
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable stereoinversion in DNA-catalyzed olefin cyclopropanation via cofactor modification†
Jingya
Hao
ab,
Wenhui
Miao
ab,
Shengmei
Lu
a,
Yu
Cheng
ab,
Guoqing
Jia
a and
Can
Li
 *a
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Zhongshan Road 457, Dalian 116023, China. E-mail: canli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 101408, China
First published on 11th May 2021
Abstract
The assembly of DNA with metal-complex cofactors can form promising biocatalysts for asymmetric reactions, although catalytic performance is typically limited by low enantioselectivities and stereo-control remains a challenge. Here, we engineer G-quadruplex-based DNA biocatalysts for an asymmetric cyclopropanation reaction, achieving enantiomeric excess (eetrans) values of up to +91% with controllable stereoinversion, where the enantioselectivity switches to −72% eetrans through modification of the Fe-porphyrin cofactor. Complementary circular dichroism, nuclear magnetic resonance, and fluorescence titration experiments show that the porphyrin ligand of the cofactor participates in the regulation of the catalytic enantioselectivity via a synergetic effect with DNA residues at the active site. These findings underline the important role of cofactor modification in DNA catalysis and thus pave the way for the rational engineering of DNA-based biocatalysts.
Introduction
Cyclopropane motifs feature in many natural products and medicinal agents1 which constitute versatile intermediates for the total synthesis of therapeutic compounds.2 As these compounds are in high demand, significant effort has been devoted to the development of cyclopropane synthesis, in particular via the use of hemoprotein enzymes engineered via directed evolution. Cytochrome P450 (ref. 3 and 4) and myoglobin5–8 enzymes have been evolved to catalyze asymmetric olefin cyclopropanations with excellent performance.9–14 This method has been applied to the synthesis of drug molecules15,16 and natural product scaffolds.17,18Despite this progress, the developed biocatalytic protocols are generally restricted to protein enzyme engineering.19 The discovery of the catalytic functions of nucleic acids20,21 has expanded the breadth of biocatalytic protocols to include RNA and DNA catalysts, initiating the pursuit of nucleic acid–based enzymes. DNA possesses inherent advantages as a catalyst. A catalytic sequence can be entirely identified from a random sequence population and folds into its practical tertiary structure spontaneously. A number of asymmetric reactions, especially Lewis acid-catalyzed reactions, have been successfully realized using DNA-based biocatalysts resulting in remarkable performances.22–31 Recently, the Roelfes32 and Sen33 groups expanded the scope of reactions catalyzed by DNA-based biocatalysts to include olefin cyclopropanation. Although the enantioselectivities achieved were moderate, this has paved the way for DNA-catalyzed carbene transfer reactions. Therefore, designing a DNA-based biocatalyst that catalyzes cyclopropanation in high enantiomeric excess (ee) remains a challenge, especially to achieve an ee greater than 90%.34
DNA catalysis is opening a promising avenue for biosynthesis, but the catalytic scope and performance of DNA catalysts still need improvement in comparison with catalysts based on protein enzymes. Considering the great role played by the cofactor ligand in the first-coordination-sphere in catalytic reactions,35–40 cofactor modification, which is as powerful as directed evolution41–44 but seriously disregarded in the field of DNA catalysis, is introduced for the development of DNA-based biocatalysts. Here, we report a cyclopropanation reaction catalyzed by a G-quadruplex (G4)–Fe-porphyrin biocatalyst that results in enantioselectivity as high as 91%. By tuning the N-methyl position of Fe-porphyrin from the para- to the ortho-position, the catalytic enantioselectivity of the reconstituted G4–Fe-porphyrin biocatalyst reverses to −72% eetrans. Complementary spectral, nuclear magnetic and isothermal titration characterization studies reveal that the stereo-divergence of the product mainly arises from the participation of the porphyrin ligand in the regulation of the enantioselectivity. This work succeeds in diversifying the functionality of DNA-based biocatalysts via cofactor modification, highlighting the great potential for cofactor modification in the field of DNA-based biocatalyst engineering.
