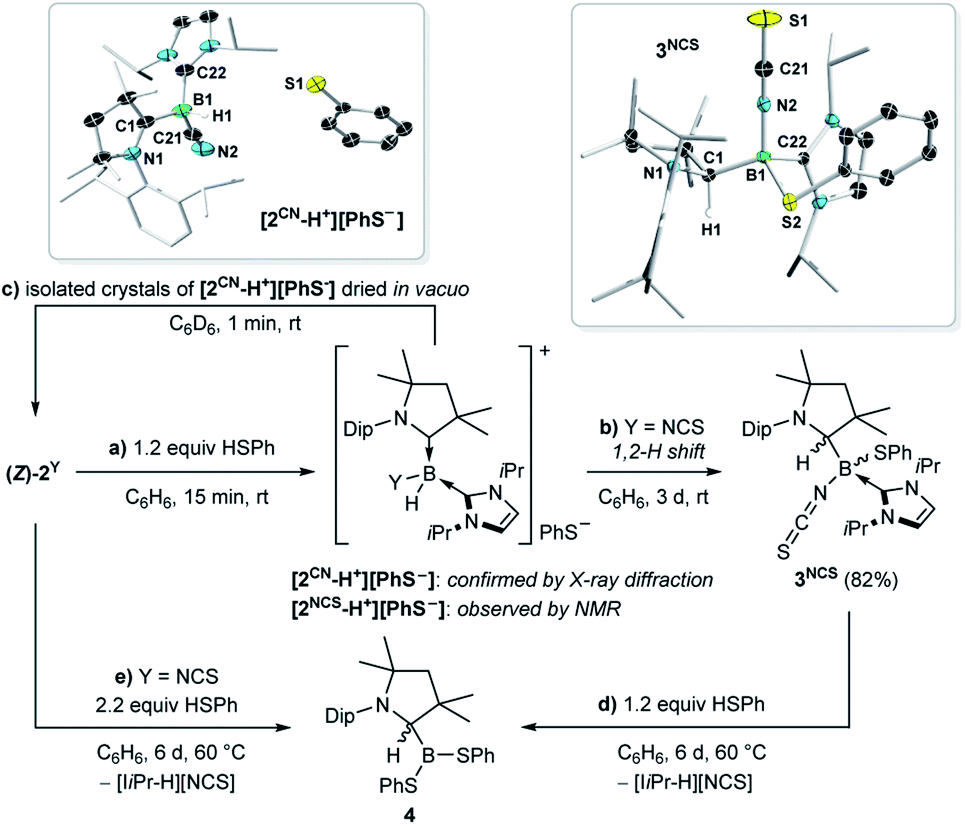 | ||
| Scheme 4 Reversible and irreversible protonation of (Z)-2CN and (Z)-2NCS, respectively, with PhSH, and crystallographically-derived molecular structures of [2CN-H+][PhS−] and 3NCS. Thermal ellipsoids drawn at 50% probability level. Ellipsoids on the CAAC ligand periphery and hydrogen atoms omitted for clarity, except for the boron-bound hydride in [2CN-H+] and C1-bound proton in 3NCS. | ||
In contrast, the analogous reaction of (Z)-2CN with 1.2 equiv. PhSH proceeded cleanly to the hydroboronium species [2CN-H+] (δ11B = −30.3 ppm), which was identified by X-ray diffraction analysis (Scheme 4a). To our surprise, however, isolated crystals of [2CN-H+][PhS−] dried in vacuo and redissolved in C6D6 yielded a yellow solution, the NMR data of which matched (Z)-2CN (Scheme 4c). Moreover, isolated crystals of [2CN-H+][PhS−] washed with hexane, left to dry in the glovebox at atmospheric pressure for one hour and redissolved in C6D6 yielded a solution of 2CN with only a small amount of residual PhSH, the rest having evaporated (see Fig. S37–S40†). While heating a suspension of [2CN-H+][PhS−] in d8-toluene showed that the concentration of 2CN and PhSH in solution increased steadily from 25 to 100 °C, the lack of solubility of the boronium salt in suitable NMR solvents prevented a quantitative analysis of the acid–base equilibrium.
