
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntrinsic photogeneration of long-lived charges in a donor-orthogonal acceptor conjugated polymer†
Jordan
Shaikh
a,
Daniel G.
Congrave
 b,
Alex
Forster
b,
Alessandro
Minotto
c,
Franco
Cacialli
c,
Timothy J. H.
Hele
a,
Thomas J.
Penfold
d,
Hugo
Bronstein
*b and
Tracey M.
Clarke
*a
b,
Alex
Forster
b,
Alessandro
Minotto
c,
Franco
Cacialli
c,
Timothy J. H.
Hele
a,
Thomas J.
Penfold
d,
Hugo
Bronstein
*b and
Tracey M.
Clarke
*a
aDepartment of Chemistry, University College London, Christopher Ingold Building, London WC1H 0AJ, UK. E-mail: tracey.clarke@ucl.ac.uk
bDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: hab60@cam.ac.uk
cDepartment of Physics, University College London, Gower Street, London WC1E 6BT, UK
dChemistry – School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
First published on 10th May 2021
Abstract
Efficient charge photogeneration in conjugated polymers typically requires the presence of a second component to act as electron acceptor. Here, we report a novel low band-gap conjugated polymer with a donor/orthogonal acceptor motif: poly-2,6-(4,4-dihexadecyl-4H-cyclopenta [2,1-b:3,4-b′]dithiophene)-alt-2,6-spiro [cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-fluorene]-2′,7′-dicarbonitrile, referred to as PCPDT-sFCN. The role of the orthogonal acceptor is to spatially isolate the LUMO from the HOMO, allowing for negligible exchange energy between electrons in these orbitals and minimising the energy gap between singlet and triplet charge transfer states. We employ ultrafast and microsecond transient absorption spectroscopy to demonstrate that, even in the absence of a separate electron acceptor, PCPDT-sFCN shows efficient charge photogeneration in both pristine solution and film. This efficient charge generation is a result of an isoenergetic singlet/triplet charge transfer state equilibrium acting as a reservoir for charge carrier formation. Furthermore, clear evidence of enhanced triplet populations, which form in less than 1 ps, is observed. Using group theory, we show that this ultrafast triplet formation is due to highly efficient, quantum mechanically allowed intersystem crossing between the bright, initially photoexcited local singlet state and the triplet charge transfer state. Remarkably, the free charges that form via the charge transfer state are extraordinarily long-lived with millisecond lifetimes, possibly due to the stabilisation imparted by the spatial separation of PCPDT-sFCN's donor and orthogonal acceptor motifs. The efficient generation of long-lived charge carriers in a pristine polymer paves the way for single-material applications such as organic photovoltaics and photodetectors.
1. Introduction
Charge photogeneration in organic photovoltaics generally requires a bulk heterojunction containing both an electron donor and acceptor, in order to overcome the binding energy of the photoexcited exciton. This is the case even for the highest efficiency organic photovoltaic devices, which incorporate novel non-fullerene acceptors.1,2 Although highly efficient charge photogeneration can be achieved using this bulk heterojunction configuration, it is accompanied by a host of other issues: energy losses,3 morphology variations,4 miscibility,5 stability,6 and added fabrication complexity. It is therefore desirable to target organic optoelectronics comprised of a single-material (pristine) active layer. However, pristine conjugated polymers typically do not possess high levels of charge photogeneration,7 nor the long charge carrier lifetimes required. Instead, any charges that do form tend to undergo rapid geminate recombination on ps–ns timescales.8,9 Furthermore, many pristine conjugated polymers exhibit solely triplet formation.10,11Triplet states are a key species that commonly form during the operation of organic electronic devices such as light emitting diodes and solar cells.12 Although traditionally seen as a loss mechanism in organic photovoltaics (OPV), the unique properties of triplets are increasingly being manipulated to enhance device efficiencies through strategies such as singlet fission13–15 and up-conversion.16,17 Triplets are also of significant value in organic light-emitting diodes (OLEDs)18 and bio-applications.19 Indeed, interest in triplet states has been mounting owing to their recently discovered prevalence in highly efficient non-fullerene acceptor OPV blends.20 Of particular importance, therefore, is the interplay between triplets and charges.
There are three primary mechanisms by which donor or acceptor triplet states form in an organic solar cell: intersystem crossing (ISC), back electron transfer from a spin-mixing charge transfer (CT) state,21 and via bimolecular recombination of free carriers.22 The rate of ISC is dictated by the size of the spin–orbit coupling and the energy gap between the singlet and triplet states involved, ΔEST. Spin–orbit coupling depends on either the presence of heavy atoms and/or changes in orbital angular momentum, whereas ΔEST depends on the exchange energy. ΔEST is typically large for conjugated polymers, on the order of 0.7 eV.12 A large exchange energy is not ideal for an organic solar cell as it creates deep triplet levels on either the donor or acceptor that can lead to preferential triplet formation instead of the desired charge carrier formation.8 Conversely, a small exchange energy has the potential advantage of allowing triplet recycling via reverse intersystem crossing (RISC).23 Reducing ΔEST is also important for thermally activated delayed fluorescence (TADF), a key strategy for enhancing OLED efficiencies.24–26
Pursuing strategies familiar to these TADF small molecules, we previously synthesised conjugated polymers with significantly reduced ΔEST.27 This was accomplished by changing the standard alternating donor–acceptor copolymer structure to a situation where the electron acceptor moiety is located physically orthogonal to the donor backbone. This configuration achieved a significant reduction in the spatial overlap between the frontier molecular orbitals, substantially decreasing ΔEST whilst still maintaining a conjugated backbone. Indeed, enhanced triplet formation and TADF was observed. However, these TADF polymers lacked the charge photogeneration abilities required for photovoltaic applications.
In our current work, we demonstrate that by changing the polymer backbone to a lower bandgap motif, the functionality can be switched from light emission to charge photogeneration in the pristine polymer. Poly-2,6-(4,4-dihexadecyl-4H-cyclopenta [2,1-b:3,4-b′]dithiophene)-alt-2,6-spiro [cyclopenta[2,1-b:3,4-b′]dithiophene-4,9′-fluorene]-2′,7′-dicarbonitrile, referred to as PCPDT-sFCN (structure shown in Scheme 1), is comprised of an electron rich donor backbone in combination with a dicyano-substituted spirobifluorene, which is located orthogonal to the main backbone. The CPDT backbone used here is well known for its role as the donor component in the intensively studied low band gap D-A polymer PCDTBT.28–30
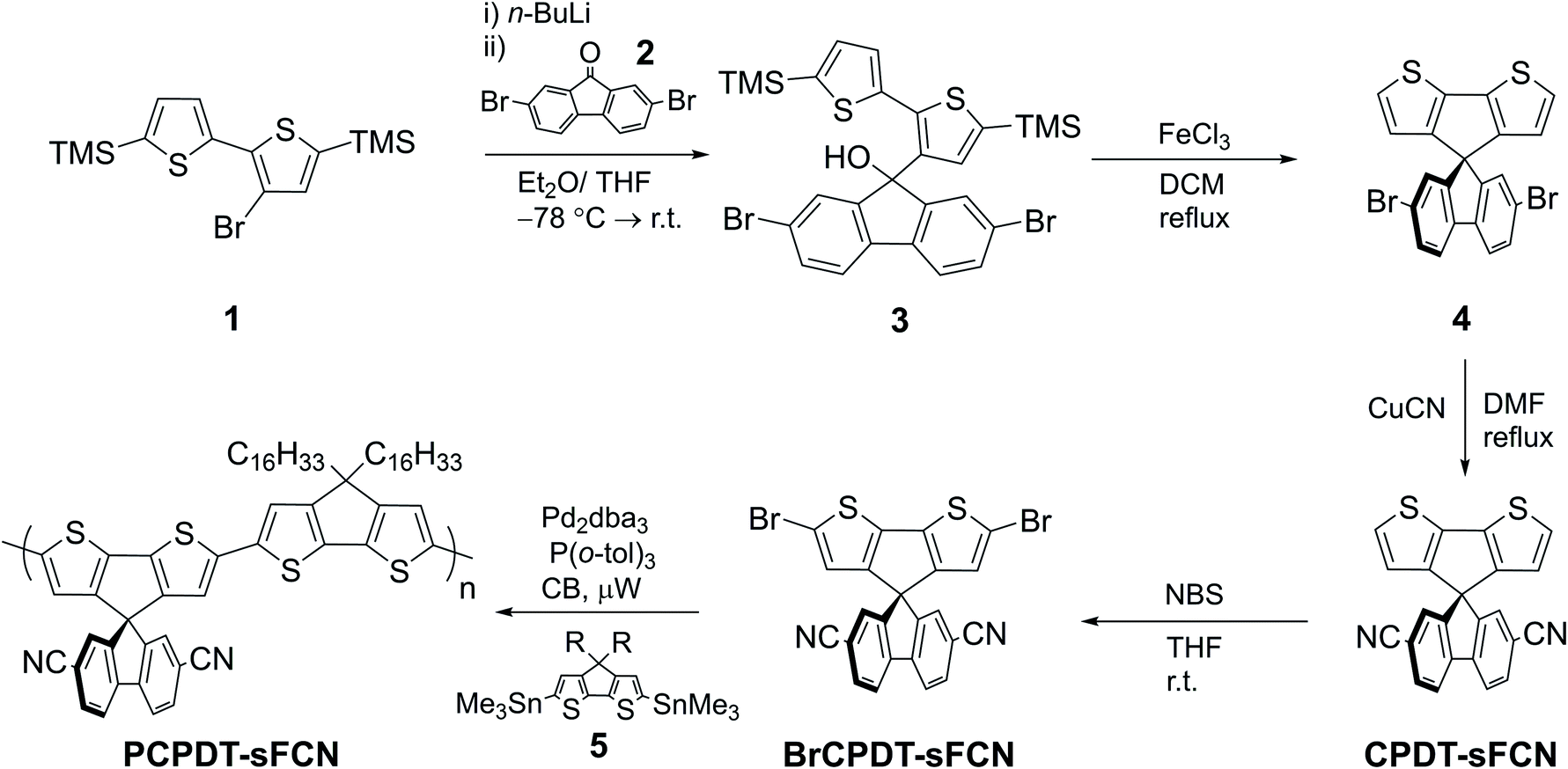 | ||
| Scheme 1 Synthesis of PCPDT-sFCN. | ||
Transient absorption spectroscopy (TAS) measurements of PCPDT-sFCN show unusually high charge carrier yields and lifetimes in both pristine solution and film, a result of isoenergetic spin-mixing singlet/triplet CT states acting as a reservoir for charge carrier formation. These CT states dissociate into extraordinarily long-lived free charges due to the spatial separation of PCPDT-sFCN's donor and orthogonal acceptor motifs. This behaviour is very unusual for a pristine conjugated polymer and offers a new synthetic strategy for future improvements in single-material optoelectronic applications such as OPV and photodetectors.
