DOI:
10.1039/D1SC00905B
(Edge Article)
Chem. Sci., 2021,
12, 7924-7929
Acrylic boronate: a multifunctional C3 building block for catalytic synthesis of rare organoborons and chemoselective heterobifunctional ligations†
Received
13th February 2021
, Accepted 26th April 2021
First published on 26th April 2021
Abstract
A novel C3 acylboron building block; acrylic boronate was successfully prepared and its versatility for catalytic synthesis of several previously inaccessible organoborons is described. Cross-metathesis and Pd-catalyzed coupling of acrylic boronate enabled two complementary routes to highly functionalized α,β-unsaturated acylborons and two new types of conjugated borylated products: α,β,γ,δ-unsaturated and bis-α,β unsaturated acylborons. The synthetic application of α,β-unsaturated acylborons was demonstrated for the first time, thereby providing a general and highly regioselective route to medicinally important 3-boryl pyrazoles. Acrylic boronate also provided a unique bis-electrophilic platform for rapid and chemoselective labeling of cysteines with acylboron tags which are potentially useful for site-selective functionalization and orthogonal ligation of proteins.
Introduction
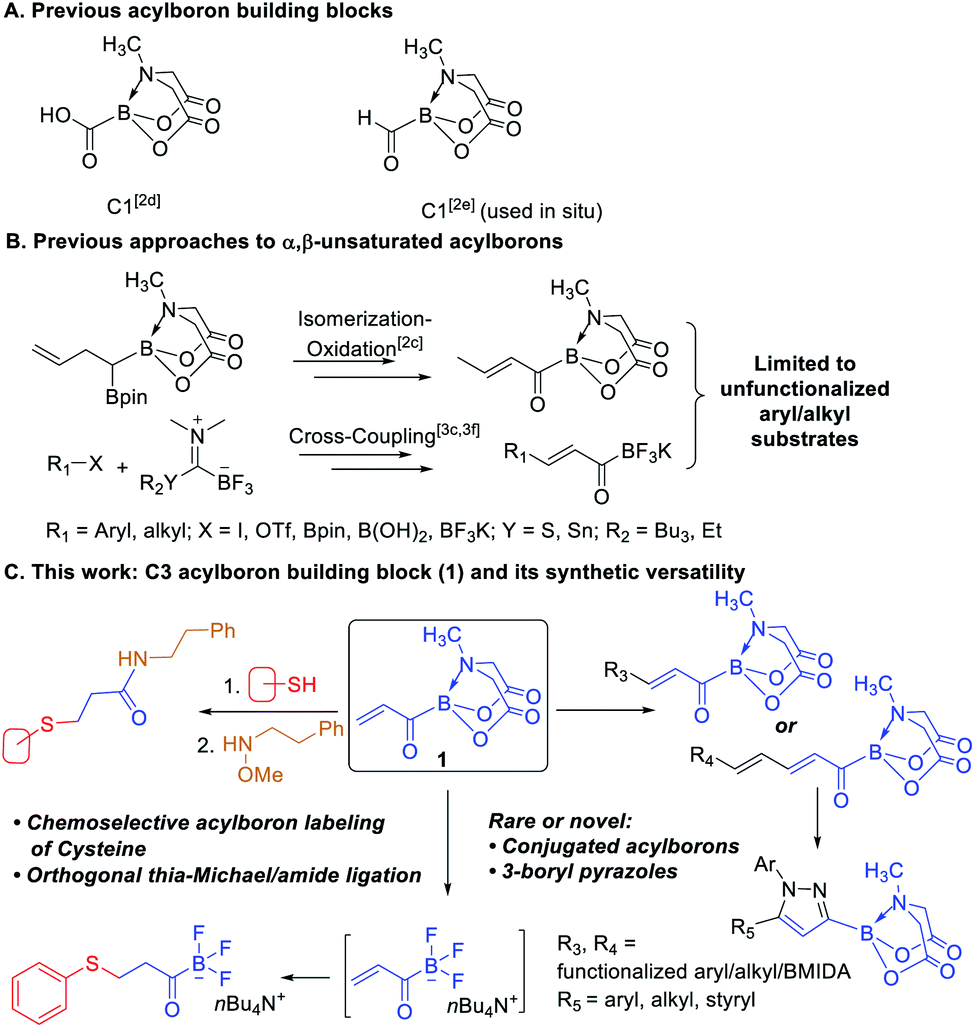
Acylborons represent an underexplored class of compounds whose existence and remarkable synthetic utility is beginning to emerge only recently.1 During the past few years, several elegant methods have been described for synthesis of MIDA acylboronates2 and potassium acyl trifluoroborates (KATs).3 These organoborons are valuable reagents for fast and chemoselective amide-bond forming reactions4 and preparation of other useful organoborons.2a,f Despite these advances, there are still significant challenges that hamper the broader investigation and application of acylborons. For instance, only two types of acylboron-based building blocks have been described and both represent a C1 scaffold having a combination of boronate and a carboxy or formyl groups (Scheme 1A).2d,e While these reagents have enabled interesting and useful transformations, the merger of other powerful functional groups with the boronate motif remains untapped. On the other hand, C3 building blocks such as acrylates have made a tremendous impact in organic synthesis, polymer/materials chemistry and biology due to their versatile chemical and physicochemical properties.5 In the course of our ongoing work2c on acylborons, we became interested to develop a C3 building block having acrylic and boronate motifs directly connected to each other. We envisioned that this acrylic boronate (1) would open-up opportunities to access novel acylborons and other organoborons that would be difficult to prepare using existing methods.
