
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTransition-metal-like bonding behaviors of a boron atom in a boron-cluster boronyl complex [(η7-B7)-B-BO]−†
Wen-Juan
Tian‡
 a,
Wei-Jia
Chen‡
b,
Miao
Yan
a,
Rui
Li
a,
Zhi-Hong
Wei
a,
Teng-Teng
Chen
b,
Qiang
Chen
a,
Hua-Jin
Zhai
*a,
Si-Dian
Li
*a and
Lai-Sheng
Wang
*b
a,
Wei-Jia
Chen‡
b,
Miao
Yan
a,
Rui
Li
a,
Zhi-Hong
Wei
a,
Teng-Teng
Chen
b,
Qiang
Chen
a,
Hua-Jin
Zhai
*a,
Si-Dian
Li
*a and
Lai-Sheng
Wang
*b
aNanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, China. E-mail: hj.zhai@sxu.edu.cn; lisidian@sxu.edu.cn
bDepartment of Chemistry, Brown University, Providence, Rhode Island 02912, USA. E-mail: lai-sheng_wang@brown.edu
First published on 3rd May 2021
Abstract
Boron displays many unusual structural and bonding properties due to its electron deficiency. Here we show that a boron atom in a boron monoxide cluster (B9O−) exhibits transition-metal-like properties. Temperature-dependent photoelectron spectroscopy provided evidence of the existence of two isomers for B9O−: the main isomer has an adiabatic detachment energy (ADE) of 4.19 eV and a higher energy isomer with an ADE of 3.59 eV. The global minimum of B9O− is found surprisingly to be an umbrella-like structure (C6v, 1A1) and its simulated spectrum agrees well with that of the main isomer observed. A low-lying isomer (Cs, 1A′) consisting of a BO unit bonded to a disk-like B8 cluster agrees well with the 3.59 eV ADE species. The unexpected umbrella-like global minimum of B9O− can be viewed as a central boron atom coordinated by a η7-B7 ligand on one side and a BO ligand on the other side, [(η7-B7)-B-BO]−. The central B atom is found to share its valence electrons with the B7 unit to fulfill double aromaticity, similar to that in half-sandwich [(η7-B7)-Zn-CO]− or [(η7-B7)-Fe(CO)3]− transition-metal complexes. The ability of boron to form a half-sandwich complex with an aromatic ligand, a prototypical property of transition metals, brings out new metallomimetic properties of boron.
1. Introduction
The electron deficiency of boron leads to unusual structures and bonding in bulk boron,1 boron compounds,2–4 and size-selected boron clusters.5–8 One interesting emerging property that has been recognized in recent years is the “metallomimetic” properties of boron,9 manifested most dramatically in a stable borylene dicarbonyl complex, in which two CO ligands are coordinated to a monovalent boron via donor–acceptor bonds.10 The formation of carbonyl complexes is one of the most prototypical chemical properties of transition metals, because of their partially filled d-shells. The partially-filled 2p orbitals endowed boron with similar properties. Even though borane compounds are well-known Lewis acids to form donor–acceptor complexes due to the electron deficiency of boron, carbonyls in the new boron metallomimetics have been found to display similar chemical reactivities to those in transition metal carbonyls.11,12 Another iconic chemical property of transition metals is the formation of sandwich or half-sandwich complexes with aromatic arene ligands.13 The interactions between the d orbitals and the π orbitals of the aromatic ligands provide exceptional stabilities for the transition-metal sandwich compounds.14 In the current article, we report a boron-cluster boronyl complex, [(η7-B7)-B-BO]−, featuring a central boron atom that exhibits the two most important bonding properties of a transition metal atom, i.e., complexation by a CO analogue of the boronyl ligand (BO−) and an arene-analog of the doubly aromatic B7 motif.During the past two decades, extensive joint experimental and theoretical investigations have revealed that most size-selected boron cluster monoanions Bn− possess 2D structures for n = 3–38, 40–42,5–8 even though 3D borospherene cages have been observed for B40− and B39−,15 minor seashell-like isomers observed for B28− and B29−,16 and a well-defined bilayer global minimum observed recently for B48−.17 The most interesting finding is the chemical bonding in the planar boron clusters, which is characterized by both delocalized σ and π bonds.18 The delocalized π bonds in the planar boron clusters are found to be similar to those in polycyclic aromatic hydrocarbons, leading to the concept of hydrocarbon analogues of boron clusters.19 One of the most prototypical planar boron cluster is B9−, which was found to have a D8h wheel-like structure with a central B atom inside a B8 ring (B©B8).20 Chemical bonding analyses revealed that the B9− cluster is bonded by eight two-center two-electron (2c–2e) σ bonds on its periphery, whereas the central boron atom is bonded with the B8 ring via three delocalized 9c–2e σ bonds and three delocalized 9c–2e π bonds, making B9− a doubly aromatic system since both the σ and π electron systems satisfy the (4N + 2) Hückel rule. It was further found that the central B atom can be replaced by a transition metal to form a novel class of doubly aromatic borometallic wheel clusters, M©Bn− for n = 8–10.21 In fact, the central B atom in the D8h B9− cluster can be viewed as having transition-metal-like bonding properties. The B8 cluster was found to have a D7h wheel structure with a triplet ground state.20a Adding two electrons to B8 resulted in a closed-shell doubly aromatic B82− species,22 similar to B9−. Subsequently, the B7− cluster was found to have a triplet C6v concave structure because the B6 ring is too small to host the central B atom.23 Adding two electrons to B7− led to a closed-shell doubly aromatic B73− unit, first realized in the half-sandwich B7Pr cluster, i.e. [(η7-B7)-PrIII].24 A number of binary clusters containing the aromatic B73− unit have been computed recently.25
Here we report a joint photoelectron spectroscopy (PES) and quantum chemical study of the B9O− cluster. Temperature-dependent PES experiments suggested that two isomers contributed to the well-resolved photoelectron spectra. Global minimum searches found that the most stable isomer of B9O− has an unexpected 3D umbrella-like structure 1 (C6v, 1A1), along with two other low-lying isomers, a disk-like structure with a BO unit bonded to the edge of a 2D B8 cluster 2 (Cs, 1A′) and a double-chain-shaped structure 3 (Cs, 1A′). The simulated spectra of structures 1 and 2 together agree well with the experimental data. The global minimum C6v umbrella structure of B9O− consists of an unprecedented central B atom being sandwiched by a BO boronyl ligand and a η7-B7 aromatic ligand, i.e. [(η7-B7)-B-BO]−. Comparisons of the bonding in the C6v B9O− with that in [(η7-B7)-Zn-CO]−, [(η7-B7)-Fe(CO)3]− and [(η7-B7)-Pr] provide evidence for transition-metal-like bonding behaviors by the central B atom, which shares its valence electrons with the B7 ligand to achieve double aromaticity.