2. Results
2.1. Synthesis
The synthesis of PCPDT-sFCN is outlined in Scheme 1. After lithium-halogen exchange, 1 was treated with 2,7-dibromo-9-fluorenone (2) to afford the tertiary alcohol 3. The TMS protecting groups incorporated into 1 are crucial to prevent side reactions during this step. Subsequent treatment of 3 with the Lewis acid FeCl3 enables dehydrative ring closure to form the spiro centre alongside deprotection of the TMS groups in one-pot, affording 4. The electron withdrawing CN groups were next installed by a Rosemund von Braun reaction to obtain CPDT-sFCN, before bromination with NBS provided the final polymer precursor BrCPDT-sFCN. PCPDT-sFCN was then prepared via the microwave-assisted Stille polycondensation of BrCPDT-sFCN with the stannylated CPDT monomer 5 in chlorobenzene. PCPDT-sFCN was obtained in number average (Mn) and weight average (Mw) molecular weights of 6.4 and 8.2 kDa, respectively. The comonomer 5, with large hexadecyl alkyl chains, was selected to ensure the high solubility of PCPDT-sFCN in common aromatic and chlorinated organic solvents. Despite the relatively low molecular weight of the material, the similarity of the absorption maxima and onset (Fig. 1) to simple PCPDT homo and co-polymers suggests that PCPDT-sFCN has reached its effective conjugation length.31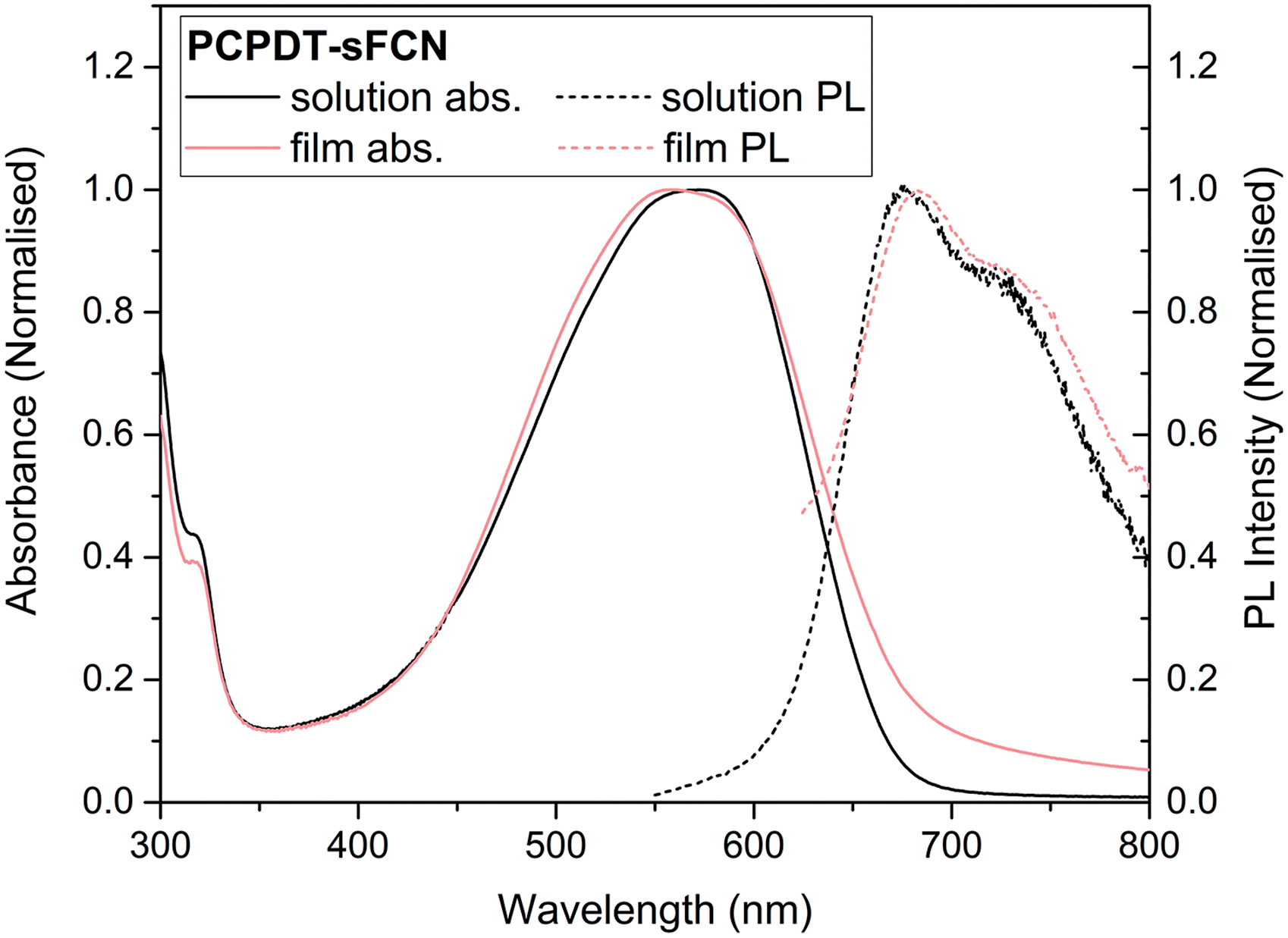 | ||
| Fig. 1 Normalised ground state absorbance and photoluminescence spectra of pristine PCPDT-sFCN in chlorobenzene solution (50 μg mL−1) and thin film. Spectra have been normalised to their respective peak maxima. | ||
2.2. Absorbance and photoluminescence
Fig. 1 shows the normalised ground state absorbance of both the pristine PCPDT-sFCN thin film and its neat chlorobenzene solution (a comparison to the individual monomer units is in Fig. S1, ESI†). PCPDT-sFCN displays a broad absorption band at 550 nm, likely due to a π→π* transition along the conjugated thiophene backbone, as this would have a much higher oscillator strength than a CT absorption. We will term the π–π* state positioned along the thiophene backbone as a “local exciton” (LE). However, it is possible that a weak CT absorption from the donor to the orthogonal acceptor moiety is found beneath or to the red of the broad 550 nm peak.1 The 550 nm absorption does not show prominent vibronic structure, illustrating a large energetic dispersity that may indicate a broad distribution of polymer conformations. This inhomogeneous broadening is found to be slightly more pronounced in the film, thus pointing to increased disorder in the solid phase.Following optical excitation with 532 nm (2.33 eV), the pristine PCPDT-sFCN in chlorobenzene solution was found to exhibit a relatively broad emission (Fig. 1), with a maximum situated at 680 nm and a shoulder at 720 nm. The measured photoluminescence (PL) quantum yields for PCPDT-sFCN were very low, even in solution (<5%). This implies highly efficient deactivation of the initially formed π–π* state, by such processes as ISC and/or internal conversion to a lower energy “dark” 1CT state. The corresponding pristine PCPDT-sFCN film is found to exhibit a very similar photoluminescence profile to the solution, albeit slightly redshifted, exhibiting a maximum at 685 nm with the additional shoulder now found at 730 nm.
2.3. TC-SPC
Time-correlated single-photon counting (TC-SPC) measurements were carried out on the pristine PCPDT-sFCN solution as shown in Fig. 2 (kinetics are shown in Fig. S2†). The PL decays are short-lived and multiexponential, with the longest decay time constant not exceeding 0.7 ns. The short-lived PL is consistent with the low PL quantum yields measured. The PL decay is also found to be faster in the pristine film when compared to its solution, which is a common occurrence due to increased non-radiative decay pathways in the solid phase.28,32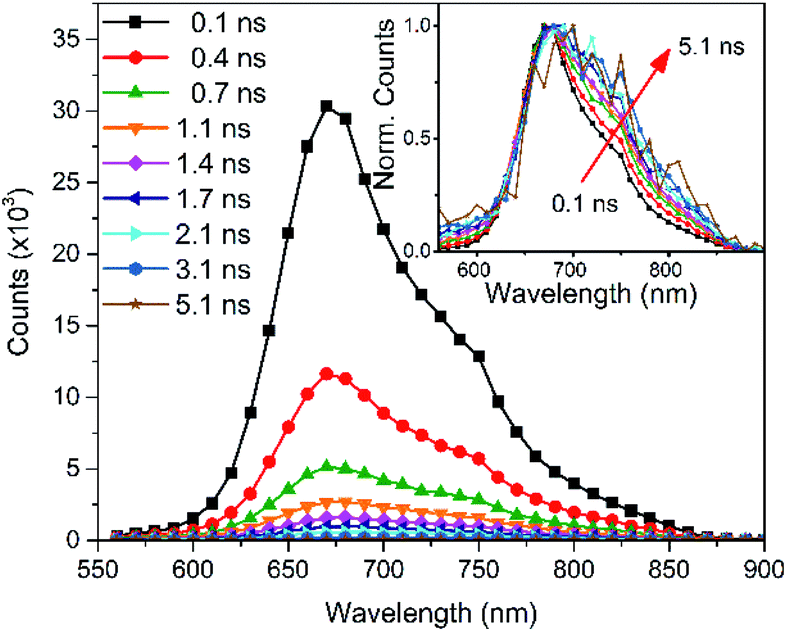 | ||
| Fig. 2 Time-correlated single photon counting PL spectra of the PCPDT-sFCN solution (chlorobenzene, 50 μg mL−1) obtained with an excitation wavelength of 467 nm. | ||
An examination of the PCPDT-sFCN solution PL spectra over time revealed a change on early nanosecond timescales, with the 720 nm shoulder becoming increasingly prominent with time relative to the 680 nm peak. This observation may suggest that either the vibronic structure and thus excited state structure is changing on nanosecond timescales (which is unlikely), or that the 720 nm peak belongs to a different species that decays with a slower rate. Given the red-shifted nature and similarity to other examples in the literature33,34 – including other orthogonal acceptor polymers27 – this could indicate the presence of very weak CT exciton emission at 720 nm. This possible presence of an intramolecular CT state will be explored further in the ps-TAS section.
