
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bond-induced selectivity of a head-to-head photo-dimerisation of dialkynylanthracene – access to tetradentate Lewis acids†
Philipp
Niermeier
 ,
Kristina A. M.
Maibom
,
Jan-Hendrik
Lamm
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
,
Kristina A. M.
Maibom
,
Jan-Hendrik
Lamm
,
Beate
Neumann
,
Hans-Georg
Stammler
and
Norbert W.
Mitzel
*
Universität Bielefeld, Fakultät für Chemie, Lehrstuhl für Anorganische Chemie und Strukturchemie (ACS), Centre for Molecular Materials (CM2), Universitätsstr. 25, D-33615 Bielefeld, Germany. E-mail: mitzel@uni-bielefeld.de; Web: https://www.uni-bielefeld.de/fakultaeten/chemie/ag/ac3-mitzel/
First published on 6th May 2021
Abstract
Using 2-hydroxypropyl-protecting groups, 1,8-dialkynylanthracene photo-dimers were prepared in head-to-head-configuration by UV irradiation on a multi-gram scale. In non-polar solvents, the combination of non-covalent hydrogen bonds and π–π-interactions induces the formation of the syn-isomer in up to 85% yield. Instead, more polar solvents or irradiation of unprotected 1,8-diethynylanthracene led to formation of the corresponding anti-isomer in large excess. Cleavage of the protecting groups under basic conditions affords a rigid hydrocarbon skeleton with four directional functions. This was used as a building block for a tetradentate boron Lewis acid. Its applicability as a host for Lewis-base substrates was demonstrated by the formation of adducts with various nitrogen bases. Adduct formation with hydrazine leads to impressive networks between the tetraboron host and the substrate molecules.
Introduction
The growing field of main group poly-Lewis acids (PLAs) finds an increasing number of applications in anion sensing1/transport,2 small molecule recognition3 and catalysis.4 Actually, the majority of PLAs are based on group-13-element functions.3d–f,5Depending on size and geometry of their cavity, tailor-made difunctional PLAs with rigid organic backbones were demonstrated to bind different substrates with high selectivity. Bisboranes with short-spacing 1,2-phenylenediyl (I) or 1,8-naphthalenediyl frameworks (II) are most suitable for the μ(1,1)-chelation of anions such as hydride,6 fluoride,7 chloride8 or azide,7d,8c while larger 1,8-biphenylenediyl (III) and 1,8-triptycenediyl derivatives (IV) are capable of forming μ(1,2)-chelates with substrates having adjacent binding sites such as cyanide or hydrazine.3e

Larger cavities (ca. 5 Å) based on functionalised 1,8-diethinylanthracenes allow the formation of stable μ(1,3)-adducts with pyrimidine.3f,9 However, while these difunctional receptors are comparatively simple, those with more than two binding sites are rather scarce.10 Tetradentate derivatives, with a doubled number of functions, are in principle accessible in the form of the corresponding head-to-head anthracene photo-dimers B instead of their monomers A (Scheme 1).11 However, the main problem associated with photo-dimerising 1,8-functional anthracenes is the formation of a mixture of two isomers, syn (head-to-head) and anti (head-to-tail). Owing to steric and electronic parameters, the head-to-tail isomer is usually formed in large excess.12
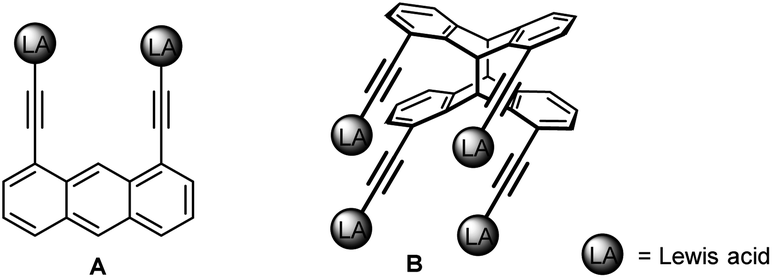 | ||
| Scheme 1 Bidentate (A) and tetradentate (B) Lewis acids with directed functions based on 1,8-diethynylanthracene (1) and its syn-photo-dimer syn-2. | ||
Recently, we reported 1,8-dialkynylanthracenes linked in positions 10 and 10′ by short hydrocarbon chains (CH2)n (n = 2,3; C, Scheme 2).13 Their irradiation with UV light led to a selective head-to-head arrangement of the substituents (D). However, the required bisanthracene precursors are available only in very limited quantities and this clearly hinders their use for further developments in the direction of PLAs.
 | ||
| Scheme 2 Restrained photo-“dimerization” of a 10,10′-linked bisanthracenes C leads to syn-photo-“dimers” D. | ||
Instead of covalent bonding, non-covalent interactions such as hydrogen bonds and π,π-interactions could also have the desired effects on the isomer ratio during photo-dimerisation. In the past, it has been already shown for some anthracenecarboxylic acids, that non-covalent interactions can affect the syn/anti ratio during photo-cyclodimerisation. To this purpose, various template strategies were applied using cyclodextrines,14a cucurbituriles14b or photoresponsive organogels.14c In addition, two anthracenecarboxylic acid molecules can be preorganised to a kind of template complex by, for example, diamines14d or β-aminoalcohols.14e However, a selective head-to-head photocyclomerization without the use of additives leading to a precursor for the design of further spatially defined systems on a gram scale has not yet become known.
Results and discussion
To investigate the photo-chemistry of 1,8-diethynylanthracene (1; Scheme 3), we now performed UV irradiation experiments on the NMR scale in various deuterated solvents. We found, that at constant concentrations of 1 (3.33 mg mL−1), the fraction of syn-2 was about 10% greater in high-polarity solvents such as DMSO or DMF (≈32%) than in non- or low-polar solvents such as benzene or chloroform (21%; see Table S1, ESI†). In DMF, however, the fraction of syn-2 was significantly lower at higher concentrations of 1. | ||
| Scheme 3 The head-to-head photo-dimer syn-2 and the head-to-tail photo-dimer anti-2 are obtained upon UV irradiation of 1,8-diethynylanthracene (1). | ||
For the preparation of syn-2 on a gram scale we irradiated solutions of monomer 1 in a photo-reactor in degassed DMF or dichloromethane for several days. Their similar polarities and identical molecular weights made the separation of both isomers difficult. Only small amounts of syn-2 per run could be separated by column chromatography on silica gel or preparative HPLC, leaving the bulk as an inseparable mixture of the two compounds. The combination of crystallisation and precipitation proved to be a more efficient method of purification (see Experimental section). Regardless of the solvent and the method of purification, syn-2 could be isolated in this way in only about 18% yield. This, together with the very time- and solvent-consuming work, prompted us to develop new and more sophisticated strategies to improve the chemoselectivity for the syn-isomer. We focused on investigating the influence of the alkyne protection group concerning the syn/anti ratio upon UV irradiation. For this purpose, we synthesised 1,8-di(3-methyl-3-hydroxy-1-butynyl)anthracene (5)15 by Sonogashira–Hagihara cross-coupling starting from 1,8-diiodoanthracene (3)16 (Scheme 4). By slight modification of the reaction conditions, the yield was significantly increased from 46% to 95%. Anthracene-1,8-diyl bis(trifluoromethanesulfonate) (4)17 as starting material afforded 5 in only 81% yield, however, this route is favourable due to the good accessibility of 4.