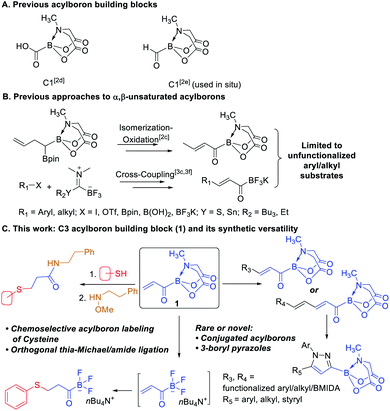 |
| | Scheme 1 Previously reported acylboron building blocks (A); previous routes to α,β-unsaturated acylborons (B); novel acylboron building block and its synthetic versatility (C). | |
Recently, we reported a mild and chemoselective oxidation approach to acylborons including the first example of an α,β-unsaturated acyl boron.2c Thereafter, Bode and coworkers described another route to acylborons and α,β-unsaturated KATs via Stille3c or Liebeskind–Srogl-type3f cross-coupling of KAT transfer reagents (Scheme 1B). We hypothesized that the acrylic boron building block (1) could provide a general and catalytic route to highly functionalized α,β-unsaturated as well as the unprecedented α,β,γ,δ-unsaturated acylborons via Cross-Metathesis (CM) or Heck coupling of 1 with readily available alkenes and aryl/vinyl halides respectively (Scheme 1C). We were also intrigued by the potential of 1 to provide a versatile platform for accessing other underexplored organoborons. Herein, we report the development of acrylic boronate (1) as a novel C3 building block and demonstrate its utility (Scheme 1C) to achieve the following benefits: (i) catalytic synthesis of previously inaccessible functionalized α,β-unsaturated as well as α,β,γ,δ-unsaturated acylborons from readily available substrates, (ii) one-pot route to rare 3-boryl pyrazoles and (iii) access to a new type of heterobifunctional bioconjugation agent capable of converting nucleophilic cysteines chemoselectively into electrophilic acylboron-based handles.
Results and discussion
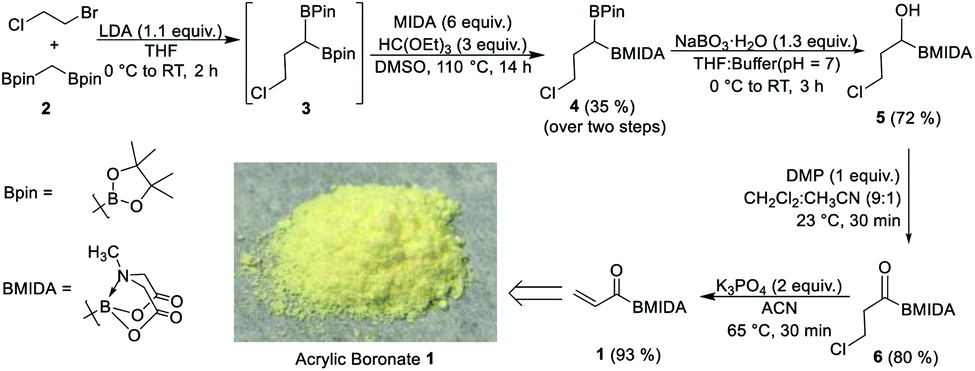
Our strategy to prepare acrylic boronate (1) involved β-halo acyl boron (6) as a strategic intermediate (Scheme 2). We reasoned that base-induced dehydrohalogenation of this key acylboron could provide access to 1. MIDA boronates are known to be unstable in the presence of strong bases;6 we hypothesized that the acyl boron (6) would activate its α-hydrogen for elimination reaction under mildly basic conditions. Motivated by the above retrosynthetic logic, we converted commercially available 1-chloro-2-bromoethane and bis[(pinacolato)boryl]methane (2, ESI, Scheme S1†) into the symmetrical diboron 3 which provided the unsymmetrical diboron (4) via boron ligand exchange. 4 was found to be compatible for chemoselective oxidation2c to give β-halo acyl boron (6). To accomplish the dehydrochlorination of 6, several organic and inorganic bases were screened (ESI, Table S1†). We were delighted to find that the acrylic boronate (1) could be obtained in high yield using K3PO4 as a base within 30 min and the pure product can be isolated as a bench-stable solid by filtration of the crude reaction mixture. Furthermore, 1 can be prepared on a 70 mmol scale without an appreciable decrease in yield by using a slightly modified protocol for workup/purification of 4 (ESI, Scheme S2†). It is worthwhile to mention that this scale-up procedure involves only two steps that require column chromatography.