2.4. Microsecond transient absorption spectroscopy
Microsecond transient absorption spectroscopy was performed, since μs timescales are ideal for investigating the presence of triplet states. It is easier to characterise triplets on μs–ms timescales as there are fewer competing excited states and diffusion-controlled O2 quenching measurements can be performed. Excitation of the pristine PCPDT-sFCN solution results in the initial formation of a broad transient species in the NIR, with its peak situated at 920 nm, as shown in Fig. 3a and b. However, by 100 μs the spectrum evolves to reveal the presence of a new even longer-lived species, with its peak centred at 1020 nm. An examination of the μs-TA kinetics at 980 nm reveals a multicomponent decay (Fig. 4a), consisting of an initial exponential component until ∼40 μs, followed by a power law decay at longer times. The presence of two decay phases is consistent with the observed spectral evolution and the close proximity of the two transitions, where the initial exponential component relates to the 920 nm band and the power law component to the 1020 nm band.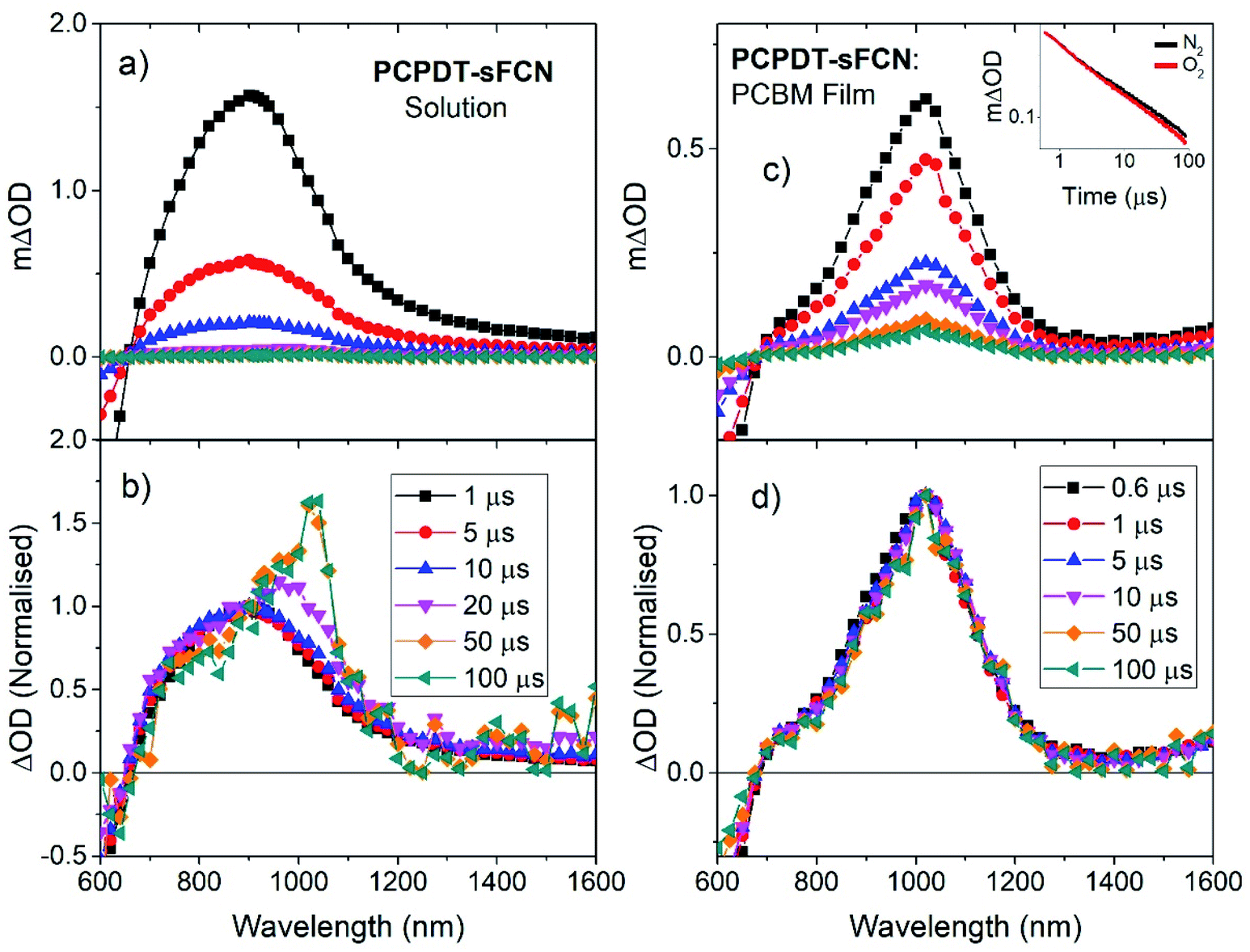 | ||
Fig. 3 (a) Microsecond transient absorption spectrum of PCPDT-sFCN in (a and b) chlorobenzene solution (50 μg mL−1) and (c and d) PCPDT-sFCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PC60BM 1:1 thin film. Obtained with an excitation wavelength of 530 nm, a probe wavelength of 980 nm, and excitation densities of 40 μJ cm−2 and 16 μJ cm−2 respectively. The lower panels (b and d) show the same spectral data normalised to 900 nm, illustrating the presence of a new species at later time scales in the case of the pristine solution. Inset in (c): microsecond transient absorption kinetics of a PCPDT-sFCN:PC60BM 1:1 thin film under N2 and O2 atmospheres, illustrating an O2 independent decay that is indicative of polarons. :PC60BM 1:1 thin film. Obtained with an excitation wavelength of 530 nm, a probe wavelength of 980 nm, and excitation densities of 40 μJ cm−2 and 16 μJ cm−2 respectively. The lower panels (b and d) show the same spectral data normalised to 900 nm, illustrating the presence of a new species at later time scales in the case of the pristine solution. Inset in (c): microsecond transient absorption kinetics of a PCPDT-sFCN:PC60BM 1:1 thin film under N2 and O2 atmospheres, illustrating an O2 independent decay that is indicative of polarons. | ||
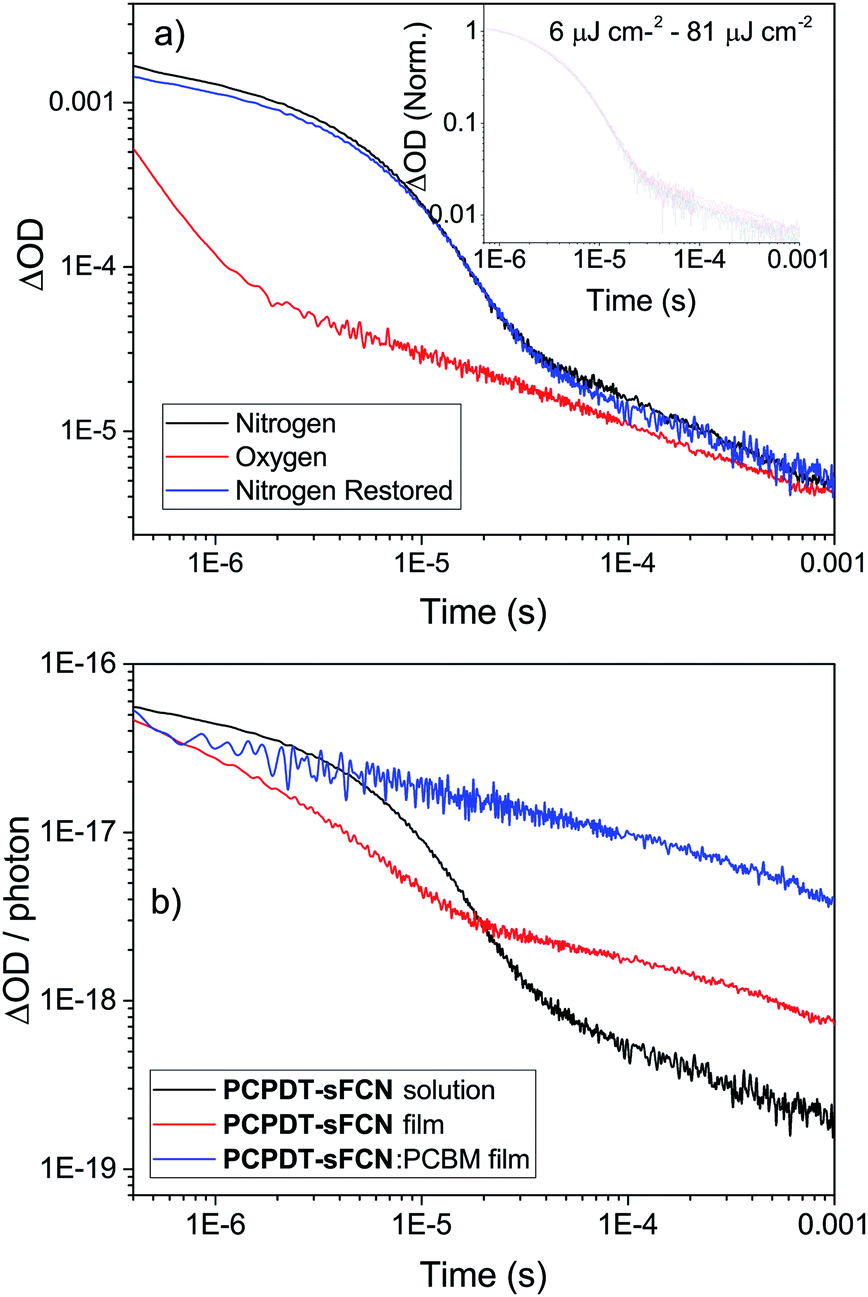 | ||
| Fig. 4 (a) μs-TA kinetics of pristine PCPDT-sFCN in chlorobenzene solution (50 μg mL−1), excited with a pump wavelength of 532 nm, using a probe wavelength of 980 nm, and an excitation energy of 20 μJ cm−2. The inset shows the independence of both decay phases with respect to excitation density. Under nitrogen, the triplets can be observed ∼400 ns to 30 μs and polarons ∼30 μs to >1 ms. (b) Normalised microsecond transient absorption kinetics of pristine PCPDT-sFCN solution (50 μg mL−1), pristine PCPDT-sFCN film, and a PCPDT-sFCN:PC60BM 1:1 blend film, probed at 980 nm. The decay data have been normalised to the number of photons absorbed at the exciting wavelength. | ||
The initial exponential component at 920 nm, which dominates the TA spectrum at early times, was analysed by subtracting the power law from the overall data, revealing the presence of a decay that could be fitted with a stretched exponential (ΔOD ∝ e(−t/τ)β), where ΔOD is the change in optical density. Excitation density dependent studies show an invariance of these kinetics with fluence (Fig. 4a inset and Fig. S3a†), indicating that the dispersive exponential behaviour does not stem from second-order processes such as triplet–triplet annihilation. The average distribution parameter of β = 0.87 ± 0.03 indicates that the exponential decay is close to first order (mono-exponential) kinetics, but that a degree of energetic distribution is present. This close-to-monoexponential behaviour is consistent with an assignment of the 920 nm band to triplets, but the presence of a small component of stretched exponential decay indicates that these triplets are located in a dispersive environment,35,36 even in solution (such as coiled or tangled polymer chains).