Results and discussion
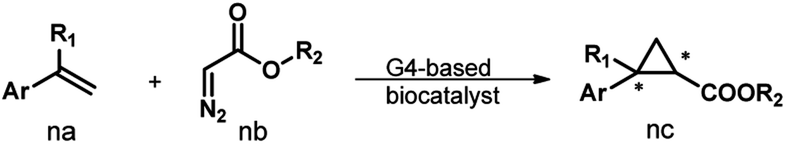
Fig. 1a illustrates the design of the G4-based biocatalysts. Three Fe-meso-tetra-(N-methylpyridyl)porphyrins with para- (FeTMPyP4), meta- (FeTMPyP3), and ortho- (FeTMPyP2) N-methyl substituents were chosen as parallel cofactors to investigate the effect of the first-coordination-sphere on the catalytic performance. The non-covalent binding of the cofactors with mA9A G4 (d[G2T2G2TGAG2T2G2A]), a thrombin binding aptamer (TBA) variant, formed the G4-based biocatalysts. The assembled biocatalysts were then tested in a reaction between styrene and ethyl diazoacetate (EDA), which results in a chiral cyclopropane product (Fig. 1b). | ||
| Fig. 1 (a) The assembly of the G4-based biocatalysts. (b) The asymmetric cyclopropanation reaction investigated in this work. | ||
Table 1 lists the results obtained using the Fe-porphyrins and their corresponding G4 biocatalysts. The use of the free FeTMPyPn (n = 4, 3, 2) cofactor as the catalyst led to relatively low activities and no chiral induction (Table 1, entries 1–3). The biocatalysts (mA9A-FeTMPyPn, n = 4, 3, 2) assembled from FeTMPyPn and mA9A G4 significantly improved the catalytic activities with turnover frequencies (TOF) increased about 10-fold when compared to those of the free FeTMPyPns. More surprisingly, mA9A-FeTMPyP2 induces the inversion of the enantioselectivity relative to that of mA9A-FeTMPyP4 from +74% to −46% (Table 1, entries 4–6). To further understand the complementary chiral induction mechanism, circular dichroism (CD), nuclear magnetic resonance (NMR), fluorescence titration and other characterization methods were then performed.
| Entry | Catalyst | % product conversion | TOF (h−1) | % eetransb |
|---|---|---|---|---|
a The experiments were carried out with styrene (30 mM), EDA (10 mM), DNA (12.5 μM), and FeTMPyPn (n = 2, 3, 4) (12.5 μM) in 10 mM potassium phosphate buffer (pH 7.0) under argon at 4 °C for 2 h, unless otherwise specified. Product conversions and ee values are based on the areas of HPLC peaks as compared to an internal standard. The diastereomeric ratios of the products ranged from 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 97:3. All data are the average of 3 attempts, reproducibility ±5%.
b (R,R)–(S,S). :10 to 97:3. All data are the average of 3 attempts, reproducibility ±5%.
b (R,R)–(S,S).
|
||||
| 1 | FeTMPyP4 | 5 | 16 | 0 |
| 2 | FeTMPyP3 | 6 | 20 | 0 |
| 3 | FeTMPyP2 | 4 | 11 | 0 |
| 4 | mA9A-FeTMPyP4 | 46 | 184 | 74 |
| 5 | mA9A-FeTMPyP3 | 36 | 144 | 63 |
| 6 | mA9A-FeTMPyP2 | 27 | 108 | −46 |
The CD spectra in Fig. 2a show that the antiparallel G4 signatures of mA9A, featuring two positive peaks at 245 nm and 295 nm, and one negative peak at 265 nm, are still present in the mA9A-FeTMPyPn (n = 4, 3, 2) catalysts. But of note is that the CD spectrum of mA9A-FeTMPyP2 shows a specifically induced CD signal (ICD) at 420 nm. Only when an asymmetric conformation is formed can an ICD signal be induced. DNA does not absorb at 420 nm, but FeTMPyP2 does (Fig. 2b). Therefore, the ICD signal can be attributed to FeTMPyP2, whose planar symmetry is broken due to the interaction with mA9A.