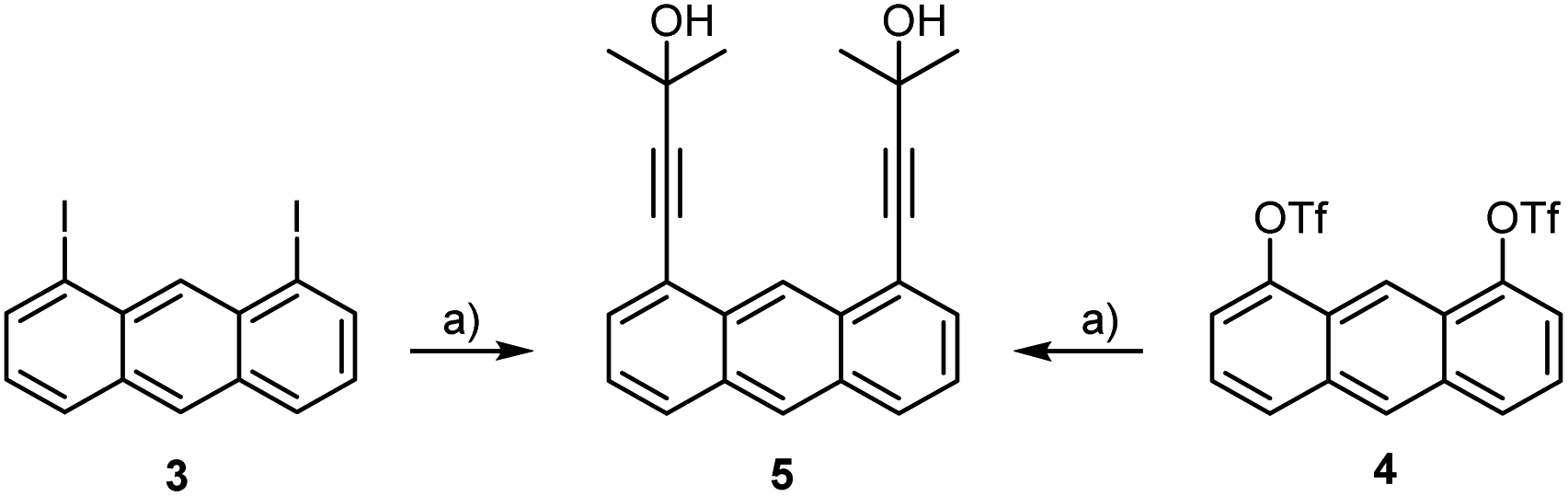 | ||
| Scheme 4 Synthesis of dialkynylanthracene 5 starting from 3 (95%) or 4 (81%), respectively; a) 2-methylbut-3-yn-2-ol, PdCl2(PPh3)2, CuI, diisopropylamine, reflux. | ||
The molecular structure of 5 in the crystalline state (Fig. 1) shows an almost parallel arrangement of two anthracene monomers and the formation of intermolecular hydrogen bonds between the hydroxyl groups. The relatively short distances between the anthracene rings indicate the presence of π–π-interactions. Most likely, this combination of hydrogen bonds and π-stacking18 is the reason for the peculiar arrangement of the anthracene monomers, in which all alkynyl-substituents are oriented in the same direction.
 | ||
| Fig. 1 Molecular structure of dialkynylanthracene 5 in the crystal. Displacement ellipsoids are drawn at 50% probability level. Hydrogen atoms, except those of OH groups, are omitted for clarity. Dotted blue lines indicate hydrogen bonds. For selected bond lengths and angles, see Fig. S18† (ESI). | ||
A similar pre-orientation of the anthracene molecules during dimerization in solution should lead to a preferential formation of the corresponding syn-photo-dimer syn-6 (Scheme 5).
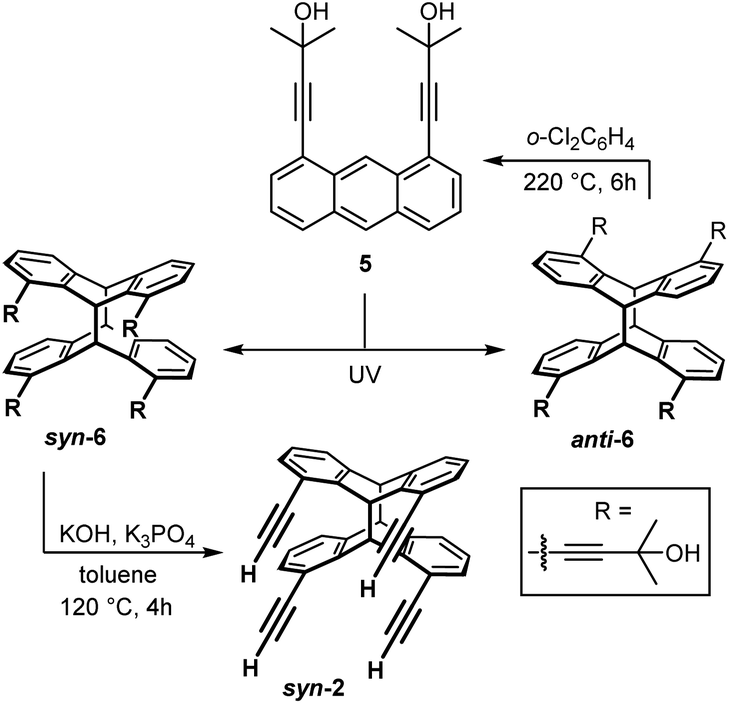 | ||
| Scheme 5 Photo-dimerization of dialkynylanthracene 5. The desired head-to-head isomer syn-6 was converted to syn-2 by cleavage of the hydroxypropyl-protecting groups; anti-6 can be thermally reconverted into its monomers 5. | ||
In order to prove this hypothesis, UV irradiation experiments were performed in analogy to the NMR experiments described above for 1,8-diethynylanthracene (1). The results are shown in Table 1. Compared to the photo-dimerization of 1 the influence of the solvent is much stronger and the polarity of the solvent has the contrary effect on the syn/anti ratio. Thus, syn-6 is formed with less than 10% in very polar solvents such as DMSO, but with over 80% when using non-polar solvents such as cyclohexane (C6D12) or methylcyclohexane (C7D14). The syn/anti ratio remains unaltered upon increasing the concentration of 5 from 4.0 to 20.0 mg mL−1.
| Solvent | c [mg mL−1] | t [h] | syn-6 [%] | anti-6 [%] |
|---|---|---|---|---|
| DMSO-d6 | 4.00 | 15 | 8 | 92 |
| DMF-d7 | 4.00 | 9 | 8 | 92 |
| CD3CN | 4.00 | 9 | 19 | 81 |
| Acetone-d6 | 4.00 | 3 | 16 | 84 |
| THF-d8 | 4.00 | 9 | 13 | 87 |
| MeOD | 4.00 | 9 | 11 | 89 |
| CD2Cl2 | 4.00 | 9 | 47 | 53 |
| CDCl3 | 4.00 | 9 | 47 | 53 |
| C6D6 | 4.00 | 3 | 51 | 49 |
| Cyclohexane-d12 | 4.00 | 9 | 85 | 15 |
| Cyclohexane-d12 | 20.0 | 25 | 85 | 15 |
| Methylcyclohexane-d14 | 4.00 | 9 | 84 | 16 |
| Methylcyclohexane-d14 | 20.0 | 25 | 84 | 16 |
The preorganisation of monomers 5 in non-polar solvents by hydrogen bonding and π-stacking prior to UV irradiation causes the preferential formation of syn-6. The selectivity for the formation of syn-6 is lower in benzene, chloroform and methylene chloride on the one hand, but much higher in saturated hydrocarbons such as cyclohexane or methylcyclohexane. A possible explanation is a stronger solvation of the anthracene units by dispersion forces in the first group of solvents, leading to dimers without stacked anthracene units, i.e. a less perfect preorganization. In contrast, in saturated hydrocarbon solvents, π-stacking between anthracene units is more dominant, leading to a preferential formation of syn-6. In turn, in more polar solvents, the solvent molecules interact with the hydroxyl groups of 5 and cleave off the preorganised dimers, resulting in the preferential formation of anti-6 for steric and electronic reasons. At this point, it is important to mention that monomer 5 can be recovered from anti-6 in thermal cycloreversion reactions by simply heating solutions in o-dichlorobenzene (Scheme 5).
For the conversion of monomer 5 on a preparative scale (≥5 g), all further experiments were carried out in cyclohexane using a photo-reactor operating at 350 nm. While the NMR experiments led to a complete conversion of 5 after several hours, the multi-gram reaction required irradiation for almost two weeks. After twelve days, small amounts of 5 were still detected by 1H NMR spectroscopy. To ensure complete conversion of the reactant, dichloromethane was added and the reaction mixture was irradiated for further two days. After removing the solvent under reduced pressure, syn-6 was obtained in good yield of 82% by column chromatography. Small amounts of anti-6 (<10%) were subsequently eluted with higher polar solvents. This head-to-tail isomer is almost completely insoluble in dichloromethane and other nonpolar solvents, which is due to its chain structure with intra- and intermolecular hydrogen bonds in the solid state (Fig. 2, right).
 | ||
| Fig. 2 Molecular structures of the photo-dimers syn-6 (left) and anti-6 (right, two molecules of an infinite chain) in the crystalline state. Displacement ellipsoids are drawn at the 50% probability level. Solvent molecules and hydrogen atoms (except OH groups), are omitted for clarity. Dotted lines indicate hydrogen bonds. For selected bond lengths and angles, see Fig. S21 (syn-6) and Fig. S22 (anti-6), respectively (ESI).† | ||
Apparently, these non-polar rods can only be fragmented by very polar solvents, to compensate for the loss of two hydrogen bonds per monomer. In contrast, syn-6 features four OH groups each interacting with a neighbouring one by intramolecular hydrogen bonds (Fig. 2 left); this saturates the polar groups, and none of these hydrogen bonds have to be cleaved upon dissolution of syn-6, which explains its better solubility in non-polar solvents.