2. Experimental and theoretical methods
2.1 Photoelectron spectroscopy
The experiment was carried out using a magnetic-bottle PES apparatus equipped with a laser vaporization cluster source, details of which have been described before.6,26 The B9O− clusters were produced by laser vaporization of a mixed Bi/10B target. The B9O− clusters were formed in the nozzle due to residual oxygen impurity on the target surface. The clusters were formed in a large waiting room nozzle,6,26 which were entrained by a He carrier gas seeded with 5% Ar and cooled via supersonic expansion. After passing a skimmer, negatively-charged clusters were extracted perpendicularly from the collimated cluster beam into a time-of-flight mass spectrometer for size analyses. The B9O− clusters of interest were mass-selected and decelerated before photodetachment. Two photon energies were used in the PES experiment: 266 nm (4.661 eV) from a Nd:YAG laser and 193 nm (6.424 eV) from an ArF excimer laser. Photoelectron spectra were calibrated using the known spectra of Bi−. The resolution of the PES apparatus was ΔEk/Ek ≈ 2.5%, i.e. ∼25 meV for 1 eV electrons.As shown previously,6,27 the cluster cooling efficiency in our experiment depends on their residence times inside the large waiting room nozzle. The clusters that leave the nozzle later are colder than those that leave earlier. Thus, the cluster temperatures can be controlled to a limited degree by selecting the appropriate timing for cluster extraction into the time-of-flight mass spectrometer, as shown in the spectra in Fig. S1.†
2.2 Theoretical methods
The global minimum search was conducted at the density-functional theory (DFT) level using the Coalescence Kick (CK) algorithm,28 aided with manual structural constructions. About 4000 stationary points for B9O−, and the same number of points for B9O, were probed in the CK search. Candidate low-lying structures were then fully optimized at the B3LYP/6-311++G(d,p)29 and PBE0/6-311++G(d,p)30 levels, respectively. Adiabatic (ADEs) and vertical (VDEs) detachment energies were calculated at the B3LYP level for the ground state transition and at the time-dependent B3LYP (TD-B3LYP)31 level for higher VDEs. Single-point coupled-cluster calculations (CCSD(T))32,33 were also performed at the B3LYP geometries to refine the ground state ADEs and VDEs and to further evaluate the relative energies of the low-lying structures. The ADE was calculated as the energy difference between the optimized anion and neutral for each isomer, whereas the first VDE was calculated as the energy difference between the anion and neutral at the anion geometry. Higher VDEs were calculated at the B3LYP level using TD-DFT. Chemical bonding was analyzed using both MOs and adaptive natural density partitioning (AdNDP).34 Orbital compositions were analyzed using Muitiwfn.35 All calculations were carried out using Gaussian 09.363. Experimental results
The photoelectron spectra of B9O− were obtained at two photon energies, 266 nm and 193 nm, as shown in Fig. 1. The 266 nm spectrum (Fig. 1a) displays three well-resolved bands. Band X′ at the low binding energy side is relatively broad with a VDE of 3.69 eV measured from the band maximum. There are hints of vibrational structures in band X′, but they are not well resolved.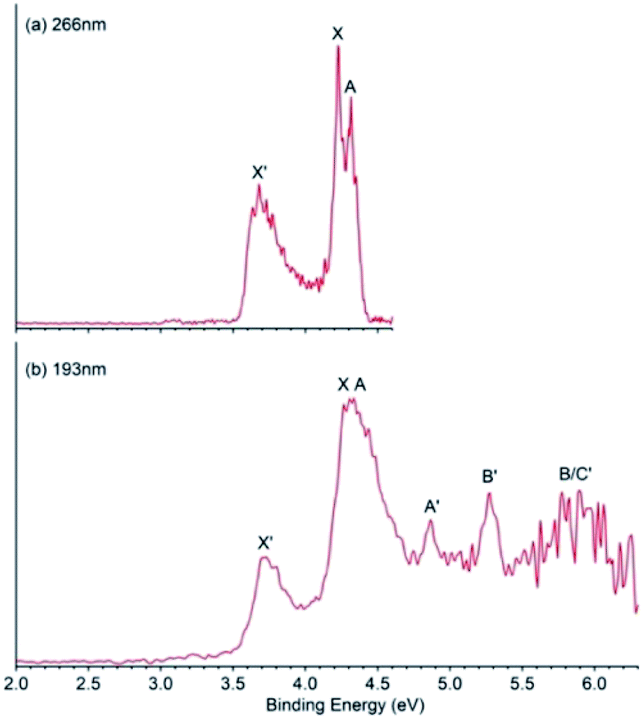 | ||
| Fig. 1 Photoelectron spectra of B9O− at (a) 266 nm (4.661 eV) and (b) 193 nm (6.424 eV). | ||
Hence, we determined the ADE by drawing a straight line at its leading edge and then adding the instrumental resolution. The ADE so measured is 3.59 eV, which also represents the electron affinity (EA) of B9O. Band X at a VDE of 4.22 eV and band A at a VDE of 4.31 eV are relatively sharp and closely spaced. The ADE for band X was estimated to be 4.19 eV from its leading edge. At 193 nm (Fig. 1b), several more PES bands were observed beyond 4.7 eV, including two well-resolved bands A′ (VDE: 4.86 eV) and B′ (VDE: 5.28 eV). The signal-to-noise ratios in the higher binding energy range are poor, but a broad band around ∼5.9 eV is discernible which may contain multiple detachment transitions. It is labeled as B/C′ for the sake of discussion.