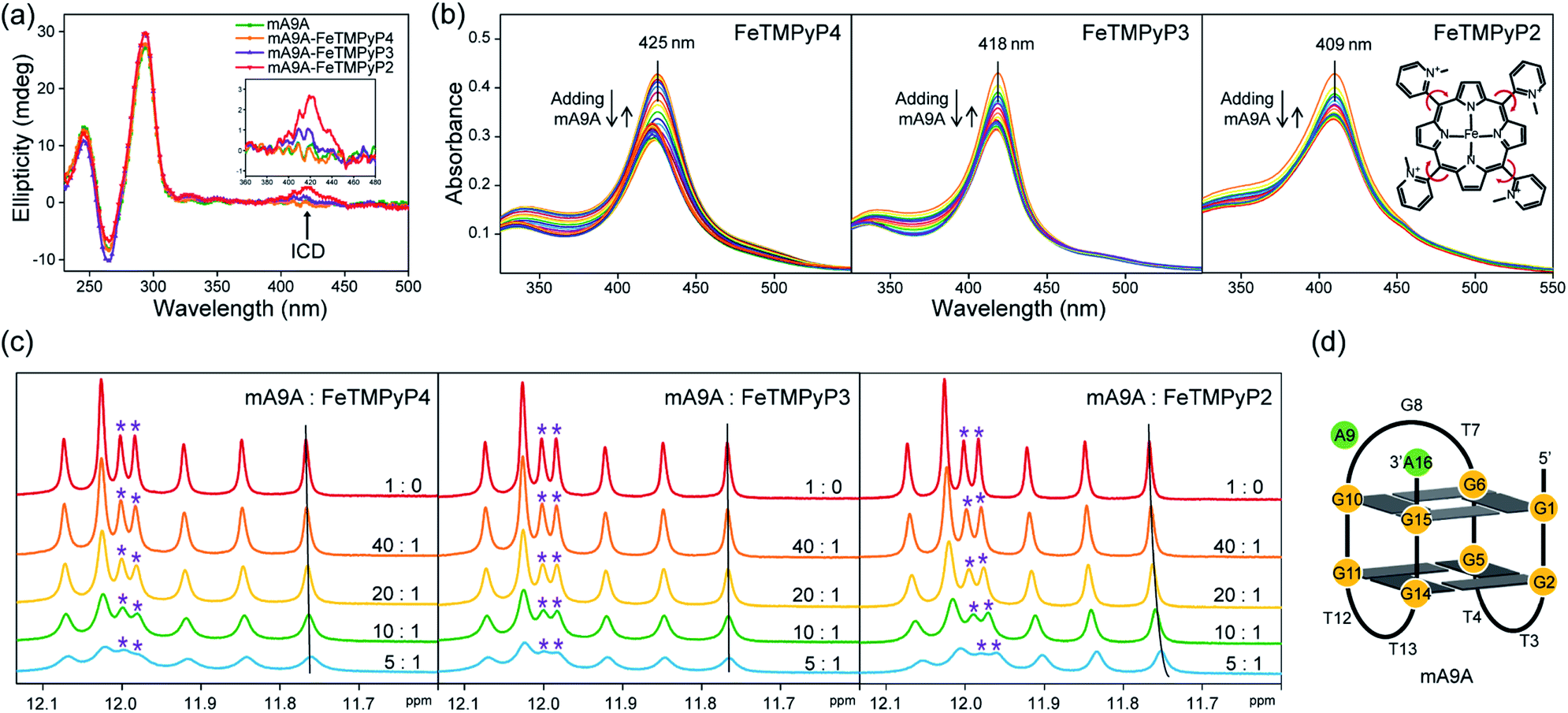 | ||
| Fig. 2 (a) CD spectra of mA9A and mA9A-FeTMPyPn (n = 4, 3, 2) (inset: a detailed view of the ICD signals). (b) UV-vis absorption spectra of FeTMPyPn (n = 4, 3, 2) when titrated with mA9A. (c) NMR spectra of mA9A over the range of 11.5–12.5 ppm when titrated with FeTMPyPn (n = 4, 3, 2). (d) The G-quadruplex structure of mA9A. | ||
The ultraviolet-visible (UV) absorption spectra (Fig. 2b) of mA9A-FeTMPyPn further support the CD results. The spectra of FeTMPyPn (n = 4, 3, 2) exhibit broad Soret absorption bands at around 420 nm. As the N-methyl group varies from the para- to the ortho-position, the Soret band shows a blue shift, indicating that FeTMPyP2 has decreased electronic conjugation relative to the other porphyrins. Due to the steric hindrance of the 2-N-methyl group, the bond between the pyridine ring and the porphyrin ring of FeTMPyP2 has a tendency to rotate, causing the pyridine ring and the porphyrin ring to be non-co- planar (Fig. 2b, right inset), which explains the generation of an ICD signal in mA9A-FeTMPyP2. When well-folded mA9A is added to FeTMPyPn, the Soret bands first fall to a minimum and then slightly rise for all cofactors. This suggests that FeTMPyPn (n = 4, 3, 2) have a similar external stacking mode on mA9A, rather than intercalating between two G-quartets.45–47 Moreover, binding with FeTMPyP4 and FeTMPyP3 makes mA9A more stable according to UV melting experiments (Fig. S1†), while binding with FeTMPyP2 does not.