The hydrocarbon syn-2 was obtained from syn-6 in up to 85% yield by elimination of acetone under basic conditions (Scheme 5), applying a protocol by Schmidt et al.19Fig. 3 shows different views on the molecular structure of syn-2, determined by single-crystal X-ray diffraction. All four substituents point in the same direction. The alkynyl-moieties are nearly linear. The alkyne protons are separated by approximately 3.7 and 5.1 Å (see Fig. 3). The bonds between the bridgehead carbon atoms are 1.62 Å, which is in the characteristic range for 9,10:9′,10′-anthracene photo-dimers.20
 | ||
| Fig. 3 Different views on the molecular structure of photo-dimer syn-2 in the crystalline state. Displacement ellipsoids are drawn at 50% probability level. Hydrogen atoms, except those of the ethynyl groups, are omitted for clarity. The values shown are averaged distances between the ethynyl protons. For selected bond lengths and angles, see Fig. S19 (ESI).† | ||
Having established a much simplified access to larger amounts of syn-2, we investigated its conversion to tetrafunctional Lewis acids, following our recent work on poly-Lewis acid chemistry.3f The terminal alkyne functions of syn-2 were first converted by quantitative stannylation followed by tin–boron exchange with chlorodiphenylborane (Scheme 6).
 | ||
| Scheme 6 Synthesis of tetraborane 8 in two steps from precursor syn-2. | ||
Tetra-Lewis-acid 8 was obtained in a very good yield of 95%. It is a moisture-sensitive compound, but otherwise storable under inert gases under cooling to −30 °C for some weeks, but not for unlimited time.
To demonstrate its applicability as a receptor for small molecules, 8 was reacted with different nitrogen bases. Applying four equivalents of pyridine, ammonia (gas) or hydrazine, the formation of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts (9, 10 and 11) with one guest molecule complexed by each of the boron atoms was observed. Their molecular structures are displayed in Fig. 4 and 5. All structures show extremely distorted alkynyl-substituents due to the large steric overload at the binding sites. This results in strongly bent alkynyl functions. The Caryl–C
:1 adducts (9, 10 and 11) with one guest molecule complexed by each of the boron atoms was observed. Their molecular structures are displayed in Fig. 4 and 5. All structures show extremely distorted alkynyl-substituents due to the large steric overload at the binding sites. This results in strongly bent alkynyl functions. The Caryl–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C angles in 9 are 174.8(3), 167.7(3), 165.9(3) and 164.6(3)°, i.e. three out of four are markedly smaller than 170°. An ideal C–CC angle of 180° is seldom found in real compounds, but such large deviation as found in 8 usually occur only in situations of intense steric repulsion. For comparison, 1,8-bis(diphenylboranyl-ethynyl)anthracene,3f the formal monomer of the dimer form 8, has corresponding C–CC angles of 178.1(3) and 177.6(3)°. Even two trimethylsilyl groups in 1,8-bis(trimethylsilyl-ethinyl)anthracene3f lead to only moderate distortions of 178(6)° in the gas phase by electron diffraction and 174.4(1) and 178.5(1)° in the crystal.21
C angles in 9 are 174.8(3), 167.7(3), 165.9(3) and 164.6(3)°, i.e. three out of four are markedly smaller than 170°. An ideal C–CC angle of 180° is seldom found in real compounds, but such large deviation as found in 8 usually occur only in situations of intense steric repulsion. For comparison, 1,8-bis(diphenylboranyl-ethynyl)anthracene,3f the formal monomer of the dimer form 8, has corresponding C–CC angles of 178.1(3) and 177.6(3)°. Even two trimethylsilyl groups in 1,8-bis(trimethylsilyl-ethinyl)anthracene3f lead to only moderate distortions of 178(6)° in the gas phase by electron diffraction and 174.4(1) and 178.5(1)° in the crystal.21
 | ||
| Fig. 4 Molecular structure of the fourfold pyridine adduct 9 (8·4Py) in the crystalline state. Displacement ellipsoids are drawn at 15% probability level. Hydrogen atoms are omitted for clarity. For selected bond lengths and angles, see Fig. S23 (ESI).† | ||
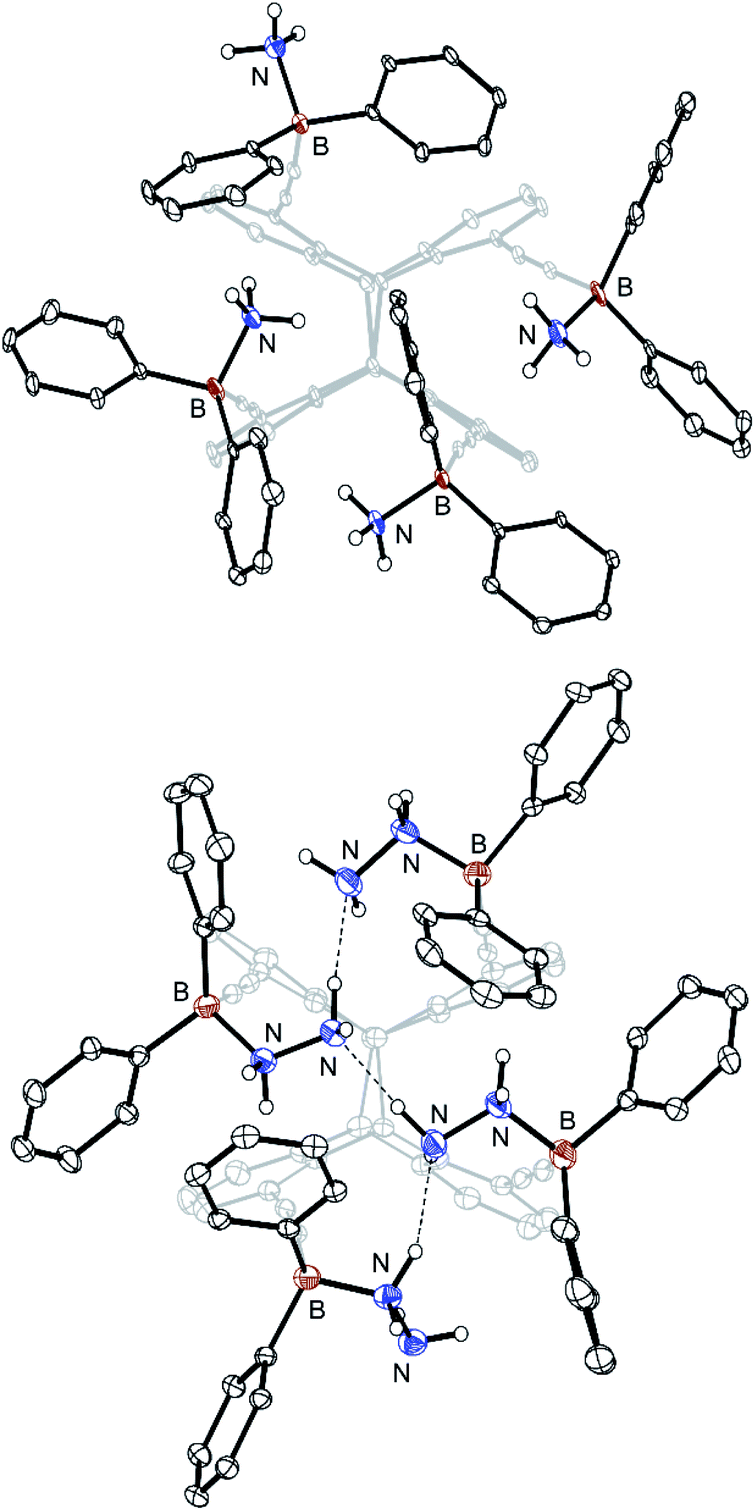 | ||
| Fig. 5 Molecular structure of the tetra-ammonia adduct 10 (8·4NH3; above) and the tetra-hydrazine adduct 11 (8·4N2H4; below) in the crystalline state. Displacement ellipsoids are drawn at 30% (for C) and 50% (for B and N) probability levels. Carbon-bound hydrogen atoms and solvent molecules are omitted for clarity. Dotted lines indicate hydrogen bonds. For selected bond lengths and angles, see Fig. S24† (10) and Fig. S26 (11), respectively (ESI).† | ||
The intramolecular B⋯B distances range from 5.71(1) to 9.17(1) Å for 9, 4.95(1) to 8.34(1) Å for 10 and 5.22(1) to 7.93(1) Å for 11, respectively. The B–N bonds are between 1.62(1) and 1.65(1) Å and therefore above the sum of the covalence radii (1.55 Å (ref. 22)).