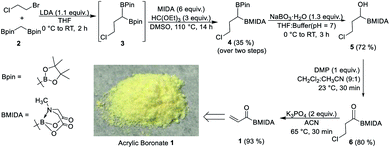 |
| | Scheme 2 Synthesis of acrylic boronate 1. | |
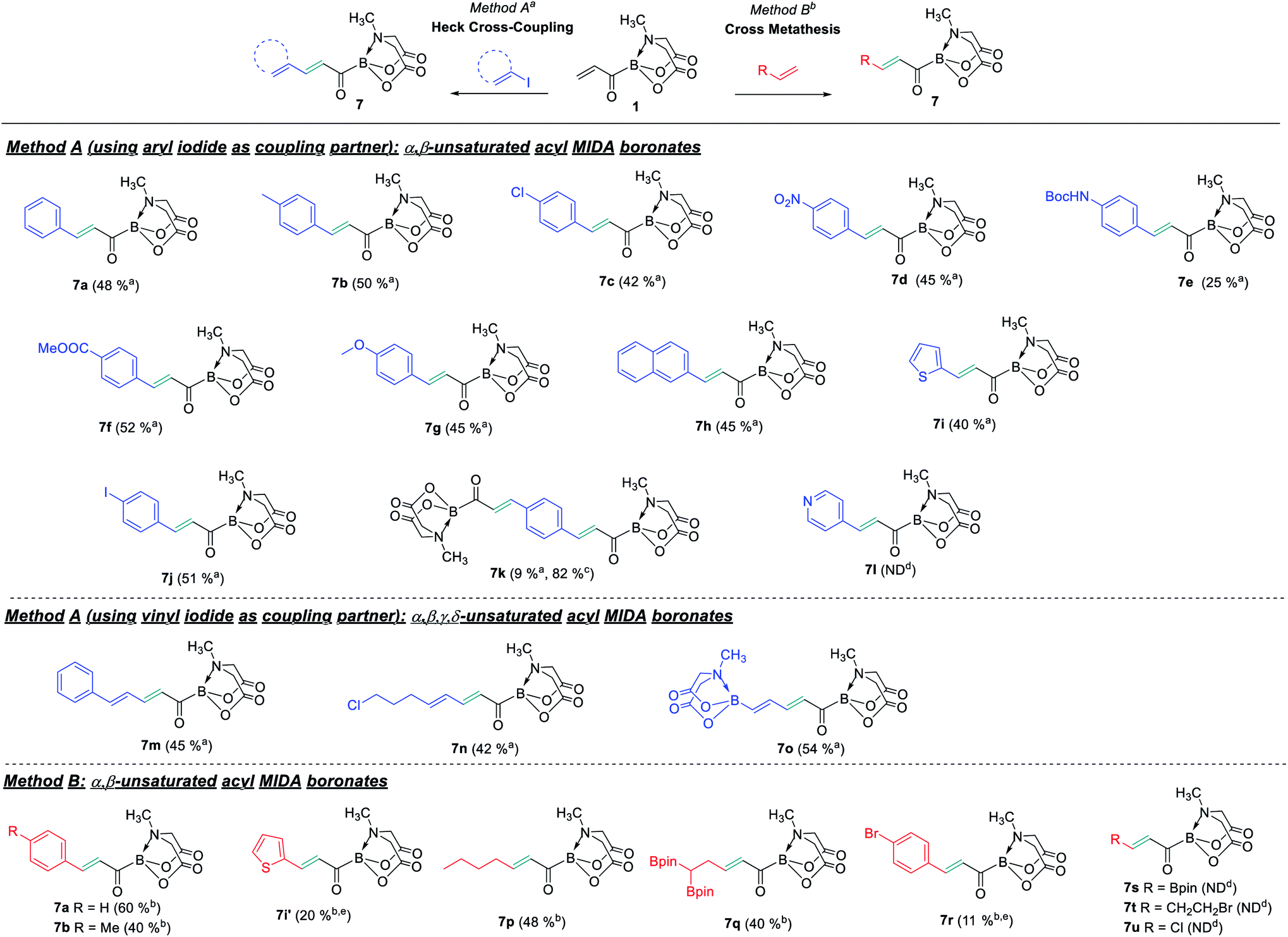
With the acrylic boronate (1) in hand, we investigated its utility as a C3 building block for rapid and catalytic access to diverse acylborons. A screen of reaction conditions (ESI, Table S2†) revealed 1 to be a competent substrate for Heck-type Pd-catalyzed cross-coupling and Pd(OAc)2/AgOAc system provided the best reaction performance (ESI, Table S2†). Next, we tested the scope of this reaction using a variety of aryl/heteroaryl and vinyl halides (Scheme 3). We were pleased to find that both electron-rich and electron-poor aryl halides were compatible, thereby, allowing installation of valuable synthetic handles such as chloro (7c), NO2 (7d), NHBoc (7e) and ester (7f). Heterocyclic motif such as thiophene was tolerated (7i), however, 4-iodopyridine was not a suitable coupling partner (7l). Interestingly, the use of 1,4-diiodobenzene as coupling partner afforded 4-iodoaryl substituted product (7j) and the unprecedented bis-α,β unsaturated acylboron (7k). It is worthwhile to mention that the previous reports provided α,β-unsaturated acylborons having mainly unsubstituted alkyl or aromatic groups.2c,3c,f Significantly, the acrylic boronate-based approach also furnished a straightforward entry to α,β,γ,δ-unsaturated acylborons (7m–7o) via cross-coupling with readily available vinyl iodides. To the best of our knowledge, this is the first example of acylborons containing α,β,γ,δ-unsaturated functional groups. Furthermore, the mild reaction conditions allowed the use of vinyl halo-boronates leading to the installation of synthetically useful vinyl BMIDA motif on the acylboron (7o).
