
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExcited state character of Cibalackrot-type compounds interpreted in terms of Hückel-aromaticity: a rationale for singlet fission chromophore design†
Weixuan
Zeng
 a,
Ouissam
El Bakouri
b,
Dariusz W.
Szczepanik
*cd,
Hugo
Bronstein
*ae and
Henrik
Ottosson
*b
a,
Ouissam
El Bakouri
b,
Dariusz W.
Szczepanik
*cd,
Hugo
Bronstein
*ae and
Henrik
Ottosson
*b
aDepartment of Chemistry, University of Cambridge, Cambridge, CB2 1EW, UK. E-mail: hab60@cam.ac.uk
bDepartment of Chemistry – Ångström Laboratory, Uppsala University, 751 20 Uppsala, Sweden. E-mail: henrik.ottosson@kemi.uu.se
cDepartment of Theoretical Chemistry, Faculty of Chemistry, Jagiellonian University, Gronostajowa, 2, 30-387 Kraków, Poland. E-mail: dariusz.szczepanik@uj.edu.pl
dInstitut de Quìmica Computacional i Catàlisi, Departament de Química, Universitat de Girona, C/ Maria Aurèlia Capmany, 69, 17003 Girona, Catalonia, Spain
eCavendish Laboratory, University of Cambridge, Cambridge, CB3 0HE, UK
First published on 25th March 2021
Abstract
The exact energies of the lowest singlet and triplet excited states in organic chromophores are crucial to their performance in optoelectronic devices. The possibility of utilizing singlet fission to enhance the performance of photovoltaic devices has resulted in a wide demand for tuneable, stable organic chromophores with wide S1–T1 energy gaps (>1 eV). Cibalackrot-type compounds were recently considered to have favorably positioned excited state energies for singlet fission, and they were found to have a degree of aromaticity in the lowest triplet excited state (T1). This work reports on a revised and deepened theoretical analysis taking into account the excited state Hückel-aromatic (instead of Baird-aromatic) as well as diradical characters, with the aim to design new organic chromophores based on this scaffold in a rational way starting from qualitative theory. We demonstrate that the substituent strategy can effectively adjust the spin distribution on the chromophore and thereby manipulate the excited state energy levels. Additionally, the improved understanding of the aromatic characters enables us to demonstrate a feasible design strategy to vary the excited state energy levels by tuning the number and nature of Hückel-aromatic units in the excited state. Finally, our study elucidates the complications and pitfalls of the excited state aromaticity and antiaromaticity concepts, highlighting that quantitative results from quantum chemical calculations of various aromaticity indices must be linked with qualitative theoretical analysis of the character of the excited states.
Introduction
Singlet exciton fission in organic molecules has the potential to significantly improve the efficiency of silicon-based photovoltaics through the reduction of thermalization losses. By absorbing a single higher energy photon, bringing a molecule to a singlet excited state, and converting this state into two equivalent lower energy triplet excited states one can potentially increase the single-junction photovoltaic limit up to 35%.1,2 In order to achieve this, strict energetic conditions must be met; the lowest triplet (T1) energy levels must be >1.11 eV (the bandgap of silicon) and the singlet energy level must be ∼2 × T1 (so that singlet fission can occur). The development of design rules that allow for the identification and synthesis of new singlet fission chromophores is therefore arguably one of the most important challenges in functional organic materials research today. There have been several proposed design rules including the use of diradicaloid systems,3–5 the assessment of the diradicaloid character of a chromophore,6,7 and the manipulation of aromaticity to engineer the excited states,8–12 which has led to a diverse range of structures in the hunt for improved singlet fission materials.13–15 However, the fact that linear acenes remain the most successful chromophore for use in singlet fission photovoltaics, despite their well-documented instability,16,17 demonstrates that more work must be done to understand the underlying design principles.Our ambition is to develop refined design rules based on aromaticity as this can facilitate the identification of novel ways for the tailoring of molecules that function as chromophores for singlet fission photovoltaics. Aromaticity comes in different forms, e.g., Hückel-,18 Möbius-,19,20 Baird-,17–29 and spherical aromaticity.30,31 Of relevance for the study presented herein are Hückel-aromaticity of closed-shell cycles with 4n + 2π-electrons and Baird-aromaticity of cycles with 4nπ-electrons in their triplet ππ* states.22,25,32,33 According to quantum chemical calculations the Baird-aromaticity concept can also be extended to the lowest singlet excited state of simple annulenes,34–36 supported by spectroscopic observations.27,32,37,38 Lately, the concepts have been utilized to rationalize a range of photophysical and photochemical properties and processes.
Cibalackrot-type compounds (indolonaphthyridines), some of which can be proved to undergo singlet fission, were recently considered by one of us to have a degree of aromaticity in the lowest triplet excited state (T1).9 The basis for the conclusions was the observation of negative nucleus independent chemical shifts (NICS) in the pyrrole rings, which were interpreted as indicative of some influence of 4π-electron pyrrole dicationic rings. Based on this finding it was suggested that these compounds were Baird-type aromatics. The remarkable photostability of the Cibalackrot chromophore, alongside its facile tuneability, also makes it an attractive candidate for use in singlet fission photovoltaics. Another recent example of a singlet fission material which, based on computations, was suggested to have Baird-aromatic character in its triplet state is dipyrrolonaphthyridinedione (DPND).10 In this context, it can be noted that compounds with 4nπ-electron cycles have been reported to show high photostability both in excited triplet and singlet states,32,39 a feature that could be traced to excited state Baird-aromatic character.19
Yet, even though the predominant form of aromaticity in the first ππ* excited states is the Baird-type aromaticity of 4nπ-electron cycles there are caveats. For example, a ππ* triplet state can be of Hückel–Baird hybrid type as earlier found for a quinoid compound (TMTQ) in its T1 state.40,41 The T1 state of such a compound has a central unit simultaneously influenced by both Hückel-aromaticity and Baird-aromaticity. Also, electronically excited states can be described by pure Hückel-type aromaticity, as observed for the charge-transfer state of a 6-aminosubstituted fulvene-based molecular motor where a 6π-electron cyclopentadienyl anionic ring makes an important contribution.42 With this background it is obvious that the excited state aromaticity concept has its complications and pitfalls, and it should be important to outline these as precisely as possible so as to enable a more efficient design of molecules with targeted properties. Thus, we would here like to report on a deepened and broadened analysis of the aromatic character of Cibalackrot-type compounds in their T1 states. Are Cibalackrot-type compounds Hückel-aromatic in their T1 states?
We argue that with a proper description of the excited state aromatic character of Cibalackrot-type compounds we can, in a second part of this study, go further and make use of qualitative arguments to design new chromophores based on this scaffold. We outline the basic scope and limitations of this chromophore as well as various derivatives in the context of its application in singlet fission materials. In addition, the exploration could serve as a guide on how to interpret and apply the concepts of excited state aromaticity and antiaromaticity, avoiding the pitfalls that undoubtedly exist. A more comprehensive and proper view on how various forms of aromaticity and antiaromaticity impact on the excited state energies of molecules should enable a more rational and efficient development of optically active compounds for, e.g., singlet fission photovoltaics.
Results and discussion
Focusing on the Cibalackrot-type compounds, we first dissected the effects of aromaticity in both the ground state (S0) and the T1 state of the parent Cibalackrot molecule (CIBA). An improved understanding of the role of aromaticity in its S0 and T1 states is core to an improved strategy for further design of singlet fission chromophore candidates based on this molecule. Then, with such an understanding, routes on how to select substituents and how to further alter the Cibalackrot core (e.g., by benzannelation) so as to manipulate the excited state energy levels for singlet fission are presented. Finally, we reveal how the knowledge gained can be utilized to tune the excitation energies of DPND derivatives. The analysis is based on computed aromaticity indices, including nucleus independent chemical shifts scans (NICS-XY) and NICS values at 1.7 Å (NICS(1.7)zz),43,44 plots of the anisotropy of the induced current density (AICD),45 the geometric harmonic oscillator model of aromaticity (HOMA),46 and the electronic aromatic fluctuation (FLU)47 and multicentre (MCI) indices.48Parent Cibalackrot
In the description of CIBA we label the various rings as shown in Fig. 1. Fused rings are labelled with two letters so that, e.g., AB and CC′ represent the perimeters of the indole-like 9-membered ring and the central 10-membered ring, respectively. For the identification of Cibalackrot-type compounds with suitable features for singlet fission it is crucial to understand the aromaticity effects in both the S0 and the T1 states as the T1 energy (E(T1)) can be linked to the difference in the total counts of aromatic cycles in the two states.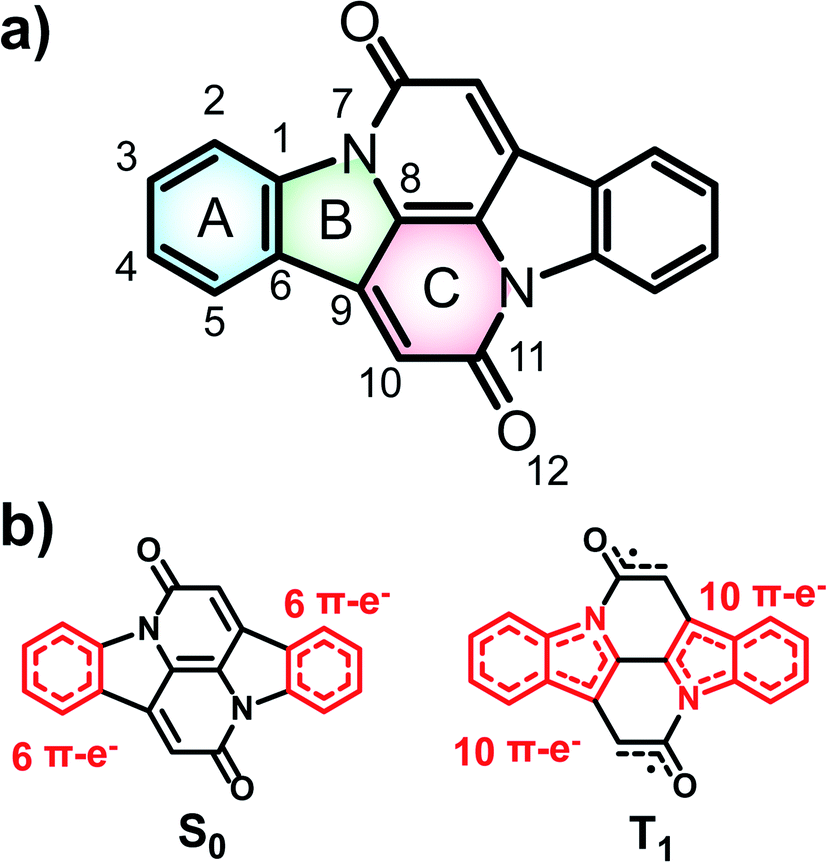 | ||
| Fig. 1 Chemical structure of indolonaphthyridine-6,13-dione (CIBA), the relevant atoms and rings in symmetric positions are signed with an apostrophe. Hückel-aromatic units are marked as red in the lower panel: two benzene units in the S0 state and two indole units in the T1 state. | ||
In the ground state (S0), the two benzene rings (A/A′) with six π-electrons should have marked Hückel-aromatic character (Fig. 1). This is supported by all aromaticity indices; the magnetic indices, i.e., the π-electron-only AICD plots and the NICS-XY scans reveal diamagnetic ring currents and negative NICS values reaching −5.8 ppm (Fig. 2), the geometric HOMA index is 0.924, and the electronic FLU and MCI indices (0.003 and 0.053, respectively) all reveal aromatic character. In contrast, rings B and B′ display markedly positive NICS values, suggesting antiaromatic character, but the AICD plot does not reveal paratropic ring currents. Also, the HOMA value of rings B and B′ (0.269) corresponds to a nonaromatic situation. The two indole-units (AB and A′B′) of CIBA in S0 have some moderate aromatic character according to HOMA (0.629) but not so according to NICS-XY and AICD.