In particular, adduct 11 shows an interesting structural motif with three intramolecular hydrogen bonds [N⋯H: 2.0(1) Å]23 between the four hydrazine molecules (Fig. 5, below). All N–N bonds have an identical length of 1.46(1) Å. The B–N–N bond angles range from 112.3(3) to 118.0(3)°. It is noteworthy that 11 formed selectively from benzene, regardless whether 8 was converted with two or four equivalents of hydrazine. However, using CD2Cl2 as the solvent instead, stepwise titration of 8 (0.2 eq. steps) with a total of two equivalents of hydrazine led to the formation of a different crystalline complex 12 (Fig. 6).
 | ||
| Fig. 6 Molecular structure of the penta-hydrazine adduct 12 ([8·5N2H4]n) in the crystalline state. Displacement ellipsoids are drawn at 30% probability level. Carbon-bound hydrogen atoms are omitted for clarity. Symmetry codes: −¼ + y, ¾ − x, −¼ + z for E′ and ¾ − y, ¼ + x, ¼ + z for E′′. Hydrogen bonds, indicated by dotted lines, are in a normal range [1.91(1)–2.11(1) Å; except for N(4)⋯H(6A) with 2.33(3) Å].22 For selected bond lengths and angles, see Fig. S27 (ESI).† | ||
12 is the result of a highly dynamic process, as the 1H NMR spectra (Fig. S11, ESI†) show that a large number of host-guest complexes are present at any time. X-Ray diffraction studies provide the formula [8·5N2H4]n with one hydrazine molecule bridging two units of the tetra-hydrazine adduct 11 in chains. This indicates a solvent dependency of the adduct formation.
In addition to the host–guest experiments mentioned above, several attempts were made to obtain 1:1 complexes of the tetra-Lewis-acid 8 and guest-compounds to demonstrate that the four boron atoms can act cooperatively and bind substrates with one or more Lewis base sites. However, no reaction was observed, when 8 was converted with tetrakis(dimethylamino)ethylene, (Me2N)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(NMe2)2, or with dimethoxymethane, MeOCH2OMe. Mono- and dianionic substrates, such as H–, F–, Cl–, CN–, SCN–, C2O42− and phthalate, C6H4-1,2-(CO2−)2, generated from the corresponding Na, K or PPh4 salts, anthracene-1,8-dicarboxylate or by tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF), respectively, as well as cyanamide, NC–NH2, and 1,2,4,5-tetrazine led to a complete decomposition of the tetra-boron compound 8. Adding tetramethylethylenediamine (TMEDA), tetramethylmethylenediamine (TMMDA), 1H-tetrazole and 1,3-diazine (pyrimidine), we observed unselective complexation reactions, whereas polymerization of the mixture occurred upon addition of the corresponding 1,4-derivative pyrazine.
C(NMe2)2, or with dimethoxymethane, MeOCH2OMe. Mono- and dianionic substrates, such as H–, F–, Cl–, CN–, SCN–, C2O42− and phthalate, C6H4-1,2-(CO2−)2, generated from the corresponding Na, K or PPh4 salts, anthracene-1,8-dicarboxylate or by tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF), respectively, as well as cyanamide, NC–NH2, and 1,2,4,5-tetrazine led to a complete decomposition of the tetra-boron compound 8. Adding tetramethylethylenediamine (TMEDA), tetramethylmethylenediamine (TMMDA), 1H-tetrazole and 1,3-diazine (pyrimidine), we observed unselective complexation reactions, whereas polymerization of the mixture occurred upon addition of the corresponding 1,4-derivative pyrazine.
Conclusions
Essentially, we have established a large-scale and high-yield access to tetraboron Lewis acids based on the head-to-head photo-dimer of 1,8-diethynylanthracene. The key step was to exploit molecular preorientation through hydrogen bonding between 2-hydroxypropyl protecting groups as well as π-stacking between anthracene units, and to use non-polar solvents to favour the formation of such precomplexes. The ability of the new poly-Lewis acid with four directional boron functions to bind and organise a series of small nitrogen bases, encourages us to exploit the cooperativity of the four boron acid sites for other suitable substrates. The photo-dimer syn-6 can also find applications as a hydrocarbon building block for the design of other spatially defined tetrafunctional systems.Experimental
General
The syntheses of 1,8-diethynylanthracene (1),9,24 1,8-diiodoanthracene (3),16 anthracene-1,8-diyl bis(trifluoromethanesulfonate) (4)17 and chlorodiphenylborane25 were performed according to literature protocols. 2-Methylbut-3-yn-2-ol (from Alfa Aesar), copper(I) iodide and potassium phosphate (both from Acros Organics), potassium hydroxide (from Carl Roth), diisopropylamine (from Merck), bis(triphenylphosphine)palladium(II) dichloride (from Fluorochem) and ammonia (from Linde) were used without further purification. (Dimethylamino)trimethylstannane (from Sigma Aldrich) was used after condensation. Pyridine (from VWR Chemicals) and hydrazine (from Fisher Scientific) were dried over potassium hydroxide and used after distillation. All reactions using organometallic reagents were carried out under an anhydrous, inert atmosphere of nitrogen using standard Schlenk techniques in dry solvents (THF dried over potassium, n-hexane dried over LiAlH4, dichloromethane dried over CaH2, benzene dried over P4O10). Photo-dimerisation reactions were performed either using a UVGL-25 Compact UV Lamp (4 W, 230 V, 365 nm) for NMR experiments or using a Rayonet photo reactor [16 lamps (14 W, 230 V), 350 nm (broadband)] for experiments in multi-gram scale. Column chromatography was performed on silica gel 60 (0.04–0.063 mm). NMR spectra were recorded on a Bruker Avance III 500, a Bruker Avance III 500 HD and a Bruker Avance 600 (at 298 K unless otherwise specified). The chemical shifts (δ) were measured in ppm with respect to the solvent [CDCl3: 1H NMR δ = 7.26 ppm, 13C NMR δ = 77.16 ppm; CD2Cl2: 1H NMR δ = 5.32 ppm, 13C NMR δ = 54.00 ppm, C6D6: 1H NMR δ = 7.16 ppm, 13C NMR δ = 128.06 ppm; THF-d8: 1H NMR δ = 1.73 and 3.58 ppm, 13C NMR δ = 25.37 and 67.57 ppm; o-Cl2C6D4 (from deutero): 1H NMR δ = 6.93 and 7.19 ppm] or referenced externally (11B:BF3·OEt2, 119Sn:SnMe4). EI mass spectra were recorded using an Autospec X magnetic sector mass spectrometer with EBE geometry (Vacuum Generators, Manchester, UK) equipped with a standard EI source. ESI mass spectra were recorded using an Esquire 3000 ion trap mass spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with a standard ESI source.