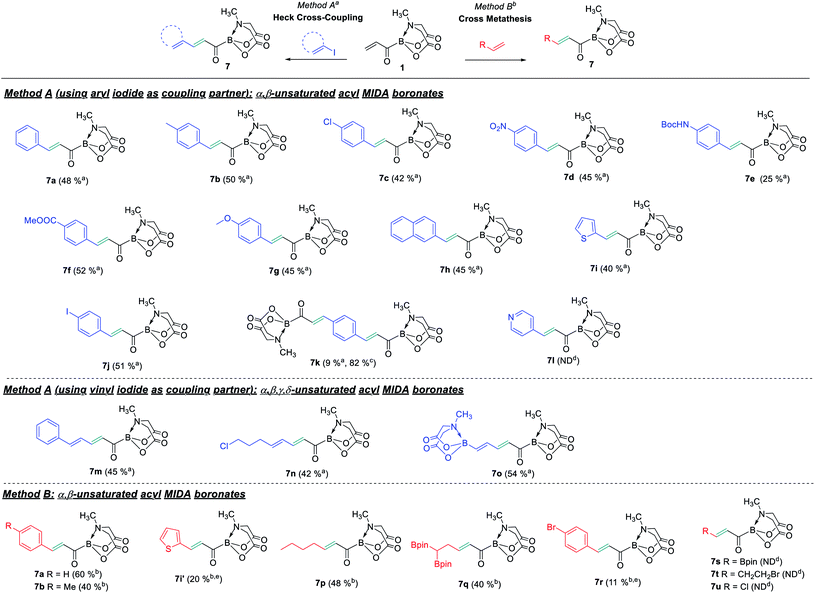 |
| | Scheme 3 Acrylic boronate (1) as a platform for catalytic synthesis of functionalized conjugated acylborons. a1, halide partner (1.5 equiv.), AgOAc (1.5 equiv.), Pd(OAc)2 (5 mol%), CH3CN, 80 °C, 14 h; b1, vinyl partner (2 equiv.), Hoveyda–Grubbs catalyst 2nd (15 mol%), Cl(CH2)2Cl (0.2 M), 55 °C, 14 h; c1 (3 equiv.), 1,4-diiodobenzene (1 equiv.), Pd(OAc)2 (15 mol%), AgOAc (2.5 equiv.), CH3CN, 80 °C, 18 h; dnot detected; einseparable mixture of product and unreacted 1, determined by NMR. | |
To expand the synthetic utility of 1, we tested the compatibility of 1 towards various Cross-Metathesis (CM) reaction conditions in the presence of commercially available stilbene or styrene. Encouragingly, the desired CM product was obtained in 60% yield using Hoveyda–Grubbs catalyst under mild conditions (7a, Scheme 3). Other α,β-unsaturated acylborons having aromatic or alkyl substituents could also be obtained (7b, 7p, Scheme 3). Further, this strategy allowed the preparation of acylboron having the synthetically useful geminal diboron group (7q) which would be difficult to install using existing methods. The CM of 1 with some terminal olefins led to incomplete conversion and an inseparable mixture of desired product and unreacted 1 was obtained after column chromatography (7i′, 7r, Scheme 3 and ESI, Table S4†). The installation of Bpin (7s), alkyl halide (7t) or chloro group (7u) was not feasible using the CM approach.
