
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed multisubstituted allylation of the chelation-assisted C–H bond of (hetero)arenes with cyclopropenes†
Kuppan
Ramachandran
and
Pazhamalai
Anbarasan
 *
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai, 600036, India. E-mail: anbarasansp@iitm.ac.in; Web: https://home.iitm.ac.in/anbarasansp/
First published on 15th September 2021
Abstract
Cyclopropenes are highly strained three-membered carbocycles, which offer unique reactivity in organic synthesis. Herein, Cp*CoIII-catalyzed ring-opening isomerization of cyclopropenes to cobalt vinylcarbene has been utilized for the synthesis of multisubstituted allylarenes via directing group-assisted functionalization of C–H bonds of arenes and heteroarenes. Employing this methodology, various substituents can be introduced at all three carbons of the allyl moiety with high selectivity. The important highlights are excellent functional group tolerance, multisubstituted allylation, high selectivity, gram scale synthesis, removable directing group, and synthesis of cyclopenta[b]indoles. In addition, a potential cobaltocycle intermediate was identified and a plausible mechanism is also proposed.
Introduction
Allylated arenes and heteroarenes are prevalent structural motifs found in various natural products and biologically important molecules.1 Synthetically, substituted allyl groups serve as an excellent handle to increase the complexity of a molecule with simple synthetic operations. These allyl groups are traditionally introduced into arenes via either the Friedel–Crafts allylation of electron rich arenes2 or reaction of prefunctionalized organometallic reagents with allyl electrophiles.3 These methods have only limited substrate scope and require sensitive organometallic reagents. In contemporary organic synthesis, traditional methods are wisely replaced with the transition metal catalyzed direct allylation of directing group-assisted C–H bonds of (hetero)arenes.4 In the past few decades, precious transition metals such as Pd,5 Ru,6 Rh,7 Ir,8etc.9 were efficiently utilized for the allylation of various C–H bonds. Recently, Cp*CoIII based catalysts have emerged as efficient alternatives to the precious metal catalysts in allylation10 and other C–H bond functionalization reactions.11 In addition, due to the small size and hard nature of cobalt catalysts, complementary reactivity was observed in a number of other transformations. All these allylation methods utilize either allylic alcohol derivatives or vinyl-epoxides, -cyclic carbonates and -cyclopropanes, allenes and others (Scheme 1a). Most of these transformations offer access to simple allylated and mono-substituted allylated arenes. Nevertheless, methods for introduction