To obtain further structural information on the mA9A-FeTMPyPn catalysts, we conducted an NMR characterization of the systems. The NMR spectrum of mA9A shows seven discrete peaks over the range of 11.5–12.5 ppm (Fig. 2c), which can be assigned to the eight guanine imino protons (H1) of the G-quartet. These signals indicate the formation of a two-layer antiparallel G4 structure in the potassium phosphate buffer (Fig. 2d), like that of TBA.48,49 NMR titration experiments (Fig. 2c) where mA9A is added to the three FeTMPyPn (n = 4, 3, 2) cofactors show similar line broadening and reductions in peak intensity. However, there are two adjacent peaks around 12.0 ppm (peaks labelled with *) with different relative declines, indicating that the coordination modes between mA9A and the different Fe-porphyrin cofactors vary slightly. Moreover, the addition of FeTMPyP2 causes an obvious upfield peak-shift, which is attributed to the strong electronic shielding effect arising from the porphyrin ligand stacking upon the G-quartet.50 Therefore, in combination with the CD and UV-vis results, we find that FeTMPyP2 adjusts to an optimal conformation through bond rotation and distortion, thereby forming a tight π–π stacking mode with mA9A and creating a strong electronic shielding effect on the imino protons of the G-quartet.
A fluorescence-based binding assay was then implemented to locate the catalytic sites.51 Since UV titration had determined the external stacking mode of the FeTMPyPn (n = 4, 3, 2) cofactors on mA9A, the 5′ end (5′FAM-mA9A) and loop 1 (int′FAM-mA9A) of the two G-quartets of mA9A were labelled separately with the fluorophore 5-carboxyfluorescein (FAM). The fluorescence of FAM can be suppressed by the presence of FeTMPyPn (Fig. 3a and S2†). By adding FeTMPyPn dropwise to the FAM-labelled mA9A, we obtained the titration quenching curves (Fig. S3†). The fitted apparent equilibrium dissociation constants (Kdapp) at the different FAM-labelled sites, shown in Fig. 3b, indicate that all the FeTMPyPn (n = 4, 3, 2) cofactors prefer binding at the 5′, 3′ end of the G-quartet. Considering that the ratio of mA9A and FeTMPyPn is greater than 1:1 when assembling the DNA-based biocatalyst and that the eetrans values for the catalytic cyclopropanation can be maintained at the highest level, the preferential binding site of FeTMPyPn with mA9A is regarded as the active center for chiral regulation. Therefore, according to the fluorescence-based binding assay, FeTMPyPn binding at the 5′, 3′-end of the G-quartet of mA9A constructs the active catalytic site of mA9A-FeTMPyPn. The Kdapp of FeTMPyP2 is the lowest at 38 nM, compared to 70 nM for FeTMPyP3 and 52 nM for FeTMPyP4, indicating that FeTMPyP2 binds the strongest with mA9A.
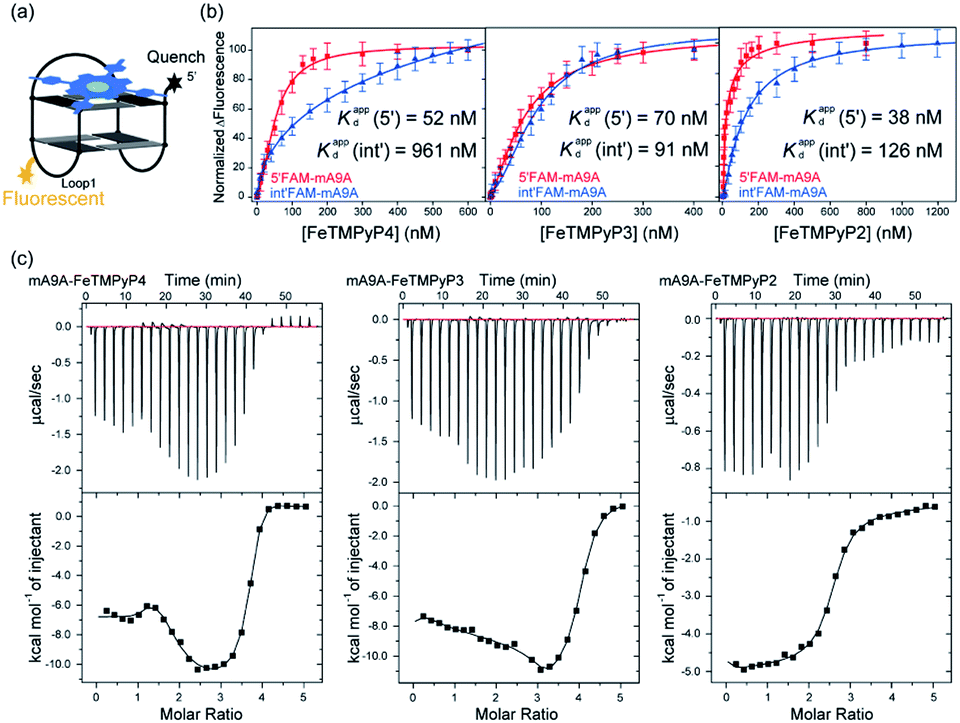 | ||