Even though the PES features of B9O− are well resolved, the spectra appear more complicated in comparison to those of B8− or B9−,20 suggesting possible existence of multiple isomers as observed for those of B7−.23 As shown previously,6,27 we could tune the cluster temperatures to a limited degree by varying the residence time of the clusters inside the nozzle. Indeed, as we varied the experimental conditions, we found that the relative intensities of the spectral features changed, providing direct evidence of the existence of isomers in the cluster beam. As shown in Fig. S1,† the relative intensities of bands X′, A′, and B′ increased under hotter source conditions, suggesting they came from a slightly higher energy isomer of B9O− with an EA of 3.59 eV for the corresponding neutral B9O isomer. Bands X and A and possibly some features around 5.9 eV should belong to a lower energy isomer of B9O− with an EA of 4.19 eV for the corresponding B9O isomer. The temperature-dependent data are critical for the spectral assignments and comparison with theoretical calculations. All the observed VDEs are given in Table 1, where they are compared with the theoretical results to be discussed below.
| Features | VDE (exp)a | Final state and electronic configuration | VDE (theo) |
|---|---|---|---|
| a The estimated experimental uncertainty is ±0.02 eV. | |||
| C 6v B 9 O − (1) | |||
| X | 4.22 | 2E1{…(6a1)2(1b1)2(3e1)4(4e1)3} | 4.21 |
| A | 4.31 | 2E1{…(6a1)2(1b1)2(3e1)4(4e1)3} | 4.37 |
| B | ∼5.9 | 2E1{…(6a1)2(1b1)2(3e1)3(4e1)4} | 5.84 |
| 2E1{…(6a1)2(1b1)2(3e1)3(4e1)4} | 5.85 | ||
| 2B1{…(6a1)2(1b1)1(3e1)4(4e1)4} | 6.25 | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| C s B 9 O − (2) | |||
| X′ | 3.69 | 2A′′{…(9a′)2(4a′′)2(10a′)2(5a′′)2(11a′)2(6a′′)1} | 3.50 |
| A′ | 4.86 | 2A′′{…(9a′)2(4a′′)2(10a′)2(5a′′)2(11a′)1(6a′′)2} | 4.76 |
| B′ | 5.28 | 2A′{…(9a′)2(4a′′)2(10a′)2(5a′′)1(11a′)2(6a′′)2} | 5.17 |
| C′ | ∼5.9 | 2A′′{…(9a′)2(4a′′)2(10a′)1(5a′′)2(11a′)2(6a′′)2} | 5.79 |
|
|||
| C s B 9 O − (3) | |||
| X′ | 3.69 | 2A′{…(2a′′)2(11a′)2(12a′)2(13a′)2(3a′′)2(14a′)1} | 3.56 |
| A′ | 4.86 | 2A′′{…(2a′′)2(11a′)2(12a′)2(13a′)2(3a′′)1(14a′)2} | 4.79 |
| B′ | 5.28 | 2A′{…(2a′′)2(11a′)2(12a′)2(13a′)1(3a′′)2(14a′)2} | 5.35 |
| C′ | ∼5.9 | 2A′{…(2a′′)2(11a′)2(12a′)1(13a′)2(3a′′)2(14a′)2} | 5.54 |
| 2A′{…(2a′′)2(11a′)1(12a′)2(13a′)2(3a′′)2(14a′)2} | 5.65 | ||
4. Theoretical results
Our global minimum searches identified several low-lying isomers, as shown in Fig. 2. Other higher-lying optimized geometries and their relative energies at the B3LYP,29 PBE0,30 and single-point CCSD(T)//B3LYP32,33 levels are given in Fig. S2 and S3 in the ESI.† As shown previously,37–39 all boron-rich oxide clusters contain the highly stable boronyl (BO) ligand,40 which is isoelectronic with CN. Similarly, we found that the three lowest-lying isomers of B9O0/− all contain the BO unit bonded to a B8 cluster (Fig. 2). The global minimum of B9O− (1) at all three levels of theory is a closed-shell 3D umbrella-like structure with C6v symmetry. The second lowest-lying isomer (2) consists of a disk-like B8 cluster bonded to a BO unit, which is 0.13 eV higher in energy than the C6v structure at the B3LYP level, 0.79 eV higher at the PBE0 level, and 0.62 eV higher at the CCSD(T) level. The next isomer (3) is 0.12 eV higher in energy than the C6v global minimum at the B3LYP level, but much higher in energy at the PBE0 level by 1.14 eV and the CCSD(T) level by 0.85 eV.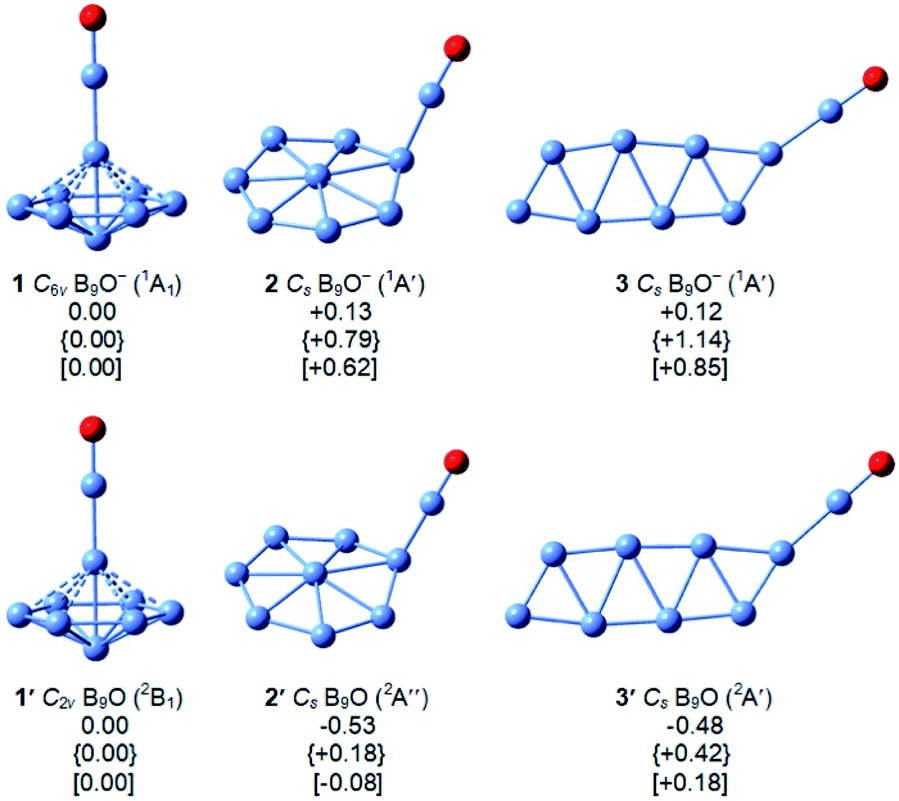 | ||
| Fig. 2 The structures of the three lowest-lying isomers of B9O− (1–3) and B9O (1′–3′) at the B3LYP/6-311++G(d,p) level. Their relative energies in eV are given at the B3LYP/6-311++G(d,p), PBE0/6-311++G(d,p) (in curly brackets), and single-point CCSD(T)//B3LYP/6-311++G(d,p) (in square brackets) levels, respectively. | ||
The low-lying isomers of the B9O neutral are similar to those of the anions (Fig. 2), except that the energy differences between the isomers become much smaller. In fact, the neutral isomers 1′ and 2′ are almost isoenergetic at the CCSD(T) level. The detailed structural parameters at the B3LYP level for the three low-lying structures of B9O0/− are given in Fig. S4.† The umbrella-like global minimum (1′) is distorted to C2v symmetry due to the Jahn–Teller effect.