of allyl group containing substitution at allylic and alkene carbons are rather limited because most of these reactions involve an initial 1,2-migratory insertion of a C–metal bond into the alkene of the allylating reagent, which is highly controlled by the steric factor. Hence, it is highly desirable to develop a complementary approach for the multisubstituted allylation of C–H bonds utilizing a unique allylating reagent, which would involve an unconventional mode of allylation.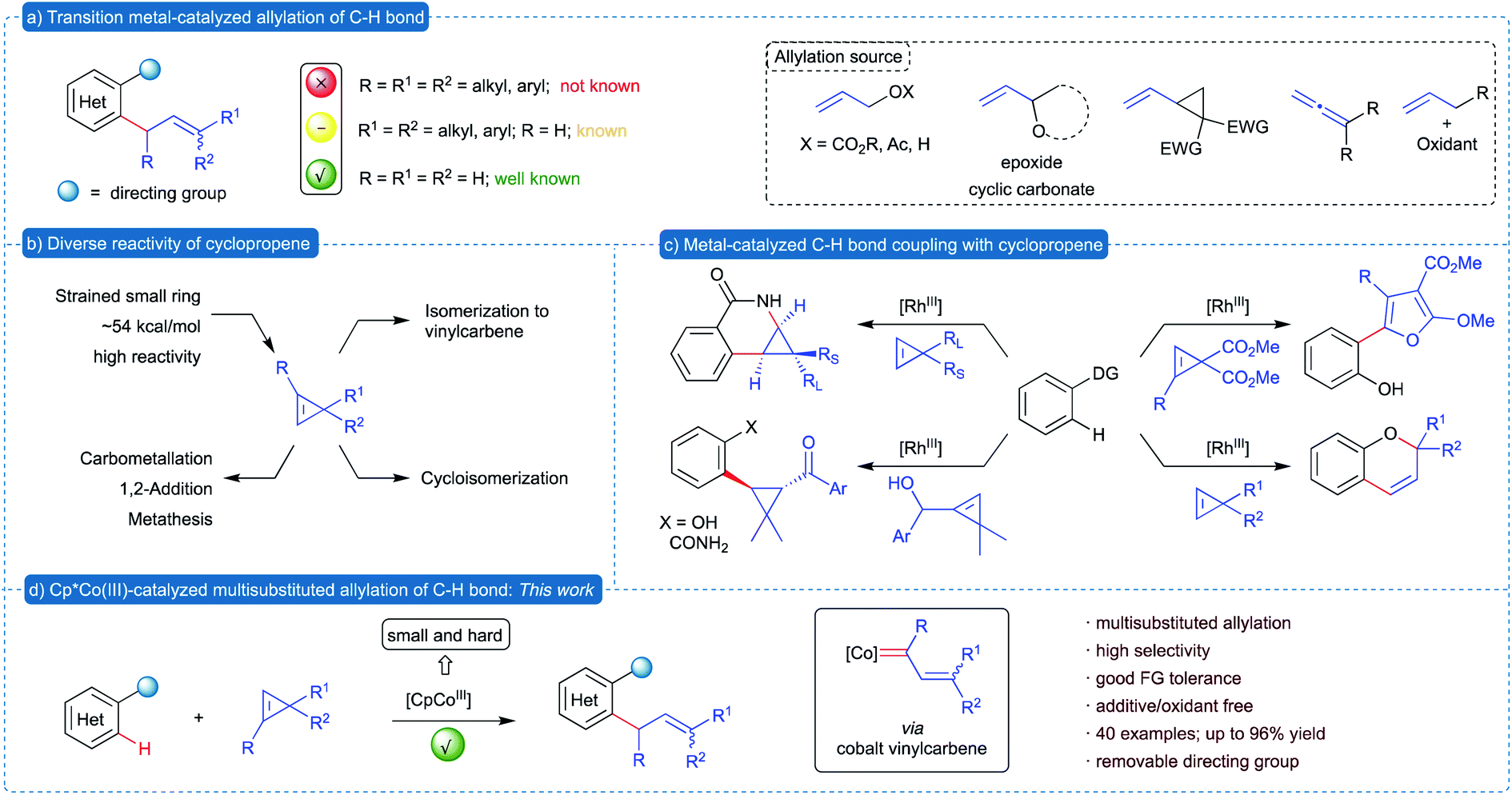 | ||
| Scheme 1 Transition metal-catalyzed C–H bond allylation and reaction of cyclopropenes. | ||
Substituted cyclopropenes constitute an important class of building blocks in organic synthesis.12 Due to their high ring strain energy, cyclopropenes demonstrated versatile and exceptional reactivity, which are efficiently exploited in various transformations. In general, the reactivity of cyclopropenes can be classified into two types: (1) reaction of alkenes without ring-opening such as 1,2-addition and carbometallation, and (2) reaction accompanied by ring opening13 such as cycloisomerization, metathesis and reaction of vinyl carbenes (Scheme 1b). On the other hand, application of cyclopropenes in transition metal-catalyzed C–H bond functionalization of (hetero)arenes is in its infancy.14 The known transformations involve cyclopropanation of arenes via addition to alkene15 or cycloisomerization to heterocycles employing Rh(III)-based catalysts16 (Scheme 1c). However, transition metal-catalyzed ring-opening of cyclopropenes to vinylcarbenes17 and its application in C–H bond functionalization is yet to be explored.
We have been extensively involved in the in situ generation of metalcarbenes from various precursors18 and its application in the construction of various complex carbo- and heterocycles through C–H bond functionalization.19 In continuation of our work on metalcarbenes and encouraged by the unique reactivity of cyclopropenes, we envisioned the utilization of cyclopropenes as a non-diazo precursor of vinylcarbenes in transition metal-catalyzed C–H bond functionalization for the synthesis of allylated (hetero)arenes. Due to the high reactivity of cyclopropenes, we intended to introduce substituents at all three carbons of the allyl moiety, which is otherwise difficult to achieve. We herein report the Cp*CoIII-catalyzed multisubstituted allylation of directing group-assisted C–H bonds of (hetero)arenes, such as indoles and pyrroles, exploiting substituted cyclopropenes as the allylating reagent.20 This method represents the first example of multisubstituted allylation of (hetero)arenes, where substituents are introduced at all three carbons of the allyl group with high selectivity control.