| Fig. 3 (a) A schematic diagram of the fluorescence quenching equilibrium dissociation binding assay. (b) Binding curves determined using fluorescence quenching titration. (c) ITC profiles for the binding of FeTMPyPn (n = 4, 3, 2) with mA9A. | ||
Isothermal titration calorimetry (ITC) provides information on intermolecular interactions by recording the heat discharged or consumed during a bimolecular reaction. Fig. 3c shows that there are several distinct exothermic processes during the titration of mA9A with FeTMPyP4 and FeTMPyP3, indicating complicated multiple binding behaviours between FeTMPyP4, FeTMPyP3 and mA9A. As explained in the above section, although mA9A enables the binding of multiple iron porphyrins, the strongest binding sites were the catalytic sites for the chiral regulation of the cyclopropanation reaction. The titration curves were fitted using a sequential binding sites model to calculate the binding parameters. The highest affinities between FeTMPyPn and mA9A were quantified to be 33 nM (n = 4), 50 nM (n = 3) and 14 nM (n = 2), coinciding with the trend of Kdapp as measured using fluorescence titration. This further supports the conclusion that the 5′, 3′-end of the G-quartet of mA9A constructs the active catalytic sites. Given that FeTMPyP2 has flexible peripheral groups (according to UV spectra), the tight assembly with mA9A can be attributed to the conformational transition of the TMPyP2 ligand, which is also confirmed by the CD spectra.
NMR, UV and fluorescence titration studies indicate that fine-tuning the N-methyl position of the cofactor does not change the preference of FeTMPyPn for binding at the 5′, 3′-end of the G-quartet of mA9A. This suggests that a similar active DNA pocket for cyclopropanation catalysis is provided by all the mA9A-FeTMPyPn (n = 4, 3, 2) catalysts (Fig. 4a). The ligands TMPyP4 and TMPyP3 are planar symmetric molecules that are unable to induce chirality in the catalytic process of the mA9A-FeTMPyP4 and mA9A-FeTMPyP3 catalysts. It is the deoxynucleotide residues at the 5′, 3′-end of the G-quartet of mA9A that hold the iron porphyrin carbene (IPC) intermediate in a certain orientation and define the configuration of the product (Fig. 4b). Nevertheless, mA9A-FeTMPyP3 shows a similar but lower enantioselectivity than mA9A-FeTMPyP4. Considering the similar binding strengths of FeTMPyP3 with the two G-quartets of mA9A (Fig. 3b), the reduction in enantioselectivity can be reasonably attributed to the multi-site binding behaviour of FeTMPyP3. In contrast to mA9A-FeTMPyP4 and mA9A-FeTMPyP3, the symmetry breaking of the porphyrin ring in TMPyP2 allows it to participate in chiral regulation. This, in synergy with the deoxynucleotide residues, induces the formation of the cyclopropane product with the opposite configuration to that catalyzed by mA9A-FeTMPyP4 (Fig. 4b).