Given the above-mentioned difficulties in assessing the relative Brønsted basicity of 2CN and 2NCS experimentally, gas-phase proton affinity (PA) calculations were performed (see ESI† for details). The calculated gas-phase PA for PhS− (339.4 kcal mol−1) is in good agreement with the experimental value of 349.0 ± 2 kcal mol−1.24 The PAs calculated for 2CN and 2NCS, 267.9 and 268.3 kcal mol−1, respectively, are identical within the error of the calculation. While at first sight these results seem in contradiction with the experimental observations of reversible protonation for 2CN and irreversible protonation for 2NCS, the latter is only rendered irreversible by the subsequent B-to-C hydrogen shift and nucleophilic attack of PhS− at boron. The higher PA of PhS− confirms that the thiolate is more basic than 2CN and 2NCS, as suggested by the reversibility of the protonation of 2CN by PhSH. Furthermore, the PAs of 2CN and 2NCS are comparable to those of the superbases CsOH (267 kcal mol−1)25 and the unsaturated NHCs, 1,3-dialkyl/diarylimidazol-2-ylidenes (262–275 kcal mol−1).26
Conclusions
We have shown that cyano- and isothiocyanatoborylenes of the form (CAAC)(NHC)BY (Y = CN 2CN, NCS 2NCS) are easily synthesised by the twofold reduction of (CAAC)BBr2Y in the presence of the NHC ligand. Computational analyses show that the HOMO of these borylenes is delocalised over the entire [(N–C)CAAC–B–Y] π framework, with a major π-bonding contribution at the B–CCAAC bond. While the borylene centre bears a slightly negative partial charge the main negative charge is located at the terminal CN nitrogen and NCS sulphur atoms, respectively. As a consequence, these compounds act as neutral nitrogen-/sulphur-centred rather than boron-centred donors towards group 6 carbonyls, generating the corresponding CN→M and NCS→M adducts, respectively, under photolytic conditions. The electron-rich borylene centre of 2Y, however, can undergo reversible electrochemical and chemical one-electron oxidation to the corresponding [2Y˙+] radical cation. The spin density of the latter is delocalised over the [(N–C)CAAC–B–Y] π framework, with a relatively large spin density at boron (0.388 for Y = CN; 0.295 for Y = NCS). Finally, both borylenes act as boron-centred Brønsted bases toward relatively weakly acidic thiophenol (pKa(DMSO) = 10.3). Whereas the protonation of 2NCS at boron is driven by a B-to-CCAAC hydride shift and irreversible nucleophilic attack of PhS− at boron, the protonation of 2CN to the corresponding boronium species is fully reversible. Calculations show that 2CN and 2NCS have in fact similar proton affinities (ca. 268 kcal mol−1), of the same order as the inorganic superbase CsOH or unsaturated NHCs. The unprecedented reversibility of the protonation for 2CN is of particular interest as reversible small molecule activation is promising for potential catalytic applications of these species.Author contributions
H. B. supervised the study. S. H. and D. E. carried out the synthetic work. I. K. carried out and analysed the EPR and CV experiments. S. H., A. R. and M. H. carried out the X-ray crystallographic analyses. F. F. and A. V. carried out the computational studies. M. A., S. H. and F. F. prepared the manuscript and the ESI. All authors read and commented on the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft for financial support. S. H. is grateful for a doctoral fellowship from the Studienstiftung des deutschen Volkes. F. F. thanks the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and the Alexander von Humboldt (AvH) Foundation for a Capes-Humboldt postdoctoral fellowship. A. V. thanks the University of Sussex for financial support.Notes and references
- (a) P. L. Timms, Acc. Chem. Res., 1973, 6, 118 CrossRef CAS; (b) P. L. Timms, J. Am. Chem. Soc., 1967, 89, 1629 CrossRef CAS; (c) J. W. C. Johns, F. A. Grimm and R. F. Porter, J. Mol. Spectrosc., 1967, 22, 435 CrossRef CAS.