Accurate mass ESI measurements were recorded using an Agilent 6220 time-of-flight mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) in extended dynamic range mode equipped with a dual ESI-source, operating with a spray voltage of 2.5 kV, or using a Q-IMS-TOF mass spectrometer Synapt G2Si (Waters, Manchester, UK) in resolution mode, interfaced to a nano-ESI ion source. Elemental analyses were performed with a CHNS elemental analyser HEKATECH EURO EA. The numbering scheme for NMR assignments of the anthracenes (Scheme 7) and their photo-dimers is based on IUPAC guidelines.
 | ||
| Scheme 7 Numbering scheme for NMR spectroscopic assignments. | ||
Synthetic procedures
Method a. A suspension of 1,8-diiodoanthracene (3; 1.23 g, 2.86 mmol) and 2-methylbut-3-yn-2-ol (1.4 mL, 14 mmol) in diisopropylamine (50 mL) was degassed by four freeze–pump–thaw cycles. During the last cycle PdCl2(PPh3)2 and CuI (three spatula tips of each compound) were added to the frozen mixture. The suspension was heated to reflux for 7 d. After quenching with saturated NH4Cl-solution (70 mL), dichloromethane (80 mL) was added. The aqueous layer was separated and extracted with dichloromethane (5 × 30 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated under reduced pressure. Column chromatography (∅ = 3 cm, l = 10 cm, eluent: dichloromethane) afforded 5 as a yellow solid. Yield: 935 mg (95%); Rf = 0.1.
Method b. A suspension of anthracene-1,8-diyl bis(trifluoromethanesulfonate) (4; 12.0 g, 25.3 mmol) and 2-methylbut-3-yn-2-ol (16.0 mL, 164 mmol) in diisopropylamine (220 mL) was degassed by four freeze–pump–thaw cycles. During the last cycle PdCl2(PPh3)2 (0.9 g, 5 mol%) and CuI (0.45 g, 9 mol%) added to the frozen mixture. The suspension was heated to reflux for 3 d. After quenching with saturated NH4Cl-solution (200 mL) and water (200 mL), the aqueous layer was separated and extracted with dichloromethane (5 × 50 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated under reduced pressure. Column chromatography (∅ = 5 cm, l = 12 cm, eluent: dichloromethane) afforded 5 as a yellow solid. Later fractions were contaminated with 2,7-dimethylocta-3,5-diyne-2,7-diol, which could be removed by sublimation (2 d, 50 °C, 5 × 10−3 mbar). Total yield: 7.05 g (81%); Rf = 0.1.
Analytical data. 1H NMR (500 MHz, CDCl3): δ = 9.40 (s, 1H, H9), 8.38 (s, 1H, H10), 7.95 (d, 3JH,H = 8.5 Hz, 2H, H4/H5), 7.63 (dd, 3JH,H = 6.9 Hz, 4JH,H = 0.8 Hz, 2H, H2/H7), 7.40 (dd, 3JH,H = 8.5, 6.9 Hz, 2H, H3/H6), 3.54 (s, 2H, OH), 1.75 (s, 12H, CH3) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 131.5, 131.5, 130.0 (C2/C7), 128.9 (C4/C5), 127.3 (C10), 125.1 (C3/C6), 124.1 (C9), 121.1, 99.9 (ArC
C), 80.3 (ArCC), 66.2 (COH), 31.8 (CH3) ppm. MS (EI, 70 eV): m/z [assignment] = 342.1 [M]+. MS (ESI, positive ions): m/z [assignment] = 360.3 [M + NH4]+, 365.2 [M + Na]+. HRMS (ESI): calcd for C24H22O2Na+: 365.15120; measured: 365.1510; dev. [ppm]: 0.55, dev. [mmu]: 0.20.
General procedure for photo-dimerisation NMR studies. In an NMR tube, small amounts of the anthracene monomer (1 or 5) were dissolved or suspended in the degassed deuterated solvent, adjusting the desired concentrations. The mixtures were irradiated with UV light (365 nm) until no more signals of the reactant could be observed in the 1H NMR spectrum, respectively. If the product partially precipitated, the solvent was removed under reduced pressure and a 1H NMR spectrum of the residue was recorded in DMSO-d6 due to better solubility. The results concerning the syn/anti ratio are given in Table S1† (for 1) and Table 1 (see above, for 5).
Photo-dimerisation of 1,8-diethynylanthracene (1). A solution of 1,8-diethynylanthracene (1; 5.33 g, 23.6 mmol) in N,N-dimethylformamide (150 mL) was degassed by four freeze–pump–thaw cycles. Using a UV reactor, the mixture was irradiated with UV light (350 nm) for 5 d. The solvent was evaporated under reduced pressure and the residue washed with dichloromethane (3 × 10 mL). The beige solid was recrystallised twice from 1,2-dichloroethane (approx. 400 mL) to remove parts of the poorly soluble head-to-tail isomer anti-2 (2.30 g). The solvent of the combined filtrates was evaporated under reduced pressure and the residue dissolved in dichloromethane (350 mL). After addition of n-pentane (650 mL), a solid precipitated. Removing the solvent by filtration and air-drying of the residue afforded syn-2 (262 mg) as a light beige solid. By multiple repetition of recrystallisation and precipitation (up to five times), the yields of syn-2 (969 mg; 18%) and anti-2 (3.52 g; 66%) could be significantly increased.
NMR data for syn-2. 1H NMR (500 MHz, CDCl3): δ = 7.08 (d, 3JH,H = 7.7 Hz, 4H, H2/H7), 6.90 (d, 3JH,H = 7.4 Hz, 4H, H4/H5), 6.80 (t, 3JH,H = 7.6 Hz, 4H, H3/H6), 6.10 (s, 2H, H9), 4.53 (s, 2H, H10), 3.30 (s, 4H, C
CH) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 145.5, 143.5, 130.2 (C2/C7), 127.5 (C4/C5), 125.8 (C3/C6), 120.7, 81.9 (CCH), 81.3 (CCH), 53.4 (C9), 47.4 (C10) ppm.
NMR data for anti-2. 1H NMR (500 MHz, CDCl3): δ = 7.11 (dd, 3JH,H = 7.4 Hz, 4JH,H = 0.8 Hz, 4H, H4/H5), 7.01 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.2 Hz, 4H, H2/H7), 6.82 (t, 3JH,H = 7.6 Hz, 4H, H3/H6), 5.74 (d, 3JH,H = 11.1 Hz, 2H, H9), 4.68 (d, 3JH,H = 11.1 Hz, 2H, H10), 3.36 (s, 4H, C
CH) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 145.0, 143.3, 129.9 (C2/C7), 126.9 (C4/C5), 126.0 (C3/C6), 120.5, 82.4 (CCH), 80.8 (CCH), 51.7 (C9), 48.1 (C10) ppm.
Photo-dimerisation of 1,8-di(3-methyl-3-hydroxy-1-butynyl)anthracene (5). A suspension of 1,8-di(3-methyl-3-hydroxy-1-butynyl)anthracene (5; 5.00 g, 14.6 mmol) in degassed cyclohexane (240 mL) was irradiated with UV light (350 nm) in a UV reactor for 12 d. After addition of degassed dichloromethane (50 mL) the mixture was irradiated for further 2 d. The solvent was evaporated under reduced pressure. Column chromatography (∅ = 5 cm, l = 14 cm, eluent: dichloromethane) afforded syn-6 as a colourless solid. Yield: 4.10 g (82%); Rf = 0.3.
Small amounts of the head-to-tail isomer anti-6 could be obtained by subsequent elution of the chromatography column with N,N-dimethylformamide. Larger amounts of anti-6 are available by irradiating solutions of 5 in other solvents. Thus, reacting 1,8-di(3-methyl-3-hydroxy-1-butynyl)anthracene (5; 5.00 g, 14.6 mmol) in a degassed n-pentane/benzene mixture (160 mL/80 mL) for 6 d afforded both syn-6 (3.18 g, 64%) and anti-6 (1.40 g, 28%) after purification by column chromatography.
Analytical data for syn-6. 1H NMR (500 MHz, CDCl3): δ = 6.95 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.2 Hz, 4H, H2/H7), 6.85 (dd, 3JH,H = 7.3 Hz, 4JH,H = 1.0 Hz, 4H, H4/H5), 6.77 (t, 3JH,H = 7.5 Hz, 4H, H3/H6), 6.01 (s, 2H, H9), 5.95 (s, 4H, OH), 4.51 (s, 2H, H10), 1.73 (s, 12H, CH3), 1.65 (s, 12H, CH3) ppm. 13C{1H} NMR (125 MHz, CDCl3): δ = 144.8, 143.6, 129.1 (C2/C7), 126.7 (C4/C5), 125.5 (C3/C6), 121.5, 97.6 (ArC
C), 80.5 (ArCC), 65.6 (COH), 53.6 (C10), 47.4 (C9), 32.1 (CH3), 31.5 (CH3) ppm. MS (ESI, positive ions): m/z [assignment] = 707.3 [M + Na]+. HRMS (ESI): calcd for C48H44O4Na+: 707.31318; measured: 707.3118; dev. [ppm]: 1.95, dev. [mmu]: 1.38.