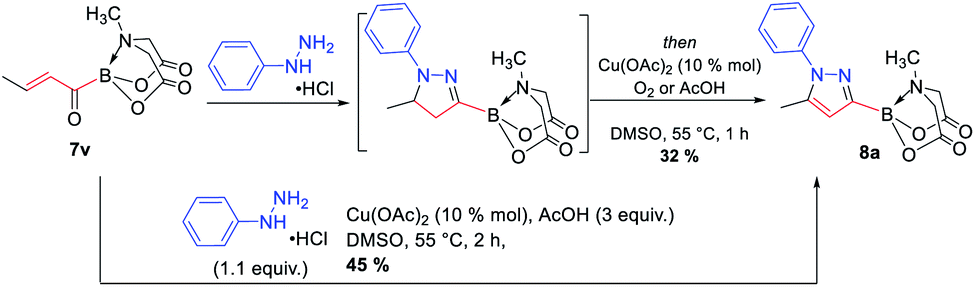
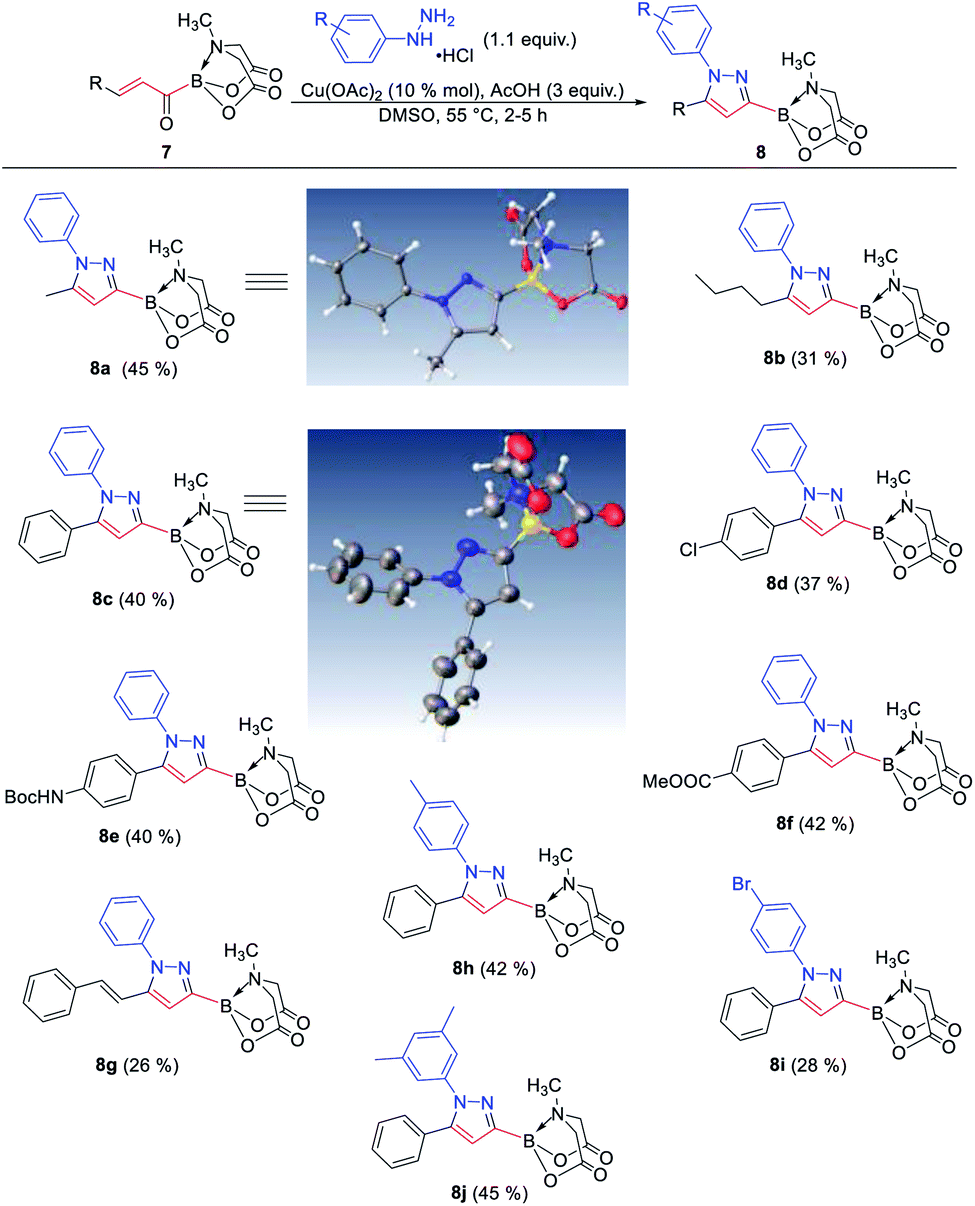
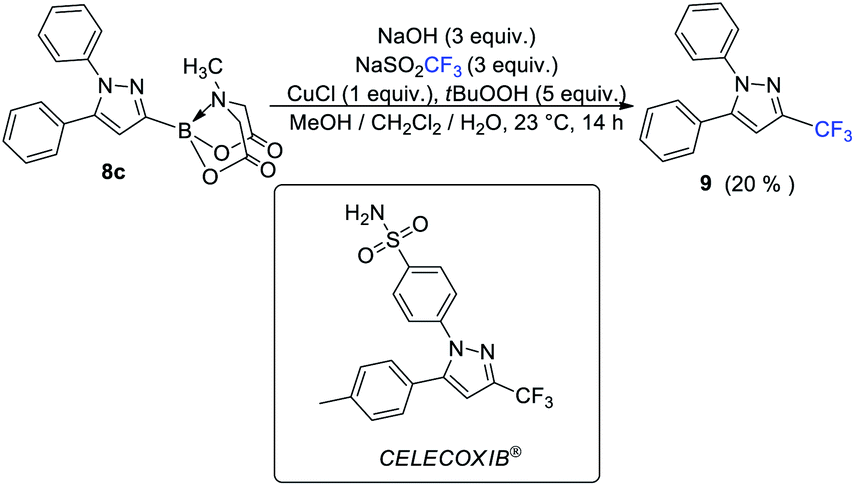
The availability of a series of α,β- and α,β,γ,δ-unsaturated acylborons motivated us to investigate their synthetic utility. In particular, we wondered if these conjugated acylborons could be used as linchpins to access boryl substituted heterocycles that would be difficult to prepare using conventional methods. For instance, C3-borylation of pyrazoles is difficult to accomplish using current approaches. Halogenation–borylation or C–H borylation of pyrazoles leads to only 4-boryl products,7 presumably due to the increased electrophilicity at the C4 position. Further, these methods are not compatible with substrates having electrophilic groups. The 1,3-dipolar cycloaddition of N-arylsydnones with ethynyl MIDA boronate gives a mixture8a of 3- and 4-boryl pyrazoles besides requiring very high temperature (165–180 °C) or electron withdrawing substituents (para-nitro) on the N-phenyl group or unsubstituted C-5 position to favor C-3 borylation.8b We envisioned that α,β-unsaturated acylborons could provide the 3-boryl pyrazoles in a regioselective fashion via cyclocondensation–oxidation reaction with aryl hydrazines.8c Thus, 7v was heated in the presence of phenylhydrazine hydrochloride to give the pyrazoline which was oxidized into the desired 3-boryl pyrazole (Scheme 4). The regioselectivity of this reaction was confirmed by X-ray analysis. Further screening of reaction conditions (ESI, Table S5†) revealed that this boryl-heterocyclization could be accomplished in one-pot using catalytic Cu(OAc)2 in the presence of acetic acid (Scheme 4). This optimized condition was compatible with α,β-unsaturated acylboron substrates having alkyl, substituted aryl as well as styryl groups (8a–8g, Scheme 5). Furthermore, several substituted phenylhydrazines (8h–8j) were viable reaction partners, thereby, opening up a general and highly regioselective route to rare 3-boryl pyrazoles. These 3-boryl pyrazoles are expected to be valuable reagents for late-stage diversification of bioactive compounds. For instance, 3-fluoroalkyl pyrazoles (e.g. Celecoxib, Scheme 6) are anti-inflammatory drugs9 and the 18F labelled 3-fluoroalkyl pyrazoles have attracted interest as PET (Positron Emission Tomography) imaging agents.10 On the other hand, trifluoromethylation of aryl/heteroarylboronic acids is a powerful method due to its remarkable suitability for radiofluorination.11 Thus, it would be desirable to establish bench stable 3-boryl pyrazoles as substrates for late-stage fluorination/fluoroalkylation. To this end, an analog of Celecoxib was successfully prepared (9, 20% unoptimized yield, Scheme 6) via hydrolysis of BMIDA followed by Cu-promoted12 trifluoromethylation of the resulting boronic acid in the same pot.