- (a) H. F. Bettinger, J. Am. Chem. Soc., 2006, 128, 2534 CrossRef CAS PubMed; (b) C. A. Thompson, L. Andrews, J. M. L. Martin and J. El-Yazal, J. Phys. Chem., 1995, 99, 13839 CrossRef CAS; (c) L. Andrews, P. Hassanzadeh, J. M. L. Martin and P. R. Taylor, J. Phys. Chem., 1993, 97, 5839 CrossRef CAS.
- Selected examples: (a) M. Ito, N. Tokitoh, T. Kawashima and R. Okazaki, Tetrahedron Lett., 1999, 40, 5557 CrossRef CAS; (b) W. J. Grigsby and P. P. Power, J. Am. Chem. Soc., 1996, 118, 7981 CrossRef CAS; (c) A. Meller, U. Seebold, W. Maringgele, M. Noltmeyer and G. M. Sheldrick, J. Am. Chem. Soc., 1989, 111, 8299 CrossRef CAS; (d) B. Pachaly and R. West, Angew. Chem., Int. Ed. Engl., 1984, 23, 454 CrossRef.
- Selected reviews: (a) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197 RSC; (b) D. Vidovic, G. A. Pierce and S. Aldridge, Chem. Commun., 2009, 1157 RSC; (c) H. Braunschweig, C. Kollann and F. Seeler, Transition Metal Borylene Complexes, in Contemporary Metal Boron Chemistry I. Structure and Bonding, ed. T. B. Marder and Z. Lin, Springer, Berlin, Heidelberg, 2008, vol. 130 Search PubMed.
- (a) M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231 CrossRef PubMed; (b) M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10282 CrossRef CAS PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed.
- (a) U. S. D. Paul, M. J. Krahfuß and U. Radius, Chem. Unserer Zeit, 2018, 53, 212 CrossRef; (b) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed.
- (a) A. Hofmann, M.-A. Légaré, L. Wüst and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 9776 CrossRef CAS PubMed; (b) M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896 CrossRef PubMed; (c) H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802 CrossRef CAS PubMed.
- (a) J. Böhnke, M. Arrowsmith and H. Braunschweig, J. Am. Chem. Soc., 2018, 140, 10368 CrossRef PubMed; (b) A. Stoy, J. Böhnke, J. O. C. Jimenéz-Halla, R. D. Dewhurst, T. Thiess and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 5947 CrossRef CAS PubMed; (c) M. Arrowsmith, J. Böhnke, H. Braunschweig and M. A. Celik, Angew. Chem., Int. Ed., 2017, 56, 14287 CrossRef CAS PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837 RSC.
- S. K. Sarkar, M. M. Siddiqui, S. Kundu, M. Ghosh, J. Kretsch, P. Stollberg, R. Herbst-Irmer, D. Stalke, A. C. Stückl, B. Schwederski, W. Kaim, S. Ghorai, E. D. Jemmis and H. W. Roesky, Dalton Trans., 2019, 48, 8551 RSC.
- M. Arrowsmith, J. I. Schweizer, M. Heinz, M. Härterich, I. Krummenacher, M. C. Holthausen and H. Braunschweig, Chem. Sci., 2019, 10, 5095 RSC.
- M. Arrowsmith, D. Auerhammer, R. Bertermann, H. Braunschweig, G. Bringmann, M. A. Celik, R. D. Dewhurst, M. Finze, M. Grüne, M. Hailmann, T. Hertle and I. Krummenacher, Angew. Chem., Int. Ed., 2016, 55, 14464 CrossRef CAS PubMed.