Analytical data for anti-6. 1H NMR (500 MHz, THF-d8): δ = 7.06 (dd, 3JH,H = 7.3 Hz, 4H, H4/H5), 6.80 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.2 Hz, 4H, H2/H7), 6.72 (t, 3JH,H = 7.5 Hz, 4H, H3/H6), 5.74 (d, 3JH,H = 11.0 Hz, 2H, H9), 5.09 (s, 4H, OH), 4.66 (d, 3JH,H = 11.0 Hz, 2H, H10), 1.72 (s, 12H, CH3), 1.62 (s, 12H, CH3) ppm. 13C{1H} NMR (125 MHz, THF-d8): δ = 145.5, 144.7, 129.4 (C2/C7), 126.8 (C4/C5), 126.3 (C3/C6), 122.7, 100.4 (ArC
C), 80.7 (ArCC), 65.5 (COH), 53.0 (C10), 49.4 (C9), 32.4 (CH3) ppm. MS (ESI, positive ions): m/z [assignment] = 707.4 [M + Na]+. HRMS (ESI): calcd for C48H44O4Na+: 707.31318; measured: 707.31285; dev. [ppm]: 0.47, dev. [mmu]: 0.33.
:1) afforded syn-2 as a colourless solid. Rf = 0.8. Small scale: Converting syn-6 (50 mg, 73 μmol), K3PO4 (94 mg, 0.44 mmol) and KOH (25 mg, 0.45 mmol); yield (syn-2): 28 mg (85%). Large scale: Converting syn-6 (2.78 g, 4.06 mmol), K3PO4 (5.25 g, 24.7 mmol) and KOH (1.40 g, 25.0 mmol); yield (syn-2): 1.44 g (78%).
General procedure for thermal cycloreversion. In a Young-NMR tube small amounts (3–5 mg) of anti-2 or anti-6 were dissolved or suspended in degassed 1,2-dichlorobenzene-d4 (0.5 mL). The mixtures were heated in a sand bath to 220 °C and the reaction progress was monitored by 1H NMR spectroscopy. After 6 h, anti-6 was completely converted into its monomer 5. The cycloreversion of anti-2 (to give its monomer 1) is stopped at a ratio of approx. 1
:1 (dimer:monomer). After several days slow decomposition was observed. The 1H NMR spectroscopic data are given in Table S3 (ESI).†
Syn-photo-dimer of 1,8-bis[(trimethylstannyl)ethynyl]anthracene (7). (Dimethylamino)trimethylstannane (1.4 mL, 8.6 mmol) was added to a suspension of the photo-dimer syn-2 (800 mg, 1.77 mmol) in THF (15 mL) and the mixture was stirred at 60 °C for 3 h. Evaporating all volatile compounds under reduced pressure and drying the residue in vacuo quantitatively afforded 7 as a light beige solid. 1H NMR (500 MHz, C6D6): δ = 7.18 (dd, 3JH,H = 7.4 Hz, 4JH,H = 1.7 Hz, 4H, H2/H7), 6.59 (dd, 3JH,H = 7.4 Hz, 4JH,H = 1.7 Hz, 4H, H4/H5), 6.56 (t, 3JH,H = 7.4 Hz, 4H, H3/H6), 6.40 (s, 2H, H9), 4.01 (s, 2H, H10), 0.45 (s, 36H, CH3) ppm. 13C{1H} NMR (125 MHz, C6D6): δ = 144.7, 144.1, 132.1 (C2/C7), 126.9 (C4/C5), 125.5 (C3/C6), 122.9, 109.6 (C
CSn), 96.9 (CCSn), 53.8 (C10), 48.9 (C9), −6.8 (CH3) ppm. 119Sn{1H} NMR (186 MHz, C6D6): δ = −66.0 ppm. Elemental analysis calcd (%) for C48H52Sn4: C 52.23, H 4.75; found: C 52.54, H 4.37.
Syn-photo-dimer of 1,8-bis[(diphenylboranyl)ethynyl]anthracene (8). Chlorodiphenylborane (0.7 mL, 4 mmol) was added to a suspension of tetrastannane 7 (971 mg, 0.88 mmol) in n-hexane (12 mL) at 0 °C. The mixture was slowly warmed to ambient temperature and stirred for 2 d. The suspension was centrifuged (2000 rpm, 40 min) and the supernatant solution removed with a syringe. The crude product was washed with n-hexane (2 × 10 mL) and benzene (2 mL) by centrifugation (2000 rpm, 40 min) and removing the supernatant solution, respectively. Drying the residue in vacuo afforded tetraborane 8 as a light grey solid. Yield: 926 mg (95%). 1H NMR (500 MHz, C6D6): δ = 7.99 (dd, 3JH,H = 7.6 Hz, 4JH,H = 1.7 Hz, 16H, Ph-Hortho), 7.25 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.3 Hz, 4H, H2/H7), 6.95–7.02 (m, 24H, Ph-Hmeta/Ph-Hpara), 6.82 (s, 2H, H9), 6.73 (dd, 3JH,H = 7.3 Hz, 4JH,H = 1.2 Hz, 4H, H4/H5), 6.66 (t, 3JH,H = 7.5 Hz, 4H, H3/H6), 4.16 (s, 2H, H10) ppm. 13C{1H} NMR (125 MHz, C6D6): δ = 144.2, 144.2, 141.4 (Ph-Cipso), 138.8 (Ph-Cortho), 132.4 (C2/C7), 131.9 (Ph-Cpara), 128.4 (C4/C5), 127.8 (Ph-Cmeta), 126.2 (C3/C6), 122.7, 120.0 (C
CB), 106.3 (CCB), 53.9 (C10), 50.0 (C9) ppm. 11B{1H} NMR (160 MHz, C6D6): δ = 43.7 (bs) ppm. Elemental analysis calcd (%) for C84H56B4: C 91.01, H 5.09, B 3.90; found: C 86.96, H 5.39, B 3.69.
Synthesis of the pyridine adduct 9 (8·4Py). Pyridine (15 μL, 0.19 mmol) was added to a solution of tetraborane 8 (50 mg, 45 μmol) in dichloromethane (1.0 mL). After a few seconds, a colourless solid precipitated. The mixture was stirred for 2.5 h at ambient temperature. The precipitate was isolated by filtration, washed with dichloromethane (0.5 mL) and dried in vacuo. 9 was obtained as a colourless crystalline solid, bearing one solvent molecule dichloromethane per formula unit. Yield: 57 mg (84%). 1H NMR (600 MHz, C6D6): δ = 8.70–8.75 (m, 8H, Py-Hortho), 7.45 (dd, 3JH,H = 7.5 Hz, 4JH,H = 1.4 Hz, 4H, H2/H7), 7.36–7.42 (m, 16H, Ph-Hortho), 7.01–7.10 (m, 24H, Ph-Hmeta/Ph-Hpara), 6.77 (t, 3JH,H = 7.4 Hz, 4H, H3/H6), 6.74 (dd, 3JH,H = 7.2 Hz, 4JH,H = 1.2 Hz, 4H, H4/H5), 6.65 (s, 2H, H9), 6.52–6.57 (m, 8H, Py-Hmeta), 6.47–6.52 (m, 4H, Py-Hpara), 4.21 (s, 2H, H10) ppm. 13C{1H} NMR (150 MHz, C6D6, 333 K): δ = 151.2 (Ph-Cipso), 146.8 (Py-Cortho), 144.8, 143.4, 139.5 (Py-Cpara), 134.7 (Ph-Cortho), 132.4 (C2/C7), 127.7 (Ph-C), 125.9 (Ph-C), 125.7 (C4/C5), 125.2 (Py-Cmeta), 100.8 (C
CB), 55.2 (C10), 50.7 (C9) ppm. Signals not observed or assignable for CCB, C2/C7 and one of the quaternary carbon atoms. Due to the very low solubility of the compound, a 11B NMR spectrum could not be recorded. Elemental analysis calcd (%) for C104H76B4N4·CH2Cl2: C 83.52, H 5.21, N 3.71; found: C 81.64, H 5.49, N 3.56.