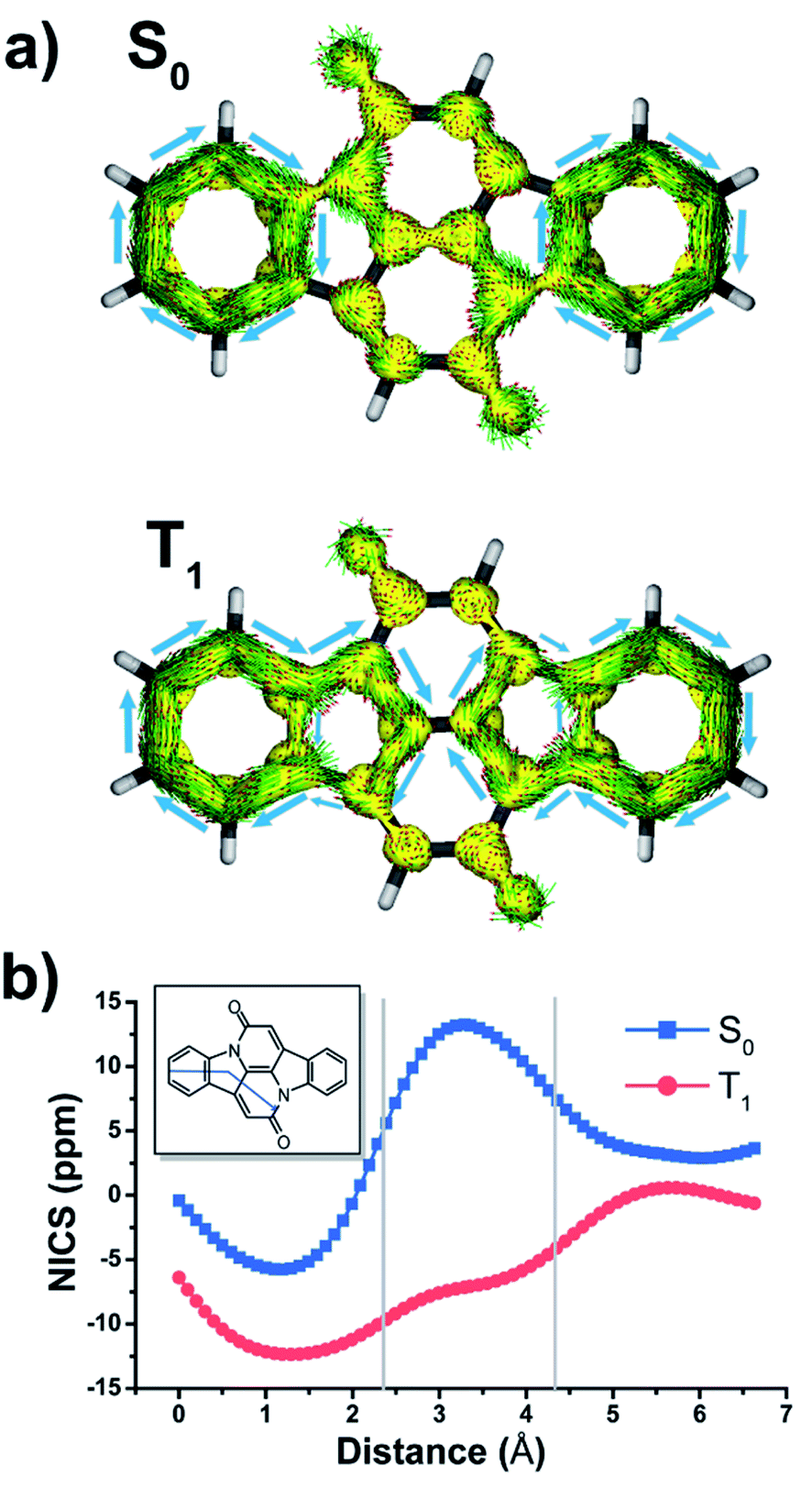 | ||
| Fig. 2 (a) π-Electron ring currents according to AICD and (b) π-NICS-XY scan in the S0 and T1 states of CIBA calculated at GIAO/(U)B3LYP/6-311+G(d,p)//(U)B3LYP/6-311+G(d,p) level. | ||
According to Fig. 2, the CC′ moiety of CIBA in its S0 state should resemble 1,5-dihydro-1,5-naphthyridine-2,6-dione (NARID, Fig. S1 and S2†). In S0, NARID has NICS values reaching down to −4.4 ppm while the CC′ moiety in CIBA has NICS values at approximately −2 ppm, indicating nonaromatic character. This is corroborated by the AICD plot which does not display any significant ring current. The FLU values of the two compounds are similar (0.033 in NARID and 0.038 in the CC′ moiety of CIBA), although the HOMA differ because the CC′ unit in CIBA has a HOMA of 0.104 while it is 0.410 in NARID. Still, the CC′ moiety of CIBA in its S0 state is best described as nonaromatic (Table 1).
| S0 | T1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | B | C | AB | CC′ | A | B | C | AB | CC′ | |
| π-NICS(1.7)zz | −5.8 | 13.2 | 3.3 | — | — | −12.3 | −7.0 | 0.5 | — | — |
| FLU | 0.003 | 0.026 | 0.032 | 0.013 | 0.038 | 0.005 | 0.016 | 0.033 | 0.007 | 0.033 |
| ΔFLUαβ/FLU | — | — | — | — | — | 0.307 | −0.180 | −0.820 | −0.181 | −0.881 |
| MCI | 0.053 | 0.007 | 0.009 | — | — | 0.048 | 0.011 | 0.005 | — | — |
| HOMA | 0.924 | 0.269 | 0.146 | 0.629 | 0.104 | 0.898 | 0.493 | 0.225 | 0.754 | 0.072 |
When going to the T1 state of CIBA the aromatic character shifts between the rings when compared to the S0 state. According to both NICS-XY and AICD the diamagnetic ring currents now expand from the 6π-electron A/A′ rings to the 10π-electron indole-like AB/A′B′ units. The changes in the HOMA values of various cycles also indicate interesting trends. First, the HOMA of the AB and A′B′ units in T1 is high (0.754) revealing an enhanced aromatic character when compared to S0. The HOMA values of the strongly aromatic A/A′ benzene rings decrease minutely while the HOMA of the pyrrole rings increases substantially. The electronic indices also reveal an increased aromatic character of the AB/A′B′ units as the FLU value in the T1 state is lowered when compared to S0. Also, the MCI value of the pyrrole rings is raised (0.011) while that of the two benzene rings is reduced modestly. Yet, is this aromatic character in the T1 state of Baird-type or of Hückel-type but with a different extension (localization) than in the S0 state?
To explore the type of aromaticity of the AB unit in the T1 state of CIBA we analysed the ratios ΔFLUαβ/FLU between the difference in spin-separated FLU values (ΔFLUαβ = FLUα − FLUβ) and the FLU. As noted above, the FLU value of the AB/A′B′ units in the T1 state is lower than in the S0 state (0.007 vs. 0.013, respectively), indicating an increase in the aromatic character. Now, the value of ΔFLUαβ/FLU clarifies whether a cycle is Baird-aromatic or Hückel-aromatic because ΔFLUαβ is zero or negligible for a Hückel-aromatic cycle while it has a nonzero value for a Baird-aromatic cycle.40,41 As references, indole and pyrrole in their closed-shell Hückel-aromatic S0 states have ΔFLUαβ/FLU values which are exactly zero while the Baird-aromatic indole and pyrrole dications in their triplet states have calculated values of 2.34 and 4.02, respectively (Table S1†). We now find that the ΔFLUαβ/FLU ratios of the AB/A′B′ moieties and the B/B′ rings of T1 state CIBA are low (−0.180 and −0.186, respectively), supporting an interpretation of the T1 state in terms of Hückel-aromaticity. Furthermore, the cumulated atomic charges, obtained from a natural population analysis, in the AB/A′B′ and B/B′ rings are −0.418 and −0.400e, respectively. Finally, the aromatic character of the indole-like AB moiety is very similar to those of the separate (Hückel-aromatic) indole molecule in S0 (Fig. S3a and b†). Combined this makes it clear that the indole units of CIBA in its T1 state are predominantly Hückel-aromatic and not Baird-aromatic.