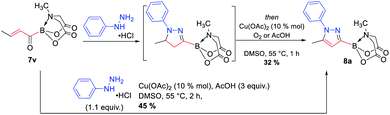 |
| | Scheme 4 Conversion of α,β-unsaturated acylboron into 3-boryl pyrazole via sequential or one-pot condensation–oxidation. | |
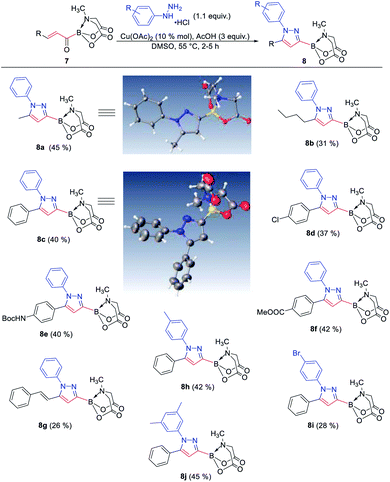 |
| | Scheme 5 Regioselective synthesis of diverse 3-boryl pyrazoles. X-ray crystal structures of two representative boryl pyrazoles (8a, 8c) are also shown. | |
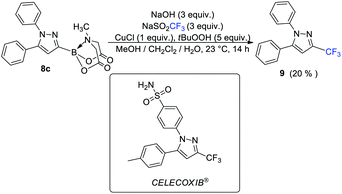 |
| | Scheme 6 Conversion of 3-boryl pyrazole into Celecoxib analog via late-stage trifluoromethylation. | |
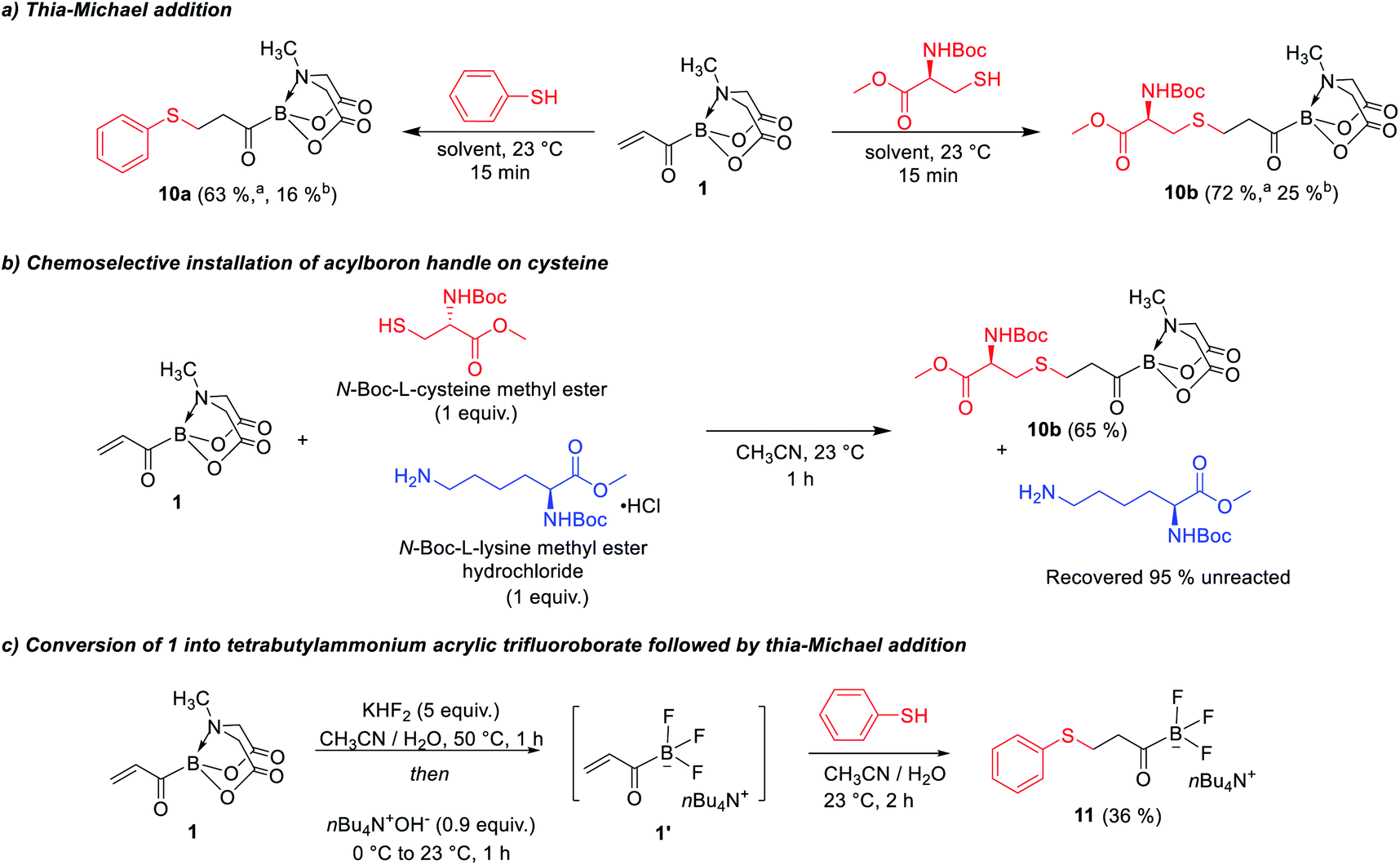
We were also intrigued by the potential of acrylic boronate and its derivatives as a new class of heterobifunctional bioconjugation agents (Scheme 7). Thiol-selective conjugate addition on maleimides is a powerful bioconjugation technique.13 The installation of reactive handles on cysteines is an important strategy for site-selective functionalization of proteins and formation of protein–protein conjugates. There has also been considerable interest in development of bifunctional13b,14 thiol-reactive reagents that convert cysteines into electrophilic handles to enable downstream reactions with nucleophilic amino acids or fluorescent probes. Previous reports have described various bis-electrophilic reagents,13,14 however, these approaches often provide unnatural linkages between the cysteine and the second nucleophile. We became interested to investigate the utility of acrylic boronate 1 as a novel bis-electrophilic platform that enables natural bond-forming thioether and amide ligations. Thus, 1 was treated with a thiol or cysteine and we were pleased to observe that the Michael addition was completed within 15 min (Scheme 7a) yielding the β-thioether acylborons. Furthermore, reaction of an equimolar mixture of cysteine and lysine with 1 exclusively provided the thia-Michael product along with unreacted lysine, thereby indicating the suitability of 1 for site selective modification of cysteine (Scheme 7b). The thioether acylborons (10a and 10b) were viable substrates for rapid amide ligations,4bvia reaction with O–Me hydroxylamine (ESI, Scheme S5†). To further investigate the utility of acrylic boronate (1) as a potential bioconjugation agent, we studied the conversion of 1 into the respective trifluoroborate salt followed by thia-Michael addition or amide ligation. The KHF2 mediated conversion of 1 into the respective acrylic trifluoroborate salt4b and subsequent thiol addition was feasible (confirmed by mass spectrometry). However, purification of the resulting β-thioether potassium acyl trifluoroborate was challenging because these potassium salts are not amenable to column chromatography. To overcome this challenge, we developed an alternative strategy, wherein, 1 was converted into the tetra-n-butyl ammonium (TBA) acrylic trifluoroborate (1′, Scheme 7c) via treatment with KHF2 followed by counterion exchange with tetra-n-butyl ammonium hydroxide in the same pot. A simple aqueous workup of the reaction mixture followed by thiol addition successfully provided the desired β-thioether TBA acyl trifluoroborate (11, Scheme 7c) which was stable to silica gel column chromatography. In a complementary approach, we also confirmed the feasibility of converting 1 into the respective potassium acyl trifluoroborate salt followed by amide ligation in the same-pot (ESI, Scheme S7†). Taken together, the above results provide acrylic boronates as promising bifunctional reagents for bioconjugation applications via thia-Michael and amide conjugation. In addition, the developed mild and diversity-oriented access to a range of functionalized α,β-(7a–7j, 7p, 7q)/α,β,γ,δ-(7m–7o)/bis-α,β (7k) unsaturated acylborons (Scheme 3) provides a versatile platform to fine-tune the thia-Michael/amide ligation reactivity (via modulating the electrophilicity of acryloyl group) and the linker length for specific chemical biology applications.