Synthesis of the ammonia adduct 10 (8·4NH3). Gaseous ammonia (570 μmol) was added to a frozen solution of tetraborane 8 (150 mg, 135 μmol) in benzene (1.0 mL) by condensation. The mixture was slowly warmed to ambient temperature. The resulted suspension was stirred overnight and the precipitate was isolated by filtration. It was washed with n-hexane (0.5 mL) and benzene (0.5 mL) and dried in vacuo. 10 was obtained as a colourless crystalline solid, bearing one solvent molecule benzene per formula unit. Yield: 113 mg (67%). 1H NMR (500 MHz, CD2Cl2): δ = 7.34–7.39 (m, 8H, Ph-Hortho), 7.24–7.29 (m, 8H, Ph-Hmeta), 7.13–7.23 (m, 12H, Ph-Hmeta/Ph-Hpara), 7.15 (d, 3JH,H = 7.6 Hz, 4H, H2/H7), 7.11 (d, 3JH,H = 6.8 Hz, 8H, Ph-Hortho), 7.03 (t, 3JH,H = 7.3 Hz, 4H, Ph-Hpara), 6.94 (d, 3JH,H = 6.9 Hz, 4H, H4/H5), 6.84 (t, 3JH,H = 7.5 Hz, 4H, H3/H6), 6.10 (s, 2H, H9), 4.59 (s, 2H, H10), 3.05 (bs, 12H, NH3) ppm. 13C{1H} NMR (125 MHz, CD2Cl2): δ = 150.9 (Ph-Cipso), 144.6, 143.9, 133.3 (Ph-Cortho), 132.3 (Ph-Cortho), 131.4 (C2/C7), 128.4 (Ph-Cmeta), 128.3 (Ph-Cmeta), 127.0 (Ph-Cpara), 126.6 (Ph-Cpara), 126.6 (C4/C5), 125.9 (C3/C6), 123.7, 109.8 (Ph-Cipso), 100.2 (C
CB), 54.4 (C10, superimposed by the solvent signal), 49.6 (C9) ppm. No signal observed for CCB. 11B NMR (160 MHz, CD2Cl2): δ = −9.4 (s) ppm. Elemental analysis calcd (%) for C84H68B4N4·C6H6:C 86.14, H 5.94, N 4.46; found: C 84.17, H 5.69, N 4.53.
Synthesis of the hydrazine adduct 11 (8·4N2H4). Hydrazine (16 μL, 0.50 mmol) was added to a solution of tetraborane 8 (125 mg, 113 μmol) in benzene (1.0 mL). After a few seconds, a colourless solid precipitated. The mixture was stirred for 3 h at ambient temperature. The precipitate was isolated by filtration. It was washed with benzene (1.0 mL) and dried in vacuo. 11 was obtained as a colourless crystalline solid. Yield: 102 mg (73%). 1H NMR (500 MHz, CD2Cl2): δ = 7.10–7.26 [m, 44H, (Ph-H, 40H; H2/H7, 4H)], 6.97 (d, 3JH,H = 7.1 Hz, 4H, H4/H5), 6.90 (t, 3JH,H = 7.5 Hz, 4H, H3/H6), 5.72 (s, 2H, H9), 5.41 (d, 3JH,H = 11.2 Hz, 4H, BNH), 4.60 (s, 2H, H10), 4.33 (d, 3JH,H = 11.2 Hz, 4H, BNH), 3.65 (bs, 8H, BNNH2) ppm. 13C{1H} NMR (125 MHz, CD2Cl2): δ = 148.2 (Ph-Cipso), 144.3, 143.1, 133.5 (Ph-Cortho), 133.1 (Ph-Cortho), 131.5 (C2/C7), 128.4 (Ph-Cmeta), 128.3 (Ph-Cmeta), 127.1 (Ph-Cpara), 126.9 (C4/C5), 126.4 (C3/C6), 123.0, 101.4 (C
CB), 54.7 (C10), 50.1 (C9) ppm. No signal observed for CCB. 11B NMR (160 MHz, CD2Cl2): δ = −5.8 (s) ppm. Elemental analysis calcd (%) for C84H72B4N8:C 81.58, H 5.87, N 9.06; found: C 80.36, H 5.79, N 8.77.
Author contributions
P. Niermeier: synthesis, data analysis, writing the manuscript. K. A. M. Maibom: synthesis. J.-H. Lamm: synthesis, data analysis, writing the manuscript. B. Neumann: determination of SCXRD structures, XRD data analysis. H.-G. Stammler: determination of SCXRD structures, XRD data analysis. N. W. Mitzel: writing, review and editing the manuscript, supervision, project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank all laboratory assistants, Dipl. Ing. Klaus-Peter Mester, Marco Wißbrock and Dr Andreas Mix for recording NMR spectra, Dr Jens Sproß and Heinz-Werner Patruck for measuring mass spectra, Barbara Teichner for performing elemental analyses, as well as Dr Anna Schwartzen for fruitful discussions. This work was funded by Deutsche Forschungsgemeinschaft (DFG, grant number MI 477/25-3, project number 248859450) and the Priority Program SPP1807 “Control of London dispersion in molecular chemistry” (grant MI477/28-2, project no. 271386299).Notes and references
- (a) M. H. Lee and F. P. Gabbaï, Inorg. Chem., 2007, 46, 8132–8138 CrossRef CAS PubMed; (b) J. K. Day, C. Bresner, N. D. Coombs, I. A. Fallis, L.-L. Ooi and S. Aldridge, Inorg. Chem., 2008, 47, 793–804 CrossRef CAS PubMed; (c) P. Chen and F. Jäkle, J. Am. Chem. Soc., 2011, 133, 20142–20145 CrossRef CAS PubMed; (d) M. Hirai and F. P. Gabbaï, Angew. Chem., Int. Ed., 2015, 54, 1205–1209 ( Angew. Chem. , 2015 , 127 , 1221–1225 ) CrossRef CAS.
- (a) M. E. Jung and H. Xia, Tetrahedron Lett., 1988, 29, 297–300 CrossRef CAS; (b) N. Chaniotakis, K. Jurkschat, D. Müller, K. Perdikaki and G. Reeske, Eur. J. Inorg. Chem., 2004, 2283–2288 CrossRef CAS; (c) Z. Yan, Z. Zhou, Y. Wu, I. A. Tikhonova and V. B. Shur, Anal. Lett., 2005, 38, 377–388 CrossRef CAS; (d) S. Benz, M. Macchione, Q. Verolet, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2016, 138, 9093–9096 CrossRef CAS PubMed.
- (a) M. Austin, K. Gebreyes, H. G. Kuivila, K. Swami and J. A. Zubieta, Organometallics, 1987, 6, 834–842 CrossRef CAS; (b) F. P. Gabbaï, A. Schier, J. Riede and M. J. Hynes, Chem. Commun., 1998, 897–898 RSC; (c) A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC; (d) J. Horstmann, M. Hyseni, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2017, 56, 6107–6111 ( Angew. Chem. , 2017 , 129 , 6203–6207 ) CrossRef CAS PubMed; (e) C.-H. Chen and F. P. Gabbaï, Chem. Sci., 2018, 9, 6210–6218 RSC; (f) P. Niermeier, S. Blomeyer, Y. K. J. Bejaoui, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2019, 58, 1965–1969 ( Angew. Chem. , 2019 , 131 , 1985–1990 ) CrossRef CAS PubMed.
- (a) T. Ooi, M. Takahashi and K. Maruoka, J. Am. Chem. Soc., 1996, 118, 11307–11308 CrossRef CAS; (b) S. N. Kessler, M. Neuburger and H. A. Wegner, Eur. J. Org. Chem., 2011, 3238–3245 CrossRef CAS; (c) S. N. Kessler, M. Neuburger and H. A. Wegner, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS PubMed; (d) M. Hirai, J. Cho and F. P. Gabbaï, Chem.–Eur. J., 2016, 22, 6537–6541 CrossRef CAS PubMed; (e) S. Benz, J. Mareda, C. Besnard, N. Sakai and S. Matile, Chem. Sci., 2017, 8, 8164–8169 RSC; (f) S. Benz, J. López-Andarias, J. Mareda, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2017, 56, 812–815 ( Angew. Chem. , 2017 , 129 , 830–833 ) CrossRef CAS PubMed.