5. Comparison between experiment and theory
To confirm the global minimum of B9O− and help assign the two observed isomers in the experiment, we calculated the ADEs and VDEs for the three low-lying isomers of B9O− at the B3LYP and single-point CCSD(T) levels of theory. The ground state ADE and VDE for each isomer are compared with the experimental data in Table 2. We found that both B3LYP and CCSD(T) gave similar results. The calculated ADE and VDE for the umbrella isomer 1 are significantly higher than those for isomers 2 and 3. The computed ADE/VDE values of 4.15/4.30 eV for isomer 1 at the CCSD(T) level are in excellent agreement with the experimental observation of 4.19/4.22 eV for the X band. On the other hand, the computed first ADE/VDE for isomers 2 and 3 are similar and both [3.45/3.58 eV for 2 and 3.48/3.62 eV for 3 at the CCSD(T) level] seem to agree well with the experimental observation of 3.59/3.69 eV for the X′ band. Thus, even though isomer 3 is higher in energy, the first ADE and VDE are not sufficient to rule it out.| Isomers | Final state | ADE (theo) | VDE (theo) | ADE (exp) | VDE (exp) | ||
|---|---|---|---|---|---|---|---|
| B3LYP | CCSD(T) | B3LYP | CCSD(T) | ||||
| 1, C6v | 2A | 4.08 | 4.15 | 4.21 | 4.30 | 4.19 ± 0.04 | 4.22 ± 0.02 |
| 2, Cs | 2A′′ | 3.38 | 3.45 | 3.50 | 3.58 | 3.59 ± 0.04 | 3.69 ± 0.02 |
| 3, Cs | 2A′ | 3.44 | 3.48 | 3.56 | 3.62 | ||
The higher VDEs for all three isomers are compared with the experimental data in Table 1, with the final electron configurations and electronic states of the corresponding neutrals indicated. The valence molecular orbital (MO) pictures are shown in Fig. S5–S7† for isomers 1–3, respectively. Because all three isomers are closed-shell, detachment from each occupied MO is expected to give a single PES band, resulting in relatively simple photoelectron spectra and facilitating comparisons with the experimental data. Each detachment channel was fitted with a unit-area Gaussian with a width of 0.04 eV to produce a simulated spectrum. The simulated spectra for isomers 1–3 using the data at the B3LYP level of theory are compared with the 193 nm spectrum in Fig. 3. The calculated VDEs for the first two detachment channels of isomer 1 are close to each other, consistent with the X and A bands. The calculated VDEs for the third and fourth detachment channels of isomer 1 are almost identical and they must contribute to the broad band at ∼5.9 eV. The fifth detachment channel with a computed VDE of 6.25 eV is near the cutoff energy of the 193 nm spectrum, where the spectral signal-to-noise ratios were poor. The overall good agreement between the simulated spectrum for isomer 1 and the observed PES bands X, A, and B provides considerable credence for the C6v umbrella structure as the global minimum of B9O−.
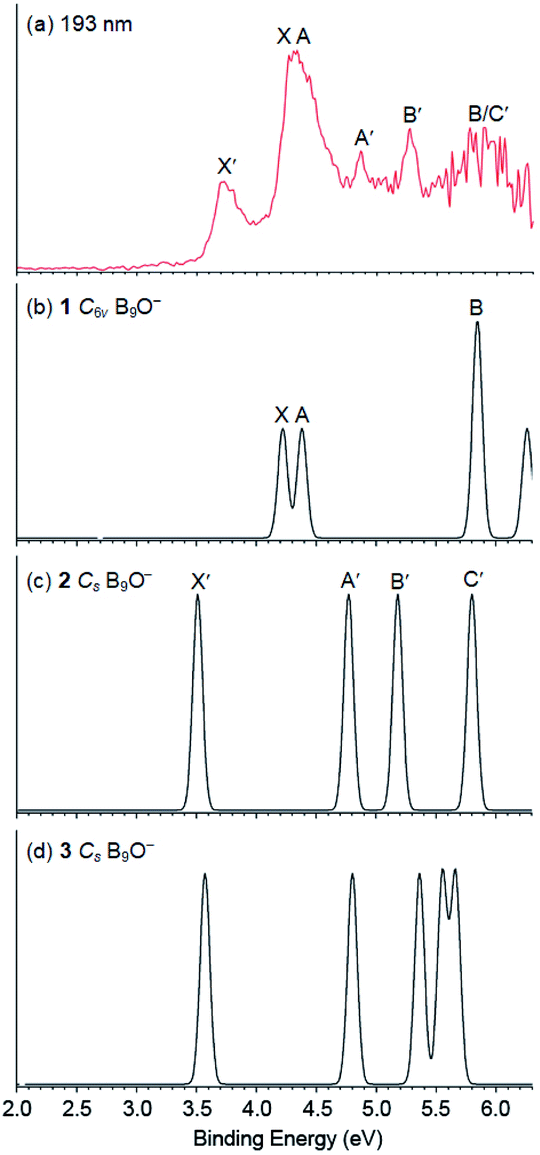 | ||
| Fig. 3 Comparison of (a) the photoelectron spectrum of B9O− at 193 nm with the simulated spectra of (b) C6v (1), (c) Cs (2) and (d) Cs (3) at the TD-B3LYP level. The simulated spectra were obtained by fitting the calculated VDEs with unit-area Gaussian functions of 0.04 eV width. | ||
As shown in Table 1 and Fig. 3, the first three detachment channels of both isomers 2 and 3 agree well with the observed X′, A′, and B′ bands. The fourth detachment channel of isomer 2 is also in good agreement with the broad feature at ∼5.9 eV. However, the computed VDEs for the fourth and fifth detachment channels of isomer 3 do not agree with the experimental observation and thus it can be ruled out as the carrier for the observed X′, A′, and B′ bands. This conclusion is also consistent with the relatively high energy of isomer 3. Hence, both the experimental and theoretical results indicate that the co-existing low-lying isomer in the cluster beam of B9O− should be isomer 2.