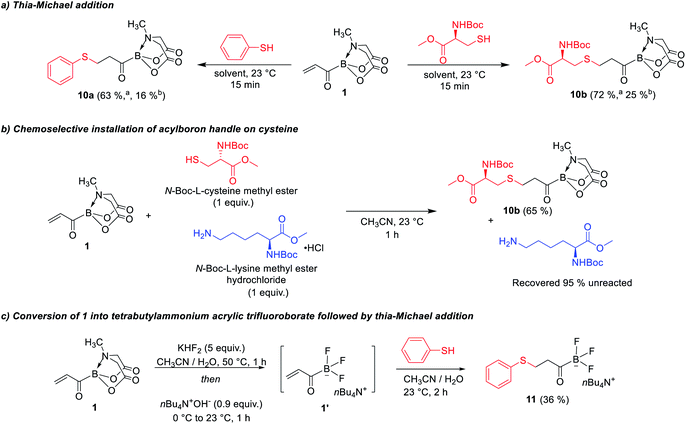 |
| | Scheme 7 Applications of acrylic boronate as a heterobifunctional ligation reagent. aUsing CH3CN as solvent; busing CH3CN/H2O (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as solvent. :1) as solvent. | |
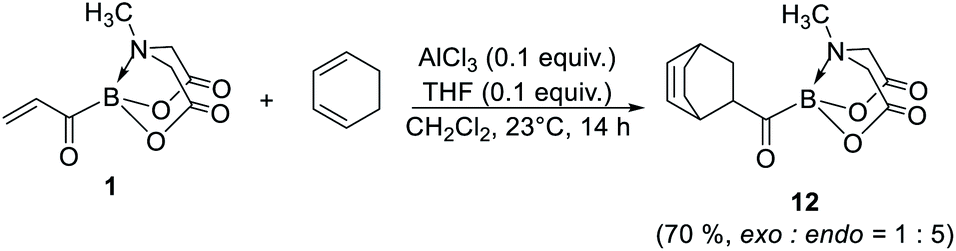
Finally, 1 was also found to be a competent dienophile for Diels–Alder reaction (12, Scheme 8). Importantly, this reaction provides a promising route to bicyclic acylborons which would be difficult to access using currently available approaches.
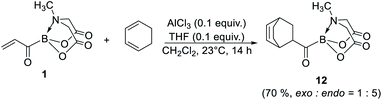 |
| | Scheme 8 Synthetic utility of 1 to access bicyclic acylboron via 4 + 2 cycloaddition. | |
Conclusions
In summary, we have developed acrylic boronate as a novel C3 building block that combines the synthetic versatility of acryloyl group and the unique reactivity of acylboron into a single chimeric functional group. Acrylic boronate enabled a mild, catalytic and general route to several previously inaccessible functionalized α,β-unsaturated acylborons via Pd-catalyzed coupling or cross-metathesis using readily available aryl/vinyl iodides and olefins as coupling partners. Furthermore, the methodology provided access to two new types of acylborons: α,β,γ,δ and bis-α,β unsaturated acylborons. The synthetic utility of α,β-unsaturated acylborons was demonstrated for the first time by engaging them in one-pot cyclocondensation–oxidation to afford rare 3-boryl pyrazoles in a highly regioselective fashion. These 3-boryl-heterocycles allowed a late-stage fluoroalkylation approach to Celecoxib analog which has potential application for synthesis of PET agents. In another new application of these conjugated acylborons, we show that acrylic boronates are promising heterobifunctional agents for fast and chemoselective installation of acylboron handles on cysteine which opens-up opportunities for dual functionalization and conjugation of peptides/proteins via orthogonal thioether and amide ligations.
Author contributions
SL and AS designed the study. Experimental work was performed by SL and LW. AS supervised the work. All authors co-wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Support for this research from NIH-NIGMS (R15GM135891) and Stevens Institute of Technology is gratefully acknowledged. We thank Dr Athula Attygalle and Zhaoyu Zheng for HRMS analysis. Single crystal X-ray diffraction studies were performed by Dr Manju Rajeswaran and Dr Daniel Paley in the Shared Materials Characterization Laboratory at Columbia University.
References
-
(a) F. K. Scharnagl, S. K. Bose and T. B. Marder, Org. Biomol. Chem., 2017, 15, 1738–1752 RSC;
(b) D. Wu, J. Taguchi, M. Tanriver and J. W. Bode, Angew. Chem., Int. Ed., 2020, 59, 16847–16858 CrossRef CAS PubMed.