- S. Hagspiel, M. Arrowsmith, F. Fantuzzi, A. Vargas, A. Rempel, A. Hermann, T. Brückner and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 6446 CrossRef CAS PubMed.
- (a) L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893 RSC; (b) L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436 CrossRef CAS PubMed.
- H. Wang, L. Wu, Z. Lin and Z. Xie, J. Am. Chem. Soc., 2017, 139, 13680 CrossRef CAS PubMed.
- (a) M. Arrowsmith, J. Böhnke, H. Braunschweig, H. Gao, M.-A. Légaré, V. Paprocki and J. Seufert, Chem.–Eur. J., 2017, 23, 12210 CrossRef CAS PubMed; (b) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327 CrossRef CAS PubMed; (c) D. P. Curran, A. Boussonnière, S. J. Geib and E. Lacôte, Angew. Chem., Int. Ed., 2012, 51, 1602 CrossRef CAS PubMed; (d) Y. Wang and G. H. Robinson, Inorg. Chem., 2011, 50, 12326 CrossRef CAS PubMed; (e) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044 CrossRef CAS PubMed.
- (a) R. E. Bachman and K. H. Whitmire, Inorg. Chem., 1995, 34, 1542 CrossRef CAS; (b) F. Biesemeier, K. Harms and U. Müller, Z. Kristallogr.–New Cryst. Struct., 2014, 218, 419 Search PubMed; (c) J. Reibenspies, D. Darensbourg and E. Atnip, Z. Kristallogr.–Cryst. Mater., 1994, 209, 379 CrossRef CAS.
- A. Trummal, L. Lipping, I. Kaljurand, I. A. Koppel and J. Leito, J. Phys. Chem. A, 2016, 120, 3663 CrossRef CAS PubMed.
- F. G. Bordwell and D. L. Hughes, J. Org. Chem., 1982, 47, 3224 CrossRef CAS.
- (a) S. Hagspiel, M. Arrowsmith, F. Fantuzzi, A. Hermann, V. Paprocki, R. Drescher, I. Krummenacher and H. Braunschweig, Chem. Sci., 2020, 11, 551 RSC; (b) D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066 RSC; (c) S. Wuertemberger-Pietsch, H. Schneider, U. Radius and T. B. Marder, Chem.–Eur. J., 2016, 22, 13032 CrossRef CAS PubMed; (d) M. R. Momeni, E. Rivard and A. Brown, Organometallics, 2013, 32, 6201 CrossRef CAS.
- A. K. Chandra, P.-C. Nam and M. T. Nguyen, J. Phys. Chem. A, 2003, 107, 9182 CrossRef CAS.
- (a) E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS; (b) M. P. Drapeau, A. Tlili, Y. Zaid, D. Toummini, F. O. Chahdi, J.-M. Sotiropoulos, T. Ollevier and M. Taillefer, Chem.–Eur. J., 2018, 24, 17449 CrossRef PubMed; (c) B. A. Trofimov, E. Y. Schmidt, I. A. Ushakov, N. V. Zorina, E. V. Skital’tseva, N. I. Protsuk and A. I. Mikhaleva, Chem.–Eur. J., 2010, 16, 8516 CrossRef CAS PubMed.
- R. Tonner, G. Heydenrych and G. Frenking, ChemPhysChem, 2008, 9, 1474 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR, IR, UV-vis, CV, EPR and details of the computational analyses. CCDC 2071470–2071477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01580j |
| ‡ All attempts to synthesise the analogous complex 2NCS-Mo only led to intractable mixtures of unidentifiable compounds. |
| § Whereas (Z)-2CN-Mo could be isolated cleanly by fractional crystallisation, the slow and partial isomerisation of (Z)-2CN-W to (E)-2CN-W in solution, even at room temperature, prevented the clean isolation of either isomer. |
| This journal is © The Royal Society of Chemistry 2021 |