6. Discussion
6.1 The structures and chemical bonding of B9O−
The 3D umbrella global minimum structure for B9O− is both surprising and unprecedented. Several boron monoxide clusters have been studied experimentally before and they are all found to consist of a planar boron cluster motif bonded to a BO unit on the periphery due to the exceptional stability of the boronyl unit.39 The boron cluster motifs in these monoxide clusters are usually related to the most stable pure boron clusters, such as isomer 2 for B9O−, which we expected to be the global minimum. It should be pointed out that the structures of the B9O cluster were studied previously using DFT methods.38 The most stable structure reported was similar to isomer 3 of the current study both as a neutral and anion. Two other low-lying structures were also identified in previous studies, one is similar to the fourth isomer of B9O− (Cs, 1A′) which is 0.22 eV above the C6v global minimum at the B3LYP level and the other one is similar to the eleventh isomer of B9O− (C2v, 1A1) which is 1.45 eV above the global minimum at the B3LYP level (Fig. S2†). Even though these structures are low-lying isomers in the neutral (Fig. S3†), they are significantly higher in energy in the anion.The most unusual structural feature of the umbrella global minimum of B9O− is the central boron atom. It forms a σ bond with the BO unit, but its bonding to the B7 unit is unprecedented. The B–B distances between this atom and the periphery of the B7 unit are 1.91 Å, significantly longer than the B–B distance (1.72 Å) between the apex B atom of the B7 unit and the six peripheral B atoms (Fig. S4†). To understand the stability and bonding of the umbrella structure of B9O−, we carried out AdNDP analyses,34 as shown in Fig. 4. The triple bond in the BO unit (Fig. 4a), the σ bond between the central B and the BO unit (Fig. 4d), and the six 2c–2e bonds for the periphery of the B7 motif (Fig. 4b) are clearly revealed. The most interesting bonding features are the two sets of three delocalized σ and π bonds (Fig. 4c and e), which are almost identical to those in the B73− motif found in the [(η7-B7)-Pr] cluster.24 This observation suggests that the central B atom shares its valence electrons with the B7 unit to fulfill double aromaticity. Thus, it is the exceptional stability of the doubly aromatic B73− system that gives rise to the extraordinary stability of the unexpected umbrella global minimum for B9O−. The extremely high electron binding energy (4.19 eV) of the umbrella structure indicates its high electronic stability. The HOMO (e1) of the C6v B9O− (Fig. S5†) is doubly degenerate and corresponds to two orbitals responsible for the σ aromaticity. Thus, the removal of an electron from the HOMO not only induces a symmetry breaking in neutral B9O (C2v) due to the Jahn–Teller effect, but also decreases the σ aromaticity in the B7 motif, which explains why the stability of the umbrella structure in the neutral (1′ in Fig. 2) is reduced relative to isomers 2′ and 3′. Even though charge transfer is not expected from the central B atom to the B7 unit, the umbrella structure of B9O− can be approximately viewed as an η7-B73− ligand coordinated to a BII center or [(η7-B7)-BII-BO]−.
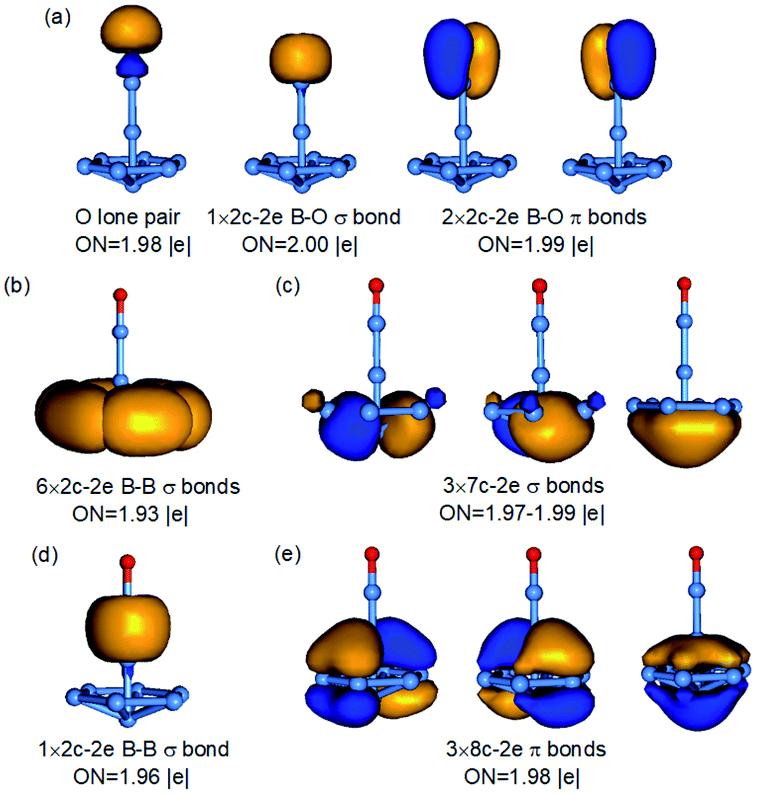 | ||
| Fig. 4 AdNDP bonding analyses for the C6v B9O− global minimum. The different bonding elements are given in (a–e). ON is the occupation number. | ||
We also analyzed the bonding in isomers 2 and 3 of B9O− using AdNDP, as displayed in Fig. S8 and S9,† respectively. Each can be described as a BO unit bonded to a B8 motif. In isomer 2, the disk-shaped B8 motif is similar to the global minimum D7h B8.20 The B8 unit still maintains σ aromaticity, but it is π antiaromatic with only two π bonds, consistent with its structural distortion. Isomer 3 contains two delocalized σ and two delocalized π bonds, which can be viewed as doubly antiaromatic in agreement with the fact that it is not energetically competitive relative to isomers 1 and 2.