 | ||
| Fig. 4 (a) A schematic diagram of the assembly of mA9A-FeTMPyPn (n = 4, 3, 2). (b) Schematic diagrams of the active centres of mA9A-FeTMPyP4 and mA9A-FeTMPyP2. | ||
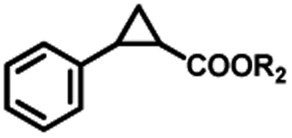
To extend the substrate scope of the mA9A-FeTMPyPn catalyzed cyclopropanation reaction, a series of olefins and diazoesters were investigated (Table 2). All three mA9A-FeTMPyPn (n = 4, 3, 2) catalysts show obvious substrate specificities. The catalytic enantioselectivities of mA9A-FeTMPyP4 and mA9A-FeTMPyP3 toward olefin substrates with substituents on the phenyl group are reduced (entries 1 vs. 2–6). The enlargement of the diazoester functional group from ethyl (Et) to –CCH3(i-Pr)2 (i-Pr is the abbreviation for isopropyl) enables a significant enhancement in the eetrans values to 91% (entries 1 vs. 8–10). However, the use of a –CH(Cy)2 (Cy is the abbreviation for cyclohexyl, entry 11) group does not result in an enhancement. Several studies have reported that the modification of diazoester substituents causes a significant impact on the activities and selectivities of carbene transfer reactions.6,52 The G4–Fe-porphyrin-catalyzed cyclopropanation has been characterized to proceed through a catalytic IPC intermediate.34 Diazoester reagents attack the active [Fe] center to form an IPC intermediate and release one molecule of N2. An appropriate substituent on the diazoester reagent can coordinate with the deoxynucleotide residues (especially dA9) through hydrophobic interactions to directly determine the conformation and properties of the IPC intermediate. Therefore, a –CCH3(i-Pr)2 substituent promotes IPC convergence to a single well-defined orientation, and the steric hindrance of the deoxynucleotide residues allows one face of the IPC to be exposed to the olefin, while keeping the other inaccessible, resulting in high enantioselectivity. For mA9A-FeTMPyP2, variation in the substituents on the phenyl group of the olefin increases the enantioselectivity to −72% eetrans (entry 6), but the catalytic cyclopropanation activities are generally lower than those of the other two biocatalysts. Although the three mA9A-FeTMPyPn (n = 4, 3, 2) catalysts show different responses to substrate variation, they are all trans product-selective with trans/cis ratios of more than 86:14, and almost nonselective toward the R1-substituted olefin substrate (entry 7).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Product | mA9A-FeTMPyP4 | mA9A-FeTMPyP3 | mA9A-FeTMPyP2 | ||||||
| % product conversion | TOF (h−1) | % eetransb | % product conversion | TOF (h−1) | % eetransb | % product conversion | TOF (h−1) | % eetransb | ||
|
a The experiments were carried out with styrene (30 mM), EDA (10 mM), DNA (12.5 μM), and FeTMPyPn (n = 2, 3, 4) (12.5 μM) in 10 mM potassium phosphate buffer (pH 7.0) under argon at 4 °C, 2 h, unless otherwise specified. Product conversions and ee values are based on the areas of HPLC peaks compared to an internal standard. The diastereomeric ratios of the products ranged from 86:14 to 97:3. All data are the average of 3 attempts, reproducibility ±5%.
b (R,R)–(S,S).
|
||||||||||

|
||||||||||
| 1 | 1c, R = 4-H | 46 | 184 | 74 | 36 | 144 | 63 | 27 | 108 | −46 |
| 2 | 2c, R = 4-Me | 36 | 144 | 70 | 35 | 140 | 60 | 25 | 100 | −45 |
| 3 | 3c, R = 4-OMe | 55 | 220 | 58 | 46 | 184 | 52 | 26 | 104 | −46 |
| 4 | 4c, R = 4-Cl | 20 | 80 | 65 | 19 | 76 | 53 | 24 | 96 | −50 |
| 5 | 5c, R = 4-F | 23 | 92 | 66 | 17 | 68 | 41 | 29 | 116 | −61 |
| 6 | 6c, R = 3, 4-F | 24 | 86 | 54 | 20 | 80 | 40 | 27 | 108 | −72 |
| 7 |
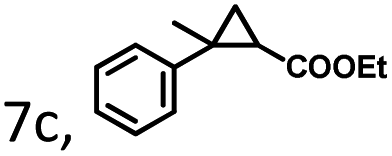
|
39 | 156 | 17 | 35 | 90 | 10 | 30 | 120 | −3 |
| 0.5 | ||||||||||
|
||||||||||
| 8 | 8c, R2 = t-Bu | 39 | 186 | 82 | 35 | 140 | 60 | 18 | 72 | −35 |
| 9 | 9c, R2 = CH(i-Pr)2 | 47 | 188 | 85 | 41 | 164 | 68 | 23 | 92 | −35 |