To confirm this triplet assignment, oxygen sensitivity experiments were performed. As shown in Fig. 4a, the presence of O2 is found to affect the two decay components very differently. The initial exponential decay (the 920 nm feature) is significantly reduced in both lifetime and amplitude, as expected for a triplet state, and thus verifies this assignment. Importantly, the observed oxygen quenching was found to be reversible when the sample was re-purged with N2, thus eliminating the possibility of the signal quenching deriving from oxygen-based degradation of the sample.
The long-lived TA at 1020 nm is likely to derive from a charged species (i.e. radical cation or anion), as the power law decay exhibited at later times (>40 μs) is generally observed for bimolecular recombination of long-lived, free charges (polarons) in donor/acceptor thin films.37,38 Fitting a power-law to the long-lived component of the pristine PCPDT-sFCN solution kinetic decays yields an average exponent of α = 0.38, a similar value to many polymer/acceptor blend films.29,39 This indicates an aggregated, amorphous environment for these PCPDT-sFCN charges in solution, which leads to relatively energetically deep trap states. The oxygen quenching experiment (Fig. 4a) confirms the assignment of the 1020 nm band to charges. This longer-lived power law component is significantly less affected by the O2; indeed, the induced reduction of the triplet component reveals more of the power law decay. The gradient of the power law is unchanged in the presence of O2, which is consistent with its assignment to a charged species. However, an important observation is that the amplitude of the power law decreases, suggesting a reduction in the population of charge carriers. This reduction is significant (∼40% at 100 μs) and reproducible, and a return to the inert atmosphere restores the charge carrier population to its original amplitude. This observation suggests that the mechanism by which the charge carriers form involves a triplet intermediate species. This observed recombination behaviour has previously been reported by Ohkita et al. for a polythiophene derivative, although this was for a PC60BM blend film and not a pristine material in solution.40 In addition, the charges are extraordinarily long-lived in the PCPDT-sFCN solution, with a lifetime on the order of milliseconds.
TA spectroscopy on μs timescales was then applied to the pristine PCPDT-sFCN film (Fig. S3†), for a comparison to the solution. A very similar spectral evolution over time was observed for the film, with the initial oxygen-sensitive triplet at ca. 960 nm decaying to reveal the longer-lived charges at 1010 nm. In addition to the red-shift of the triplet from 920 nm in solution to 960 nm in the film, a closer examination of the kinetics and relative amplitudes of the two components reveals differences to the solution behaviour. Fig. 4b shows the film kinetics at 980 nm, which reveals a multicomponent decay profile that is similar in nature to what was observed in the solution phase (the subtle differences are discussed in the ESI†). In contrast to the solution, however, the relative triplet population in the film is considerably reduced, while the charge population is greater. The presence of additional intermolecular interactions in the film may promote charge carrier formation or stabilise the charges, prolonging their lifetime.
To investigate the polaron formation in PCPDT-sFCN further, a blend film of PCPDT-sFCN with the well-known acceptor PC60BM was examined (Fig. 3c, d and 4b). PC60BM was chosen because it has been well-studied and its excited species transitions are known;41,42 furthermore, its transient species have low oscillator strengths and thus the PCPDT-sFCN features will dominate. Fig. 3c shows the μs-TA spectrum obtained for a PCPDT-sFCN:PC60BM 1:1 blend. The spectrum at 1 μs is found to exhibit the polaron transition at 1020 nm, consistent with that observed on the late μs timescales for the pristine PCPDT-sFCN film and solution. In addition, no oxygen sensitivity is observed (Fig. 3c inset), as expected for a polaron. As such, the singlet exciton must be quenched by the PC60BM on ultrafast timescales in the blend, which significantly inhibits triplet formation from taking place. This also has the additional consequence of a much higher polaron yield in the blend film compared to the pristine film and solution, by a factor of 5 and 20 respectively (Fig. 4b).
2.5. Picosecond transient absorption spectroscopy
To help understand the observed PL and μs-TAS results, ps-TAS was employed. Fig. 5 shows the TA spectra obtained for PCPDT-sFCN in solution and film by exciting with a pump wavelength of 520 nm. The ground state bleach contribution can be seen in the region 450–690 nm for both samples, while the region from 690–780 nm correlates well with the spectral emission region of PCPDT-sFCN and is ascribed to stimulated emission. At early times prior to 1 ps, a broad photoinduced absorption can be observed at 1300 nm, with a prominent shoulder at ca. 1000 nm. The transition at 1300 nm is found to undergo a red shift of ∼30 meV within 10 ps in the solution, but no such red-shift is observed in the film. By 6 ns the 1300 nm transition has completely decayed in both film and solution, while the 1000 nm shoulder is resolved into a long-lived species at 960 nm. The 1300 nm band is assigned to the singlet exciton due to its short-lived nature (most likely a thiophene-based π–π* local exciton (LE)). The assignment of this band to the local PCPDT-sFCN singlet exciton is consistent with the observed spectral red-shift, which can therefore be attributed to vibrational relaxation, afforded by the energetic disorder.43 The red-shift was not observed in the film due to the more rapid vibrational relaxation enabled by the condensed phase.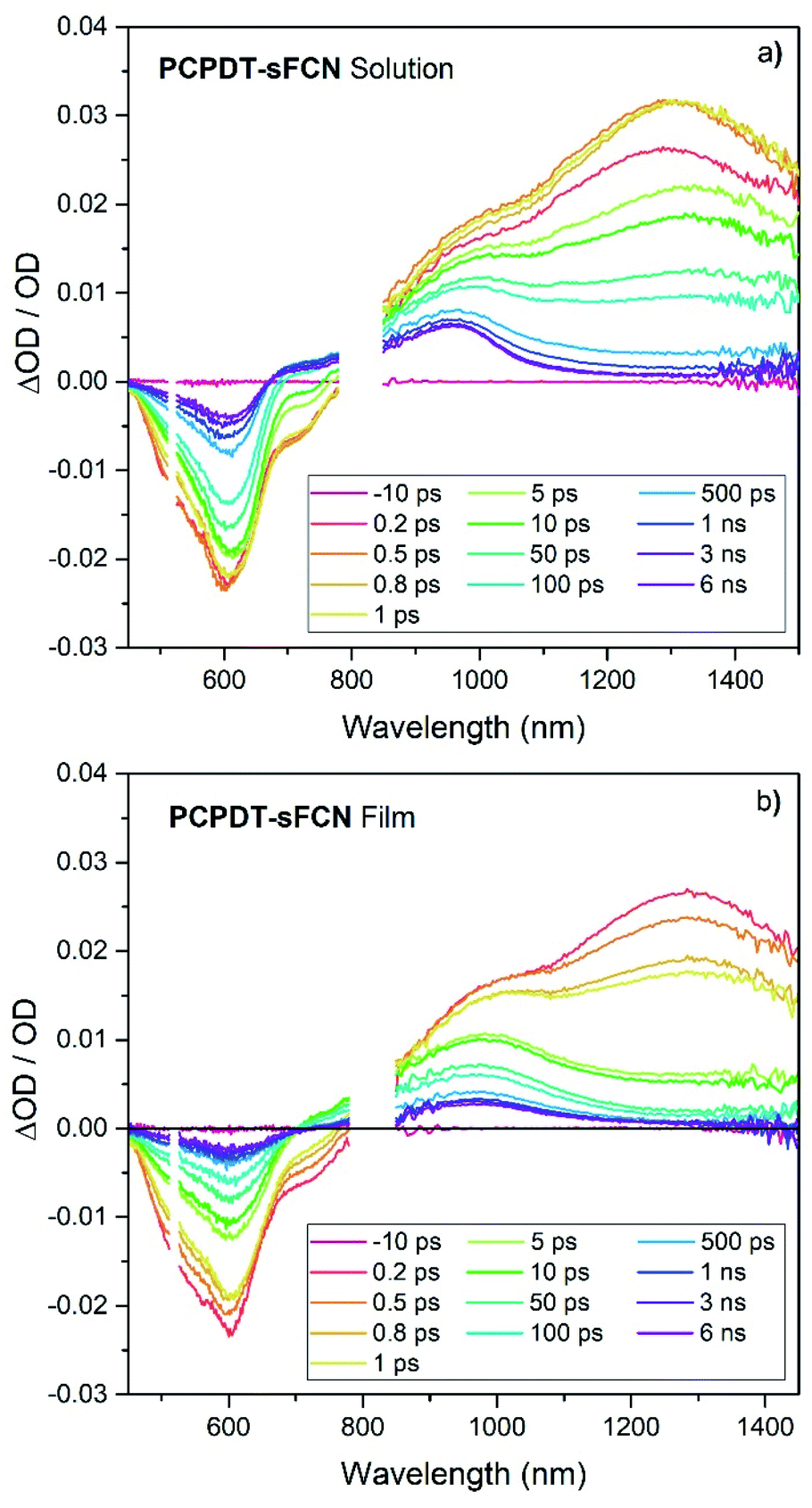 | ||
| Fig. 5 Picosecond transient absorption spectrum of PCPDT-sFCN in (a) chlorobenzene solution (50 μg mL−1) and (b) thin film, excited with a pump wavelength of 520 nm and an excitation density of 50 μJ cm−2. Spectra have been corrected for the absorbance at the exciting wavelength. | ||
The 960 nm ps-TA band is assigned to the PCPDT-sFCN triplet because it is identical in position and spectral shape compared to the triplet absorption in the μs-TA spectrum, as shown in Fig. 6a. Due to this strong resemblance, a likely candidate for the triplet observed in the ps-TAS is therefore the T1 state, assuming rapid internal conversion down the triplet manifold. Interestingly, the 960 nm band reached its maximum population by 1 ps, irrespective of phase (solution/film), and this suggests that the PCPDT-sFCN triplet is generated on ultrafast timescales. This is an unusual finding and will be addressed in detail later. It was also observed that the PCPDT-sFCN triplet spectrum at 6 ns for the film was broader than in solution, particularly on the red side between 1000–1100 nm (Fig. S4†), whereas the singlet state spectral width remained the same from solution to film. This 1000–1100 nm region is where the PCPDT-sFCN polaron was observed in the μs-TA spectrum. Given that higher charge carrier populations were observed in the film compared to the solution (Fig. 4b), this implies that polarons and/or CT states are also observed in the film ps-TA spectra.