-
(a) Z. He, P. Trinchera, S. Adachi, J. D. St Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2012, 51, 11092–11096 CrossRef CAS PubMed;
(b) M. L. Lepage, S. Lai, N. Peressin, R. Hadjerci, B. O. Patrick and D. M. Perrin, Angew. Chem., Int. Ed., 2017, 56, 15257–15261 CrossRef CAS PubMed;
(c) S. Lin, L. Wang, N. Aminoleslami, Y. Lao, C. Yagel and A. Sharma, Chem. Sci., 2019, 10, 4684–4691 RSC;
(d) A. Holownia, C.-H. Tien, D. B. Diaz, R. T. Larson and A. K. Yudin, Angew. Chem., Int. Ed., 2019, 131, 15292–15297 CrossRef;
(e) Y. M. Ivon, I. V. Mazurenko, Y. O. Kuchkovska, Z. V. Voitenko and O. O. Grygorenko, Angew. Chem., Int. Ed., 2020, 59, 18016–18022 CrossRef CAS;
(f) D.-H. Tan, Y.-H. Cai, Y.-F. Zeng, W.-X. Lv, L. Yang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2019, 131, 13922–13926 CrossRef;
(g) C. F. Lee, A. Holownia, J. M. Bennett, J. M. Elkins, J. D. St Denis, S. Adachi and A. K. Yudin, Angew. Chem., Int. Ed., 2017, 56, 6264–6267 CrossRef CAS PubMed;
(h) S. Adachi, S. K. Liew, C. F. Lee, A. Lough, Z. He, J. D. St Denis, G. Poda and A. K. Yudin, Org. Lett., 2015, 17, 5594–5597 CrossRef CAS PubMed.
-
(a) A. M. Dumas and J. W. Bode, Org. Lett., 2012, 14, 2138–2141 CrossRef CAS PubMed;
(b) J. Taguchi, T. Ikeda, R. Takahashi, I. Sasaki, Y. Ogasawara, T. Dairi, N. Kato, Y. Yamamoto, J. W. Bode and H. Ito, Angew. Chem., Int. Ed., 2017, 56, 13847–13851 CrossRef CAS PubMed;
(c) D. Wu, N. A. Fohn and J. W. Bode, Angew. Chem., Int. Ed., 2019, 58, 11058–11062 CrossRef CAS PubMed;
(d) J. Taguchi, T. Takeuchi, R. Takahashi, F. Masero and H. Ito, Angew. Chem., Int. Ed., 2019, 58, 7299–7303 CrossRef CAS PubMed;
(e) L.-J. Cheng, S. Zhao and M. Neal, Angew. Chem., Int. Ed., 2020, 60, 2094–2098 CrossRef PubMed;
(f) A. Schuhmacher, S. J. Ryan and J. W. Bode, Angew. Chem., Int. Ed., 2021, 60, 3918–3922 CrossRef CAS PubMed.
-
(a) H. Noda, G. Erős and J. W. Bode, J. Am. Chem. Soc., 2014, 136, 5611–5614 CrossRef CAS PubMed;
(b) H. Noda and J. W. Bode, Chem. Sci., 2014, 5, 4328–4332 RSC;
(c) A. O. Gálvez, C. P. Schaack, H. Noda and J. W. Bode, J. Am. Chem. Soc., 2017, 139, 1826–1829 CrossRef;
(d) C. J. White and J. W. Bode, ACS Cent. Sci., 2018, 4, 197–206 CrossRef CAS.
-
(a) A. Rybak and M. A. R. Meier, Green Chem., 2007, 9, 1356–1361 RSC;
(b)
T. Ohara, T. Sato, N. Shimizu, G. Prescher, H. Schwind, O. Weiberg, K. Marten, H. Greim, T. D. Shaffer and P. Nandi, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2020, pp. 1–21 Search PubMed;
(c) A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed;
(d) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS;
(e) Y. Dong, J. B. Matson and K. J. Edgar, Biomacromolecules, 2017, 18, 1661–1676 CrossRef CAS.
- E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed.
-
(a) M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287–4299 CrossRef CAS PubMed;
(b) E. Yamamoto, S. Ukigai and H. Ito, Chem. Sci., 2015, 6, 2943–2951 RSC.
-
(a) T. Delaunay, M. Es-Sayed, J.-P. Vors, N. Monteiro and G. Balme, Chem. Lett., 2011, 40, 1434–1436 CrossRef CAS;
(b) D. L. Browne, J. F. Vivat, A. Plant, E. Gomez-Bengoa and J. P. A. Harrity, J. Am. Chem. Soc., 2009, 131, 7762–7769 CrossRef CAS PubMed;
(c) Y. R. Huang and J. A. Katzenellenbogen, Org. Lett., 2000, 2, 2833–2836 CrossRef CAS PubMed.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347–1365 CrossRef CAS PubMed.
- J. Prabhakaran, M. D. Underwood, R. V. Parsey, V. Arango, V. J. Majo, N. R. Simpson, R. Van Heertum, J. J. Mann and J. S. D. Kumar, Bioorg. Med. Chem., 2007, 15, 1802–1807 CrossRef CAS PubMed.
-
(a) P. Novák, A. Lishchynskyi and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 7767–7770 CrossRef PubMed;
(b) P. Ivashkin, G. Lemonnier, J. Cousin, V. Grégoire, D. Labar, P. Jubault and X. Pannecoucke, Chem.–Eur. J., 2014, 20, 9514–9518 CrossRef CAS PubMed.
- Y. Ye, S. A. Künzi and M. S. Sanford, Org. Lett., 2012, 14, 4979–4981 CrossRef CAS PubMed.
-
(a) P. Ochtrop and C. P. R. Hackenberger, Curr. Opin. Chem. Biol., 2020, 58, 28–36 CrossRef CAS PubMed;
(b) J. M. J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem.–Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- A. L. Baumann, S. Schwagerus, K. Broi, K. Kemnitz-Hassanin, C. E. Stieger, N. Trieloff, P. Schmieder and C. P. R. Hackenberger, J. Am. Chem. Soc., 2020, 142, 9544–9552 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2061438 and 2061439. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00905b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*