
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole of rare-earth elements in enhancing bioelectrocatalysis for biosensing with NAD+-dependent glutamate dehydrogenase†
Lihao
Guan
a,
Fei
Wu
*b,
Guoyuan
Ren
a,
Jialu
Wang
a,
Xiaoti
Yang
b,
Xiaohua
Huang
 c,
Ping
Yu
b,
Yuqing
Lin
*a and
Lanqun
Mao
bd
c,
Ping
Yu
b,
Yuqing
Lin
*a and
Lanqun
Mao
bd
aDepartment of Chemistry, Capital Normal University, Beijing 100048, China. E-mail: linyuqing@cnu.edu.cn
bBeijing National Laboratory for Molecular Science, Key Laboratory of Analytical Chemistry for Living Biosystems, Institute of Chemistry, University of Chinese Academy of Sciences, Beijing 100049, China. E-mail: wufei317@iccas.ac.cn
cNational and Local Joint Engineering Research Center of Biomedical Functional Materials, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China
dCollege of Chemistry, Beijing Normal University, Beijing 100875, China
First published on 9th September 2021
Abstract
Dehydrogenases (DHs) are widely explored bioelectrocatalysts in the development of enzymatic bioelectronics like biosensors and biofuel cells. However, the relatively low intrinsic reaction rates of DHs which mostly depend on diffusional coenzymes (e.g., NAD+) have limited their bioelectrocatalytic performance in applications such as biosensors with a high sensitivity. In this study, we find that rare-earth elements (REEs) can enhance the activity of NAD+-dependent glutamate dehydrogenase (GDH) toward highly sensitive electrochemical biosensing of glutamate in vivo. Electrochemical studies show that the sensitivity of the GDH-based glutamate biosensor is remarkably enhanced in the presence of REE cations (i.e., Yb3+, La3+ or Eu3+) in solution, of which Yb3+ yields the highest sensitivity increase (ca. 95%). With the potential effect of REE cations on NAD+ electrochemistry being ruled out, homogeneous kinetic assays by steady-state and stopped-flow spectroscopy reveal a two-fold enhancement in the intrinsic reaction rate of GDH by introducing Yb3+, mainly through accelerating the rate-determining NADH releasing step during the catalytic cycle. In-depth structural investigations using small angle X-ray scattering and infrared spectroscopy indicate that Yb3+ induces the backbone compaction of GDH and subtle β-sheet transitions in the active site, which may reduce the energetic barrier to NADH dissociation from the binding pocket as further suggested by molecular dynamics simulation. This study not only unmasks the mechanism of REE-promoted GDH kinetics but also paves a new way to highly sensitive biosensing of glutamate in vivo.
Introduction
Dehydrogenases (DHs) are essential for catalyzing many oxidative reactions in living biosystems and have been an important part of the bioelectrocatalytic family to be frequently applied in biofuel cells,1–6 biosensors,7–12 and biosynthesis.13–15 The performances of DHs-based bioelectronics involve the enzymatic turnover of substrates with coenzymes and electrochemical regeneration of coenzymes, which are thus determined by the intrinsic enzyme kinetics of DHs and the electron transfer kinetics between enzymes and electrode surface.16–18 There are a list of strategies to improve the bioelectrocatalytic activity by promoting interfacial electron transfer kinetics, such as tuning the electronic properties of the electrode materials,12,19,20 selecting appropriate mediators for shuttling electron transfer,21 engineering nanostructured interfaces or protein structures to regulate enzyme conformation/orientation on the electrode surface.8,22–25 In addition, chemical regulation of enzyme kinetics is the other efficient means in promoting the complete turnover coupling electrode process, usually by biogenic ions (e.g., Ca2+, Mg2+ and Zn2+) or molecules (e.g., ATP).26 In recent years, rare-earth elements (REEs), non-necessary trace elements for most living organisms on earth, have emerged as new modulators for several enzymes like methanol dehydrogenases,27–29 and their physiological effects are attracting increasing attention.30,31Glutamate is the primary excitatory neurotransmitter involved in brain functions such as cognition, memory and learning.32–35In vivo biosensing of glutamate is thus of great significance to understand the physiological role of glutamate. Besides the frequently explored glutamate oxidase,36 glutamate dehydrogenase (GDH) catalyzing NAD(P)+-dependent glutamate oxidation has received growing interest as the bioelectrocatalytic unit in developing oxygen-insensitive glutamate biosensors. Being a hexamer of identical 501-residue polypeptide chains, GDH contains two functional domains respectively for glutamate binding and NAD+ binding, with a flexible “antenna” coupling the distinct domains through a conformational switch during catalytic turnover (Fig. 1).37–39 The two-substrate enzymatic reaction undergoes an ordered bi–bi scheme,40 so the bioelectrocatalytic performance is not only determined by glutamate dynamics but also heavily dependent on NAD+ association and NADH dissociation. In this regard, ions and molecules that have an effect on the enzyme conformation or binding dynamics at the active site are potential modulators of GDH-based biosensors. A number of biogenic GDH activators and inhibitors have indeed been reported,38 but are unexplored in an electrochemical scheme. Gao et al. made the pioneering discovery that REEs in trivalent forms can regulate the bioelectrocatalytic activity of GDH41,42 through a “hormesis effect”, i.e., REEs at low concentrations can activate GDH, while inhibit GDH at high concentrations.29,43 This finding implies that REEs in low quantities may be efficient enzyme promoters to improve the sensitivity of GDH-based glutamate biosensors, while the underlying mechanism needs further clarification.