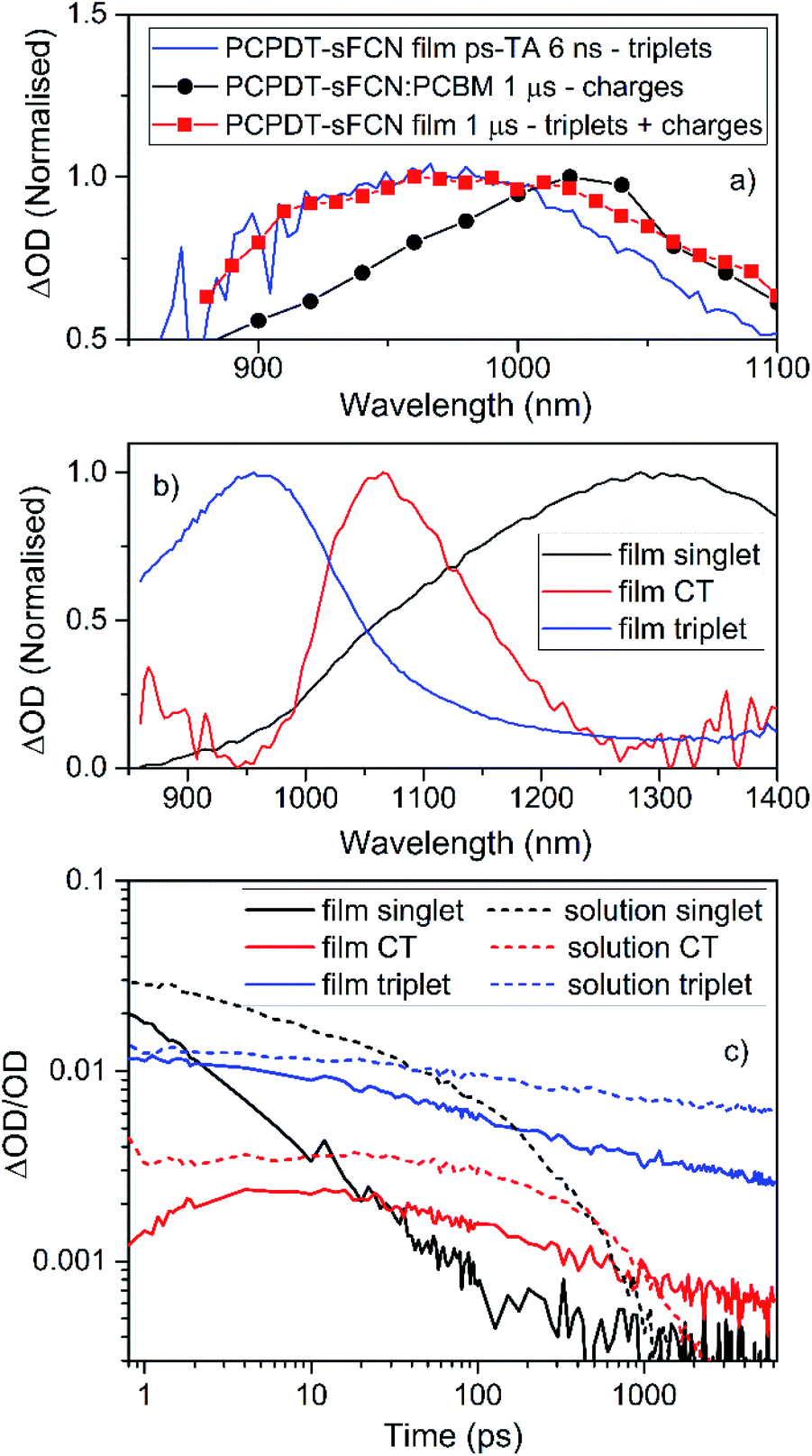 | ||
| Fig. 6 (a) A comparison of the PCPDT-sFCN film μs-TA and ps-TA raw data, showing that the 6 ns ps-TA band at 950 nm clearly matches the triplet at 950 nm in the μs-TA data. The PCPDT-sFCN film at 1 μs also clearly has a contribution from charge carriers, as evidenced by the comparison to the fullerene blend, which exhibits primarily charges. (b) Spectral components obtained by global analysis of the picosecond transient absorption data of the PCPDT-sFCN film, excited with a pump wavelength of 520 nm and an excitation density of 50 μJ cm−2. The solution data (Fig. S5†) is very similar. (c) Decay kinetics for each ps-TA spectral component extracted using global analyses for both the PCPDT-sFCN solution (chlorobenzene, 50 μg mL−1) and film. | ||
Global analyses were performed to extract kinetic information from the ps-TA spectra. The presence of three spectral components was extracted for both film and solution (Fig. 6b and S5†). These components were observed at 960, 1080, and 1300 nm. The transitions at 960 and 1300 nm, which are clearly observable in the raw spectral data, have been assigned to absorption by the PCPDT-sFCN triplet and singlet states respectively. The third transition located at 1080 nm, which was only apparent from the raw spectral data of the film, is assigned to a CT state. This CT state assignment is supported by the observation of a red-shifted, weak, CT state-like fluorescence in the time resolved PL spectra on very early nanosecond timescales. Furthermore, the 1080 nm band is at a very similar spectral position compared to the charges observed in the μs-TA spectra (1060 nm), and it is well-known that CT state and fully separated charges possess virtually identical absorption spectra.8,44,45 It should be pointed out that while this CT state component is relatively weak in magnitude, it is unlikely to be due to an artifact of the global analysis related to the singlet exciton red-shift with time: this would not have led to the observed population growth of this component. Furthermore, the film shows no singlet exciton red-shift over time, and it also possesses the third GA component.
The GA kinetics are shown in Fig. 6c. The singlet exciton shows complex multi-exponential kinetics rather than the expected mono-exponential kinetics, even at low excitation densities. The lack of excitation density dependence (Fig. S6†) shows that annihilation effects are not present and so the unusual need for multi-exponential fits for these decays reflects PCPDT-sFCN's energetic dispersity and the presence of multiple intrinsic decay pathways. This behaviour is observed in both solution and film, although the film does show faster singlet decay kinetics, a result of the condensed phase quenching the excited states more rapidly. The singlet exciton lifetime of <23 ps in the film is at least an order of magnitude faster than many other conjugated polymers which possess a similar optical band gap of 1.8–1.9 eV,32,46 a reflection of the ultrafast deactivation via triplet formation and/or internal conversion to a lower energy intramolecular CT state.
The GA kinetics show a growth in the CT state population (Fig. 6c), particularly for the film, reaching a maximum at approximately 5 ps for the PCPDT-sFCN film, and 10–15 ps for the solution. The rise times for the CT states match one of the decay time constants in the respective singlet state decay (1 ps for the film and 8 ps for the solution), suggesting that the CT state is formed directly via internal conversion of the initially photogenerated 1π–π* state. In solution, the CT state possesses monomolecular, excitation density independent decay dynamics,43,47 yielding a lifetime of τ = 550 ± 20 ps. This geminate recombination is another indication that it is indeed the CT state that is being observed, rather than free charges. In the film, however, the CT state shows non-first order decay kinetics, suggesting the presence of multiple decay pathways. Furthermore, the CT state is much longer-lived in the film than in the solution, which is consistent with both its appearance in the raw data at 6 ns and also the higher free charge population observed on the later μs timescales. A possible reason for these observations could be that the initially formed intramolecular CT state is more able to easily dissociate onto adjacent polymer chains in the film, thereby both prolonging the lifetime of the charges and also providing an additional decay pathway for the CT state.
We now turn our attention to the triplet exciton. The triplet first appears in the raw ps-TA data as a strong shoulder at ca. 1000 nm at ultrafast times, which is revealed as a distinct band at 960 nm at longer times. As such, it is identical to the PCPDT-sFCN film triplet observed in the μs-TA data (Fig. 6a). Note that little vibronic structure was observed in the singlet exciton component extracted from the global analysis (Fig. 6b), and this suggests that the 1000 nm shoulder in the raw data at early times is unlikely related to a vibronic transition of the singlet. Furthermore, the charged species component at 1080 nm is too far to the red and too weak (by almost an order of magnitude, Fig. 6c) to be significantly contributing to the initial strong shoulder at ca. 1000 nm. As such, it appears that triplet formation in PCPDT-sFCN does indeed occur on ultrafast sub-ps timescales. This is the case for both solution and film, indicating that the additional intermolecular interactions present in the film do not impede the ultrafast triplet formation, suggesting an intrinsic behaviour rather than a morphological/conformational dependent process. In both cases the maximum triplet population is reached prior to ∼1 ps. This implies that the triplet is populated on instrument limited timescales (<200 fs). The triplet exciton shows faster decay kinetics in the film compared to the solution (Fig. 6c), as was observed for the singlet. This is consistent with the smaller triplet population observed in the μs-TA data in the film compared to the solution. Again, multi-exponential kinetics indicate that the triplet possesses multiple decay pathways. However, this time the decay kinetics do depend on excitation density (on these early times: on μs timescales the triplet decay is excitation density independent). This could relate to mechanisms such as triplet–triplet annihilation or triplet-charge annihilation.
3. Discussion
Upon excitation the main photoproduct of the pristine PCPDT-sFCN, be it in solution or the solid phase, is highly likely to be the 1π–π* state (the ‘local’ singlet exciton (LE), distributed along the polymer backbone), rather than a lower energy CT state involving the orthogonal spirobifluorene unit. Local singlet excitons are well known to have considerably higher oscillator strengths than CT states, simply because of greater orbital overlap. A CT state is also evident for PCPDT-sFCN, but the ps-TAS results show the growth of this CT state on ps timescales, considerably slower than the local singlet exciton formation. In addition to the PCPDT-sFCN singlet, the PCPDT-sFCN triplet is also found generated on ultrafast timescales in both solution and thin film. Given the expansion of conjugated polymers into bio-applications,48 such efficient and rapid triplet formation is useful for photocaging and photodynamic therapy purposes. The ultrafast triplet formation, while unusual, is not unprecedented. It has also been observed for small molecule diketopyrrolopyrrole/fullerene blends as a result of ultrafast spin-mixing and back recombination.8The ultrafast timescales on which the PCPDT-sFCN triplet is generated is akin to singlet fission.49 However, singlet fission is highly unlikely in PCPDT-sFCN. Singlet fission has an energy requirement that 2ET1 ≤ ES1 must be met.50 Assuming that any singlet fission must occur within the local excited state manifolds (the singlet and triplet CT states will certainly be too close together energetically in an orthogonal system for singlet fission), the lowest 1LE state of PCPDT-sFCN experimentally lies at 1.94 eV. For singlet fission to occur, this means the lowest 3LE state would have to be located at ≤0.97 eV and we know that this is not the case: the efficient oxygen quenching of the PCPDT-sFCN triplet necessitates its energy being greater than that of the oxygen singlet state (0.98 eV).