| 10 | 10c, R2 = CCH3(i-Pr)2 | 41 | 164 | 91 | 39 | 156 | 64 | 27 | 108 | −31 |
| 11 | 11c, R2 = CH(Cy)2 | 42 | 168 | 36 | 40 | 160 | 25 | 29 | 116 | −11 |
Conclusions
In conclusion, stereo-divergence of G4 biocatalyst-catalyzed olefin cyclopropanation was achieved via cofactor modification. By tuning the N-methyl substituent of the porphyrin ligand in the cofactor from the para- to the ortho-position, the self-assembled G4–Fe-porphyrin biocatalysts are able to switch the enantioselectivity of the reaction from +91% to −72% eetrans. CD, NMR, ITC, and other characterization studies reveal that the porphyrin ligand cooperating with the deoxynucleotide residues gives the IPC intermediate a single well-defined configuration and results in a specific enantiopreference. This finding is down to the rational design of DNA-based biocatalysts through cofactor modification, a method which serves as an effective way to regulate the catalytic performance of DNA-based biocatalysts.Author contributions
J. Y. Hao conceived of the presented idea. J. Y. Hao, W. H. Miao, and S. M. Lu planned the experiments. J. Y. Hao and W. H. Miao carried out the experiments. J. Y. Hao, W. H. Miao, S. M. Lu, Y. Cheng, and C. Li contributed to the interpretation of the results. J. Y. Hao, S. M. Lu, and C. Li wrote the manuscript. C. Li supervised the project. All authors contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Key R&D Program of China (2017YFB0702800) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB17000000). We thank Dr Ruotian Chen for his help with revising the manuscript. We thank Xianghui Liu and Kai Wang for helpful discussion. We thank A/Prof. Xuanjun Ai and Dr Ting Yang for their help with the NMR and ITC experiments.Notes and references
- H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS PubMed.
- P. S. Coelho, Z. J. Wang, M. E. Ener, S. A. Baril, A. Kannan, F. H. Arnold and E. M. Brustad, Nat. Chem. Biol., 2013, 9, 485 CrossRef CAS PubMed.
- P. S. Coelho, E. M. Brustad, A. Kannan and F. H. Arnold, Science, 2013, 339, 307 CrossRef CAS PubMed.
- M. Bordeaux, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2015, 54, 1744 CrossRef CAS PubMed.
- V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 2512 CrossRef CAS PubMed.
- P. Bajaj, G. Sreenilayam, V. Tyagi and R. Fasan, Angew. Chem., Int. Ed., 2016, 55, 16110 CrossRef CAS PubMed.
- A. Iffland, S. Gendreizig, P. Tafelmeyer and K. Johnsson, Biochem. Biophys. Res. Commun., 2001, 286, 126 CrossRef CAS PubMed.
- R. Patel, Adv. Synth. Catal., 2001, 343, 527 CrossRef CAS.
- R. E. Cobb, N. Sun and H. Zhao, Methods, 2013, 60, 81 CrossRef CAS PubMed.
- A. Tinoco, V. Steck, V. Tyagi and R. Fasan, J. Am. Chem. Soc., 2017, 139, 5293 CrossRef CAS PubMed.
- A. L. Chandgude, X. K. Ren and R. Fasan, J. Am. Chem. Soc., 2019, 141, 9145 CrossRef PubMed.
- A. M. Knight, S. B. J. Kan, R. D. Lewis, O. F. Brandenberg, K. Chen and F. H. Arnold, ACS Cent. Sci., 2018, 4, 372 CrossRef CAS PubMed.
- X. K. Ren, N. Y. Liu, A. L. Chandgude and R. Fasan, Angew. Chem., Int. Ed., 2020, 59, 21634 CrossRef CAS PubMed.
- K. Huber, B. Hamad and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2011, 10, 255 CrossRef CAS PubMed.
- K. E. Hernandez, H. Renata, R. D. Lewis, S. B. Jennifer Kan, C. Zhang, J. Forte, D. Rozzell, J. A. McIntosh and F. H. Arnold, ACS Catal., 2016, 6, 7810 CrossRef CAS PubMed.
- R. M. Atkinson and K. S. Ditman, Clin. Pharmacol. Ther., 1965, 6, 631 CrossRef CAS PubMed.
- M. J. S. RenéCsuk and Y. von Scholz, Tetrahedron: Asymmetry, 1996, 7, 3505 CrossRef.
- A. Z. Khan, M. Bilal, T. Rasheed and H. M. N. Iqbal, Chin. J. Catal., 2018, 39, 1861 CrossRef CAS.
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595 CrossRef CAS PubMed.
- G. F. J. Ronald and R. Breaker, Chem. Biol., 1994, 1, 223 CrossRef.