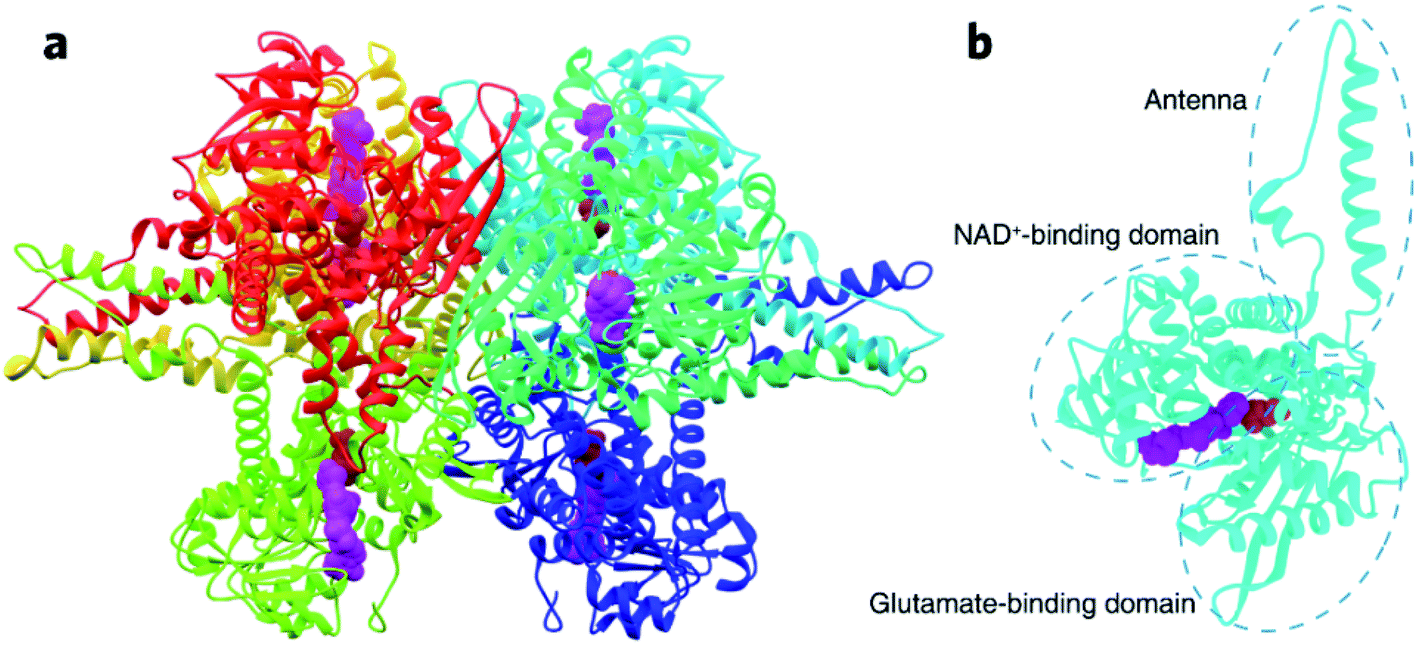 | ||
| Fig. 1 Crystal structure of GDH from bovine liver (PDB: 1HWY). (a) The entire hexamer of GDH with bound NAD+ (represented by purple spheres) and glutamate (represented by red spheres). (b) GDH subunit consisting of the NAD+-binding domain, glutamate-binding domain, and connecting antenna. | ||
Here, we report an investigation of the role of trivalent REEs (i.e., Yb3+, La3+ and Eu3+) in promoting electrochemical oxidation of glutamate catalysed by NAD+-dependent GDH. The presence of REEs at low micromolar levels in the electrolyte amplified current responses of the GDH-based biosensor to glutamate titrations by maximally 100%, mainly through accelerating the rate-determining NADH release. Protein conformational analysis and molecular dynamics (MD) simulation revealed that REE coordination to GDH can induce partial backbone reconfiguration into a more compact state and may assist NADH dissociation. Our efforts provide an allosteric mechanism for REE-enhanced GDH kinetics and bioelectrocatalysis, and bring up a new class of promoters for constructing highly sensitive glutamate biosensors for in vivo analysis.
Results and discussion
Role of REEs in bioelectrocatalysis of NAD+-dependent GDH
The overall bioelectrocatalytic cycle of NAD+-dependent GDH can be divided into two processes, the chemical process where GDH catalyzes oxidative deamination of glutamate to α-ketoglutarate while reducing NAD+ to NADH, and the electrode process where NADH is electro-oxidized back to NAD+ for the subsequent cycle. In order to probe the role of REEs during the cycle, we first performed electrochemical characterizations to unravel the impact of REEs on the bioelectrocatalytic performance of NAD+-dependent GDH. The GDH-based electrochemical glutamate biosensors were designed with glassy carbon electrodes (GCEs) modified by adsorbed GDH and methylene green (MG), the electrocatalyst for NADH oxidation at 0.0 V (vs. Ag/AgCl) to regenerate NAD+, and tested by amperometric titrations in artificial cerebrospinal fluid (aCSF, pH 8.5) containing NAD+ in the absence or presence of Yb3+, La3+ or Eu3+. As shown in Fig. 2a, the oxidation current of the GDH-based biosensor increases upon glutamate titrations, demonstrating the successful build-up of the electrode-coupled glutamate oxidation catalytic cycle. In REE-free aCSF the glutamate sensing sensitivity is 16.6 nA μM−1 cm−2. While in the presence of Yb3+, La3+ or Eu3+, the sensitivity increases by 94.6% (32.3 nA μM−1 cm−2), 88.6% (31.3 nA μM−1 cm−2) or 16.9% (19.4 nA μM−1 cm−2), respectively (Fig. 2b).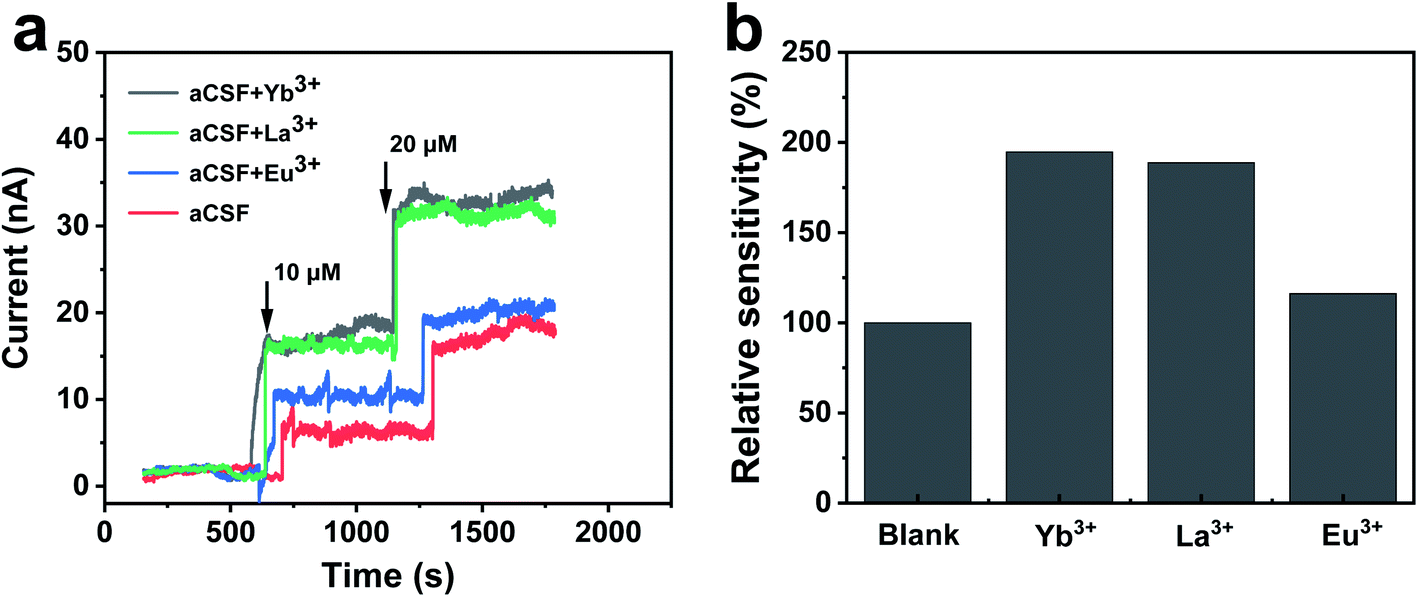 | ||