Alternatively, the ultrafast triplet formation could be occurring via singlet–triplet ISC between the initially excited 1LE and the 3CT states.51,52 This ISC process can be highly efficient, particularly in the case where the energy gap between the states is small and the acceptor's molecular orbitals are found near perpendicular to the donor's: the spin-flip can be coupled to a large change in orbital angular momentum and therefore conserve overall angular momentum.53 This ISC process has been observed previously in small molecule and polymeric D-o-A systems and has been invoked to describe the mechanism via which they undergo TADF.54
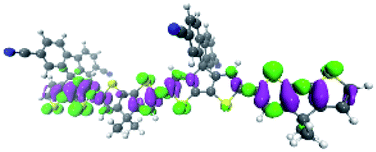
To explore this ISC possibility further, we investigated PCPDT-sFCN computationally. Due to the large size of the polymer, computation of the electronic structure of an entire polymer chain is not feasible, and instead the singlet and triplet manifolds were calculated utilising time density functional theory (TD-DFT) for a dimeric unit of the PCPDT-sFCN polymer (CAM-B3LYP/def2-TZVP). The results from the calculations (Table 1, Fig. 7) do indeed show a local singlet exciton (S2(LE)) at higher energy than the singlet charge transfer state (S1(CT)). The calculated S2 energy of 2.16 eV is in the same range as the experimentally determined optical band gap of 1.94 eV, and the calculated S1–S2 gap of 0.15 eV is similar to the difference between the π–π* emission and weak CT emission observed in the PL spectra (0.18 eV). The calculated triplet manifold indicates the T1 state to be a local π–π* triplet, while the T3 state is CT in character and virtually isoenergetic with the S1 (1CT) state.
| State | Type | Energy (eV) | Symmetry | Density difference |
|---|---|---|---|---|
| S1 | CT | 2.01 | B 2 |
|
| S2 | LE | 2.16 | B 1 |
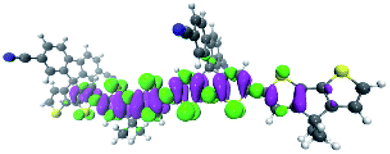
|
| T1 | LE | 1.30 | B 1 |
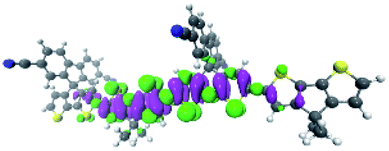
|
| T2 | LE | 1.71 | B 1 |
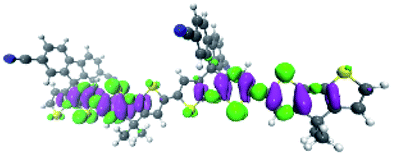
|
| T3 | CT | 2.00 | B 2 |
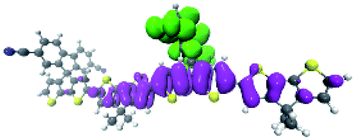
|
| T4 | LE | 2.11 | B 1 |
|
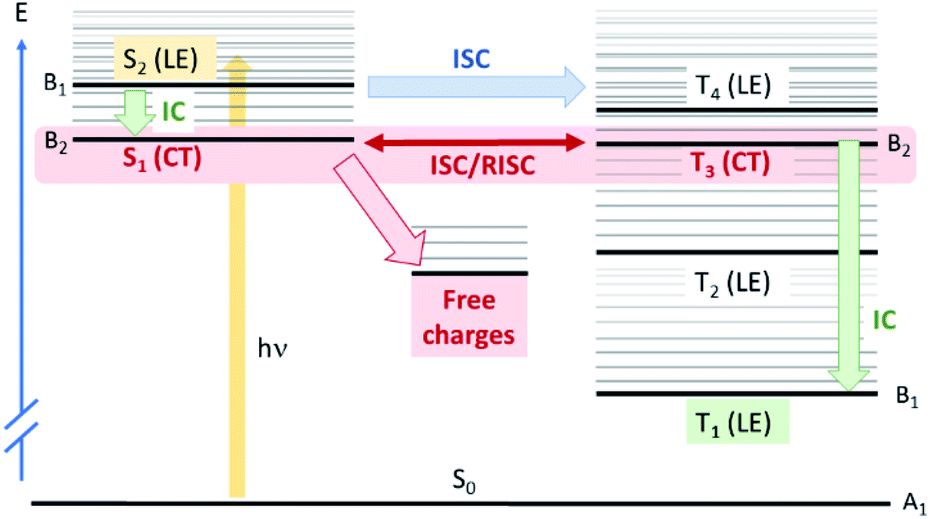 | ||
| Fig. 7 Schematic representation of the calculated electronic states and possible different photophysical pathways for PCPDT-sFCN. States that are experimentally observed are highlighted with a solid colour background. The initially photoexcited species is the allowed S2(LE) state. From here, ultra-fast quantum mechanically allowed intersystem crossing (ISC) can occur to the triplet manifold, mainly T3(CT). Note that this ISC is shown as a horizontal transition to an isoenergetic triplet state: the vibrationally excited T3(CT) state. ISC to T4 is less likely as it is symmetry forbidden. The T3(CT) is in a rapid ISC/RISC equilibrium with the isoenergetic S1(CT) state, from which free charges form. In addition, vibrational relaxation and internal conversion (IC) within the triplet manifold results in the formation of T1(LE), which is observed in the TAS. Finally, the photoexcited S2(LE) can also undergo IC to form S1(CT). | ||
Now that we have established the nature and energetics of the pertinent excited states, we return to our ISC hypothesis. We can use theoretical arguments to explain the observed photophysics of PCPDT-sFCN. The full polymer in a thin film will usually be amorphous and therefore have little symmetry. However, locally the polymer has C2v symmetry, with the C2 axis passing through the quaternary spiro carbon (Fig. S7†). The HOMO of PCPDT-sFCN therefore transforms as A2 symmetry, the LUMO of the thiophene units as B2 and the LUMO of the fluorene unit as B1. This means that the S1(CT) state is A2 × B1 = B2, so is y-polarized dipole allowed, but very weak due to poor orbital overlap. The S2(LE) state is A2 × B2 = B1, so is x-polarized and allowed. This gives rise to the broad absorption feature seen in the ground state absorption spectrum of PCPDT-sFCN. The T1(LE) state arising from HOMO–LUMO (thiophene) will be B1 and the T3(CT) state arising from HOMO–LUMO (fluorene) will be B2. Within this approximate model, all LE states considered here and localised along the thiophene backbone are B1 and all CT states are B2.
Concerning spin–orbit coupling, the spin orbit coupling (SOC) operator transforms as molecular rotations, which for C2v span A2, B1, and B2, but not A1. For SOC between the bright S2(LE) state and T3(CT) state, we have B1 × B2 = A2, which transforms as Rz, meaning that direct spin–orbit coupling between S2 and T3 is allowed. These results are consistent with the DFT-calculated SOC matrix elements (Table S1†), which exhibit the greatest value of 2.08 cm−1 for the S2(LE)→T3(CT) transition. As such, ISC from the initially photogenerated S2(LE) state to T3(CT) is fully quantum mechanically allowed, accounting for the ultrafast triplet formation observed experimentally for PCPDT-sFCN. Furthermore, the S2(LE) and T3(CT) states are separated by only 0.16 eV, and this will also facilitate a high rate of ISC. The presence of multiple triplet states that are energetically close to the bright S2(LE) state – particularly the T4(LE) – also enable increased ISC, as suggested by Fermi's golden rule. However, direct ISC to T4 is less likely as it is a symmetry–forbidden transition. Finally, the rate of the S2(LE)–T3(CT) transition could be enhanced via vibronic coupling between the S1(CT) and S2(LE) states, which can give rise to strong second-order coupling.55 We also apply these group theory arguments to the internal conversion processes (see ESI†).
A remarkable observation was the relatively efficient charge generation occurring in dilute PCPDT-sFCN solution (50 μg mL−1), and in the absence of a separate electron acceptor. Furthermore, these charges are extraordinarily long-lived, with millisecond lifetimes. This is highly unusual because conjugated polymers in solution often display only triplet formation,32 and for those that are capable of charge generation in solution or pristine films, these charges generally decay on nanosecond timescales due to rapid geminate recombination.9 The charge generation process in the pristine polymer solution is likely aided by the strongly intramolecular donor/acceptor nature of PCPDT-sFCN, providing significant charge-transfer character to the lowest energy singlet state. The spatial separation of electron and hole in this CT state reduces the exciton binding energy, and this, coupled to the aggregate configuration with multiple interchain interactions, allows the separated charges to form (depicted in Fig. 7). Furthermore, the strong spatial separation of hole and electron inhibits recombination, allowing the remarkably long charge carrier lifetime observed. A summary of all experimentally determined time constants for PCPDT-sFCN is shown in Fig. S8.†
The charge generation in PCPDT-sFCN is enhanced even further in the pristine film. Such efficient formation of long-lived charge carriers usually only occurs in the presence of a separate electron acceptor, with the accompanying blend morphology variations and stability concerns, additional fabrication complexity, and energy losses. In contrast, PCPDT-sFCN can overcome its own exciton binding energy to a significant extent without the need for a separate electron acceptor (although charge photogeneration is enhanced even further when an acceptor is present).
The extraordinarily long-lived charge carriers in pristine PCPDT-sFCN thus demonstrate that the orthogonal acceptor strategy has significant potential for single-material optoelectronic applications, such as organic photovoltaics and photodetectors. Indeed, a comparison of the polymer polaron decay kinetics of pristine PCPDT-sFCN in solution and well-known blends P3HT:PC60BM and PDTSiTTz:PC60BM (Fig. 8) reveals that pristine PCPDT-sFCN polarons have a very similar millisecond lifetime to the polarons in the two donor/acceptor blends. This observation is particularly remarkable because both P3HT:PC60BM and PDTSiTTz:PC60BM exhibit strong non-Langevin behaviour,56,57 with significantly supressed charge carrier recombination: as such, their polarons are significantly longer-lived than in most other donor/acceptor blends. It should be noted, however, that there are multiple other factors to consider in the pursuit of single-material devices, such as charge carrier mobility – which dictates charge transport and collection – and supressing non-radiative loss mechanisms.