The central NARID segment (CC′) of CIBA in its T1 state clearly has a nonaromatic character according to both NICS and AICD, as well as to HOMA (0.072). The spin density reveals that the triplet diradical character is mainly localized within the NARID unit. As shown in Fig. 3, 69% of the spin density is localized on the CC′ segment, of which 22% and 40% at the two carbonyl O atoms and the two Cα atoms, respectively. Also, the spin density distribution within the central moiety is almost identical with that of NARID (Fig. S2c†). This observation on the triplet diradical localization to the CC′ moiety is consistent with the description of the T1 state shown in Fig. 1. Moreover, the FLU value is rather high (0.033) indicating a nonaromatic situation. Although large ΔFLUαβ/FLU values are found for the CC′ segment in triplet state CIBA and NARID (ΔFLUαβ/FLU = −0.820 and −0.881, respectively), these values are due to the high spin density localized in this unit, and not to a Hückel–Baird hybrid character.
 | ||
| Fig. 3 (a) Spin density distribution in the T1 state of CIBA (ρ = 0.0008), and (b) EDDBH isosurfaces (ρ = 0.006) with the corresponding electron populations in the S0 state and in the T1 state (total and dissected into paired and unpaired-electron components), calculated at (U)B3LYP/6-311+G(d,p) level. | ||
The interpretation of the altered Hückel-aromatic character when going from S0 to T1, and not a gain of Baird-aromaticity, is further corroborated by the analysis of global delocalization effects (involving heavy atoms only) using the electron density of delocalized bonds (EDDBH) method (Fig. 3b).49,50 The total electron delocalization in CIBA is slightly more effective in T1 than in the S0 state (+0.244e), but this is clearly an effect of enhanced π-conjugation (not cyclic π-delocalization) by unpaired electrons exclusively within the NARID moiety including the oxygen atoms (+1.408e), which to a large degree is compensated by the overall reduction of paired-electron delocalization (−1.164e) with respect to S0. Thus, the lack of cyclic delocalization of spin-density and the actual reduction of paired-electron delocalization confirm that there is no Baird-type aromatic stabilization in CIBA in the T1 state, at least not within the indole units. Here it is noteworthy DPND singlet fission chromophore was recently analysed,51 and also for this compound there is a lack of triplet state Baird-aromatic character (see below and the ESI†), contrary to what was originally stated.10
Importantly, the S1 state of CIBA is similar to the T1 state since both states are described by single HOMO to LUMO excitations and they exhibit similar delocalization and hole–electron characters (Fig. S6 and Table S2†) along a tuning variable (Fig. 4a). With this information we postulate that one can tune the E(T1) and E(S1) in similar manners. The importance of this finding should be stressed as combined evaluations of the substituent effects only functions for species in which the T1 and S1 states are described by the same electron configuration. The opposite is illustrated by 5,10-bis(styryl)dibenzo[a,e]pentalene. This compound functions as a singlet fission chromophore,52 in contrast to the parent compound, dibenzo[a,e]pentalene, which stems from a relative stabilization of the T1 state as compared to the S1 state upon the 5,10-bis(styryl) substitution of dibenzo[a,e]pentalene (Fig. S8†) caused by a difference in the character of the T1 and S1 states.8
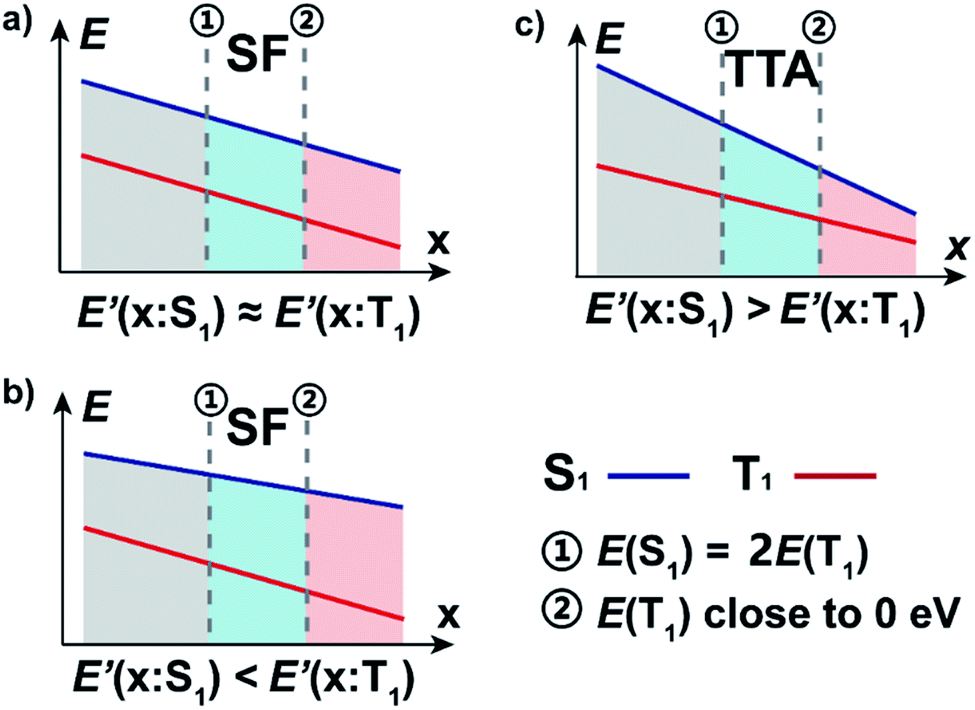 | ||
| Fig. 4 Schematic of curves of energy levels (E) vs. variable indexes, the E′ defined as the slope of E along the parameter x. There are three expected situations (a–c) each separated into three regions: (i) where aromaticity is not strong enough to ensure the stability (gray), (ii) balance regions with combination of proper energy levels and good stability (blue), and (iii) where the energy of T1 is so low that it tends to undergo severe energy loss via the internal conversion process (red). | ||
The results above show the spin density distribution and how the aromatic character is localized among the rings in the S0, T1 and S1 states. The excited state aromatic characters of CIBA are closely linked to the Hückel aromatic contribution of the indole-like AB ring while the diradical character is focused to the central CC′ ring (supporting local resonance effects between C–C and C–O bonds according to the EDDBH results). Based on this, in the second part of the study we probe how to tune the excited state energy levels by substituents, by further benzannelation, and other modifications of the Cibalackrot scaffold. The knowledge gained from the three aspects can be combined into a design approach.
Substituted Cibalackrot-type compounds
The E(T1) of the parent CIBA is 1.73 eV for vertical excitation and 1.37 eV for adiabatic excitation, and as the spin density to a significant extent is localized to the two Cα atoms it should be possible to further delocalize the radical-pair character onto the substituents through proper choice of substituents. Such enhanced spin delocalizations should lower the T1 state in energy. As the S1 state, similar as the T1 state, is described by the singly excited HOMO to LUMO excited configuration (Table S2†), the energy of the S1 state (E(S1)), could according to hypothesis above, change in energy to a similar extent as E(T1) when changing the substituents at the two Cα positions. If this holds then it should be possible to deduce a diagram with the excited state energies as functions of the spin density at the substituents as the variable by which the tuning is driven, similar as done earlier by two of us through usage of the (excited state) (anti)aromaticity as a tuning variable.8 We tested this through the placement of radical stabilizing groups at the Cα-positions. The position of the threshold where 2E(T1) = E(S1) is located will depend on the ΔE(S1 − T1) and the slopes of the trendlines for E(T1) and E(S1).The differently substituted Cibalackrots (SCIBA's) in Fig. 5, as well as further ones in Fig. S9,† were analysed. The thiophene substituted model (SCIBA1) was taken from our previous work,9 as it undergoes singlet fission. The benzene substituted model (SCIBA2) was also involved as it was reported to not undergo singlet fission.53 Our computations now reveal that SCIBA2 exhibits higher E(T1) and E(S1) compared to SCIBA1 which makes the E(S1)/E(T1) ratio smaller indicating a less efficient singlet fission process. By considering the dihedral angles between the substituents and the Cα–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds in the two compounds in the S0 state (32.4° for SCIBA1 and 45.8° for SCIBA2) it is clear that the conjugation to the phenyl groups in SCIBA2 is weaker than to the thiopheno groups in SCIBA1, and this likely contributes to a lower E(S1)/E(T1) ratio for SCIBA2. It is also possible that the different dihedral angles could influence the stacking of the compounds in the solid state, which can have effects on the formation of the key intermediate state of the singlet fission.54,55 Thus, SCIBA1 and SCIBA2 provide a connection between the theoretical results herein and experimental data on functioning singlet fission chromophores.
O) bonds in the two compounds in the S0 state (32.4° for SCIBA1 and 45.8° for SCIBA2) it is clear that the conjugation to the phenyl groups in SCIBA2 is weaker than to the thiopheno groups in SCIBA1, and this likely contributes to a lower E(S1)/E(T1) ratio for SCIBA2. It is also possible that the different dihedral angles could influence the stacking of the compounds in the solid state, which can have effects on the formation of the key intermediate state of the singlet fission.54,55 Thus, SCIBA1 and SCIBA2 provide a connection between the theoretical results herein and experimental data on functioning singlet fission chromophores.