- P. Fournier, R. Fiammengo and A. Jaschke, Angew. Chem., Int. Ed., 2009, 48, 4426 CrossRef CAS PubMed.
- D. Coquiere, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2007, 46, 9308 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem. Commun., 2012, 48, 6366 RSC.
- A. J. Boersma, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 3346 CrossRef CAS PubMed.
- Y. Li, M. Cheng, J. Hao, C. Wang, G. Jia and C. Li, Chem. Sci., 2015, 6, 5578 RSC.
- C. Wang, G. Jia, J. Zhou, Y. Li, Y. Liu, S. Lu and C. Li, Angew. Chem., Int. Ed., 2012, 51, 9352 CrossRef CAS PubMed.
- M. Cheng, Y. Li, J. Zhou, G. Jia, S. M. Lu, Y. Yang and C. Li, Chem. Commun., 2016, 52, 9644 RSC.
- A. J. Boersma, R. P. Megens, B. L. Feringa and G. Roelfes, Chem. Soc. Rev., 2010, 39, 2083 RSC.
- A. Rioz-Martinez and G. Roelfes, Curr. Opin. Chem. Biol., 2015, 25, 80 CrossRef CAS PubMed.
- G. Roelfes, Mol. BioSyst., 2007, 3, 126 RSC.
- A. Rioz-Martinez, J. Oelerich, N. Segaud and G. Roelfes, Angew. Chem., Int. Ed., 2016, 55, 14136 CrossRef CAS PubMed.
- H. Ibrahim, P. Mulyk and D. Sen, ACS Omega, 2019, 4, 15280 CrossRef CAS PubMed.
- J. Y. Hao, W. H. Miao, Y. Cheng, S. M. Lu, G. Q. Jia and C. Li, ACS Catal., 2020, 10, 6561 CrossRef CAS.
- K. Ohora, H. Meichin, L. M. Zhao, M. W. Wolf, A. Nakayama, J. Hasegawa, N. Lehnert and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 17265 CrossRef PubMed.
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945 CrossRef CAS PubMed.
- S. N. Natoli and J. F. Hartwig, Acc. Chem. Res., 2019, 52, 326 CrossRef CAS PubMed.
- E. J. Moore, V. Steck, P. Bajaj and R. Fasan, J. Org. Chem., 2018, 83, 7480 CrossRef CAS PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, ACS Catal., 2017, 7, 7629 CrossRef CAS PubMed.
- G. Sreenilayam, E. J. Moore, V. Steck and R. Fasan, Adv. Synth. Catal., 2017, 359, 2076 CrossRef CAS PubMed.
- K. A. Powell, S. W. Ramer, S. B. Del Cardayre, W. P. Stemmer, M. B. Tobin, P. F. Longchamp and G. W. Huisman, Angew. Chem., Int. Ed., 2001, 40, 3948 CrossRef CAS PubMed.
- F. H. Arnold, Acc. Chem. Res., 1998, 31, 125 CrossRef CAS.
- N. J. Turner, Trends Biotechnol., 2003, 21, 474 CrossRef CAS PubMed.
- G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791 CrossRef CAS PubMed.
- C. Wei, G. Jia, J. Yuan, Z. Feng and C. Li, Biochemistry, 2006, 45, 6681 CrossRef CAS PubMed.
- C. Wei, G. Jia, J. Zhou, G. Han and C. Li, Phys. Chem. Chem. Phys., 2009, 11, 4025 RSC.
- C. Wei, J. Wang and M. Zhang, Biophys. Chem., 2010, 148, 51 CrossRef CAS PubMed.
- R. F. Macaya, P. Schultze, F. W. Smith, J. A. Roe and J. Feigon, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3745 CrossRef CAS PubMed.
- P. Schultze, R. F. Macaya and J. Feigon, J. Mol. Biol., 1994, 235, 1532 CrossRef CAS PubMed.
- C. Lin, J. Dickerhoff and D. Yang, Methods Mol. Biol., 2019, 2035, 157 CrossRef CAS PubMed.
- M. P. Cheng, J. Y. Hao, Y. H. Li, Y. Cheng, G. Q. Jia, J. Zhou and C. Li, Biochimie, 2018, 146, 20 CrossRef CAS PubMed.
- V. Steck, D. M. Carminati, N. R. Johnson and R. Fasan, ACS Catal., 2020, 10, 10967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00755f |
| This journal is © The Royal Society of Chemistry 2021 |