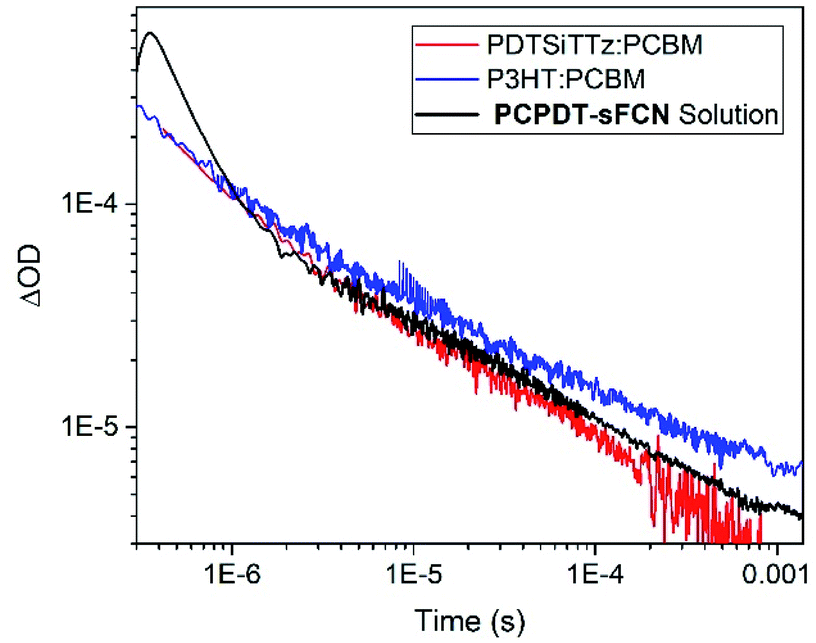 | ||
| Fig. 8 The μs TA kinetics of the PCPDT-sFCN solution under O2 atmosphere to show more of the polaron decay, compared to the polymer polaron decay kinetics of well-known non-Langevin (reduced recombination) bulk-heterojunction blends P3HT:PC60BM and PDTSiTTz:PC60BM. The PCPDT-sFCN data was measured using a pump wavelength of 532 nm, a probe wavelength of 980 nm, and an excitation energy of 20 μJ cm−2. The P3HT:PC60BM data was measured using a pump wavelength of 500 nm, a probe wavelength of 980 nm, and an excitation energy of 36 μJ cm−2. The PDTSiTTz:PC60BM data was measured using a pump wavelength of 532 nm, a probe wavelength of 1100 nm, and an excitation energy of 36 μJ cm−2. | ||
One of the key features of the donor-orthogonal-acceptor architecture (D-o-A) is the reduced spatial overlap between the frontier molecular orbitals, which is a consequence of the physical separation of the donor and acceptor units. This reduced spatial overlap has the effect of minimising the exchange energy between the singlet and triplet CT states, such that the 1CT–3CT energy gap becomes near degenerate, as observed in the calculated energy levels for PCPDT-sFCN. This allows for an equilibrium between the 1CT and 3CT states via spin-mixing, as seen for some other materials.8 Note that SOC between these two charge transfer states S1(CT) and T3(CT) is B2 × B2 = A1, and therefore formally forbidden, with a small calculated SOC matrix element of 0.02 cm−1. The question then arises of how the singlet and triplet CT states might couple, given that they have very weak direct spin–orbit coupling. However, the isoenergetic nature of these states (2.00 eV and 2.01 eV) mean that virtually any perturbation can couple them, such as nonadiabatic coupling.53
Indeed, the equilibrium between the 1CT and 3CT states may have an impact on charge photogeneration. A pertinent observation in this context is the reduction in the charge carrier population on μs timescales in the presence of oxygen. This is consistent with the presence of a 1CT/3CT equilibrium – oxygen would quench the 3CT state, thereby fewer CT states would be available for charge photogeneration. An interesting point here concerns the 3CT states formed via the equilibrium: how much do they directly contribute to charge generation? Since internal conversion through the triplet manifold must be rapid, this could act as a kinetic sink for the formation of any 3CT states. This not only implies that charge generation primarily occurs via the 1CT state, it also implies that the 1CT/3CT equilibrium must be slow, such that charge separated states can form in the significant populations we observe. In the context of the above experimental observation of reduced charge carrier populations under oxygen, this would then suggest that the quenching of 3CT states by oxygen leads to a shift in the equilibrium to the right (Le Chatelier's Principle), thereby depleting 1CT and thus reducing the number of free charges formed. A significant implication of this observation is that the slow 1CT/3CT equilibrium may be acting as a reservoir for charge carrier formation, enabling charge carriers to generate and exist in a pristine polymer long after typically expected in standard conjugated polymers. While the existence of 1CT/3CT equilibria is not new in organic electronics materials,58–60 this is, to our knowledge, the first time it has been successfully combined with an orthogonal acceptor to enable long-lived charge carrier populations, particularly in the pristine material.
4. Conclusions
A low band-gap conjugated polymer with a donor-orthogonal acceptor motif, PCPDT-sFCN, was synthesized and investigated using a combination of spectroscopic techniques. The orthogonal acceptor spatially isolated the LUMO from the HOMO, allowing isoenergetic singlet and triplet charge transfer states. A rich photophysical mechanism was uncovered with important implications for the future utilisation of the orthogonal acceptor strategy. Ultrafast triplet formation was observed, with a maximum triplet population forming prior to 1 ps: a highly unusual observation when singlet fission is not present. We show using DFT and group theory that this ultra-fast triplet formation is due to very efficient, quantum mechanically allowed intersystem crossing between the initially photoexcited local singlet exciton and the triplet charge transfer state.PCPDT-sFCN also shows remarkably long-lived charge carriers in both pristine solution and film, with lifetimes on the order of milliseconds. This occurs due to the isoenergetic singlet/triplet charge transfer state equilibrium acting as a reservoir of long-lived excited states, particularly in the condensed (film) phase. The charge transfer states are then able to dissociate into free charges relatively easily due to the spatial separation of PCPDT-sFCN's donor and orthogonal acceptor motifs, which may also assist in inhibiting bimolecular recombination. The orthogonal acceptor strategy therefore combines facile charge generation with a reservoir of excited states from which long-lived charges can be created, offering a new design rule for future improvements in optoelectronics, particularly for the development of single-material devices.
5. Experimental
5.1. Synthesis
Synthetic details for PCPDT-sFCN are available in the ESI.†5.2. Sample preparation
PC60BM was purchased from Ossila (>99% purity). Solutions were prepared via dissolving PCPDT-sFCN in spectroscopic grade chlorobenzene (Alfa Aesar) and stirring overnight at 65 °C in a glovebox, with an N2 atmosphere. Thin films were prepared via spin-coating from solution (10 μg mL−1) onto ultra-flat quartz substrates (1 mm thickness) for 90 seconds at 1600 rpm. Substrates were cleaned via separately sonicating in solutions of deionised water, acetone, and isopropanol for 15 min each. The pristine PCPDT-sFCN chlorobenzene solution was prepared with a concentration of 50 μg mL−1. The sample was also degassed by using three cycles of freeze pump thaw and sealing under a N2 atmosphere. This was achieved using a custom-made Young's tap quartz cuvette with a pathlength of 2 mm, whereby the solution was degassed on a Schlenk line and transferred to the cuvette via cannulation. Unless explicitly stated, all measurements were carried out under an inert atmosphere.5.3. Steady state absorption and PL
Absorbance spectra were recorded with a Perkin Elmer LAMBDA 365 UV/Vis spectrophotometer. Fluorescence spectra were recorded with a Horiba FluoroMax-4 spectrofluorometer, and corrected for instrument response at the exciting wavelength. Steady-state spectra were recorded at room temperature, in chlorobenzene solution (50 μg mL−1).5.4. Single-photon counting
TC-SPC measurements were carried out on a DeltaFlex TCSPC Lifetime spectrometer (Horiba). Measurements for the pristine PCPDT-sFCN solution and thin film were obtained using a laser diode with a pump wavelength of 467 nm. The instrument response function (IRF) was collected using chlorobenzene solution in a 2 mm pathlength quartz cuvette from Starna Scientific for the solution or using poly(methyl methacrylate) for the pristine film.5.5. Ultrafast TAS
Degassed solutions under inert nitrogen atmosphere were excited with a pump wavelengths of 520 nm, 5–50 μJ cm−2 pulses, generated by a commercially available optical parametric amplifier TOPAS (Light conversion) pumped by a Solstice Ti:sapphire regenerative amplifier (Newport Ltd). Changes in the optical density of the films induced by the laser excitation were probed with a second broadband pulse (830–1450 nm) generated in a sapphire crystal. A HELIOS (Ultrafast systems) transient absorption spectrometer was used for recording the dynamics of the transient absorption spectra up to 6.5 ns with an average 200 fs instrument response function.5.6. Microsecond TAS
Microsecond TAS was recorded using a 6 ns, 10 Hz Nd:YAG laser (Spectra-Physics, INDI-40-10) for the excitation pulse. The excitation wavelength of 532 nm was selected with a versaScan L-532 OPO (Newport Spectra-Physics). Excitation density was set from 2 to 80 μJ cm−2 using neutral density filters, and measured with an ES111C power meter (Thorlabs). Probe light was provided by a quartz tungsten halogen lamp (IL1, Bentham). The TA signals were recorded with Si and InGaAs photodiodes coupled to a preamplifier and an electronic filter (Costronic Electronics) connected to a Tektronix DPO4034B oscilloscope and PC. Probe wavelengths were selected with a Cornerstone 130 monochromator (Oriel Instruments) before the detector.5.7. DFT calculations
All calculations were performed using long range correct BLYP functional61 and a def2-SVP basis set62 as implemented within the ORCA quantum chemistry package.63 The range separate value w = 0.18 a−1 was determined using an optimal tuning procedure.64 The ground state geometry was optimised using DFT, while the excited S1 state geometry was optimised using time-dependent density functional theory (TDDFT) within the Tamm–Damcoff approximation.65 The environment was described using a polarisable continuum model with the parameters set to those of toluene. The spin orbit couple matrix elements between the excited states were also calculated with TDDFT using by using quasi-degenerate perturbation theory.66Author contributions
JS performed the majority of the spectroscopic work, with assistance from AM. DC and AF performed the synthesis. TJHH and TJP performed the theoretical and computational work. FC assisted in a supervisory capacity. TMC and HB conceptualised, supervised, and administered the project. All authors contributed to writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Optoelectronics Group at the University of Cambridge for sharing the software for global analysis, and the Durrant group at Imperial College London for the use of their TC-SPC and ps-TAS. TMC would like to acknowledge support from EPSRC project EP/N026411/1. HB would like to acknowledge support from EPSRC project EP/S003126/1. AM and FC would like to acknowledge support from EPSRC project EP/P006280/1. TJHH acknowledges a Royal Society University Research Fellowship URF\R1\201502.Notes and references
- Q. Yue, W. Liu and X. Zhu, J. Am. Chem. Soc., 2020, 142, 11613–11628 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65, 272–275 CrossRef CAS.
- M. S. Vezie, M. Azzouzi, A. M. Telford, T. R. Hopper, A. B. Sieval, J. C. Hummelen, K. Fallon, H. Bronstein, T. Kirchartz, A. A. Bakulin, T. M. Clarke and J. Nelson, ACS Energy Lett., 2019, 4, 2096–2103 CrossRef CAS.
- A. Guerrero and G. Garcia-Belmonte, Nano-Micro Lett., 2016, 9, 10 CrossRef PubMed.
- H. B. Naveed and W. Ma, Joule, 2018, 2, 621–641 CrossRef CAS.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2017, 16, 363–369 CrossRef CAS PubMed.
- I. G. Scheblykin, A. Yartsev, T. Pullerits, V. Gulbinas and V. Sundström, J. Phys. Chem. B, 2007, 111, 6303–6321 CrossRef CAS PubMed.
- E. Salvadori, N. Luke, J. Shaikh, A. Leventis, H. Bronstein, C. W. M. Kay and T. M. Clarke, J. Mater. Chem. A, 2017, 5, 24335–24343 RSC.
- J. M. Marin-Beloqui, K. J. Fallon, H. Bronstein and T. M. Clarke, J. Phys. Chem. Lett., 2019, 10, 3813–3819 CrossRef CAS PubMed.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, M. Heeney, S. Tierney, I. McCulloch, D. D. C. Bradley and J. R. Durrant, Chem. Commun., 2006, 3939 RSC.
- G. Grancini, M. De Bastiani, N. Martino, D. Fazzi, H. J. Egelhaaf, T. Sauermann, M. R. Antognazza, G. Lanzani, M. Caironi, L. Franco and A. Petrozza, Phys. Chem. Chem. Phys., 2014, 16, 8294–8300 RSC.
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed.
- Y. Kasai, Y. Tamai, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2015, 137, 15980–15983 CrossRef CAS PubMed.
- H. L. Stern, A. Cheminal, S. R. Yost, K. Broch, S. L. Bayliss, K. Chen, M. Tabachnyk, K. Thorley, N. Greenham, J. M. Hodgkiss, J. Anthony, M. Head-Gordon, A. J. Musser, A. Rao and R. H. Friend, Nat. Chem., 2017, 9, 1205 CrossRef CAS PubMed.
- C. Simpson, T. M. Clarke, R. W. MacQueen, Y. Y. Cheng, A. J. Trevitt, A. J. Mozer, P. Wagner, T. W. Schmidt and A. Nattestad, Phys. Chem. Chem. Phys., 2015, 17, 24826–24830 RSC.
- T. Dilbeck and K. Hanson, J. Phys. Chem. Lett., 2018, 9, 5810–5821 CrossRef CAS PubMed.
- Q. Wei, N. Fei, A. Islam, T. Lei, L. Hong, R. Peng, X. Fan, L. Chen, P. Gao and Z. Ge, Adv. Opt. Mater., 2018, 6, 1800512 CrossRef.
- D. Offenbartl-Stiegert, T. M. Clarke, H. Bronstein, H. P. Nguyen and S. Howorka, Org. Biomol. Chem., 2019, 17, 6178–6183 RSC.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, arXiv:2010.10978v1 [physics.app-ph], 2020.
- S. M. Menke and R. J. Holmes, Energy Environ. Sci., 2014, 7, 499–512 RSC.
- S. D. Dimitrov, S. Wheeler, D. Niedzialek, B. C. Schroeder, H. Utzat, J. M. Frost, J. Yao, A. Gillett, P. S. Tuladhar, I. McCulloch, J. Nelson and J. R. Durrant, Nat. Commun., 2015, 6, 6501 CrossRef CAS PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- G. Méhes, H. Nomura, Q. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed. Engl., 2012, 51, 11311–11315 CrossRef PubMed.
- L. Yu, Z. Wu, G. Xie, W. Zeng, D. Ma and C. Yang, Chem. Sci., 2018, 9, 1385–1391 RSC.
- J.-M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC.
- D. M. E. Freeman, A. J. Musser, J. M. Frost, H. L. Stern, A. K. Forster, K. J. Fallon, A. G. Rapidis, F. Cacialli, I. McCulloch, T. M. Clarke, R. H. Friend and H. Bronstein, J. Am. Chem. Soc., 2017, 139, 11073–11080 CrossRef CAS PubMed.
- N. Banerji, S. Cowan, M. Leclerc, E. Vauthey and A. J. Heeger, J. Am. Chem. Soc., 2010, 132, 17459–17470 CrossRef CAS PubMed.
- T. M. Clarke, J. Peet, A. Nattestad, N. Drolet, G. Dennler, C. Lungenschmied, M. Leclerc and A. J. Mozer, Org. Electron., 2012, 13, 2639–2646 CrossRef CAS.
- M. A. Faist, S. Shoaee, S. Tuladhar, G. F. A. Dibb, S. Foster, W. Gong, T. Kirchartz, D. D. C. Bradley, J. R. Durrant and J. Nelson, Adv. Energy Mater., 2013, 3, 744–752 CrossRef CAS.
- M. Horie, L. A. Majewski, M. J. Fearn, C.-Y. Yu, Y. Luo, A. Song, B. R. Saunders and M. L. Turner, J. Mater. Chem., 2010, 20, 4347–4355 RSC.
- S. Cook, A. Furube and R. Katoh, Energy Environ. Sci., 2008, 1, 294–299 RSC.
- D. Jarzab, F. Cordella, J. Gao, M. Scharber, H.-J. Egelhaaf and M. A. Loi, Adv. Energy Mater., 2011, 1, 604–609 CrossRef CAS.
- M. C. Scharber, C. Lungenschmied, H.-J. Egelhaaf, G. Matt, M. Bednorz, T. Fromherz, J. Gao, D. Jarzab and M. A. Loi, Energy Environ. Sci., 2011, 4, 5077–5083 RSC.
- J. Fu, N. F. Jamaludin, B. Wu, M. Li, A. Solanki, Y. F. Ng, S. Mhaisalkar, C. H. A. Huan and T. C. Sum, Adv. Energy Mater., 2019, 9, 1803119 CrossRef.
- R. J. E. Westbrook, D. I. Sanchez-Molina, D. J. Manuel Marin-Beloqui, D. H. Bronstein and D. S. A. Haque, J. Phys. Chem. C, 2018, 122, 1326–1332 CrossRef CAS.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934–20941 CrossRef CAS.
- A. F. Nogueira, I. Montanari, J. Nelson, J. R. Durrant, C. Winder, N. S. Sariciftci and C. Brabec, J. Phys. Chem. B, 2003, 107, 1567–1573 CrossRef CAS.
- T. M. Clarke, C. Lungenschmied, J. Peet, N. Drolet and A. J. Mozer, Adv. Energy Mater., 2015, 5, 1401345 CrossRef.
- H. Ohkita, S. Cook, Y. Astuti, W. Duffy, S. Tierney, W. Zhang, M. Heeney, I. McCulloch, J. Nelson, D. D. C. Bradley and J. R. Durrant, J. Am. Chem. Soc., 2008, 130, 3030–3042 CrossRef CAS PubMed.
- P. C. Y. Chow, S. Albert-Seifried, S. Gélinas and R. H. Friend, Adv. Mater., 2014, 26, 4851–4854 CrossRef CAS PubMed.
- C. Keiderling, S. Dimitrov and J. R. Durrant, J. Phys. Chem. C, 2017, 121, 14470–14475 CrossRef CAS.
- F. Etzold, I. A. Howard, R. Mauer, M. Meister, T.-D. Kim, K.-S. Lee, N. S. Baek and F. Laquai, J. Am. Chem. Soc., 2011, 133, 9469–9479 CrossRef CAS PubMed.
- S. Westenhoff, I. A. Howard, J. M. Hodgkiss, K. R. Kirov, H. A. Bronstein, C. K. Williams, N. C. Greenham and R. H. Friend, J. Am. Chem. Soc., 2008, 130, 13653–13658 CrossRef CAS PubMed.
- J. G. Müller, J. M. Lupton, J. Feldmann, U. Lemmer, M. C. Scharber, N. S. Sariciftci, C. J. Brabec and U. Scherf, Phys. Rev. B, 2005, 72, 195208–195217 CrossRef.
- T. M. Clarke, J. Peet, C. Lungenschmied, N. Drolet, X. Lu, B. M. Ocko, A. J. Mozer and M. A. Loi, J. Mater. Chem. A, 2014, 2, 12583–12593 RSC.
- S. Shoaee, M. P. Eng, E. Espildora, J. L. Delgado, B. Campo, N. Martin, D. Vanderzande and J. R. Durrant, Energy Environ. Sci., 2010, 3, 971–976 RSC.
- A. J. C. Kuehne, Adv. Biosyst., 2017, 1, 1700100 CrossRef PubMed.
- S. R. Yost, J. Lee, M. W. Wilson, T. Wu, D. P. McMahon, R. R. Parkhurst, N. J. Thompson, D. N. Congreve, A. Rao, K. Johnson, M. Y. Sfeir, M. G. Bawendi, T. M. Swager, R. H. Friend, M. A. Baldo and T. Van Voorhis, Nat. Chem., 2014, 6, 492–497 CrossRef CAS PubMed.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X. Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- M. T. Colvin, A. B. Ricks, A. M. Scott, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2012, 116, 1923–1930 CrossRef CAS PubMed.
- R. S. Minns, D. S. N. Parker, T. J. Penfold, G. A. Worth and H. H. Fielding, Phys. Chem. Chem. Phys., 2010, 12, 15607–15615 RSC.
- M. Lv, Y. Yu, M. E. Sandoval-Salinas, J. Xu, Z. Lei, D. Casanova, Y. Yang and J. Chen, Angew. Chem., Int. Ed., 2020, 59, 22179–22184 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- A. Pivrikas, G. Juška, A. J. Mozer, M. Scharber, K. Arlauskas, N. S. Sariciftci, H. Stubb and R. Österbacka, Phys. Rev. Lett., 2005, 94, 176806–176809 CrossRef CAS PubMed.
- T. M. Clarke, D. B. Rodovsky, A. A. Herzing, J. Peet, G. Dennler, D. DeLongchamp, C. Lungenschmied and A. J. Mozer, Adv. Energy Mater., 2011, 1, 1062–1067 CrossRef CAS.
- X.-K. Chen, V. Coropceanu and J.-L. Brédas, Nat. Commun., 2018, 9, 5295 CrossRef CAS PubMed.
- F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- M. Wang, T. Chatterjee, C. J. Foster, T. Wu, C.-L. Yi, H. Yu, K.-T. Wong and B. Hu, J. Mater. Chem. C, 2020, 8, 3395–3401 RSC.
- Y. Tawada, T. Tsuneda, S. Yanagisawa, T. Yanai and K. Hirao, J. Chem. Phys., 2004, 120, 8425–8433 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Neese, WIREs Computational Molecular Science, 2012, 2, 73–78 CrossRef CAS.
- J. Eng, B. A. Laidlaw and T. J. Penfold, J. Comput. Chem., 2019, 40, 2191–2199 CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- B. de Souza, G. Farias, F. Neese and R. Izsák, J. Chem. Theory Comput., 2019, 15, 1896–1904 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of polymers, additional spectroscopic measurements and theoretical calculations. See DOI: 10.1039/d1sc00919b |
| This journal is © The Royal Society of Chemistry 2021 |