6.2 Comparison of the bonding in C6v B9O− with transition metal complexes
The high stability of the doubly aromatic B73− unit, first realized in the [(η7-B7)-Pr] cluster,24 is interesting. It has been used recently to design binary clusters with alkali or alkali earth elements,25 which can transfer charges more easily to the B7 motif. In particular, the B73− ligand has been utilized to stabilize the single σ bond in Mg22+ or Zn22+ in B7M2− type clusters.25a,b The bonding mode of the η7-B7 unit to the central B atom in the C6v B9O− cluster is reminiscent of the coordination mode of arenes to transition metals in sandwich or half-sandwich complexes.13,14 Thus, the C6v B9O− cluster brings out a new metallomimetic property of boron. To further understand this metallomimetic property, we compare the bonding in the C6v B9O− cluster with that of two model metal complexes, [(η7-B7)-Zn-CO]− and [(η7-B7)-Fe(CO)3]−, as shown in Fig. 5 and S10,† respectively.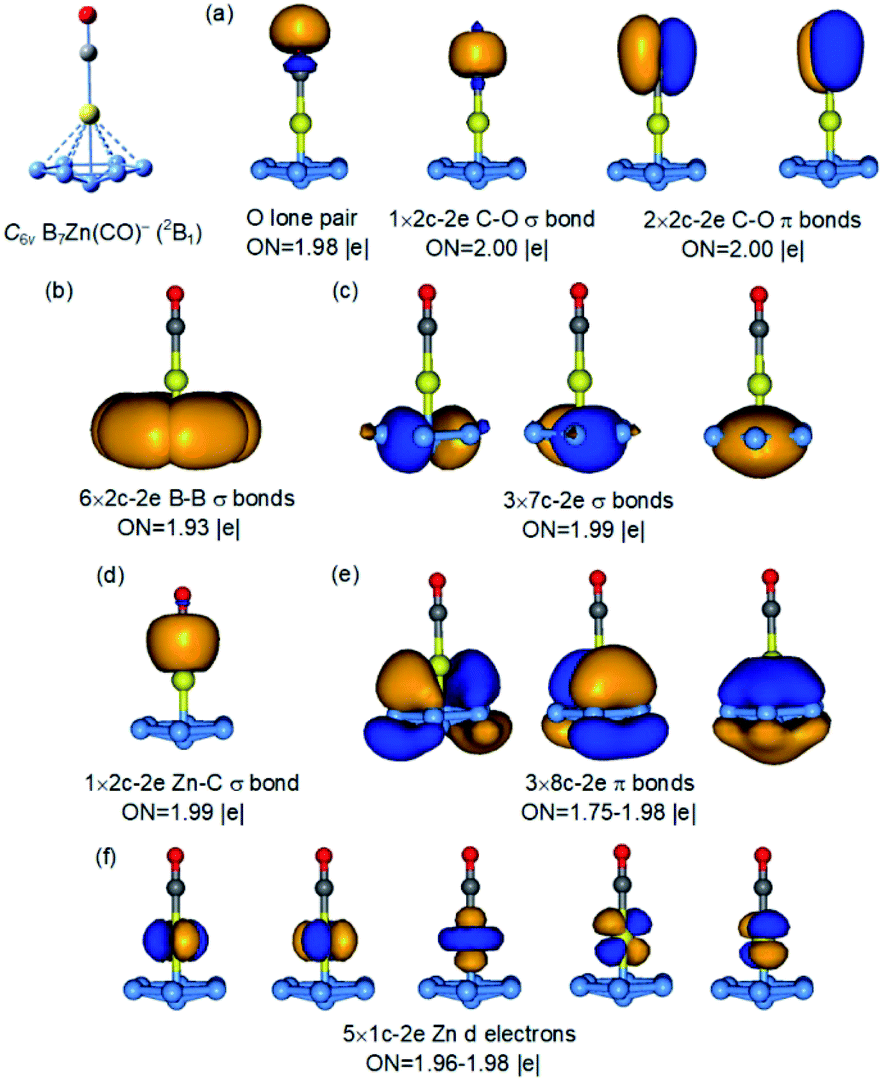 | ||
| Fig. 5 AdNDP bonding analyses for the C6v B7Zn(CO)− complex. ON is the occupation number. | ||
Because the 3d electrons of Zn do not participate in bonding, the [(η7-B7)-Zn-CO]− complex can be considered as isoelectronic to [(η7-B7)-B-BO]− and their bonding is nearly identical. As shown in Fig. S10,† the bonding between the η7-B7 motif with Fe in [(η7-B7)-Fe(CO)3]− is also similar to that in the C6v B9O− cluster (Fig. 4). In fact, the 18-electron rule is fulfilled in [(η7-B7)-Fe(CO)3]−, making it a highly stable species isoelectronic to the well-known (η6-C6H6)–Cr(CO)3 complex.14 To further demonstrate the analogy between the BO− and CO ligands,41 we also analysed the bonding in [(η7-B7)-B-CO] by replacing the BO− ligand in the C6v B9O− cluster with CO. As shown in Fig. S12,† the bonding in this model complex is identical to that in [(η7-B7)-B-BO]−. Thus, the central B atom in [(η7-B7)-B-BO]− can be considered to embody the two most important bonding properties of transition metals, the ability to form carbonyl complexes and sandwich-type complexes with aromatic arenes. While half-sandwich complexes of planar aromatic boron clusters with transition metal atoms have been observed or proposed,42,43 the discovery of the half-sandwich complex between B7 and the B atom is unprecedented, suggesting again the versatility of the chemical bonding of boron.