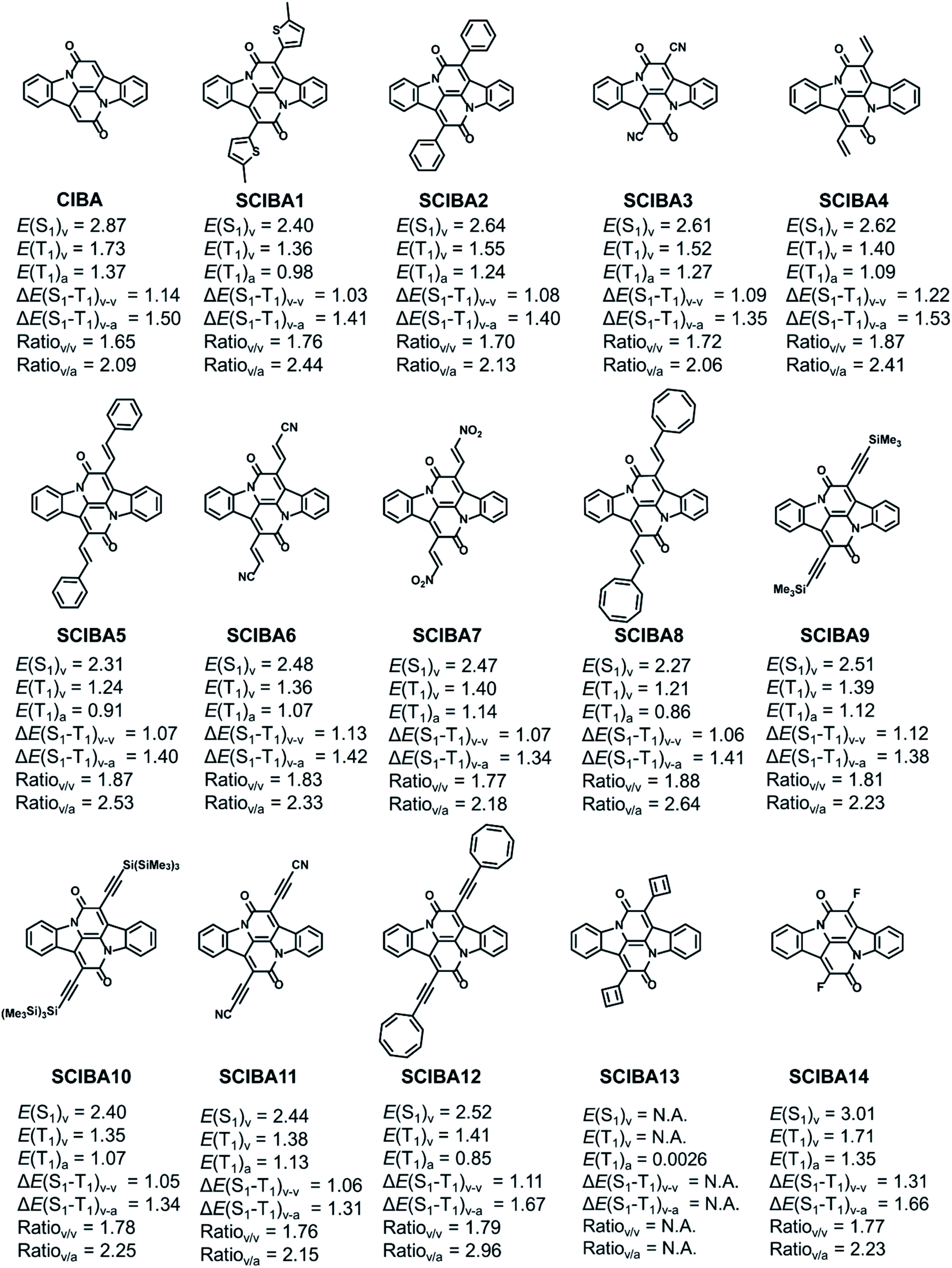 | ||
| Fig. 5 Chemical structures and relevant excitation energies (in eV) of the parent CIBA and SCIBA's. Calculations at TD-M06-2x/def2-SVP//M06-2X/def2-SVP level. | ||
We also tested cyano and vinyl substituents at the Cα atom as well as a range of different substituents with ethenyl or ethynyl moieties as spacers, including radical-stabilizing cyano, nitro, cyclooctatetraenyl (COTyl), cyclobutadienyl (CBDyl), borolyl, 2-2H-pyrazinyl, and bulky silyl groups (SCIBA3–13, 23–30, Fig. S9†). Additionally, some of the above SCIBA compounds were further substituted with cyano groups on the 3/3′ positions of the parent CIBA core (SCIBA15–22), representing the effects of different substitution strategies. These species are discussed in the ESI.† Here it should be noted that SCIBA23–30, despite being synthetically unfeasible, enable us to examine the design model towards and until the extreme point corresponding to the full radical pair localization at the substituents X.
In the SCIBAs, the spin density at the Cα atoms of the parent Cibalackrot is delocalized onto the substituents and a gradually smaller portion of the spin density is located at the central CC′ moiety (Fig. S10†). Importantly, SCIBA13 with two CBDyl substituents, despite being experimentally unrealistic, allow us to explore computationally the extreme point with the T1 state completely described as a radical pair with the unpaired spin density at the two CBDyl groups (for the function of the CBDyl groups in this regard see the ESI†).
To evaluate the potential of the new substituted Cibalackrots to undergo singlet fission, the E(T1) and E(S1) were calculated and plotted against the spin population of the substituents X based on the optimized ground state geometry and the relaxed T1 geometry of the compound, respectively (Fig. 6a and b). Linear fits with reasonable correlations were found and the higher the spin population on the substituents X, the lower E(T1) and E(S1) of the compound in question. This result agrees well with the radical delocalization approach for stabilization of the T1 states. It is also apparent that the S1 state displays a similar trend as the T1 state. Notably, none of the ratios with vertical E(T1)'s exceed 2, while all those with adiabatic E(T1)'s do. Also, the spin population on the substituent Xs are higher in the adiabatic T1 geometries compared to those of the vertical geometries, which indicates that delocalization of spin density onto the substituents X is accompanied by relaxation in the T1 state. For SCIBA1, the Cibalackrot derivative which represents a functioning singlet fission chromophore,9 the ratios are 1.76 and 2.44, respectively. Also notice, SCIBA2 exhibits smaller spin population on the Xs in both vertical and adiabatic excitation, compared to SCIBA1, which could be attributed to the different substituents and result in different singlet fission features.
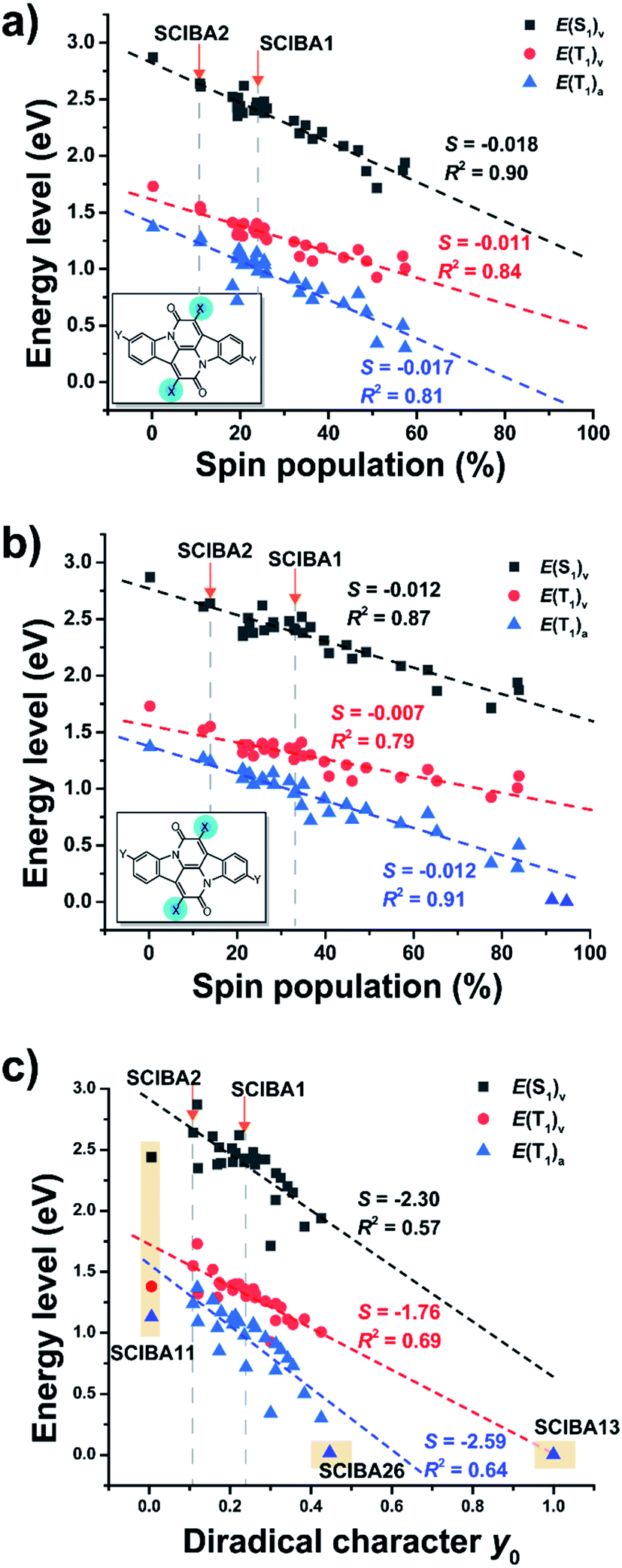 | ||
| Fig. 6 Plots of excited state energies versus (a) the spin population on the substituent X moieties based on vertical excitation, (b) on the adiabatic excitation of T1 geometries and (c) diradical character y0 of parent CIBA and SCIBAs. Subscripted ‘v’ refers to vertical excitation energies based on S0 geometries and ‘a’ refers to adiabatic excitation based on optimized T1 geometries. | ||
Thus, with SCIBA1 as an anchor we can conclude that several of the SCIBA's likely will function for singlet fission, these being SCIBA5, SCIBA8 and SCIBA12. Additional dicyano substitution at the 3/3′ positions, as in SCIBA15–22, could enhance their performance further as the E(S1) and E(T1) slightly decreased. Thus, the trend of the E(S1) and E(T1) indicates that fine-tuning via choice of substituents is a design strategy that provides an opportunity for tailoring chromophores to function in a singlet fission process. In the ESI† is also shown plots of E(T1) and E(S1) versus the spin populations at the Cα and at the CC′ moieties, respectively.