Results and discussion
N-Heterocycles, particularly indoles, are of great synthetic interest due to their relevance in biological systems. Hence, we started our investigation with allylation of indole derivatives. First, we examined the reaction of N-pyridylindole 1a and 3,3-diphenylcyclopropene 2a as the model reaction in the presence of [Cp*CoI2CO] catalyst. Based on the initial optimization (see the ESI† for more details), reaction of 1a and 2a in the presence of 10 mol% of [Cp*CoI2CO], 20 mol% of AgSbF6 and 10 mol% of NaOAc in DCE at 100 °C for 1.5 h afforded the expected allylated product 3a in 91% yield (Table 1, entry 1). In the absence of either [Cp*CoI2CO] or AgSbF6, allylation was not observed. Although 1a was recovered from the reaction mixture, 2a underwent intramolecular rearrangement to the indene derivative and was isolated in high yields (Table 1, entry 2). A similar result was observed when NaOAc was removed (Table 1, entry 3). These observations suggest that the reaction is catalyzed by Cp*CoIII, and AgSbF6 and NaOAc are essential for the observed activity of the catalyst. Replacement of NaOAc with other bases such as KOAc, NaOPiv and AgOAc gave product 3a in lower yield, reiterating the importance of NaOAc (Table 1, entries 4–6). Screening of solvents revealed that polar solvents, such as DMF, DMSO, and CH3CN that can chelate with metals, are not suitable for the present transformation (Table 1, entry 7). Also use of non-polar aromatic solvents such as toluene was not beneficial (Table 1, entry 8). Reducing the catalyst loading to 5 mol% or use of the Lewis acidic catalyst [Cp*Co(CH3CN)3](SbF6)2 in the absence of AgSbF6 gave the product 3a in reduced yield (Table 1, entries 1 and 9). Thus, 10 mol% of [Cp*CoI2CO], 20 mol% of AgSbF6 and 10 mol% of NaOAc in DCE at 100 °C were chosen as optimal conditions for the allylation of C–H bonds of indole 1 with cyclopropenes 2.|
|
||
|---|---|---|
| Entry | Conditions | Yieldb (%) |
| a Reaction conditions: 1a (1.0 equiv.), 2a (2 equiv.), [Cp*CoI2CO] (10 mol%), DCE, 100 °C, 1–2 h. b All are isolated yields. c With 5 mol% of [Cp*CoI2CO]. | ||
| 1 | Standard conditions | 91 (69)c |
| 2 | Absence of Cp*CoI2CO or AgSbF6 | NR |
| 3 | Without NaOAc | 5 |
| 4 | KOAc instead of NaOAc | 60 |
| 5 | NaOPiv instead of NaOAc | 56 |
| 6 | AgOAc instead of NaOAc | 51 |
| 7 | DMF, DMSO and CH3CN instead of DCE | NR |
| 8 | Toluene instead of DCE | 28 |
| 9 | With [Cp*Co(CH3CN)3](SbF6)2 instead of the catalyst and AgSbF6 | 47 |

After successfully establishing the allylation of the C–H bond with cyclopropene, the scope and limitation of the strategy were investigated. To begin with, various substituted indole derivatives were examined under the optimized conditions with cyclopropene 2a (Scheme 2). 5-Alkyl, aryl and halo substituted indole derivatives were readily allylated under the optimized conditions to furnish products 3a–3g in excellent yields. Electron-donating (methoxy) group was well tolerated under the reaction conditions to afford the allylated product 3h in 82% yield in 2 h. On the other hand, electron-withdrawing (cyano) group also showed reasonable compatibility and gave the vinylated product 3j in relatively lower yield after 4 h as a mixture of E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z isomers in 2:1 ratio. But the allyloxy group underwent deprotection under the reaction conditions and the corresponding hydroxy product 3i was isolated.
:Z isomers in 2:1 ratio. But the allyloxy group underwent deprotection under the reaction conditions and the corresponding hydroxy product 3i was isolated.
 | ||
| Scheme 2 Cp*Co(III)-catalysed multisubstituted allylation of chelation-assisted C–H bonds with cyclopropenes. ‡From the corresponding allyl ether. †Ratio of stereoisomers. | ||
Similar results were observed with indole substituted at the 4-position, and synthesis of allyated products 3k–3m was achieved in good yield. Importantly, sterically congested 7-ethylindole and 3-methyl indole derivatives underwent a smooth reaction to give the allylated products 3n and 3p in relatively low yields (66 and 52% yields, respectively), and the latter was isolated as a 2:1 mixture of allylated and vinylated products. Furthermore, a 7-azaindole derivative having an additional coordination site also furnished the allylated product 3o in 81% yield. Subsequently, the effect of substituents on the pyridine directing group was examined. The 4-methylpyridine derivative effectively directed the allylation to provide the product 3q in 64% yield. But the electron deficient 5-nitropyridine derivative showed poor directing ability and the formation of product 3r was observed in 30% yield. Interestingly, replacement of pyridine with pyrimidine as the directing group, which can be readily removed after the reaction, furnished the product 3s in excellent yield.
Successively, the scope and limitations of cyclopropenes were explored. Symmetrically 3,3-disubstituted cyclopropenes on reaction with 1a under the optimized conditions afforded the products 3t and 3u in excellent yields. Use of unsymmetrical 3,3-diaryl, 3-aryl-3-heteroaryl and 3-alkyl-3-aryl cyclopropenes also afforded the corresponding allylated products 3v–3y in excellent yields as a mixture of E/Z isomers with variable ratio. Interestingly, symmetrical cyclopropenes derived from dialkyl ketones, such as dicyclohexyl ketone, cyclododecanone and 4-phenylcyclohexanone, also showed high level of compatibility under the optimized conditions to deliver allylated products 3z, 3aa and 3ab in good yields. Replacement of symmetrical disubstituted cyclopropene with unsymmetrical 1,3-diphenylcyclopropene furnished the product 3ac in 92% yield with complete E-selectivity. Consequently, trisubstituted cyclopropenes were examined. Formation of allylated product 3ad having substituents at allylic and alkene carbons in moderate yield from 1,3-dimethyl-3-phenylcyclopropenes was observed. Similar observations were noted with 1,2,3-triarylcyclopropenes containing substituents at the alkene carbon, where allylated products 3ae–3ah were obtained in moderate yields. This observation suggests that the steric factor in cyclopropene significantly affects the outcome of the reaction.
Subsequently, studies were directed to understand the importance of the directing group. Thus, bisindole having different substituents at nitrogen, methyl and pyridyl, respectively, selectively afforded the allylation at pyridyl substituted indole (3ai), which supported the importance of the directing group. It is important to note that formation of C7-allylated product 3aj was observed in 33% yield when N-pyridylindoline was treated with 2a under the optimized conditions. This result further expands the scope of allylation to the C7–H bond of the indole derivative.
Having successfully studied the allylation of indole derivatives, we next focused on the allylation of pyrroles and arenes using pyridine and pyrazole as the directing group, respectively. The reaction of N-pyridylpyrrole 4a with 2a in the presence of [Cp*CoIII] under the optimized conditions furnished the allylated product 6a in 82% yield (Scheme 3). Similarly, other substituted allylpyrroles 6b–6d were obtained in moderate to good yields. Next, N-phenylpyrazole reacted efficiently with 3a under the optimized conditions and afforded vinylated product 7a in 67% yield as the sole product, instead of the corresponding allylated product. Similar results were obtained with other substituted N-arylpyrazole derivatives and gave vinylated products 7b–7e in good yields.
 | ||
| Scheme 3 Reaction of 2a with N-pyridylpyrrole 4 and N-arylpyrazole 5. | ||
After having shown the wide substrate scope, the synthetic application of the developed strategy was investigated. Gram scale synthesis of 2a and 1a furnished the allylated product 3a in comparable yield, suggesting that the reaction can be readily scaled up and the methodology is applicable to the large-scale preparation of allylated derivatives (Scheme 4a). Reaction of 3s with NaOEt furnished 2-allylated indole 9 in good yield, revealing that the employed directing group could be readily removed, and the method can be used for further synthetic manipulations (Scheme 4b).
 | ||
| Scheme 4 Synthetic utility. (a) Gram scale reaction. (b) Removal of directing group. (c) Synthesis of cyclopenta[b]indoles. (d) One-pot synthesis of cyclopenta[b]indole. | ||
Cyclopenta[b]indoles are important structural motifs widely found in bioactive natural products and pharmaceutically interesting compounds.21 We next envisioned the conversion of allylated products 3 to the corresponding cyclopenta[b]indoles 10. Thus, reaction of 3a with 30 mol% of BF3·OEt2 in DCE afforded the product 10a in 81% yield (Scheme 4c). Subsequently, various substituted cyclopenta[b]indoles 10b–j were achieved in moderate to good yields in the presence of BF3·OEt2. Importantly, Cp*CoIII-catalyzed allylation and Lewis acid mediated cyclization were successfully integrated to afford cyclopenta[b]indole 3a in 68% yield in one-pot (Scheme 4d).
We also performed preliminary investigation to understand the possible mechanism of the developed transformation. The initial competitive experiment with electronically different indoles revealed preference to electron-rich indole derivatives (Scheme 5a). This suggests the possible electrophilic metalation pathway for C–H bond functionalization with Cp*CoIII. On the other hand, electronically different cyclopropenes showed only very slight preference to electron-rich cyclopropenes, which might be due to the high reactivity of cyclopropenes. Next, a deuterium exchange experiment was performed to support the formation of the Co–C bond through C–H bond functionalization. Treatment of 1a with CD3OD under the optimized conditions, in the absence of cyclopropene, led to the recovery of 1a with a significant amount of deuteration at C2- and C7-position suggesting the potential reversible formation of the Co–C bond (Scheme 5b).
 | ||
| Scheme 5 Preliminary mechanistic investigation. (a) Competitive experiment. (b) Deuterium exchange experiment. (c) Stoichiometric reaction. | ||
Next, we focused our attention on isolating the possible transition state of C–H metalated species. Thus, equimolar ratios of 1a and Cp*CoI2CO were treated in the presence of 2 equiv. of AgSbF6 and 1 equiv. of NaOAc in DCE at 100 °C for 6 h. We noticed a clear change in the color of the reaction mixture and the formation of a solid. Isolation and characterization of the formed solid through 1H NMR showed that all five methyl groups of Cp* are not equal and one of them appeared as a doublet. Consequently, single crystal X-ray analysis of the isolated solid confirmed the formation of pyridinium salt 11 in 59% yield as a single diastereomer (Scheme 5c).22 The formation of 11 can be explained through the initial insertion, reductive elimination followed by protonation of the resultant allyl Co-species.
To isolate the potential intermediate, the stoichiometric reaction was performed at a reduced temperature (65 °C). Various attempts to isolate the complex formed was unsuccessful. Hence, the formed intermediate was analyzed by ESI-HRMS, which showed the presence of only complex 12. The mass spectrum was in complete agreement with the simulated spectra of complex 12 (see the ESI† for more details). Successively, treatment of the reaction mixture with cyclopropene 2a furnished the allylated product 3a in good yield. Interestingly, heating of complex 12 to 100 °C afforded the pyridinium salt 11 in 42% yield. These observations suggest complex 12 as a potential intermediate of the present allylation and formation of pyridinium salt 11.
Based on the preliminary mechanistic investigation, the plausible mechanism for the multisubstituted allylation is proposed (Scheme 6). The catalytic reaction starts with the formation of A from the pre-catalyst Cp*CoI2CO and AgSbF6/NaOAc through exchange of ligands. Reaction of active species A with indole derivative 1 would furnish the cyclometallated species Bvia base-assisted electrophilic C–H bond functionalization. Formation of vinylcarbene D could be explained through the initial coordination of cyclopropene 2 to B followed by rearrangement. Subsequently, 1,1-migratory insertion would give intermediate E, which on protodemetallation would give the product 3 and regenerate the active catalyst A. On the other hand, based on the substrate, intermediate E could exist as allyl-Co species E′, which on protonation would give the vinylated product. As seen in the substrate scope, a C3-allylated product could be formed via the intermolecular reaction of vinylcarbene with cyclometallated species B, which makes the C3-position more nucleophilic.
Conclusions
In conclusion, we have developed an efficient and general multisubstituted allylation of chelation-assisted C–H bond of (hetero)arenes in the presence of Cp*CoIII catalyst and substituted cyclopropenes as an efficient allylating reagent. The allylation occurs through ring-opening isomerization of cyclopropenes to vinylcarbenes followed by migratory insertion, which allows introduction of substituents in all three carbons of the allyl moiety. Various functional groups were well tolerated to afford diversely substituted allyated indoles, pyrroles and arenes in good yields and selectivity. Importantly, gram scale synthesis, removal of the directing group and synthetic manifestation to cyclopenta[b]indoles demonstrated the synthetic applicability of the present methodology. Furthermore, a potential metalated intermediate was identified through a stoichiometric reaction, which paved way to the plausible mechanism. | ||
| Scheme 6 Plausible mechanism. | ||
Data availability
All the data have been included in the ESI.†Author contributions
K. R. performed all experiments. Both authors contributed to the conception of the experiments, discussion of the results and preparation of manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank DST-SERB, New Delhi, India (Project No.: SB/SJF/2020-21/15) for financial support through SwarnaJayanti Fellowship. K. R. thanks University Grants Commission (UGC), New Delhi for fellowship.Notes and references
- (a) H. Y. Sohn, K. H. Son, C. S. Kwon, G. S. Kwon and S. S. Kang, Phytomedicine, 2004, 11, 666–672 CrossRef CAS PubMed; (b) J. Du, Z.-D. He, R.-W. Jiang, W.-C. Ye, H.-X. Xu and P. P.-H. But, Phytochemistry, 2003, 62, 1235–1238 CrossRef CAS PubMed; (c) G. Ni, Q.-J. Zhang, Z.-F. Zheng, R.-Y. Chen and D.-Q. Yu, J. Nat. Prod., 2009, 72, 966–968 CrossRef CAS PubMed.
- (a) R. M. Roberts and A. A. Khalaf, Friedel–Crafts Alkylation Chemistry: A Century of Discovery, M. Dekker, 1984 Search PubMed; (b) M. Niggemann and M. J. Meel, Angew. Chem., Int. Ed., 2010, 49, 3684–3687 CrossRef CAS PubMed.
- (a) Y. Kiyotsuka, H. P. Acharya, Y. Katayama, T. Hyodo and Y. Kobayashi, Org. Lett., 2008, 10, 1719–1722 CrossRef CAS PubMed; (b) C. Spino, M.-C. Tremblay and C. Gobdout, Org. Lett., 2004, 6, 2801–2804 CrossRef CAS PubMed; (c) N. Harrington-Frost, H. Leuser, M. I. Calaza, F. F. Kneisel and P. Knochel, Org. Lett., 2003, 5, 2111–2114 CrossRef CAS PubMed.
- (a) N. K. Mishra, S. Sharma, J. Park, S. Han and I. S. Kim, ACS Catal., 2017, 2821–2847 CrossRef CAS; (b) S. Dutta, T. Bhattacharya, D. B. Werz and D. Maiti, Chem, 2021, 7, 555–605 CrossRef CAS.
- (a) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278–15284 CrossRef CAS PubMed; (b) S. Bae, H.-L. Jang, H. Jung and J. M. Joo, J. Org. Chem., 2015, 80, 690–697 CrossRef CAS PubMed; (c) S. Fan, F. Chen and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 5918–5923 CrossRef CAS PubMed; (d) T. K. Achar, X. Zhang, R. Mondal, M. S. Shanavas, S. Maiti, S. Maity, N. Pal, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10353–10360 CrossRef CAS PubMed.
- (a) S. Oi, Y. Tanaka and Y. Inoue, Organometallics, 2006, 25, 4773–4778 CrossRef CAS; (b) M. Kim, S. Sharma, N. K. Mishra, S. Han, J. Park, M. Kim, Y. Shin, J. H. Kwak, S. H. Han and I. S. Kim, Chem. Commun., 2014, 50, 11303–11306 RSC; (c) R. Manikandan, P. Madasamy and M. Jeganmohan, Chem.–Eur. J., 2015, 21, 13934–13938 CrossRef CAS PubMed; (d) G. S. Kumar and M. Kapur, Org. Lett., 2016, 18, 1112–1115 CrossRef CAS PubMed; (e) X. Wu and H. Ji, Org. Lett., 2018, 20, 2224–2227 CrossRef CAS PubMed; (f) X.-Q. Hu, Z. Hu, A. S. Trita, G. Zhang and L. J. Gooßen, Chem. Sci., 2018, 9, 5289–5294 RSC.
- (a) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540–542 CrossRef CAS PubMed; (b) R. Zeng, C. Fu and S. Ma, J. Am. Chem. Soc., 2012, 134, 9597–9600 CrossRef CAS PubMed; (c) B. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed; (d) H. Wang, N. Schröder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386–5389 CrossRef CAS PubMed; (e) Z.-T. Jiang, J. Huang, Y. Zeng, F. Hu and Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 10626–10631 CrossRef CAS PubMed; (f) J. Xia, L. Kong, X. Zhou, G. Zheng and X. Li, Org. Lett., 2017, 19, 5972–5975 CrossRef CAS PubMed; (g) S.-S. Zhang, J.-Q. Wu, Y.-X. Lao, X.-G. Liu, Y. Liu, W.-X. Lv, D.-H. Tan, Y.-F. Zeng and H. Wang, Org. Lett., 2014, 16, 6412–6415 CrossRef CAS PubMed; (h) S. E. Korkis, D. J. Burns and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 12252–12257 CrossRef CAS PubMed; (i) Z. Qi, L. Kong and X. Li, Org. Lett., 2016, 18, 4392–4395 CrossRef CAS PubMed; (j) G. Zhu, W. Shi, H. Gao, Z. Zhou, H. Song and W. Yi, Org. Lett., 2019, 21, 4143–4147 CrossRef CAS PubMed.
- (a) Y. J. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248–4250 CrossRef CAS PubMed; (b) R. Yabe, Y. Ebe and T. Nishimura, Synthesis, 2021 DOI:10.1055/a-1477-7059.
- (a) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2011, 50, 2990–2994 CrossRef CAS PubMed; (b) N. Barsu, D. Kalsi and B. Sundararaju, Chem.–Eur. J., 2015, 21, 9364–9368 CrossRef CAS PubMed; (c) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476–3480 CrossRef CAS PubMed; (d) S. Ali, J. Huo and C. Wang, Org. Lett., 2019, 21, 6961–6965 CrossRef CAS PubMed.
- (a) D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722–17725 CrossRef CAS PubMed; (b) T. Gensch, S. Vásquez-Céspedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714–3717 CrossRef CAS PubMed; (c) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408–7412 CrossRef CAS PubMed; (d) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944–9947 CrossRef CAS PubMed; (e) Y. Bunno, N. Murakami, Y. Suzuki, M. Kanai, T. Yoshino and S. Matsunaga, Org. Lett., 2016, 18, 2216–2219 CrossRef CAS PubMed; (f) D. Kalsi, R. A. Laskar, N. Barsu, J. R. Premkumar and B. Sundararaju, Org. Lett., 2016, 18, 4198–4201 CrossRef CAS PubMed; (g) K. Ramachandran and P. Anbarasan, Eur. J. Org. Chem., 2017, 2017, 3965–3968 CrossRef CAS; (h) H. Wang, M. M. Lorion and L. Ackermann, ACS Catal., 2017, 7, 3430–3433 CrossRef CAS; (i) L. Kong, S. Yu, G. Tang, H. Wang, X. Zhou and X. Li, Org. Lett., 2016, 18, 3802–3805 CrossRef CAS PubMed; (j) D. Zell, V. Müller, U. Dhawa, M. Bursch, R. R. Presa, S. Grimme and L. Ackermann, Chem.–Eur. J., 2017, 23, 12145–12148 CrossRef CAS PubMed; (k) S. Y. Choi, H. D. Kim, J.-U. Park, S.-a. Park and J. H. Kim, Org. Lett., 2019, 21, 10038–10042 CrossRef CAS PubMed; (l) R. Tanaka, I. Tanimoto, M. Kojima, T. Yoshino and S. Matsunaga, J. Org. Chem., 2019, 84, 13203–13210 CrossRef CAS PubMed; (m) U. Dhawa, C. Tian, W. Li and L. Ackermann, ACS Catal., 2020, 10, 6457–6462 CrossRef CAS.
- (a) T. Yoshino and S. Matsunaga, Adv. Synth. Catal., 2017, 359, 1245–1262 CrossRef CAS; (b) Z.-H. Guan, M. Usman, Z.-H. Ren and Y.-Y. Wang, Synthesis, 2017, 49, 1419–1443 CrossRef; (c) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2015, 498–525 Search PubMed; (d) J. Ghorai and P. Anbarasan, Asian J. Org. Chem., 2019, 8, 430–455 CrossRef CAS; (e) S. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2017, 53, 3165–3180 RSC.
- (a) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; (b) Z.-B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534–5563 RSC; (c) M. R. Wilson and R. E. Taylor, Angew. Chem., Int. Ed., 2013, 52, 4078–4087 CrossRef CAS PubMed; (d) L. Dian and I. Marek, Chem. Rev., 2018, 118, 8415–8434 CrossRef CAS PubMed; (e) A. Archambeau, F. Miege, C. Meyer and J. Cossy, Acc. Chem. Res., 2015, 48, 1021–1031 CrossRef CAS PubMed.
- (a) T. Nakano, K. Endo and Y. Ukaji, Chem.–Asian J., 2016, 11, 713–721 CrossRef CAS PubMed; (b) D. T. H. Phan and V. M. Dong, Tetrahedron, 2013, 69, 5726–5731 CrossRef CAS; (c) S. Ma and J. Zhang, J. Am. Chem. Soc., 2003, 125, 12386–12387 CrossRef CAS PubMed; (d) H. Zhang, B. Wang, K. Wang, G. Xie, C. Li, Y. Zhang and J. Wang, Chem. Commun., 2014, 50, 8050–8052 RSC; (e) F. Miege, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2011, 50, 5932–5937 CrossRef CAS PubMed; (f) A. Archambeau, F. Miege, C. Meyer and J. Cossy, Angew. Chem., Int. Ed., 2012, 51, 11540–11544 CrossRef CAS PubMed; (g) D. Zhang, Z. Kang, J. Liu and W. Hu, iScience, 2019, 14, 292–300 CrossRef CAS PubMed; (h) P. C. Young, M. S. Hadfield, L. Arrowsmith, K. M. Macleod, R. J. Mudd, J. A. Jordan-Hore and A.-L. Lee, Org. Lett., 2012, 14, 898–901 CrossRef CAS PubMed; (i) L. H. Phun, J. Aponte-Guzman and S. France, Angew. Chem., Int. Ed., 2012, 51, 3198–3202 CrossRef CAS PubMed.
- T. A. Shah, P. B. De, S. Pradhan, S. Banerjee and T. Punniyamurthy, Chem.–Asian J., 2019, 14, 4520–4533 CrossRef CAS PubMed.
- (a) N. Semakul, K. E. Jackson, R. S. Paton and T. Rovis, Chem. Sci., 2017, 8, 1015–1020 RSC; (b) Y. Luo, H.-L. Teng, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2017, 56, 9207–9210 CrossRef CAS PubMed; (c) T. K. Hyster and T. Rovis, Synlett, 2013, 24, 1842–1844 CrossRef CAS PubMed.
- (a) H. Zhang, K. Wang, B. Wang, H. Yi, F. Hu, C. Li, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13234–13238 CrossRef CAS PubMed; (b) X. Wang, A. Lerchen, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2018, 57, 1712–1716 CrossRef CAS PubMed.
- (a) H. Zhang, B. Wang, H. Yi, Y. Zhang and J. Wang, Org. Lett., 2015, 17, 3322–3325 CrossRef CAS PubMed; (b) M. J. González, J. González, L. A. López and R. Vicente, Angew. Chem., Int. Ed., 2015, 54, 12139–12143 CrossRef PubMed; (c) R. J. Ross, R. Jeyaseelan and M. Lautens, Org. Lett., 2020, 22, 4838–4843 CrossRef CAS PubMed.
- (a) D. Yadagiri and P. Anbarasan, Chem. Sci., 2015, 6, 5847–5852 RSC; (b) D. Yadagiri, A. C. S. Reddy and P. Anbarasan, Chem. Sci., 2016, 7, 5934–5938 RSC; (c) A. C. S. Reddy, V. S. K. Choutipalli, J. Ghorai, V. Subramanian and P. Anbarasan, ACS Catal., 2017, 7, 6283–6288 CrossRef CAS; (d) A. C. S. Reddy and P. Anbarasan, J. Catal., 2018, 363, 102–108 CrossRef CAS; (e) D. Yadagiri, M. Chaitanya, A. C. S. Reddy and P. Anbarasan, Org. Lett., 2018, 20, 3762–3765 CrossRef CAS PubMed; (f) S. Rajasekar and P. Anbarasan, Org. Lett., 2019, 21, 3067–3071 CrossRef CAS PubMed; (g) A. C. S. Reddy, P. M. Reddy and P. Anbarasan, Adv. Synth. Catal., 2020, 362, 801–806 CrossRef CAS; (h) P. M. Reddy, K. Ramachandran and P. Anbarasan, J. Catal., 2021, 396, 291–296 CrossRef CAS.
- (a) D. Yadagiri and P. Anbarasan, Org. Lett., 2014, 16, 2510–2513 CrossRef CAS PubMed; (b) M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960–4963 CrossRef CAS PubMed; (c) S. Rajasekar, D. Yadagiri and P. Anbarasan, Chem.–Eur. J., 2015, 21, 17079–17084 CrossRef CAS PubMed; (d) J. Ghorai, A. C. S. Reddy and P. Anbarasan, Chem.–Eur. J., 2016, 22, 16042–16046 CrossRef CAS PubMed; (e) J. Ghorai, A. C. S. Reddy and P. Anbarasan, Chem.–Asian J., 2018, 13, 2499–2504 CrossRef CAS PubMed.
- J. Foerstner, A. Kakoschke, D. Stellfeldt, H. Butenschön and R. Wartchow, Organometallics, 1998, 17, 893–896 CrossRef CAS.
- (a) K. Stratmann, R. E. Moore, R. Bonjouklian, J. B. Deeter, G. M. L. Patterson, S. Shaffer, C. D. Smith and T. A. Smitka, J. Am. Chem. Soc., 1994, 116, 9935–9942 CrossRef CAS; (b) J. Nakazawa, J. Yajima, T. Usui, M. Ueki, A. Takatsuki, M. Imoto, Y. Y. Toyoshima and H. Osada, Chem. Biol., 2003, 10, 131–137 CrossRef CAS PubMed; (c) H. Chen, J. Bai, Z.-F. Fang, S.-S. Yu, S.-G. Ma, S. Xu, Y. Li, J. Qu, J.-H. Ren, L. Li, Y.-K. Si and X.-G. Chen, J. Nat. Prod., 2011, 74, 2438–2445 CrossRef CAS PubMed.
- CCDC2091505†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC2091505. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03476f |
| This journal is © The Royal Society of Chemistry 2021 |