- For a selection of group 13-based PLAs see: (a) D. F. Shriver and M. J. Biallas, J. Am. Chem. Soc., 1967, 89, 1078–1081 CrossRef CAS; (b) C. H. Chen and F. P. Gabbaï, Angew. Chem., Int. Ed., 2018, 57, 521–525 ( Angew. Chem. , 2018 , 130 , 530–534 ) CrossRef CAS PubMed; (c) O. Saied, M. Simard and J. D. Wuest, Organometallics, 1996, 15, 2345–2349 CrossRef CAS; (d) W. Uhl, F. Hannemann, W. Saak and R. Wartchow, Eur. J. Inorg. Chem., 1998, 921–926 CrossRef CAS; (e) W. Uhl, A. Hepp, H. Westenberg, S. Zemke, E.-U. Würthwein and J. Hellmann, Organometallics, 2010, 29, 1406–1412 CrossRef CAS; (f) W. Uhl, H. R. Bock, F. Breher, M. Claesener, S. Haddadpour, B. Jasper and A. Hepp, Organometallics, 2007, 26, 2363–2369 CrossRef CAS; (g) W. Uhl, M. Claesener, S. Haddadpour, B. Jasper and A. Hepp, Dalton Trans., 2007, 417–423 RSC; (h) P. Jutzi, J. Izundu, H. Sielemann, B. Neumann and H.-G. Stammler, Organometallics, 2009, 28, 2619–2624 CrossRef CAS; (i) W. Uhl, D. Kovert, S. Zemke and A. Hepp, Organometallics, 2011, 30, 4736–4741 CrossRef CAS; (j) M. Tschinkl, A. Schier, J. Riede and F. P. Gabbaï, Inorg. Chem., 1997, 36, 5706–5711 CrossRef CAS PubMed; (k) J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2010, 16, 11906–11914 CrossRef CAS PubMed; (l) J.-H. Lamm, P. Niermeier, A. Mix, J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Angew. Chem., Int. Ed., 2014, 53, 7938–7942 ( Angew. Chem. , 2014 , 126 , 8072–8076 ) CrossRef CAS PubMed.
- (a) H. E. Katz, J. Am. Chem. Soc., 1985, 107, 1420–1421 CrossRef CAS; (b) H. E. Katz, J. Org. Chem., 1985, 50, 5027–5032 CrossRef CAS.
- (a) V. C. Williams, W. E. Piers, W. Clegg, M. R. J. Elsegood, S. Collins and T. B. Marder, J. Am. Chem. Soc., 1999, 121, 3244–3245 CrossRef CAS; (b) S. Solé and F. P. Gabbaï, Chem. Commun., 2004, 1284–1285 RSC; (c) M. Melaïmi, S. Solé, C.-W. Chiu, H. Wang and F. P. Gabbaï, Inorg. Chem., 2006, 45, 8136–8143 CrossRef PubMed; (d) H. Zhao and F. P. Gabbaï, Organometallics, 2012, 31, 2327–2335 CrossRef CAS.
- (a) H. E. Katz, Organometallics, 1987, 6, 1134–1136 CrossRef CAS; (b) S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686–14687 CrossRef CAS PubMed; (c) J. Chai, S. P. Lewis, S. Collins, T. J. J. Sciarone, L. D. Henderson, P. A. Chase, G. J. Irvine, W. E. Piers, M. R. J. Elsegood and W. Clegg, Organometallics, 2007, 26, 5667–5679 CrossRef CAS.
- H. E. Katz, J. Org. Chem., 1989, 54, 2179–2183 CrossRef CAS.
- For tridentate PLAs see: (a) E. Weisheim, C. G. Reuter, P. Heinrichs, Yu. V. Vishnevskiy, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2015, 21, 12436–12448 CrossRef CAS; (b) E. Weisheim, A. Schwartzen, L. Kuhlmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Eur. J. Inorg. Chem., 2016, 8, 1257–1266 CrossRef; (c) E. Weisheim, L. Büker, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dalton Trans., 2016, 45, 198–207 RSC; (d) J. Tomaschautzky, B. Neumann, H.-G. Stammler, A. Mix and N. W. Mitzel, Dalton Trans., 2017, 46, 1645–1659 RSC.
- For general aspects on the scope of anthracene photo-dimerization see: (a) H.-D. Becker, Chem. Rev., 1993, 93, 145–172 CrossRef CAS; (b) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2000, 29, 43–55 RSC; (c) H. Bouas-Laurent, A. Castellan, J.-P. Desvergne and R. Lapouyade, Chem. Soc. Rev., 2001, 30, 248–263 RSC.
- (a) J.-P. Desvergne, F. Chepko and H. Bouas-Laurent, J. Chem. Soc., Perkin Trans. 2, 1978, 84 RSC; (b) P. Kissel, A. D. Schlüter and J. Sakamoto, Chem.–Eur. J., 2009, 15, 8955–8960 CrossRef CAS PubMed; (c) J.-H. Lamm, J. Glatthor, J.-H. Weddeling, A. Mix, J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Org. Biomol. Chem., 2014, 12, 7355–7365 RSC.
- P. Niermeier, J. H. Lamm, A. Mix, B. Neumann, H.-G. Stammler and N. W. Mitzel, ChemistryOpen, 2019, 8, 304–315 CrossRef CAS PubMed.
- (a) C. Yang, A. Nakamura, T. Wada and Y. Inoue, Org. Lett., 2006, 8, 3005–3008 CrossRef CAS PubMed; (b) C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inuoe, J. Am. Chem. Soc., 2008, 130, 8574–8575 CrossRef CAS PubMed; (c) A. Dawn, N. Fujita, S. Haraguchi, K. Sada and S. Shinkai, Chem. Commun., 2009, 2100–2102 RSC; (d) Y. Ito and G. Olovsson, J. Chem. Soc., Perkin Trans. 1, 1997, 127–133 RSC; (e) Y. Kawanami, S.-y. Katsumata, M. Nishijima, G. Fukuhara, K. Asano, T. Suzuki, C. Yang, A. Nakamura, T. Mori and Y. Inuoe, J. Am. Chem. Soc., 2016, 138, 12187–12201 CrossRef CAS.
- K. Zhong, Z. Wang, Y. Liang, T. Chen, B. Yin and L. Y. Jin, Supramol. Chem., 2014, 26, 729–735 CrossRef CAS.
- M. Goichi, K. Segawa, S. Suzuki and S. Toyota, Synthesis, 2005, 13, 2116–2118 Search PubMed.
- M. Servalli, N. Trapp, M. Wörle and F.-G. Klärner, J. Org. Chem., 2016, 81, 2572–2580 CrossRef CAS PubMed.
- H. Y. Lee, A. Olasz, M. Pink, H. Park and D. Lee, Chem. Commun., 2011, 47, 481–483 RSC.
- A. Smeyanov and A. Schmidt, Synth. Commun., 2013, 43, 2809–2816 CrossRef CAS.
- (a) M. Ehrenberg, Acta Crystallogr., 1966, 20, 177–182 CrossRef CAS; (b) C. H. Choi and M. Kertesz, Chem. Commun., 1997, 22, 2199–2200 RSC; (c) P. Niermeier, J.-H. Lamm, J.-H. Peters, B. Neumann, H.-G. Stammler and N. W. Mitzel, Synthesis, 2019, 51, 1623–1632 CrossRef CAS.
- A. A. Otlyotov, J.-H. Lamm, S. Blomeyer, N. W. Mitzel, V. V. Rybkin, Y. A. Zhabanov, N. V. Tverdova, N. I. Giricheva and G. V. Girichev, Phys. Chem. Chem. Phys., 2017, 19, 13093–13100 RSC.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- For the classification of hydrogen bonds see: (a) G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed; (b) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 ( Angew. Chem. , 2002 , 114 , 50–80 ) CrossRef.
- (a) M. E. Tauchert, T. R. Kaiser, A. P. V. Göthlich, F. Rominger, D. C. M. Ward and P. Hofmann, ChemCatChem, 2010, 2, 674–682 CrossRef CAS; (b) F. Vögtle, H. Koch and K. Rissanen, Chem. Ber., 1992, 125, 2129–2135 CrossRef.
- J. C. Thomas and J. C. Peters, Inorg. Chem., 2003, 42, 5055–5073 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 11B, 13C NMR spectra, UV-vis absorption spectra, crystallographic details. CCDC 1952344–1952355. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02065j |
| This journal is © The Royal Society of Chemistry 2021 |