As predicted above SCIBA13 represents the extreme case with essentially all unpaired spin density localized at the substituents, confirmed through the spin density distribution and the EDDBH analysis (Fig. S11 and S12†). The optimized open-shell singlet state of SCIBA13 is located minutely below the optimized T1 state, affording an E(T1) of 0.003 eV. Importantly, the E(T1) value of SCIBA13 falls on the trendline extrapolated from the SCIBA's with regular substituents, revealing that the design strategy functions all the way to this extreme point. Noteworthy, we refrained from calculations of the vertical excitation levels of S1 and T1 of SCIBA13 due to the difficulty of the computational approaches for the calculation of excitations of compounds with open-shell ground states.
Here it can be noted that only a limited number of substituent groups X were found to allow for spin populations in the range 70–90%, i.e., groups that allow for a predominant yet not complete localization of the two unpaired electrons onto the substituents X as for SCIBA13. After an extensive search three substituents with such features were identified, these being the 2-borolylvinyl, 3-borolylvinyl, and 2-2H-pyrazinylethynyl groups (for all compounds examined see SCIBA23–30, Fig. S13 and S14†). As noted above, most of these are not realistic, yet show that the trend sustains.
One can also ask if there are substituents that raise E(T1) and E(S1) relative to the parent CIBA? Such groups should be those that resist delocalization of spin density onto them. Through computations we find that fluoro substitution at Cα (SCIBA14) significantly raise E(S1) while E(T1) remains at a similar energy when compared to the parent CIBA. This could be assigned to the very strong electron withdrawing ability of the fluorine among the halogens, which resulted in the highest delocalization of the unpaired electrons at the carbonyl O and the Cα atoms (see ESI† for detailed discussion).
The diradical character in the S0 state (y0) has earlier been demonstrated to be an effective indicator in the search of novel candidates with potentials as singlet fission chromophores.6,7 To explore the feasibility of the diradical character-based design of the Cibalackrots, we plotted the relevant excited state energies against the y0 values in S0 state (Fig. 6c). The correlation with the linear fit is now slightly weaker than the plots based on the spin population in the T1 state. Yet, considering that the diradical character y0 is a property calculated in the S0 state while the spin population is taken at the optimal T1 state geometry, the poorer correlation can be understood. Moreover, the y0 value provides a quicker prediction of the state energies of a novel compound than the spin density in the T1 state. Unsurprisingly, the y0 value correlates best with the vertical E(T1). Notably, we excluded two compounds from the fit of E(T1) and E(S1) against y0, SCIBA11 and SCIBA13. For further discussion see the ESI (Fig. S18†).
We can now apply our earlier developed geometrical model where the E(S1) and E(T1) are plotted against a variable along which the energies vary.8 In the diagram that results the singlet fission chromophores are located towards the right side. Previously we calculated the degree of (anti)aromaticity in S0 or T1 as the variable against which E(T1) and E(S1) are plotted, yet now we use the unpaired spin density at X or Cα (Fig. S19†) or the diradical character value y0. However, this principle requires that the T1 and S1 states are (i) described similarly, i.e., by the same electron configuration except for the multiplicity difference, and (ii) that the exchange interaction is constant throughout the set of molecules explored. The latter requires that HOMO and LUMO are similarly distributed in the various compounds.
Benzannelated Cibalackrot-type compounds
Having demonstrated the effect of the extent of localisation of spin density in the T1 state to the Cα atoms on the T1 energy levels we now turn to the peripheral aromatic rings (AB/A′B′ rings) of the Cibalackrot scaffold. Since our results above suggest that the formation of the Hückel-type excited state aromatic pyrrole rings is one of the drivers for the (de)localisation of the unpaired electrons towards the Cα and carbonyl O atoms of the NARID core this indicates that by altering the aromatic character of the peripheral benzannelated moieties it should also be possible to tune the excited state energies. This could then offer two independent and complementary design handles for the manipulation of excited state energies.We first consider the size of the peripheral aromatic system by both removing (BCIBA0) and adding (BCIBA2) linearly fused benzene rings relative to CIBA. Looking at the excited state energies it is clear that the addition of linearly fused rings raises both S1 and T1 energetically which in the usual context of chromophore design would be considered counterintuitive. Yet, we can rationalise these changes by considering the degree of Hückel-aromaticity in the peripheral rings in the S0 and T1 states. In all three cases we observe a gain in aromaticity of the pyrrolic rings when going from S0 to T1. In the case of CIBA and BCIBA2 in their S0 states we observe that the benzene and naphthalene ring systems are aromatic while none of the rings in BCIBA0 are (Fig. 7, S20 and S21†). Next, also the T1 state can be understood from the perspective of Clar's sextets as each of the three compounds in this state is described by two Clar's sextets, but in CIBA and BCIBA2 they are migrating while localized in BCIBA0. Now, as BCIBA0 lacks Clar's sextets in its S0 state while CIBA and BCIBA2 have two each, it becomes obvious that the lower E(T1) of BCIBA0 can be rationalized by a relative gain in Hückel-aromaticity upon excitation to T1. The relative aromaticity gain is smaller for CIBA and BCIBA2 in T1 as their aromatic units merely expand by incorporating the pyrrole rings. In addition, there is a difference in the S0 state for the latter two compounds which impacts on E(T1) as the two migrating sextets of S0 state BCIBA2 should provide for an enhanced stability as compared to the two localized sextets of S0CIBA. Additionally, one can observe more negative NICS values in rings A and D of BCIBA2 than in ring A of CIBA (Fig. 2, S22 and Table S4†).
 | ||
| Fig. 7 (a) Chemical structure of BCIBAs, the relevant atoms and rings in symmetric positions are signed with an apostrophe. Red units represent cycles which are Clar's sextets in the S0 and T1 states, respectively. (b) Plots of excited state energies of the compounds. Subscripted ‘v’ refers to vertical excitation energies based on S0 geometries and ‘a’ refers to adiabatic excitation based on optimized T1 geometries. (c) Relative energies Eref of the BCIBA2 isomers in the S0 and adiabatic T1 states, with the S0 state of BCIBA2 as the reference. | ||
Next, we considered benzannelated CIBA isomers (BCIBA2a–e, Fig. 7 and S20†) where each has two migratory Clar's sextets in the S0 state. There is a variation in the relative energies in the S0 state, which should be due to steric congestion between the ketone units and the benzene rings. Comparing the E(T1) of the various BCIBA2 isomers it is clear that the energies decrease as one goes from a system with low Hückel-aromaticity in the T1 state such as BCIBA2 having two migratory sextets, over a system with increased aromaticity, e.g., BCIBA2a and BCIBA2b with two localized sextets and one migratory, to maximal number of Hückel-aromatic sextets which occurs in the BCIBA2c isomer having four localized sextets in their T1 states. Thus, the connectivity within the benzannelated units allows for tuning of E(T1) by nearly 0.25 eV (6 kcal mol−1). There are only small variations in the E(T1) between BCIBA2a and BCIBA2b, which are equiaromatic,56 and between the three BCIBA2c–e (also equiaromatic), which likely are due to differences in the extent of steric congestion between the benzene and ketone moieties in the T1 relative to the S0 state.
Then, when considering the tetrabenzannelated CIBA isomers (BCIBA3 and isomers) we see that the rationale still holds as the isomer with the largest increase in number of Clar's sextets when going from S0 to T1 has the lowest E(T1) (1.26 eV, Fig. S21†). BCIBA3d, on the other hand, has the smallest increase in number of Clar's sextets, and accordingly, the largest E(T1) (1.47 eV). Combined, the calculations demonstrate that through systematic manipulation of the amount of Clar's sextets in the T1vs. the S0 state we can qualitatively predict and rationalise trends in the excited state energies.
Five BCIBA molecules (including BCIBA0) were selected for a detailed analysis on how E(T1) depends on the delocalization of the unpaired electrons. The EDDBH plots reveal that the main difference among these molecules occurs at the C atoms of the two pyrrolo units as the delocalization visually varies from one compound to another, while it seems constant at the two C–CO units (Fig. S23†). There is also a correlation between the amount of delocalization and E(T1) (R2 = 0.98, Fig. S24†). All in all, we observe that the more delocalized unpaired electrons in the C atoms of the two pyrrolo units, the more stable the T1 state becomes as the conjugation in this unit increases (Table S3†).