7. Conclusions
In conclusion, we have investigated the structures and bonding of a boron monoxide cluster B9O− using photoelectron spectroscopy and theoretical calculations. Temperature-dependent experiments revealed the existence of two isomers in the photoelectron spectra, one with an EA of 3.59(4) eV and another with an EA of 4.19(4) eV for the corresponding B9O neutrals. The global minimum of B9O− is found surprisingly to have a 3D C6v umbrella-like structure, corresponding to the high EA species, whereas a low-lying isomer consisting of a BO unit bonded to the periphery of a disk-like B8 motif is found to correspond to the low EA species. The C6v global minimum can be viewed as a half-sandwich complex with the central B atom coordinated by a η7-B7 unit and a BO unit, i.e. [(η7-B7)-B-BO]−. Chemical bonding analyses showed that the central B atom shares its valence electrons with the B7 unit to fulfill double aromaticity, which provides the exceptional stability for the unprecedented 3D global minimum of the B9O− cluster. Comparison of the bonding in the C6v B9O− cluster with that in [(η7-B7)-Pr], as well as in model complexes [(η7-B7)-Zn-CO]−, [(η7-B7)-Fe(CO)3]−, and [(η7-B7)-B-CO] suggest that the central B atom in [(η7-B7)-B-BO]− behaves like transition metals in its bonding to the aromatic B7 unit and the BO− ligand.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The experimental work carried out at Brown University was supported by the U.S. National Science Foundation (CHE-2053541 to L. S. W.). Theoretical work carried out at Shanxi University was supported by the National Science Foundation of China (21873058 and 21573138 to H. J. Z.; 21720102006 to S. D. L.), the Scientific Research Startup Fund of Shanxi University (203545048 to W. J. T) and the Applied Basic Research Programs of Natural Science Foundation of Shanxi Province (201901D211147 to W. J. T).Notes and references
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640–8668 CrossRef CAS PubMed.
- W. N. Lipscomb, Science, 1977, 196, 1047–1055 CrossRef CAS PubMed.
- E. D. Jemmis, M. M. Balakrishnarajan and P. D. Pancharatna, Chem. Rev., 2002, 102, 93–144 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS.
- L. S. Wang, Int. Rev. Phys. Chem., 2016, 35, 69–142 Search PubMed.
- S. Pan, J. Barroso, S. Jalife, T. Heine, K. R. Asmis and G. Merino, Acc. Chem. Res., 2019, 52, 2732–2744 CrossRef CAS PubMed.
- T. Jian, X. Chen, S. D. Li, A. I. Boldyrev, J. Li and L. S. Wang, Chem. Soc. Rev., 2019, 48, 3550–3591 RSC.
- M.-A. Legare, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef CAS PubMed.
- (a) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, K. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed; (b) M. A. Celik, R. Sure, S. Klein, R. Kinjo, G. Bertrand and G. Frenking, Chem.–Eur. J., 2012, 18, 5676–5692 CrossRef CAS PubMed.
- H. Braunschweig, I. Krummenacher, M.-A. Legare, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed.
- H. Wang, L. Wu, Z. Lin and Z. Xie, Angew. Chem., Int. Ed., 2018, 57, 8708–8713 CrossRef CAS PubMed.
- G. Wilkinson, M. Rosenblum, M. C. Whiting and R. B. Woodward, J. Am. Chem. Soc., 1952, 74, 2125–2126 CrossRef CAS.
- E. L. Muetterties, J. R. Bleeke and E. J. Wucherer, Chem. Rev., 1982, 82, 499–525 CrossRef CAS.
- (a) H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed; (b) Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754–760 CrossRef CAS PubMed.
- (a) Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed; (b) H. R. Li, T. Jian, W. L. Li, C. Q. Miao, Y. J. Wang, Q. Chen, X. M. Luo, K. Wang, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 29147–29155 RSC.
- W. J. Chen, Y. Y. Ma, T. T. Chen, M. Z. Ao, D. F. Yuan, Q. Chen, X. X. Tian, Y. W. Mu, S. D. Li and L. S. Wang, Nanoscale, 2021, 13, 3868–3876 RSC.
- (a) A. P. Sergeeva, I. A. Popov, Z. A. Piazza, L. W. Li, C. Romanescu, L. S. Wang and A. I. Boldyrev, Acc. Chem. Res., 2014, 47, 1349–1358 CrossRef CAS PubMed; (b) A. I. Boldyrev and L. S. Wang, Phys. Chem. Chem. Phys., 2016, 18, 11589–11605 RSC.
- (a) H. J. Zhai, B. Kiran, J. Li and L. S. Wang, Nat. Mater., 2003, 2, 827–833 CrossRef CAS PubMed; (b) W. Huang, A. P. Sergeeva, H. J. Zhai, B. B. Averkiev, L. S. Wang and A. I. Boldyrev, Nat. Chem., 2010, 2, 202–206 CrossRef CAS PubMed; (c) A. P. Sergeeva, Z. A. Piazza, C. Romanescu, W. L. Li, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 18065–18073 CrossRef CAS PubMed.
- (a) H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed; (b) L. L. Pan, J. Li and L. S. Wang, J. Chem. Phys., 2008, 129, 024302 CrossRef PubMed.
- (a) A. C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2011, 50, 9334–9337 CrossRef PubMed; (b) W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed; (c) T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS PubMed; (d) A. C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef PubMed; (e) T. T. Chen, W. L. Li, H. Bai, W. J. Chen, X. R. Dong, J. Li and L. S. Wang, J. Phys. Chem. A, 2019, 123, 5317–5324 CrossRef CAS PubMed.