| Fig. 2 Effect of REEs on bioelectrocatalytic performance of NAD+-dependent GDH. (a) Amperometric current responses of the GDH-based biosensor toward titrations of 10 and 20 μM glutamate at 0.0 V vs. Ag/AgCl in aCSF containing 2 mM NAD+ in the absence or presence of 16 μM Yb3+, La3+ or Eu3+. (b) Relative sensitivity of the GDH-based glutamate biosensor. | ||
As the current response is not only an outcome of GDH catalysis but also of NAD+ regeneration on the electrode, it is necessary to clarify whether REEs promote the electrochemical oxidation of NADH. We compared cyclic voltammograms of NADH oxidation before and after the introduction of Yb3+. As depicted in Fig. S2a,† the oxidation of MG occurred at −0.10 V at the MG-modified GCE and the oxidation peak current was remarkably enhanced with the addition of 1 mM NADH, which was ascribed to the electrocatalytic oxidation of NADH by MG. After Yb3+ was introduced into the solution, the oxidation peak current remained almost the same as that without Yb3+. In addition, the effect of Yb3+ on the electrocatalytic NADH oxidation was evaluated by chronoamperometry. The addition of Yb3+ into aCSF containing NADH did not produce a recordable current response, as displayed in Fig. S2b,† revealing that REEs do not enhance the electrochemical oxidation of NADH. Consequently, it can be deduced that Yb3+ probably promotes the bioelectrocatalysis of GDH for glutamate sensing through the catalytic turnover within GDH.
After ruling out the involvement of REEs in the electrode process, we proposed that REEs may exert a promotive effect on enzymes in the chemical process. We thus conducted steady-state activity assays in a homogeneous setup to confirm our speculation. NADH production was monitored at 340 nm by UV-vis spectroscopy in a mixture of GDH, NAD+ and glutamate with or without Yb3+. As displayed in Fig. 3a, the absorbance profiles over time show a faster generation of NADH with GDH in aCSF containing Yb3+ after reactions reach the steady state (5 s after mixing). We collected data points within the beginning 10–30 s of the profiles to evaluate enzyme activity in terms of initial reaction rate. From the slope of linear fitting plots, we obtained the overall turnover rate of GDH to be 1.24 ± 0.0037 μM s−1 in the presence of 16 μM Yb3+, approximately 195% increase from 0.42 ± 0.0067 μM s−1 in blank aCSF (Fig. 3b, n = 3). As the concentration of Yb3+ doubled (32 μM), the increase in the turnover rate became smaller (1.51 times, Fig. S3†), indicating the concentration dependence of the Yb3+-induced enhancement, which can be explained by the hormesis effect.41,43,44
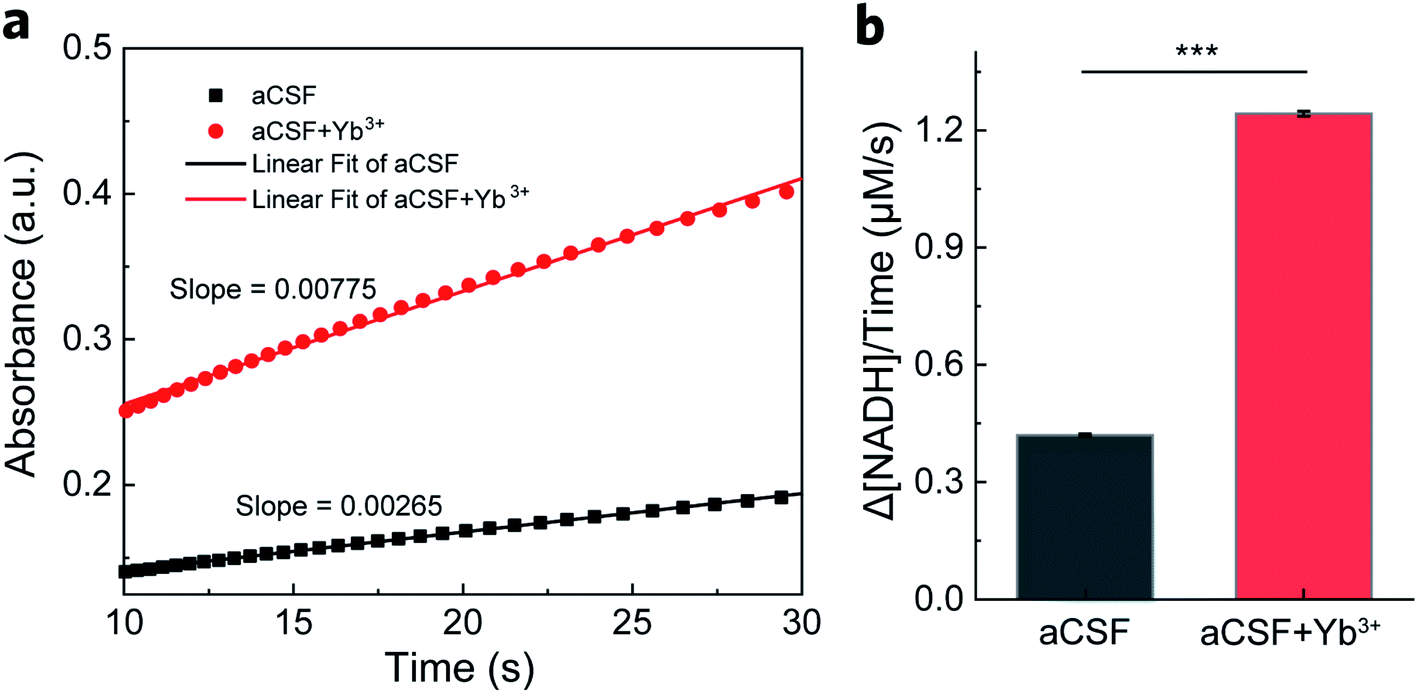 | ||
| Fig. 3 Steady-state GDH activity assay. (a) The absorbance of NADH at 340 nm generated in aCSF containing 0.1 mg mL−1 (ca. 300 nM) GDH, 250 μM NAD+ and 5 mM glutamate in the absence and presence of 16 μM Yb3+. Linear fitting of the steady-state profile during 10–30 s yields the corresponding slope for reaction rate calculation. (b) The calculated overall reaction rate of NADH production in the absence and presence of 16 μM Yb3+. Error bars represent standard error of the mean (n = 3, t-test, ***P < 0.001). | ||
Mechanism for REE-promoted NAD+-dependent GDH catalysis
The complete catalytic cycle of GDH is a sequence of four kinetic phases involving proton release, hydride transfer, intermediary complex transformation and product release as previously reported.40,45–47 Transient-state kinetic assays by stopped-flow spectroscopic measurements herein also resolved four distinguishable phases (Fig. 4a). Within 3 ms upon mixing of GDH–NAD+ and glutamate (Glu) solutions, a complex of GDH–NAD+–Glu forms as the starting point to trigger the sequential reaction process. Phase I (3–30 ms) illustrates the proton release step prior to hydride transfer. Then in phase II (30–100 ms), hydride transfer bursts between bound glutamate and NAD+ to result in the ternary complex of GDH, NADH and α-iminoglutarate, the latter accepting one molecule of H2O from Lys. 126 accompanied by a conformational change of GDH to form α-carbinolamine that subsequently deaminates into α-ketoglutarate.37,45,48 In phase III (100 ms to 4 s), glutamate slowly displaces α-ketoglutarate in the binding pocket.45,49 The final phase IV is the steady-state release of NADH that takes the longest period of time, and thus is the rate-determining step (RDS).50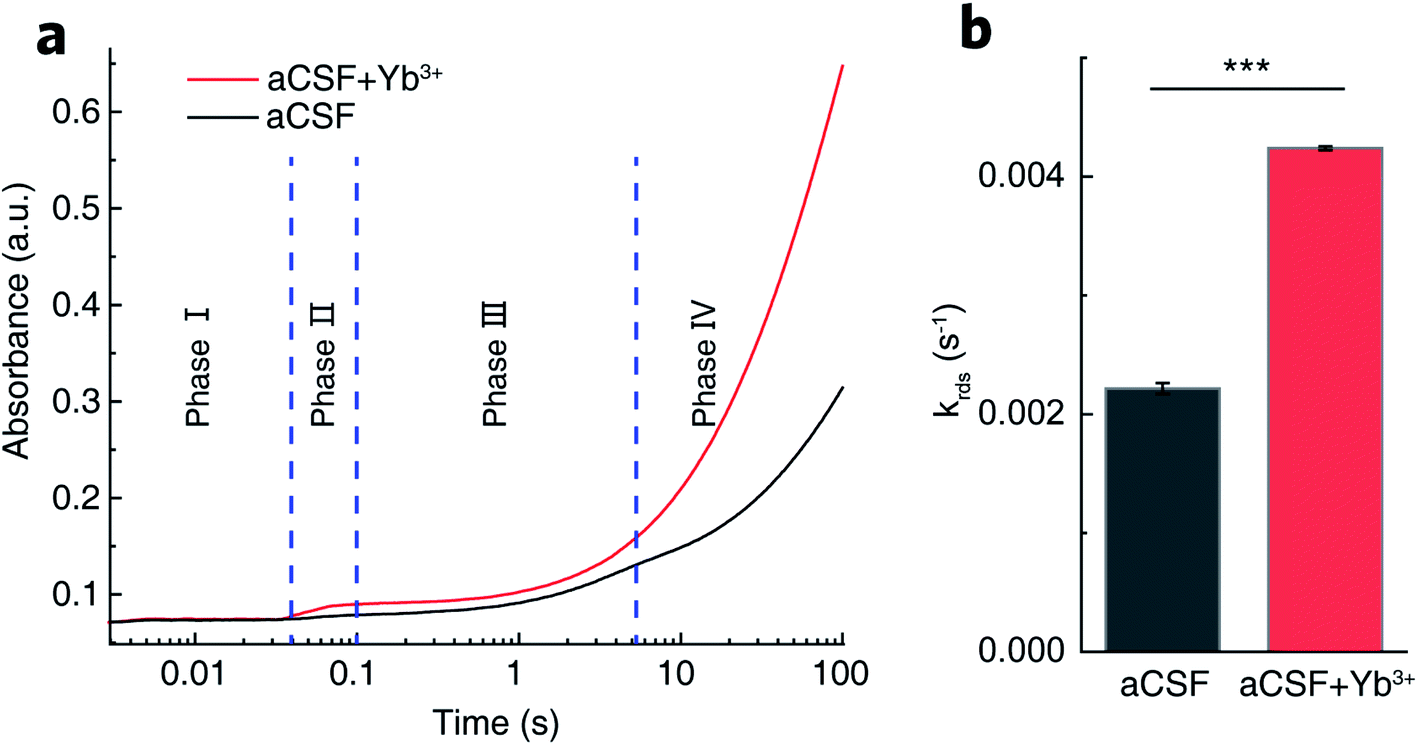 | ||
| Fig. 4 Transient-state GDH kinetics. (a) Typical absorbance profiles at 340 nm over 100 s showing four distinguishable phases of GDH-catalyzed glutamate oxidation in aCSF containing 0.1 mg mL−1 GDH, 250 μM NAD+ and 5 mM glutamate in the absence (black) and presence (red) of 16 μM Yb3+ by stopped-flow spectroscopy. (b) Rate constants of the rate-determining NADH release in phase IV obtained by fitting the absorbance profile during 10–1500 s using two consecutive one-order kinetic modeling. Error bars represent standard error of the mean (n = 3, t-test, ***P < 0.001). | ||
By fitting the curves of phase IV into a kinetic model of two continuous one-order reactions, we acquired the apparent rate constant (krds) to be (2.22 ± 0.05) × 10−3 s−1 in REE-free aCSF. With Yb3+ present, krds was determined to be (4.24 ± 0.02) × 10−3 s−1 (Fig. 4b). The kinetic enhancement on phase IV (91%) demonstrates that Yb3+ does promote NADH release. Moreover, we measured the respective rate constants for phase I (k1), II (k2) and III (k3), summarized in Table S1.† No statistical kinetic difference was observed during phase I, but k2 and k3 increased respectively by 51% and 40% in Yb3+-containing aCSF, implying that Yb3+ might also facilitate hydride transfer, intermediary complex transformation and displacement of α-ketoglutarate by glutamate.
To uncover the structural basis for the promotive effects of Yb3+ indicated by the transient-state kinetic study above, we first employed small angle X-ray scattering (SAXS) to probe possible Yb3+-induced conformational alterations in solvated GDH. The scattering intensity, I(q), measured from isotropically monodispersed GDH ligand with NAD+ in aCSF demonstrated its globular form in dilute solution (Fig. 5a). Modeling the indirect inverse Fourier transform of I(q) produced the real-space distribution profile, p(r), which describes pair distances between scattering centers of enzyme molecules (Fig. 5b).51,52 The radius of gyration (Rg) yielding size information for polymeric molecules was then extracted from p(r),53 and determined as 6.39 ± 0.19 nm for GDH in blank aCSF. After introducing Yb3+, GDH exhibited a smaller Rg of 5.66 ± 0.16 nm (Fig. 5c). Reduction in protein size excluded the probability of REE-induced enzyme aggregation while reflected higher compactness of the GDH entity, in accordance with the reported conformational modulation of proteins by REEs, such as the structural compaction in the cases of calmodulin and lanmodulin.39,54 Based on this result, we hypothesized that Yb3+ cations might coordinate electron-sufficient moieties at the active site and bring domains or local residues in closer proximity (Fig. 5d). In consequence they may reduce the spatial gap for hydride transfer and/or nucleophilic attack on α-iminoglutarate by Lys. 126-bonded H2O to form α-ketoglutarate,55 explaining the increase of k2 in phase II.
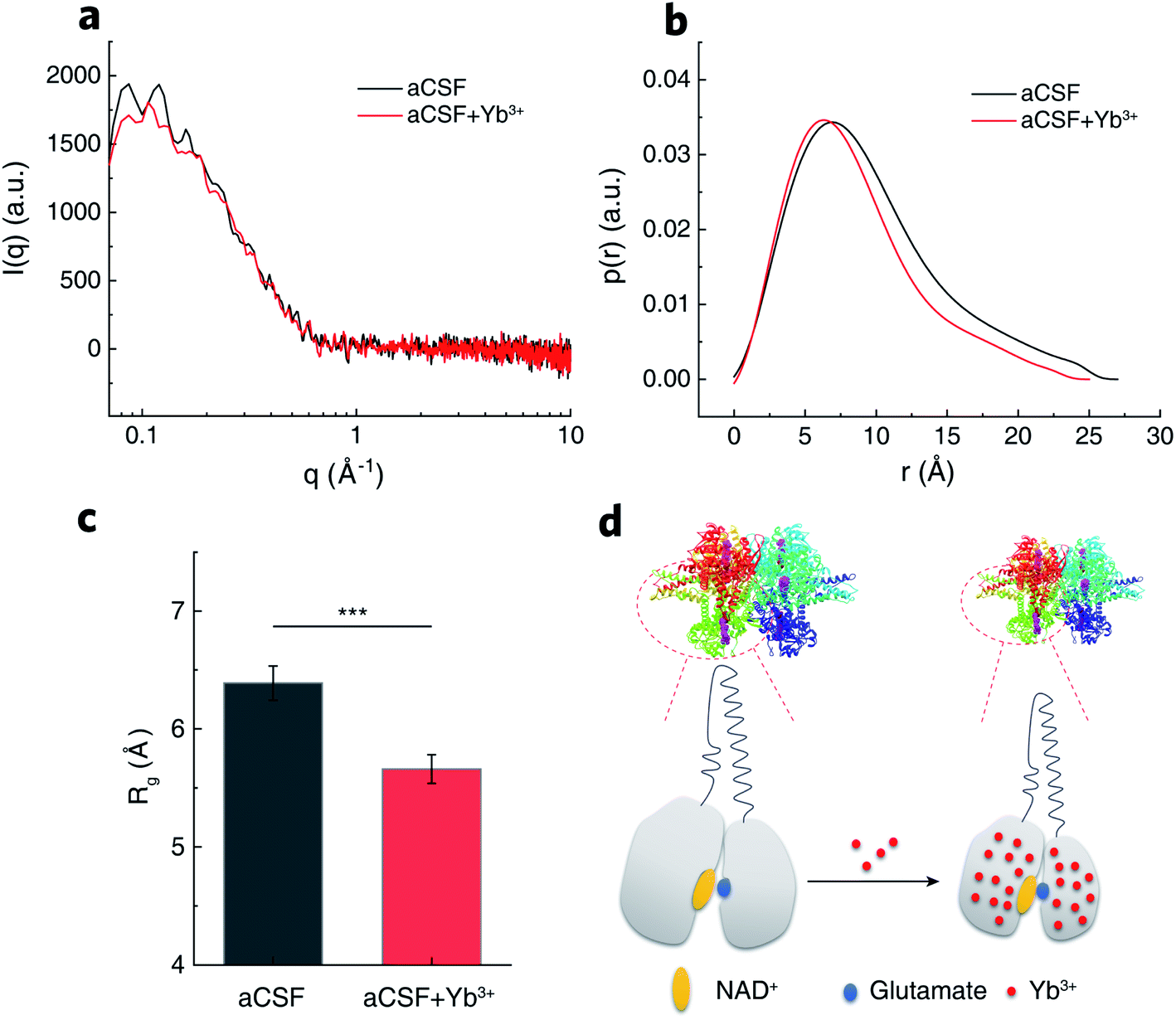 | ||
| Fig. 5 Protein conformation of GDH. (a) Scattering intensities of GDH in aCSF containing NAD+ in the absence (black) and presence (red) of Yb3+ by SAXS. (b) Corresponding pair distances distribution function obtained from (a). (c) Rg encoding protein size information calculated from (b). Error bars represent standard error of the mean (n = 8, t-test, ***P < 0.001). (d) Schematic illustration of Yb3+-induced GDH conformation change into a more compact state. | ||
To confirm our hypothesis of Yb3+ localization in GDH, we conducted attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopic investigations to probe regional transitions triggered by inner-bound Yb3+. The amide I band between 1700 and 1600 cm−1, originating from the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in protein backbones and highly sensitive to secondary structural changes,19,56–58 was selected from the full IR spectra of GDH for further deconvolution (Fig. 6a). We used fourth derivation to identify peak positions of convoluted bands related to different secondary structures (Fig. 6b). Notably, Yb3+ exerted no effect on band positions except that the vibrational peak of flexible β-sheets centered in the high-frequency vibrational range (HF-β-sheets, 1697 cm−1) was slightly shifted to 1692 cm−1 in Yb3+-treated GDH. Lower vibrational frequency indicated the loss in backbone flexibility of β-sheets that were loosely packed. This phenomenon is in line with SAXS observation of gain in backbone tightness upon Yb3+ coordination. We then resolved the secondary component bands by Gaussian fitting for proportion calculation (Fig. S4†). As shown in Fig. 6c, α-helices and β-sheets prevailed in solvated GDH with a minor distribution of turns and the unordered. Compared to the secondary composition in blank aCSF, α-helices were almost unaffected by Yb3+, but the proportion of β-sheets decreased by 6.7%. In the meantime, the proportion of β-turns increased by 9%. According to the crystal structure of GDH,35 α-helices establish the main protein scaffold, while β-sheets gather at the glutamate and NAD+-binding sites (Fig. 6d). In this regard, IR results indicated the possible localization of Yb3+ in the active site that caused a subtle transition from β-sheets to β-turns. This may lead to a plausible configuration of one Yb3+ ion coordinated to Glu. 275, Ser. 276 and Asp. 277 consecutively in a β-turn in the vicinity of bound NAD+/NADH (Fig. 6e).
O bonds in protein backbones and highly sensitive to secondary structural changes,19,56–58 was selected from the full IR spectra of GDH for further deconvolution (Fig. 6a). We used fourth derivation to identify peak positions of convoluted bands related to different secondary structures (Fig. 6b). Notably, Yb3+ exerted no effect on band positions except that the vibrational peak of flexible β-sheets centered in the high-frequency vibrational range (HF-β-sheets, 1697 cm−1) was slightly shifted to 1692 cm−1 in Yb3+-treated GDH. Lower vibrational frequency indicated the loss in backbone flexibility of β-sheets that were loosely packed. This phenomenon is in line with SAXS observation of gain in backbone tightness upon Yb3+ coordination. We then resolved the secondary component bands by Gaussian fitting for proportion calculation (Fig. S4†). As shown in Fig. 6c, α-helices and β-sheets prevailed in solvated GDH with a minor distribution of turns and the unordered. Compared to the secondary composition in blank aCSF, α-helices were almost unaffected by Yb3+, but the proportion of β-sheets decreased by 6.7%. In the meantime, the proportion of β-turns increased by 9%. According to the crystal structure of GDH,35 α-helices establish the main protein scaffold, while β-sheets gather at the glutamate and NAD+-binding sites (Fig. 6d). In this regard, IR results indicated the possible localization of Yb3+ in the active site that caused a subtle transition from β-sheets to β-turns. This may lead to a plausible configuration of one Yb3+ ion coordinated to Glu. 275, Ser. 276 and Asp. 277 consecutively in a β-turn in the vicinity of bound NAD+/NADH (Fig. 6e).
 | ||
| Fig. 6 Regional changes of GDH structure. (a) ATR-FTIR spectra of GDH pre-incubated with or without Yb3+. Dashed frame denotes the amide I band region. (b) Fourth derivation of amide I band. (c) Compositional percentages of different GDH secondary structures by integrating peak areas of deconvoluted bands. (d) Zoomed image of GDH active site with bound NADH (purple), glutamate (cyan) and residues binding NADH (red). Lys. 126 attacking glutamate intermediate and β-sheets guarding the active site are highlighted in dark blue and light blue, respectively. (e) The most plausible anchoring site for Yb3+ facing three potential ligand residues, Glu. 275, Ser. 276 and Asp. 277, which constitute a β-turn near bound NADH. | ||
As shown in stopped-flow measurements, major kinetic promotion stemmed from phase IV acceleration (91%) by Yb3+. Meanwhile, SAXS and IR characterizations suggest the possible localization of Yb3+ at the active site. Taken together, we proposed two models of the Yb3+ coordination configuration by MD simulations. Because REEs prefer an octadentate coordination,30 one hydrated Yb3+ ion is ligated to seven water molecules and the carboxylic side chain of Asp. 277 in one model (Fig. S5a†). Glu. 275 forms a hydrogen bond with one water ligand of Yb3+. In the other model (Fig. S5b†), both Asp. 277 and Glu. 275 are in the primary coordination sphere of Yb3+ with the rest occupied by six water molecules. Existence of these two types of configurations suggests the dynamic feature of Yb3+ coordination influenced by electrostatic forces and hydrogen bonding. Ser. 276 was not found involved in anchoring Yb3+ in either situation. Computed free energy over the distance describes a four-stage course of NADH dissociation from the center of the binding pocket (Fig. S5c and S6†). In the initial stage, NADH within 0.5 nm away from the binding sites (still inside the pocket) experiences comparable free energies (around 120 kJ mol−1). As NADH continues leaving the binding sites, electrostatic attractive force from Asp. 277 and/or Glu. 275 retards its leaving in terms of increased free energy. Yb3+ coordination to these negatively charged residues weakens the electrostatic attraction between the amine moiety of NADH and the side carboxylates of residues. The total free energy for NADH release was 381 kJ mol−1 and 303 kJ mol−1 in the absence and presence of Yb3+, respectively. This remarkable energetic reduction in the kinetic barrier (ca. 80 kJ mol−1) for the RDS may be the primary factor that promotes GDH catalysis.
Implication for sensitive glutamate biosensing in vivo
Knowing that REEs can efficiently enhance the bioelectrocatalytic activity of NAD+-dependent GDH, we were in a position to explore the applicability of REEs in promoting biosensing sensitivity in vivo. To do this, an online electrochemical analytical system (OECS) was constructed for continuous monitoring of glutamate as illustrated in Fig. S7a.† Two streams of aCSF solutions with different compositions (one for brain dialysate collection and the other for NAD+ and Yb3+ supply) were continuously perfused by microinjection pumps and loaded into an electrochemical flow cell equipped with GDH-MG-modified GCE as the glutamate probe. Fig. S7b† compares the online glutamate sensing results in the presence and absence of Yb3+, showing that Yb3+ dramatically enhanced the online current responses. The OECS displayed a good linear response with glutamate concentration ranging from 4 to 100 μM (I (nA) = 1.01Cglutamate (μM) + 11.36, R2 = 0.98) (Fig. S7c†). The detection limit, based on a signal-to-noise ratio of 3, was calculated to be 2.92 ± 0.11 μM. During online brain analysis, a large current increase was recorded for the brain microdialysate continuously sampled from the striatum of rats (Fig. S7d†), validating the applicability of the Yb3+-promoted, GDH-based OECS for in vivo real-time biosensing of glutamate. The basal dialysate level of glutamate in the rat striatum was determined to be 4.52 ± 0.27 μM (n = 3), consistent with the reported basal range of glutamate in extracellular space (4 to 350 μM).59–62 Taken together, the OECS equipped with REEs and GDH could be used as an effective and sensitive platform to record the basal level and dynamics of glutamate under physiological and pathological conditions in vivo.Conclusions
In summary, by investigating the interaction mechanism between Yb3+ and GDH, we have successfully demonstrated that REEs serve as allosteric promoters for bioelectrocatalysis of GDH by triggering subtle reorientation of peptide segments, consequently expediting phase coupling along with the catalytic scheme. Steady-state and transient-state spectroscopic investigations clarified the role of Yb3+ in modulating GDH kinetics and showed that Yb3+ in low quantity introduces a two-fold increase of the entire turnover rate by promoting the glutamate intermediate conversion to α-ketoglutarate and releasing of reduced coenzyme (i.e., NADH). Furthermore, the molecular basis of Yb3+ as the GDH promoter was unmasked by conformational analysis, i.e., SAXS and ATR-FTIR spectroscopy, and the results indicated that binding of Yb3+ with GDH leads to a narrower enzyme body accompanied by transitions of β-sheets to β-turns at the active site. In consequence, the total free energy for NADH release in the rate-determining step was reduced by about 80 kJ mol−1 as suggested by MD simulation. This mechanism lays the foundation for the development of new REEs-based regulatory tools for GDH-based biosensors, especially for regulating and monitoring glutamate levels in vivo toward neuromodulation and neurodegenerative diseases treatment. Furthermore, this research will shed light on a broad investigation of enzymes in the bioelectrochemistry area from a unique perspective into interactions between REEs and non-metallic NAD+-dependent DHs.Data availability
All the data have been included in the ESI.†Author contributions
L. M., Y. L. and F. W. conceived the idea for the project. L. G. purified the enzyme and conducted the experiments of stopped-flow, SAXS and ATR-FTIR measurements. G. R. and J. W. performed the electrochemical experiments. L. G., F. W. and Y. L. studied the reaction mechanism, analyzed enzyme conformation changes and designed MD simulation. L. G., F. W. and Y. L. drafted the manuscript. L. M., X. Y., P. Y. and X. H. finalized the manuscript. All authors discussed and commented on the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key Research and Development Program (Grant No. 2018YFE0200800), the National Natural Science Foundation of China (No. 22074095 for Y. Lin; No. 21790390, 21790391 and 22134002 for L. Mao; No. 21874139 and 21927804 for F. Wu), the National Basic Research Program of China (2016YFA0200104), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB30000000), Chinese Academy of Sciences (QYZDJ-SSW-SLH030), and High-level Teachers in Beijing Municipal Universities in the Period of 13th Five year Plan (CIT&TCD20190330). We are grateful to Prof. T. S. Shi (Hebei University) for help with stopped-flow experiments.Notes and references
- J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439–2461 CrossRef CAS PubMed.
- L. Xia, K. Van Nguyen, Y. Holade, H. Han, K. Dooley, P. Atanassov, S. Banta and S. D. Minteer, ACS Energy Lett., 2017, 2, 1435–1438 CrossRef CAS.
- Y. Ulyanova, M. A. Arugula, M. Rasmussen, E. Pinchon, U. Lindstrom, S. Singhal and S. D. Minteer, ACS Catal., 2014, 4, 4289–4294 CrossRef CAS.
- Y. H. Kim, E. Campbell, J. Yu, S. D. Minteer and S. Banta, Angew. Chem., Int. Ed., 2013, 52, 1437–1440 CrossRef CAS PubMed.
- C. Ji, J. Hou, K. Wang, Y. H. Ng and V. Chen, Angew. Chem., Int. Ed., 2017, 56, 9762–9766 CrossRef CAS PubMed.
- M. T. Meredith and S. D. Minteer, Annu. Rev. Anal. Chem., 2012, 5, 157–179 CrossRef CAS PubMed.
- S. Guo, P. Yu, W. Li, Y. Yi, F. Wu and L. Mao, J. Am. Chem. Soc., 2020, 142, 2074–2082 CrossRef CAS PubMed.
- Z. Guo, L. Murphy, V. Stein, W. A. Johnston, S. Alcala-Perez and K. Alexandrov, J. Am. Chem. Soc., 2016, 138, 10108–10111 CrossRef CAS PubMed.
- S. Velasco-Lozano, M. Roca, A. Leal-Duaso, J. A. Mayoral, E. Pires, V. Moliner and F. López-Gallego, Chem. Sci., 2020, 11, 12009–12020 RSC.
- L. Zhang, M. Zhou and S. Dong, Anal. Chem., 2012, 84, 10345–10349 CrossRef CAS PubMed.
- S. Tsujimura, K. Murata and W. Akatsuka, J. Am. Chem. Soc., 2014, 136, 14432–14437 CrossRef CAS PubMed.
- G. Wu, Y. Gao, D. Zhao, P. Ling and F. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 40978–40986 CrossRef CAS PubMed.
- J. Kreuzer, N. C. Bach, D. Forler and S. A. Sieber, Chem. Sci., 2015, 6, 237–245 RSC.
- F. Dong, H. Chen, C. A. Malapit, M. B. Prater, M. Li, M. Yuan, K. Lim and S. D. Minteer, J. Am. Chem. Soc., 2020, 142, 8374–8382 CrossRef CAS PubMed.
- S. V. Lozano, M. Roca, A. L. Duaso, J. A. Mayoral, E. Pires, V. Moliner and F. L. Gallego, Chem. Sci., 2020, 11, 12009–12020 RSC.
- P. Das, M. Das, S. R. Chinnadayyala, I. M. Singha and P. Goswami, Biosens. Bioelectron., 2016, 79, 386–397 CrossRef CAS PubMed.
- A. J. Gross, X. Chen, F. Giroud, C. Travelet, R. Borsali and S. Cosnier, J. Am. Chem. Soc., 2017, 139, 16076–16079 CrossRef CAS PubMed.
- I. S. Kucherenko, O. O. Soldatkin, D. Y. Kucherenko, O. V. Soldatkina and S. V. Dzyadevych, Nanoscale Adv., 2019, 1, 4560–4577 RSC.
- F. Wu, L. Su, P. Yu and L. Mao, J. Am. Chem. Soc., 2017, 139, 1565–1574 CrossRef CAS PubMed.
- M. T. D. Martino, F. Tonin, N. A. Yewdall, M. Abdelghani, D. S. Williams, U. Hanefeld, F. P. J. T. Rutjes, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Chem. Sci., 2020, 11, 2765–2769 RSC.
- F. Wu, P. Yu, X. Yang, Z. Han, M. Wang and L. Mao, J. Am. Chem. Soc., 2018, 140, 12700–12704 CrossRef CAS PubMed.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS PubMed.
- Y. Zhang, Y. Deng, C. Wang, L. Li, L. Xu, Y. Yu and X. Su, Chem. Sci., 2019, 10, 5959–5966 RSC.
- J. F. Darby, M. Atobe, J. D. Firth, P. Bond, G. J. Davies, P. O’Brien and R. E. Hubbard, Chem. Sci., 2017, 8, 7772–7779 RSC.
- Y. B. Shpigler and D. Avnir, Chem. Sci., 2020, 11, 3965–3977 RSC.
- R. C. Hudson, L. D. Ruttersmith and R. M. Daniel, Biochim. Biophys. Acta, 1993, 1202, 244–250 CrossRef CAS.
- A. Pol, T. R. M. Barends, A. Dietl, A. F. Khadem, J. Eygensteyn, M. S. M. Jetten and H. J. M. Op den Camp, Environ. Microbiol., 2014, 16, 255–264 CrossRef CAS PubMed.
- Y. W. Deng, S. Y. Ro and A. C. Rosenzweig, J. Biol. Inorg Chem., 2018, 23, 1037–1047 CrossRef CAS PubMed.
- A. McSkimming, T. Cheisson, P. J. Carroll and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 1223–1226 CrossRef CAS PubMed.
- T. Cheisson and E. J. Schelter, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- J. A. Cotruvo, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, J. Am. Chem. Soc., 2018, 140, 15056–15061 CrossRef PubMed.
- D. J. Rossi, T. Oshima and D. Attwell, Nature, 2000, 403, 316–321 CrossRef CAS PubMed.
- I. Ahmed, S. K. Bose, N. Pavese, A. Ramlackhansingh, F. Turkheimer, G. Hotton, A. Hammers and D. J. Brooks, Brain, 2011, 134, 979–986 CrossRef PubMed.
- S. C. Buckingham, S. L. Campbell, B. R. Haas, V. Montana, S. Robel, T. Ogunrinu and H. Sontheimer, Nat. Med., 2011, 17, 1269–1274 CrossRef CAS PubMed.
- K. Takikawa, D. Asanuma, S. Namiki, H. Sakamoto, T. Ariyoshi, N. Kimpara and K. Hirose, Angew. Chem., Int. Ed., 2014, 53, 13439–13443 CrossRef CAS PubMed.
- R. D. O’Neill, S.-C. Chang, J. P. Lowry and C. J. McNeil, Biosens. Bioelectron., 2004, 19, 1521–1528 CrossRef PubMed.
- T. J. Smith, P. E. Peterson, T. Schmidt, J. Fang and C. A. Stanley, J. Mol. Biol., 2001, 307, 707–720 CrossRef CAS PubMed.
- T. J. Smith and C. A. Stanley, Trends Biochem. Sci., 2008, 33, 557–564 CrossRef CAS PubMed.
- P. E. Peterson and T. J. Smith, Structure, 1999, 7, 769–782 CrossRef CAS PubMed.
- R. C. Hudson and R. M. Daniel, Comp. Biochem. Physiol. B-Biochem. Mol. Biol., 1993, 106, 767–792 CrossRef CAS.
- W. K. Xin and X. X. Gao, Analyst, 1996, 121, 687–690 RSC.
- Q.-K. Zhuang, H.-C. Dai, X.-X. Gao and W.-K. Xin, Bioelectrochemistry, 2000, 52, 37–41 CrossRef CAS PubMed.
- E. Agathokleous, M. Kitao and E. J. Calabrese, Environ. Pollut., 2018, 238, 1044–1047 CrossRef CAS PubMed.
- L. Wang, A. Lu, T. Lu, X. Ding and X. Huang, Biochimie, 2010, 92, 41–50 CrossRef CAS PubMed.
- N. Singh, S. J. Maniscalco and H. F. Fisher, J. Biol. Chem., 1993, 268, 21–28 CrossRef CAS.
- H. F. Fisher, S. Maniscalco, N. Singh, R. N. Mehrotra and R. Srinivasan, Biochim. Biophys. Acta, Proteins Proteomics, 1992, 1119, 52–56 CAS.
- E. Silverstein and G. Sulebele, Biochemistry, 1973, 12, 2164–2172 CrossRef CAS PubMed.
- M. Iwatsubo and D. Pantaloni, Bull. Soc. Chem. Biol., 1967, 49, 1563 CAS.
- A. Colen, R. Wilkinson and H. Fisher, J. Biol. Chem., 1975, 250, 5243–5246 CrossRef CAS.
- H. F. Fisher, Methods Enzymol., Elsevier, 1985, vol. 113, pp. 16–27 Search PubMed.
- C. M. Jeffries, M. A. Graewert, C. E. Blanchet, D. B. Langley, A. E. Whitten and D. I. Svergun, Nat. Protoc., 2016, 11, 2122–2153 CrossRef CAS PubMed.
- G. L. Hura, A. L. Menon, M. Hammel, R. P. Rambo, F. L. Poole, Ii, S. E. Tsutakawa, F. E. Jenney, Jr, S. Classen, K. A. Frankel and R. C. Hopkins, Nat. Methods, 2009, 6, 606–612 CrossRef CAS PubMed.
- L. Pollack, M. W. Tate, A. C. Finnefrock, C. Kalidas, S. Trotter, N. C. Darnton, L. Lurio, R. H. Austin, C. A. Batt, S. M. Gruner and S. G. J. Mochrie, Phys. Rev. Lett., 2001, 86, 4962–4965 CrossRef CAS PubMed.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo, J. Am. Chem. Soc., 2019, 141, 2857–2861 CrossRef CAS PubMed.
- J. E. Rife and W. W. Cleland, Biochemistry, 1980, 19, 2328–2333 CrossRef CAS PubMed.
- S. Krimm and J. Bandekar, Adv. Protein Chem., 1986, 38, 181–364 CrossRef CAS PubMed.
- J. Bandekar, Biochim. Biophys. Acta, Proteins Proteomics, 1992, 1120, 123–143 CAS.
- H. Yang, S. Yang, J. Kong, A. Dong and S. Yu, Nat. Protoc., 2015, 10, 382–396 CrossRef CAS PubMed.
- S. Chakraborty and C. Retna Raj, Electrochem. Commun., 2007, 9, 1323–1330 CrossRef CAS.
- A. M. Andrews, A. Schepartz, J. V. Sweedler and P. S. Weiss, J. Am. Chem. Soc., 2014, 136, 1–2 CrossRef CAS PubMed.
- Y. Yu, X. Liu, D. Jiang, Q. Sun, T. Zhou, M. Zhu, L. Jin and G. Shi, Biosens. Bioelectron., 2011, 26, 3227–3232 CrossRef CAS PubMed.
- C.-S. Yang, P.-J. Tsai, N.-N. Lin, L. Liu and J.-S. Kuo, Free Radical Biol. Med., 1995, 19, 453–459 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00193k |
| This journal is © The Royal Society of Chemistry 2021 |