Furthermore, it can be seen that the magnitude of the S1–T1 splitting is not constant across the series. For BCIBA0 we see a relatively large vertical S1–T1 splitting of 1.41 eV which diminishes to 1.04 eV in BCIBA2. Noteworthy, the HOMO and LUMO are localized similarly in these compounds, but when analysing the electron configurations in the vertically excited S1 and T1 states (Table S5†), we find that as the system becomes larger configurations in addition to the singly excited HOMO-to-LUMO excitation become important for the description of the T1 state. Hence, our assumption on a similarity between the T1 and S1 states is not entirely correct, and this is found especially when we move from BCIBA0 and CIBA to BCIBA2–3. As the two states are no longer strongly similar in character, we now observe differences in their energy gaps. This highlights that although we can rationalise and tune the excited state energies rather precisely through the understanding and modification of Hückel-aromaticity in the lowest excited states, there remains additional differences between the molecules.
Other modifications of Cibalackrot-type compounds
The CIBA scaffold can be viewed as composed of various segments which can be exchanged to other moieties, an apparent first one being a change of the two carbonyl groups to thiocarbonyls, leading to THIO-CIBA. This replacement brings down both the E(T1) and E(S1) to 0.91 and 2.33 eV, respectively, whereby the E(S1)/E(T1) ratio increases compared to the parent CIBA from 2.09 to 2.56 (Fig. S24†). Other modifications are replacements of the peripheral benzene rings with heterocycles as well as replacements of the pyrrole rings with rings that either have stronger or weaker Hückel-aromatic character. We specifically probed the replacements of the two pyrrole rings in BCIBA0 with either pyrazole or benzene rings (Fig. 8). Synthetic routes to non-benzannulated bipyrrolinones and diphenoquinones have been previously shown which would serve as an ideal starting point for the attempted synthesis of these types of compounds.57–59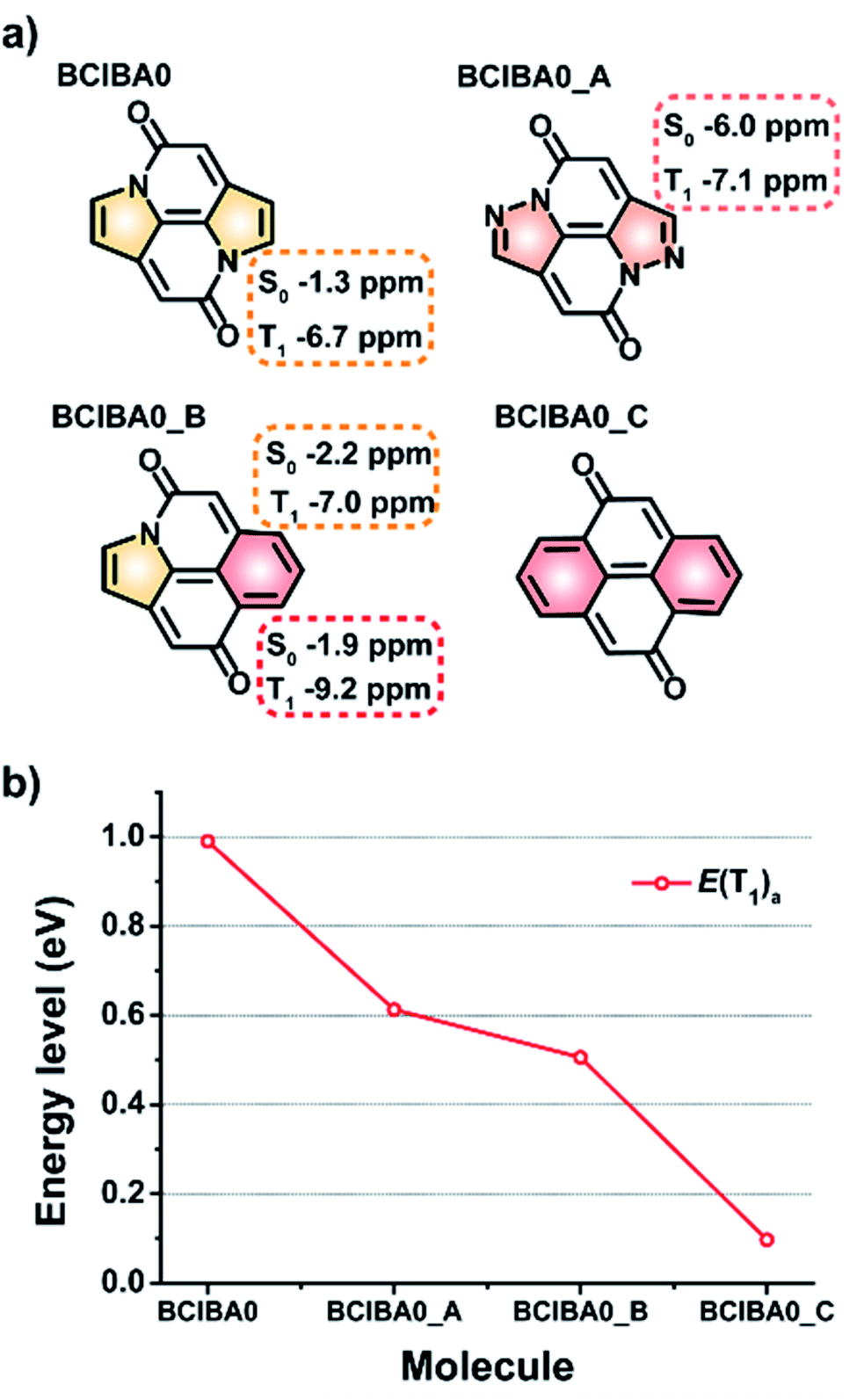 | ||
| Fig. 8 (a) BCIBA0 and its modified models (BCIBA0_X) with π-NICS(1.7)zz values marked on the modified rings in the S0 and T1 states. (b) Plots of adiabatic T1 excited state energies of the compounds. | ||
Indeed, with two benzene rings we achieve a modified BCIBA0 (BCIBA0_C) which has a negligible E(T1) as the attainment of strong Hückel-aromatic character of two benzene rings when the molecule is in T1 forces the triplet diradical character fully towards the carbonyl O and Cα atoms (Fig. S25†). With one pyrrole and one benzene ring, i.e.BCIBA0_B, the E(T1) is intermediate between BCIBA0 and BCIBA0_C. Yet, with two pyrazole rings (BCIBA0_A), having weakened Hückel-aromatic character compared to pyrrole,60 the E(T1) also moves down, contrary to the simple rationale. Clearly, there are limitations of the approach on modulating the E(T1) based on the Hückel-aromatic character of the various rings in the CIBA and BCIBA0 scaffolds. The E(T1) most obviously also depends on bonding features including aromatic character in the S0 state. This becomes further apparent as one regards imidazole rings in place of the pyrazoles (Fig. S24†).
Combination into a design approach
Having shown that the excited state energies can be tuned by (i) choice of substituents at the Cα atoms, (ii) benzannelations, or (iii) other modifications, we now asked to what extents these approaches can be combined? The BCIBA3 has a low E(T1) and the apparent question is to what extent this energy can be lowered further by attachment of substituents at the Cα position?Clearly, with two vinylcyclooctatetraene or two styryl substituents at the Cα atom, one can achieve a lowered E(T1) and a higher E(S1)/E(T1) ratio (BCIBA3_S1 and BCIBA3_S2, Fig. 9), however, a dilemma being that the E(T1) moves significantly below the ideal value of ∼1.1 eV. Instead, to preferentially raise E(S1) as compared to E(T1) one may utilize fluoro substitution at Cα, leading to BCIBA3_S3. For this compound, the adiabatic E(T1) is slightly above the 1.1 eV and the E(S1)/E(T1) ratios resemble those of SCIBA1 which functions as a singlet fission chromophore. Using computations, we have thus identified a CIBA-based chromophore with high E(T1) and high E(S1)/E(T1) ratio motivating further experimental work towards this or similar compounds.
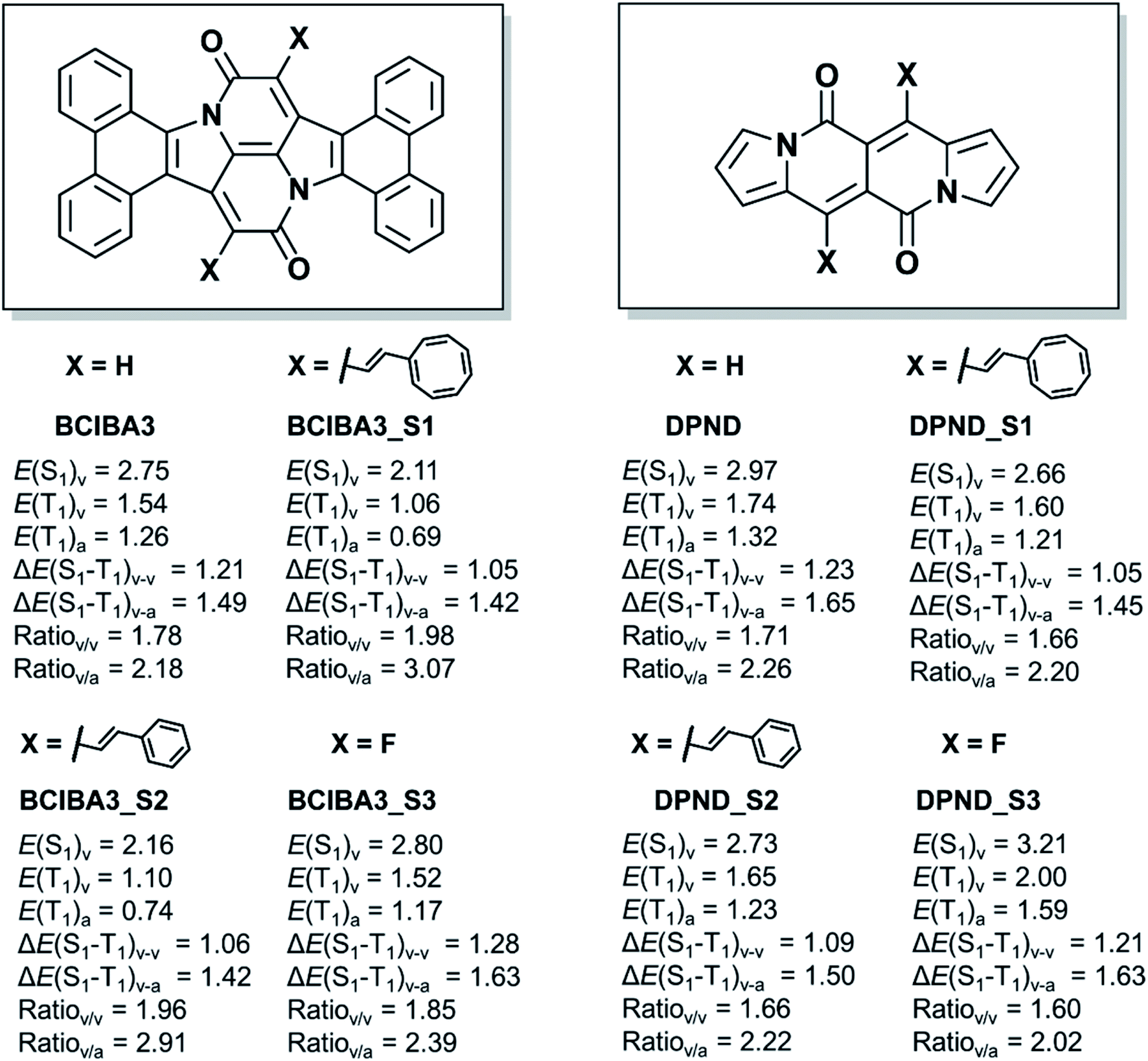 | ||
| Fig. 9 Chemical structures and relevant excitation energies (in eV) of substituted tetrabenzannelated CIBAs (BCIBA3s) as well as DPNDs. Calculations at TD-M06-2x/def2-SVP//M06-2X/def2-SVP level. | ||
Importantly, the same approach can be utilized to tailor DPNDs with targeted excited state energies. By changing the substituents at the site with highest spin density in the T1 state we can again vary the E(T1), and also the E(S1)/E(T1) ratios as seen in Fig. 9. It is likely that benzannelation also can be utilized to further tune the excited state energies of DPNDs. Thus, by identifying where the spin density is localized and which units are Hückel-aromatic one can design new singlet fission candidate chromophores.
Conclusions and outlook
Our reanalysis of the Cibalackrot scaffold revealed its Hückel aromatic characters in both the S0 and T1 states, instead of Baird-aromatic in the latter. In the triplet state we find that the pyrrolic rings gain Hückel-aromatic character relative to the S0 state and that the unpaired electrons are delocalized mainly through the central core (NARID) of the molecule. By changing the substituents at the site of greatest spin density, i.e. at the two Cα atoms, one can effectively adjust the spin density distribution on the chromophore whereby the excited state energy levels can be varied over a wide range (∼1 eV). Another means to tune the excited state energies is provided by increasing the Hückel aromatic character in the excited state relative to the ground state by elaborating on the number and type of aromatic sextets of the peripheral aromatic ring system through further benzannelation of the Cibalackrot scaffold.Taken together, we are able to present a more comprehensive understanding of the nature of the excited states in Cibalackrot-type compounds, their energies and how they can be tuned through tailoring the molecular scaffold whereby novel singlet fission chromophore candidates were identified. Here, it is noteworthy that also dipyrrolonaphthyridinedione (DPND) in its T1 state is Hückel-aromatic instead of Baird-aromatic. This is clear from a similar analysis as provided for the Cibalackrot-type molecules, and it can be rationalized by the structures of Fig. 10. Hence, by choice of substituent at the two Cβ atoms, having highest spin density, the E(T1) of DPND can be tuned.
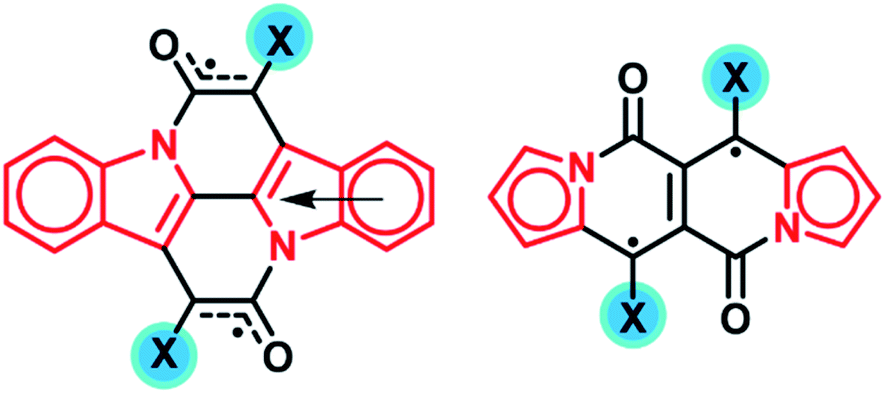 | ||
| Fig. 10 The main resonance structures of the substituted CIBA and DPND in their T1 states. The Hückel-aromatic units are marked in red while the substituents X used as handles for tuning E(T1) are marked in blue. | ||
Across the entire field of organic chromophore applications, it is paramount to be able to precisely tune excited states to advance technologies. We argue that without a detailed understanding of these states it is not possible to design new and improved chromophores that can achieve the desired properties. Indeed, tools such as changing substituents at sites of greater/smaller spin density and the manipulation of the excited state aromatic character have hardly been explored within the context of functional organic materials and could provide new and powerful tools to rationally tailor their properties. Earlier, one of us argued that Baird's rule on excited state aromaticity and antiaromaticity can be a handy back-of-an-envelope tool for such design of optically active functional materials.61 Yet, here we also note the complications, limitations and pitfalls of the excited state aromaticity and antiaromaticity concepts, where Baird-type excited state (anti)aromaticity is the most common form, yet not the only one. Polycyclic systems can be Hückel-aromatic in both the S0 state and in the lowest excited states, yet with the aromatic character distributed differently between the rings. As Hückel-aromaticity implies a larger number of paired electrons in the aromatic cycle it should, when possible, be preferred over Baird-aromaticity.41 Hence, care needs to be exercised and one needs to explore what type of aromaticity is at hand in a particular system when in its lowest triplet or singlet excited state. There is otherwise a clear risk of the overuse of the Baird-aromaticity concept to molecules to which it does not apply.
Computational methods
All S0 and T1 optimized geometries were obtained using the M06-2X62 functional together with the def2-SVP basis set.63 Basing on the optimized ground state geometries, the E(S1)'s were evaluated at TD-DFT M06-2X/def2-SVP level. The E(T1)'s were evaluated with a ΔSCF procedure at DFT M06-2X/def2-SVP level, which manually adjusts the spin multiplicities for both vertical and adiabatic geometries.64 The electron spin density distributions were analysed with Multiwfn 3.7 (ref. 65) based on the relaxed T1 state geometries.Aromaticity was evaluated in terms of the nucleus independent chemical shift (NICS),43,44 anisotropy of the induced current density (AICD)45 plots, the aromatic fluctuation index (FLU),47 and the multicenter index (MCI)48 computed at the optimized geometries on B3LYP/6-311+G(d,p) level,66–68 which had been benchmarked and widely used in evaluating aromatic criteria. NICS values were calculated at 1.7 Å above the ring centers (NICS(1.7)zz)44 using the gauge independent atomic orbital (GIAO)69 method. The π-only NICS(1.7)zz plots (π-NICS(1.7)zz) were obtained by using the sigma-model method implemented in the AROMA package70 which removes the sigma contributions. NICS-XY scans were performed using the AROMA package scanning from 1.7 Å above the plane of the molecule. AICD plots were generated with the AICD 2.0.0 program at 0.050 a.u. isosurface.45 The harmonic oscillator model of aromaticity (HOMA)46 measures the geometric aspect of aromaticity and was calculated with Multiwfn 3.7. The FLU and MCI were performed with the ESI-3D collection of programs.71 Electron delocalization has been also examined using the electron density of delocalization bond (EDDBH)49,50 at the M06-2X/def2-SVP and B3LYP/6-311+G(d,p) levels. In EDDBH calculations, Multiwfn and NBO 3.1 (ref. 72) programs have been used, where the latter was employed together with Gaussian 16.73 EDDBH surfaces have been visualized using Avogadro.74,75 Atomic charges and electronic spin densities have been calculated using quantum theory of atoms-in-molecules (QTAIM) scheme,76 which was done using the “medium quality grid” with a spacing of 0.1 Bohr.
The diradical characters (yn, n = 0, 1, 2, …, ) were calculated according to the literature method,6 with spin-projected UHF (PUHF) theory and 6-311+G(d,p) basis set.
Author contributions
H. O. and O. E. B. formulated the initial idea and developed it together with W. Z., D. W. S. and H. B. Computations were performed by W. Z., O. E. B. and D. W. S. All authors discussed the results, wrote and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. Z. is grateful to the Marie Skłodowska-Curie grant agreement No. 886066 (EXAM project) for financial support, and W. Z. and H. B. thankfully acknowledge funding by EPSRC (Grants EP/S003126/1). O. E. B. is grateful to the Wenner-Gren Foundations for a postdoctoral fellowship (UPD2018-0305), and D. W. S. acknowledges financial support by the European Union's Framework Programme for Research and innovation Horizon 2020 (2014–2020) under the Marie Skłodowska-Curie Grant Agreement No. 797335 “MulArEffect”. H. O. gratefully acknowledges financial support from The Swedish Research Council (grant 2019-05618). The computations were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC), Linköping, partially funded by the Swedish Research Council through grants 2015-04538 and 2019-05618.References
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS PubMed.
- A. Rao and R. H. Friend, Nat. Rev. Mater., 2017, 2, 17063 CrossRef CAS.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290–1299 CrossRef CAS PubMed.
- J. Wen, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2015, 137, 165–172 CrossRef CAS PubMed.
- T. Ullrich, P. Pinter, J. Messelberger, P. Haines, R. Kaur, M. M. Hansmann, D. Munz and D. M. Guldi, Angew. Chem., Int. Ed., 2020, 59, 7906–7914 CrossRef CAS PubMed.
- T. Minami and M. Nakano, J. Phys. Chem. Lett., 2012, 3, 145–150 CrossRef CAS.
- M. Nakano, Chem. Rec., 2017, 17, 27–62 CrossRef CAS PubMed.
- O. El Bakouri, J. R. Smith and H. Ottosson, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- K. J. Fallon, P. Budden, E. Salvadori, A. M. Ganose, C. N. Savory, L. Eyre, S. Dowland, Q. Ai, S. Goodlett, C. Risko, D. O. Scanlon, C. W. M. Kay, A. Rao, R. H. Friend, A. J. Musser and H. Bronstein, J. Am. Chem. Soc., 2019, 141, 13867–13876 CrossRef CAS PubMed.
- L. Wang, L. Lin, J. Yang, Y. Wu, H. Wang, J. Zhu, J. Yao and H. Fu, J. Am. Chem. Soc., 2020, 142, 10235–10239 CrossRef CAS PubMed.
- R. Kotani, L. Liu, P. Kumar, H. Kuramochi, T. Tahara, P. Liu, A. Osuka, P. B. Karadakov and S. Saito, J. Am. Chem. Soc., 2020, 142, 14985–14992 CrossRef CAS PubMed.
- J. R. Palmer, K. A. Well, J. E. Yarnell, J. M. Favale and F. N. Castellano, J. Phys. Chem. Lett., 2020, 11, 5092–5099 CrossRef CAS PubMed.
- N. V. Korovina, C. H. Chang and J. C. Johnson, Nat. Chem., 2020, 12, 391–398 CrossRef CAS PubMed.
- J. Stoycheva, A. Tadjer, M. Garavelli, M. Spassova, A. Nenov and J. Romanova, J. Phys. Chem. Lett., 2020, 11, 1390–1396 CrossRef CAS PubMed.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- B. H. Northrop, K. N. Houk and A. Maliakal, Photochem. Photobiol. Sci., 2008, 7, 1463–1468 CrossRef CAS PubMed.
- S. S. Zade, N. Zamoshchik, A. R. Reddy, G. Fridman-Marueli, D. Sheberla and M. Bendikov, J. Am. Chem. Soc., 2011, 133, 10803–10816 CrossRef CAS PubMed.
- E. Hückel, Z. Phys., 1931, 70, 204–286 CrossRef.
- H. S. Rzepa, Chem. Rev., 2005, 105, 3697–3715 CrossRef CAS PubMed.
- Z. S. Yoon, A. Osuka and D. Kim, Nat. Chem., 2009, 1, 113–122 CrossRef CAS PubMed.
- E. Miliordos, Phys. Rev. A: At., Mol., Opt. Phys., 2010, 82, 062118 CrossRef.
- N. C. Baird, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- H. Ottosson, Nat. Chem., 2012, 4, 969–971 CrossRef CAS PubMed.
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- R. Papadakis and H. Ottosson, Chem. Soc. Rev., 2015, 44, 6472–6493 RSC.
- B. Durbeej, J. Wang and B. Oruganti, ChemPlusChem, 2018, 83, 958–967 CrossRef CAS PubMed.
- J. Oh, Y. M. Sung, Y. Hong and D. Kim, Acc. Chem. Res., 2018, 51, 1349–1358 CrossRef CAS PubMed.
- C. Liu, Y. Ni, X. Lu, G. Li and J. Wu, Acc. Chem. Res., 2019, 52, 2309–2321 CrossRef CAS PubMed.
- G. Markert, E. Paenurk and R. Gershoni-Poranne, Chem.–Eur. J., 2021 DOI:10.1002/chem.202005248.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1184 CrossRef PubMed.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2000, 39, 3915–3917 CrossRef CAS PubMed.
- D. Shukla and P. Wan, J. Am. Chem. Soc., 1993, 115, 2990–2991 CrossRef CAS.
- P. Wan and E. Krogh, J. Chem. Soc., Chem. Commun., 1985, 1207–1208 RSC.
- F. Feixas, J. Vandenbussche, P. Bultinck, E. Matito and M. Solà, Phys. Chem. Chem. Phys., 2011, 13, 20690–20703 RSC.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 7303–7309 CrossRef CAS.
- P. B. Karadakov, J. Phys. Chem. A, 2008, 112, 12707–12713 CrossRef CAS PubMed.
- M. Rosenberg, H. Ottosson and K. Kilså, Phys. Chem. Chem. Phys., 2011, 13, 12912–12919 RSC.
- Y. M. Sung, M.-C. Yoon, J. M. Lim, H. Rath, K. Naoda, A. Osuka and D. Kim, Nat. Chem., 2015, 7, 418–422 CrossRef CAS PubMed.
- L. A. Paquette, M. J. Broadhurst, P. Warner, G. A. Olah and G. Liang, J. Am. Chem. Soc., 1973, 95, 3386–3387 CrossRef CAS.
- K. Jorner, F. Feixas, R. Ayub, R. Lindh, M. Solà and H. Ottosson, Chem.–Eur. J., 2016, 22, 2793–2800 CrossRef CAS PubMed.
- S. Escayola, C. Tonnelé, E. Matito, A. Poater, H. Ottosson, M. Solà and D. Casanova, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202100261.
- B. Oruganti, J. Wang and B. Durbeej, Org. Lett., 2017, 19, 4818–4821 CrossRef CAS PubMed.
- A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Chem.–Eur. J., 2014, 20, 5673–5688 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 014109 CrossRef PubMed.
- P. Bultinck, R. Ponec and S. V. Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
- D. W. Szczepanik, M. Andrzejak, K. Dyduch, E. Żak, M. Makowski, G. Mazur and J. Mrozek, Phys. Chem. Chem. Phys., 2014, 16, 20514–20523 RSC.
- D. W. Szczepanik, M. Andrzejak, J. Dominikowska, B. Pawełek, T. M. Krygowski, H. Szatylowicz and M. Solà, Phys. Chem. Chem. Phys., 2017, 19, 28970–28981 RSC.
- M. Swart, Theor. Chem. Acc., 2020, 139, 160 Search PubMed.
- Y. Wu, Y. Wang, J. Chen, G. Zhang, J. Yao, D. Zhang and H. Fu, Angew. Chem., Int. Ed., 2017, 56, 9400–9404 CrossRef CAS PubMed.
- J. L. Ryerson, A. Zaykov, L. E. A. Suarez, R. W. A. Havenith, B. R. Stepp, P. I. Dron, J. Kaleta, A. Akdag, S. J. Teat, T. F. Magnera, J. R. Miller, Z. Havlas, R. Broer, S. Faraji, J. Michl and J. C. Johnson, J. Chem. Phys., 2019, 151, 184903 CrossRef PubMed.
- Z. T. Armstrong, M. B. Kunz, A. C. Jones and M. T. Zanni, J. Phys. Chem. C, 2020, 124, 15123–15131 CrossRef CAS.
- A. Zaykov, P. Felkel, E. A. Buchanan, M. Jovanovic, R. W. A. Havenith, R. K. Kathir, R. Broer, Z. Havlas and J. Michl, J. Am. Chem. Soc., 2019, 141, 17729–17743 CrossRef CAS PubMed.
- P. W. Fowler, S. Cotton, D. Jenkinson, W. Myrvold and W. H. Bird, Chem. Phys. Lett., 2014, 597, 30–35 CrossRef CAS.
- H.-D. Becker and K. Gustafsson, Tetrahedron Lett., 1976, 17, 4883–4886 CrossRef.
- K. Hemming, A. D. Redhouse, R. K. Smalley, J. R. Thompson, P. D. Kennewell and R. Westwood, Tetrahedron Lett., 1992, 33, 2231–2234 CrossRef CAS.
- G. Pfeiffer and H. Bauer, Liebigs Ann. Chem., 1980, 564–576 CrossRef CAS.
- S. Dey, D. Manogaran, S. Manogaran and H. F. Schaefer, J. Phys. Chem. A, 2018, 122, 6953–6960 CrossRef CAS PubMed.
- H. Möllerstedt, M. C. Piqueras, R. Crespo and H. Ottosson, J. Am. Chem. Soc., 2004, 126, 13938–13939 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 Search PubMed.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295 Search PubMed.
- D. Padula, Ö. H. Omar, T. Nematiaram and A. Troisi, Energy Environ. Sci., 2019, 12, 2412–2416 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Chem. Phys., 1994, 98, 11623–11627 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- A. Stanger and A. Rahalkar, AROMA, http://schulich.technion.ac.il/Amnon_Stanger.htm Search PubMed.
- E. Matito, ESI-3D: Electron Sharing Indexes Program for 3D Molecular Space Partitioning, Institute of Computational Chemistry, Girona, 2006, http://iqc.udg.edu/%7Eeduard/esi Search PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO 3.1 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Avogadro: an open-source molecular builder and visualization tool. Version 1.2.0 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeerschd, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Plots of NICS-XY scans, AICD, EDDBH, spin density distribution and structural data. List of compounds include the following: the parent CIBA, substituted Cibalackrots, structurally altered Cibalackrot compounds and further modifications of the CIBA scaffold. See DOI: 10.1039/d1sc00382h |
| This journal is © The Royal Society of Chemistry 2021 |