- A. N. Alexandrova, H. J. Zhai, L. S. Wang and A. I. Boldyrev, Inorg. Chem., 2004, 43, 3552–3554 CrossRef CAS PubMed.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, J. Phys. Chem. A, 2004, 108, 3509–3517 CrossRef CAS.
- T. T. Chen, W. L. Li, T. Jian, X. Chen, J. Li and L. S. Wang, Angew. Chem., Int. Ed., 2017, 56, 6916–6920 CrossRef CAS PubMed.
- (a) R. Yu, J. Barroso, M. H. Wang, W. Y. Liang, C. Chen, X. Zarate, M. Orozco-Ic, Z. H. Cui and G. Merino, Phys. Chem. Chem. Phys., 2020, 22, 12312–12320 RSC; (b) W. Wang, J. Wang, C. Gong, D. Zhang and X. Zhang, Chin. J. Chem. Phys., 2020, 33, 578–582 CrossRef CAS; (c) Y. J. Wang, L. Y. Feng and H. J. Zhai, Phys. Chem. Chem. Phys., 2019, 21, 18338–18345 RSC.
- L. S. Wang, H. S. Cheng and J. Fan, J. Chem. Phys., 1995, 102, 9480–9493 CrossRef CAS.
- J. Akola, M. Manninen, H. Hakkinen, U. Landman, X. Li and L. S. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, R11297–R11300 CrossRef CAS.
- M. Saunders, J. Comput. Chem., 2004, 25, 621–626 CrossRef CAS PubMed.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- R. J. Bartlett and M. Musial, Rev. Mod. Phys., 2007, 79, 291–352 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700–3703 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207–5217 RSC.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) H. J. Zhai, Q. Chen, H. Bai, S. D. Li and L. S. Wang, Acc. Chem. Res., 2014, 47, 2435–2445 CrossRef CAS PubMed; (b) H. J. Zhai, S. D. Li and L. S. Wang, J. Am. Chem. Soc., 2007, 129, 9254–9255 CrossRef CAS PubMed; (c) S. D. Li, H. J. Zhai and L. S. Wang, J. Am. Chem. Soc., 2008, 130, 2573–2579 CrossRef CAS PubMed; (d) H. J. Zhai, J. C. Guo, S. D. Li and L. S. Wang, ChemPhysChem, 2011, 12, 2549–2553 CrossRef CAS PubMed; (e) H. J. Zhai, Q. Chen, H. Bai, H. G. Lu, W. L. Li, S. D. Li and L. S. Wang, J. Chem. Phys., 2013, 139, 174301 CrossRef PubMed; (f) M. T. Nguyen, M. H. Matus, V. T. Ngan, D. J. Grant and D. A. Dixon, J. Phys. Chem. A, 2009, 113, 4895–4909 CrossRef CAS PubMed.
- (a) M. L. Drummond, V. Meunier and B. G. Sumpter, J. Phys. Chem. A, 2007, 111, 6539–6551 CrossRef CAS PubMed; (b) T. B. Tai, M. T. Nguyen and D. A. Dixon, J. Phys. Chem. A, 2010, 114, 2893–2912 CrossRef CAS PubMed.
- (a) H. J. Zhai, C. Q. Miao, S. D. Li and L. S. Wang, J. Phys. Chem. A, 2010, 114, 12155–12161 CrossRef CAS PubMed; (b) Q. Chen, H. J. Zhai, S. D. Li and L. S. Wang, J. Chem. Phys., 2012, 137, 044307 CrossRef PubMed; (c) Q. Chen, H. Bai, H. J. Zhai, S. D. Li and L. S. Wang, J. Chem. Phys., 2013, 139, 044308 CrossRef PubMed; (d) H. Bai, H. J. Zhai, S. D. Li and L. S. Wang, Phys. Chem. Chem. Phys., 2013, 15, 9646–9653 RSC.
- H. J. Zhai, L. M. Wang, S. D. Li and L. S. Wang, J. Phys. Chem. A, 2007, 111, 1030–1035 CrossRef CAS PubMed.
- (a) Q. Zhang, W. L. Li, C. Q. Xu, M. Chen, M. Zhou, J. Li, D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 11078–11083 CrossRef CAS PubMed; (b) J. Jin, G. Wang and M. Zhou, J. Phys. Chem. A, 2018, 122, 2688–2694 CrossRef CAS PubMed.
- (a) I. A. Popov, W. L. Li, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, J. Phys. Chem. A, 2014, 118, 8098–8105 CrossRef CAS PubMed; (b) J. Czekner, L. F. Cheng, G. S. Kocheril, M. Kulichenko, A. I. Boldyrev and L. S. Wang, Angew. Chem., Int. Ed., 2019, 58, 8877–8881 CrossRef CAS PubMed.
- (a) L. Liu, D. Moreno, E. Osorio, A. C. Castro, S. Pan, P. K. Chattaraj, T. Heine and G. Merino, RSC Adv., 2016, 6, 27177–27182 RSC; (b) R. Saha, S. Kar, S. Pan, G. Martinez-Guajardo, G. Merino and P. K. Chattaraj, J. Phys. Chem. A, 2017, 121, 2971–2979 CrossRef CAS PubMed; (c) L. Zhao, X. Qu, Y. Wang, J. Lv, L. Zhang, Z. Hu, G. Gu and Y. Ma, J. Phys.: Condens. Matter, 2017, 29, 265401 CrossRef PubMed; (d) Y. Wang, X. Wu and J. Zhao, J. Cluster Sci., 2018, 29, 847–852 CrossRef CAS; (e) B. Chen, W. Sun, X. Kuang, C. Lu, X. Xia, H. Shi and G. L. Gutsev, Phys. Chem. Chem. Phys., 2018, 20, 30376–30383 RSC; (f) X. Y. Zhao, X. M. Luo, X. X. Tian, H. G. Lu and S. D. Li, J. Cluster Sci., 2019, 30, 115–121 CrossRef CAS; (g) M. Ren, S. Jin, D. Wei, Y. Jin, Y. Tian, C. Lu and G. L. Gutsev, Phys. Chem. Chem. Phys., 2019, 21, 21746–21752 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00534k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |