
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterocyclic group transfer reactions with I(III) N-HVI reagents: access to N-alkyl(heteroaryl)onium salts via olefin aminolactonization†
Anthony F.
Tierno‡
a,
Jennifer C.
Walters‡
b,
Andres
Vazquez-Lopez‡
b,
Xiao
Xiao
c and
Sarah E.
Wengryniuk
 *b
*b
aDepartment of Chemistry, Towson University, 8000 York Road, Towson, Maryland, USA 21252
bDepartment of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania, USA 19122. E-mail: sarahw@temple.edu
cThe Education Ministry Key Lab of Resource Chemistry, Shanghai Key Laboratory of Rare Earth Functional Materials, Shanghai Normal University, Shanghai 200234, China
First published on 12th April 2021
Abstract
Pyridinium and related N-alkyl(heteroaryl)onium salts are versatile synthetic intermediates in organic chemistry, with applications ranging from ring functionalizations to provide diverse piperidine scaffolds to their recent emergence as radical precursors in deaminative cross couplings. Despite their ever-expanding applications, methods for their synthesis have seen little innovation, continuing to rely on a limited set of decades old transformations and a limited subset of coupling partners. Herein, we leverage (bis)cationic nitrogen-ligated I(III) hypervalent iodine reagents, or N-HVIs, as “heterocyclic group transfer reagents” to provide access to a broad scope of N-alkyl(heteroaryl)onium salts via the aminolactonization of alkenoic acids, the first example of engaging an olefin to directly generate these salts. The reactions proceed in excellent yields, under mild conditions, and are capable of incorporating a broad scope of sterically and electronically diverse aromatic heterocycles. The N-HVI reagents can be generated in situ, the products isolated via simple trituration, and subsequent derivatizations demonstrate the power of this platform for diversity-oriented synthesis of 6-membered nitrogen heterocycles.
Introduction
N-Alkyl-pyridinium and related (heteroaryl)onium salts (1) are versatile functional handles that have applications across Nature,1 materials science,2,3 and medicinal and synthetic chemistry4 (Fig. 1). In organic chemistry, they serve as ionic liquids5 (2) and phase transfer catalysts,6 exhibit a diverse range of biological activities (3, 4), and have a long history as synthetic intermediates, an area that has seen a recent surge of new advancements. Representative of their versatile reactivity, pyridinium and related salts can undergo full or partial reductions,7–9 cycloadditions,10,11 photochemical isomerizations,12 cross couplings,13 addition of one- or two-electron heteroatom or carbon nucleophiles,8,14 and facile C–H metalations,15 and many of these include asymmetric variants16–18 (Fig. 1). The breadth of available transformations makes pyridinium salts valuable templates for accessing functionalized 6-membered aza-heterocyclic scaffolds, which are prevalent in agrochemicals, alkaloid natural products, and are the most commonly encountered heterocyclic motif in FDA approved small molecule drugs.19 In addition to manipulations of the heterocyclic ring, pyridinium salts can undergo ring openings to produce Zincke aldehydes, which have shown utility as synthetic building blocks,20 and 2,4,6-triphenylpyridinium salts have emerged as a new and powerful class of radical precursors for deaminative metal-catalyzed cross couplings (Fig. 1).21–23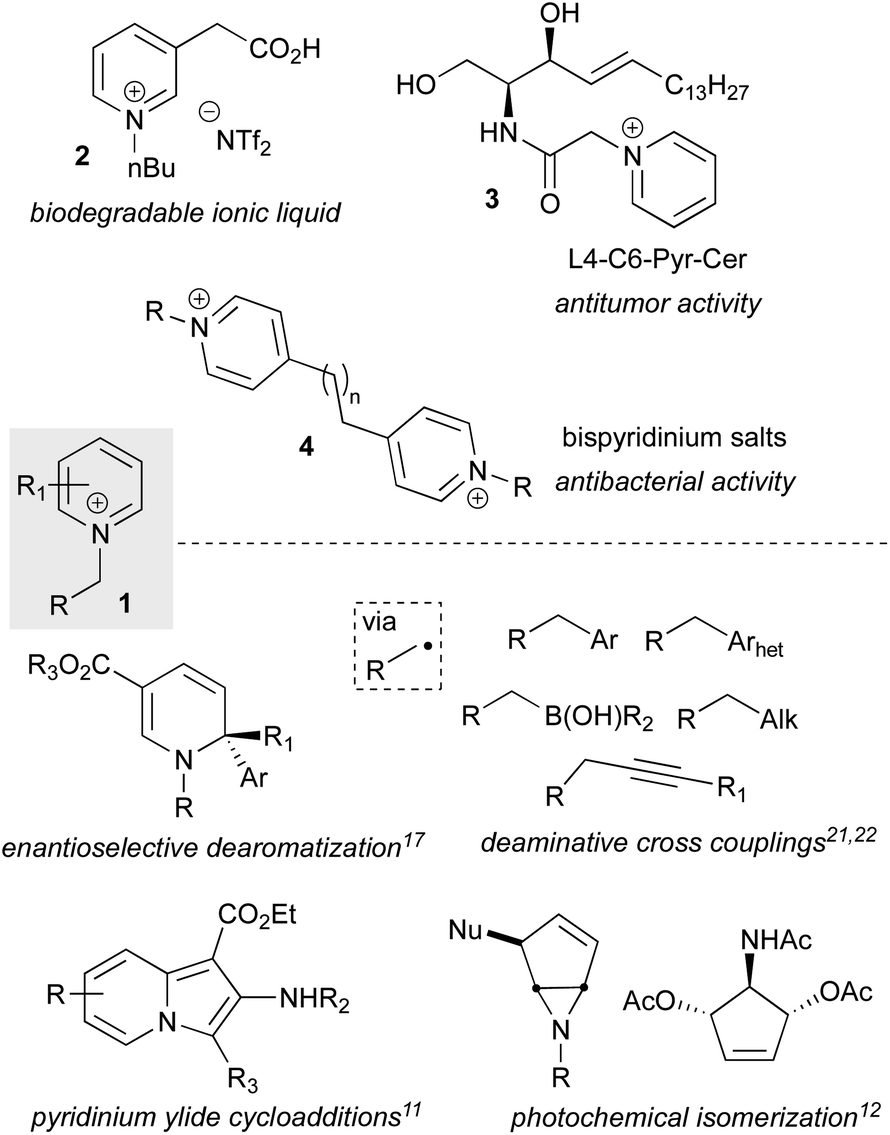 | ||
| Fig. 1 N-Alkyl pyridinium salts as the active components in materials and bioactive molecules as well as versatile synthetic intermediates. | ||
At present, N-alkyl pyridinium salts are commonly accessed either by reaction of a primary amine with an oxopyrylium or Zincke salt, or via nucleophilic substitutions of primary or activated electrophiles (Scheme 1a). While both of these strategies have been widely applied, this provides just two functional handles from which to devise a synthetic route to a pyridinium salt, and the limited scope of available oxopyrylium scaffolds renders this commonly employed approach intractable when the goal is structural diversity at the heterocycle.4,15 Recently, our laboratory and others have been exploring the synthetic applications of (bis)cationic nitrogen-ligated hypervalent iodine(III) reagents, or N-HVIs, possessing two datively bound heterocyclic ligands (8, Scheme 1b).24–32 Considering the versatile group transfer reactivity of I(III) reagents33,34 we wondered if N-HVIs could serve as “heterocyclic group transfer” (HGT) reagents to access diverse N-alkyl(heteroaryl)onium salts through incorporation of the heterocyclic ligand into a substrate of interest.35 Given the appealing features of I(III) reagents, and the modular synthesis of N-HVIs, we envisioned that this could serve as a convenient, general platform for the synthesis of diverse (heteroaryl)onium salts from alkenes, providing a novel means of accessing these valuable functional handles.
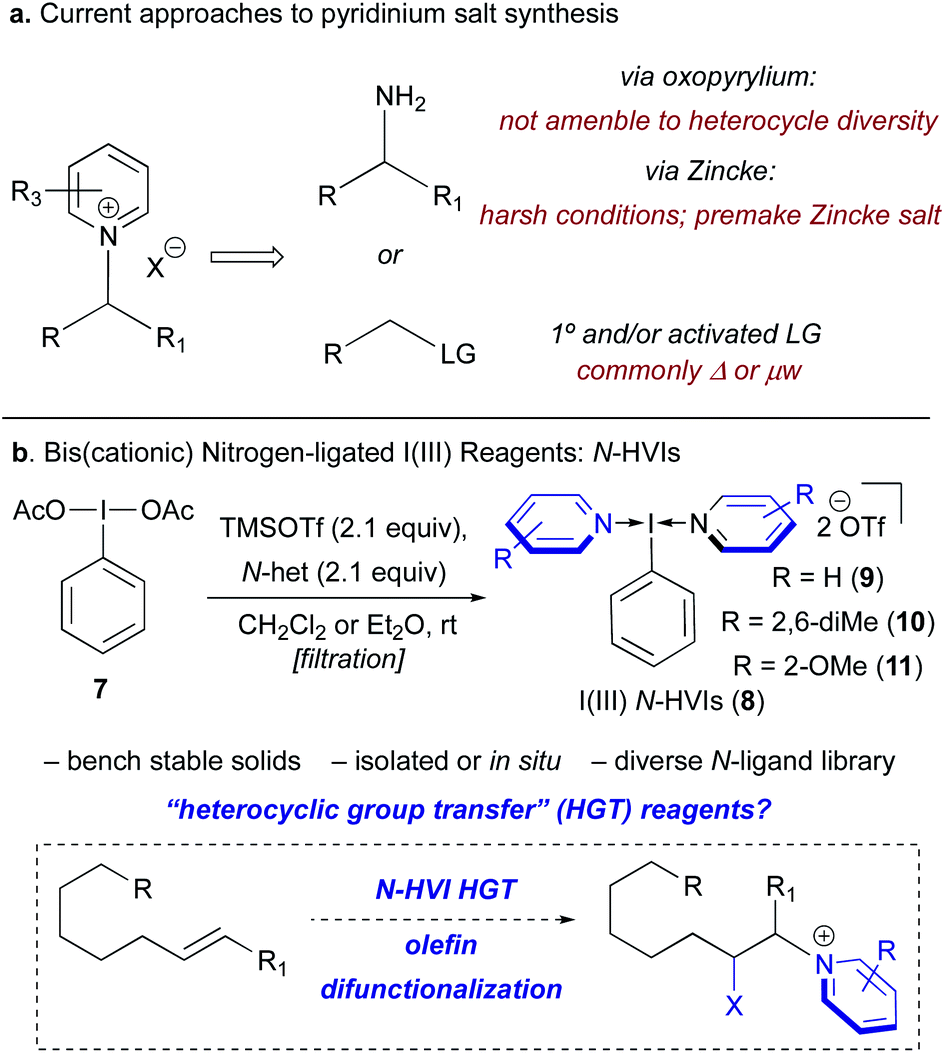 | ||
| Scheme 1 (a) Current approaches to pyridinium salt synthesis from either amines or activated electrophiles. (b) Novel approach via N-HVI HGT of alkenes to access diverse pyridinium salts. | ||
Herein, we report the first example of the heterocyclic group transfer (HGT) reactivity of I(III) N-HVIs, demonstrated in the aminolactonization of alkenoic acids to give N-alkyl(heteroaryl)onium lactones (Scheme 2),34,36 the products of which represent core scaffolds in a wide array of bioactive natural products.36 This represents the first general method for the direct conversion of alkenes to (heteroaryl)onium salts.37,38 Furthermore, this is rare example of an I(III)-mediated olefin oxyamination with an exogenous amine nucleophile and therefore represents a significant advancement in I(III)-mediated olefin functionalizations.39–42 The reactions proceed in excellent yields, under mild conditions, and are capable of incorporating a broad scope of sterically and electronically diverse aromatic heterocycles. The N-HVI reagents can be generated in situ, the products isolated via simple trituration, and subsequent derivatizations demonstrate the power of this platform for diversity-oriented synthesis of 6-membered nitrogen heterocycles. Mechanistic studies indicate the reaction proceeds via initial olefin activation followed by lactonization and subsequent intermolecular nucleophilic displacement of an (alkyl)(aryl)iodonium salt hypernucleofuge.
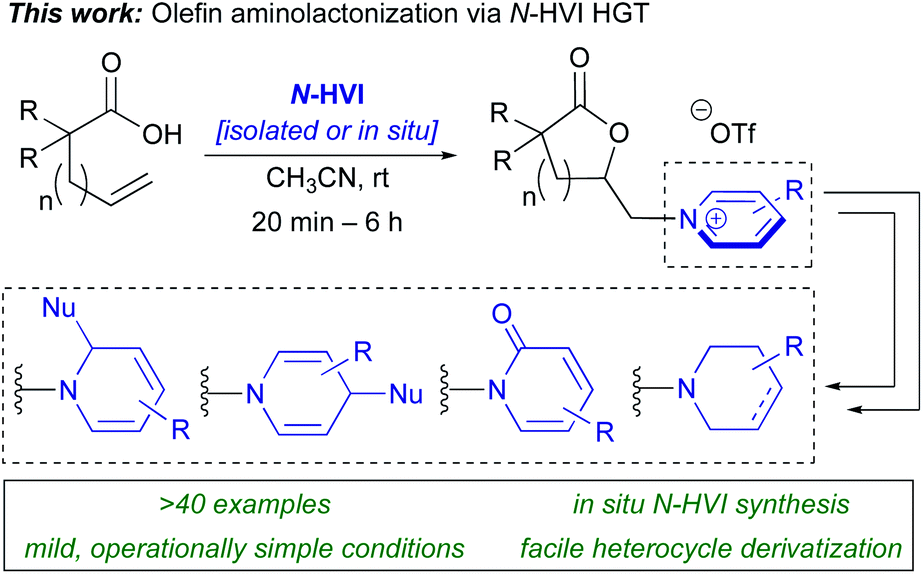 | ||
| Scheme 2 This report: N-HVI mediated HGT enabled mild, general synthesis of lactone pyridinium salts via aminolactonization. | ||
Results and discussion
To begin our studies, 2,2-diphenyl-pentenoic acid (12) was used as a model substrate along with pyridine-ligated N-HVI (Py-HVI, 9) and complete conversion to desired pyridinium lactone 13 was observed after only 20 minutes in CH3CN at room temperature (Scheme 3a). The product (13) could then be isolated via trituration with Et2O, yielding pure 13 in 96% yield. Control reactions indicated that the N-HVI was required for high yields of 13, as typical halo-lactonization or oxy-lactonization conditions using NIS, NBS, or PhI(OAc)2 in the presence of pyridine gave no conversion to the desired pyridinium lactone (see ESI† for full details). Attention then turned to maximizing the efficiency and operational simplicity of the transformation by developing an in situ protocol for the generation of N-HVIs, thereby eliminating the additional step of reagent isolation. It was found that a one-pot protocol involving sequential addition of PhI(OAc)2, TMSOTf, and pyridine in CH3CN, leading to formation of 9, followed by addition of 12, gave 13 in nearly equivalent yield of 94% (Scheme 3b). Furthermore, the reaction could be run “precaution-free”, where stringent drying of glassware and use of inert atmosphere was omitted, with minimal effect on the reaction yield (Scheme 3b).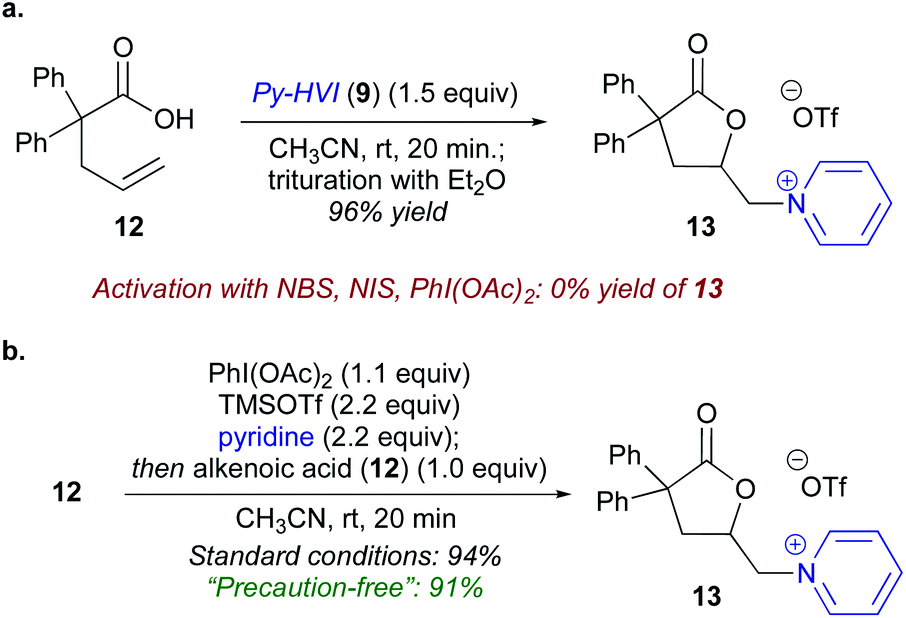 | ||
| Scheme 3 Initial reaction development and in situ N-HVI protocol. (a) Conversion of 2,2-diphenyl-pentenoic acid (12) to pyridinium lactone 13 with N-HVI. (b) Operationally simplified variants including insitu generation of N-HVIs and “precaution-free” protocol which does not rigorously exclude air or moisture. | ||
With efficient procedures in hand, the scope of the heterocycle was examined (Table 1). Substitution at the 2-position was well tolerated, with 2-Me-pyridine, 2-OMe-pyridine, and 2-OEt-pyridine all incorporated in excellent yield to give (14–16); somewhat unsurprisingly, use of sterically encumbered 2,6-lutidine led to only 21% yield of lactone 17. Turning to the 4-position of the pyridine, both electron-donating (18–23) and electron-withdrawing groups (25–28) were well tolerated and gave the corresponding pyridinium lactones in high yields, with the exception of 24, possessing a free carboxylic acid, wherein we postulate the low yield could be due to the formation of zwitterionic protonated pyridinium salts. Both pyridine (13) and 4-CN pyridine lactone 28 were also obtained under precaution free conditions; while the former saw almost no drop in yield (91%), the latter saw a decrease to 53% from 91%, reflective of the high moisture sensitivity of highly electron-deficient N-HVIs, and thus incorporation of electron-poor heterocycles would benefit from rigorously dried conditions. Pyridines possessing substitution at the 3- and 5-position were of particular interest as access to these substitution patterns on the corresponding piperidines can be challenging due to a lack of inherent activation or directing ability and in turn have limited commercial availability. 3-Acyl, 3-bromo, and 3-fluoro-pyridine, as well as 3,5-disubstituted derivatives were all found to give the pyridinium salts (29–31) in high yields. An oxidatively sensitive boronic ester was compatible with the mild conditions, yielding the versatile 3-Bpin (34) or 4-Bpin (35) pyridinium salts. Finally, we examined other aromatic azaheterocycles; benzofused derivatives including quinolines and isoquinoline could be efficiently incorporated (36–39), as well as both pi-deficient and pi-excessive diazines, including pyrazines to give 40 and 41, and N–Me-imidazole to give lactone 42.
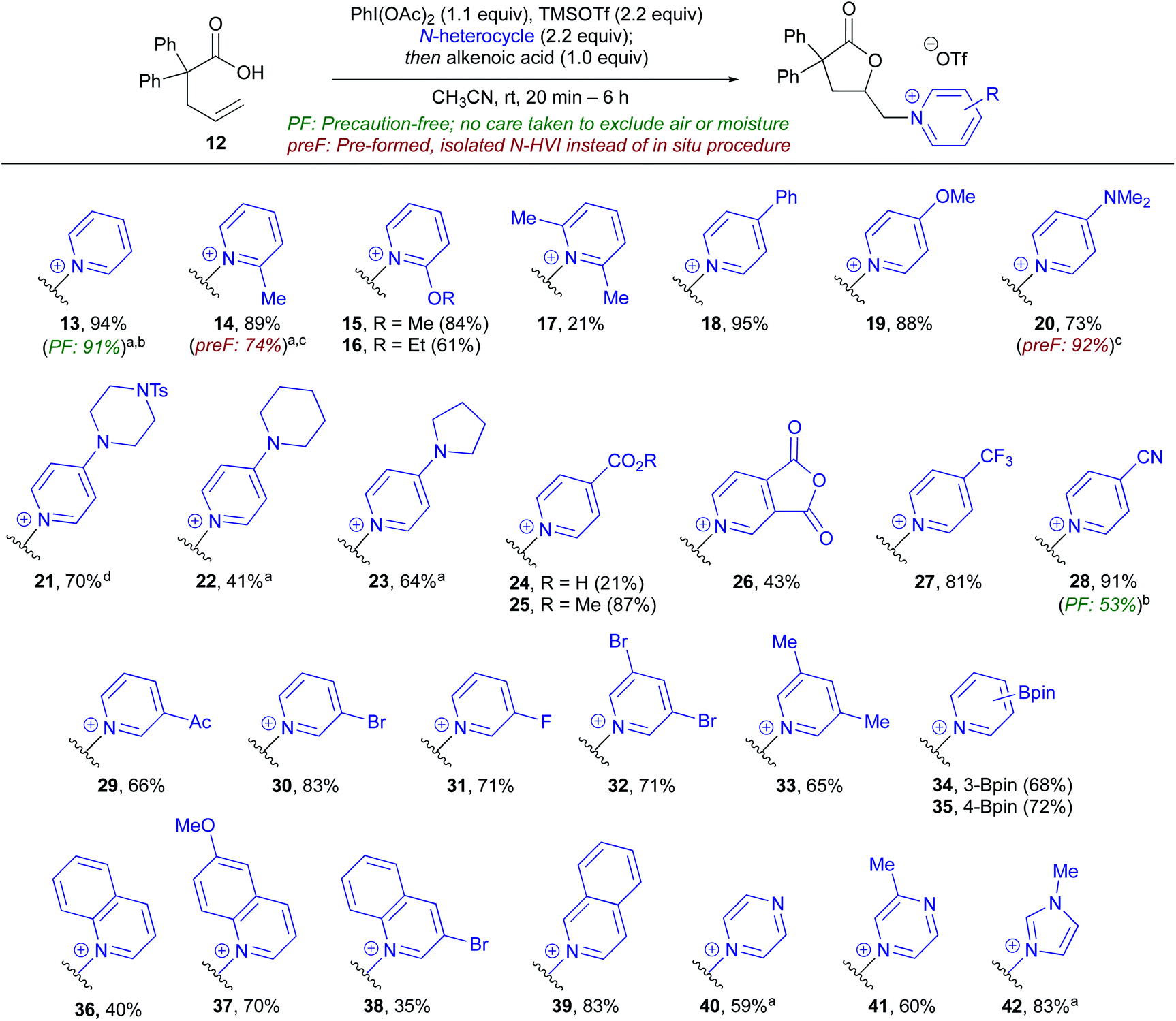
During the course of the N-heterocycle scope studies, it was found that amino acid derived pyridines 43 and 44 were unsuccessful under the standard conditions, giving no conversion to desired products 45 and 46 (Scheme 4). This was hypothesized to be due to the amino acid functionality being incompatible with formation of the N-HVI. To circumvent this issue, we envisioned an activation strategy wherein an N-HVI possessing relatively non-nucleophilic “dummy ligands” would be used for olefin activation followed by incorporation of the heterocycle of interest (Scheme 4). Not only would such a “dummy ligand” protocol allow for the incorporation of heterocycles possessing sensitive functionality, but it would also only require one equivalent of the heterocycle of interest, which is advantageous when considering use of either expensive heterocycles or those that require multi-step sequences to produce. To this end, we looked to 2,6-lut-HVI (10) as the activator due to the low nucleophilicity of 2,6-lutidine which had already translated to a low yield in pyridinium salt formation (17, Table 1). Gratifyingly, it was found that treatment of alkenoic acid 12 with 2,6-lut-HVI 10 in the presence of just one equivalent of either Boc-3-(3-pyridyl)-L-alanine methyl ester (43) or Fmoc-3-(4-pyridyl)-L-alanine methyl ester (44) under otherwise standard conditions now produced the desired pyridinium lactones (45, 46) in 71% and 41% yield respectively.
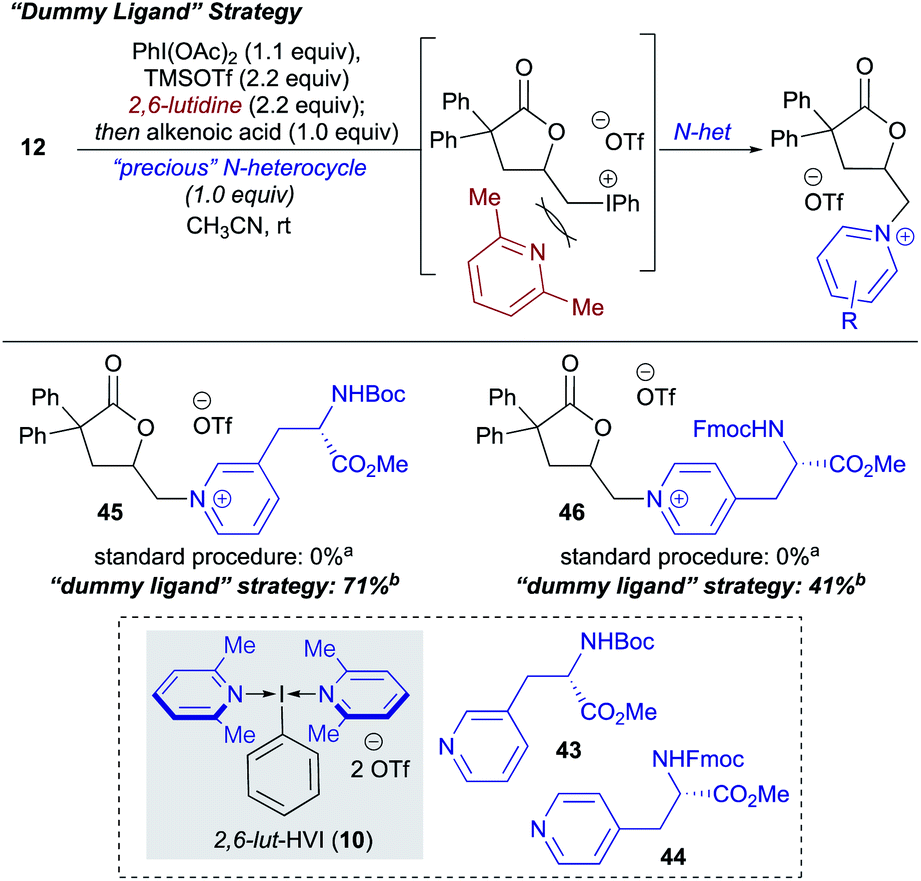 | ||
| Scheme 4 2,6-Lutidine “Dummy Ligand” strategy for the incorporation of “precious” or incompatible heterocycles. a Standard procedure as shown in Table 1 for in situ generated N-HVI HGT. bdr not determined due to lack of suitable resolution of diastereomers. | ||
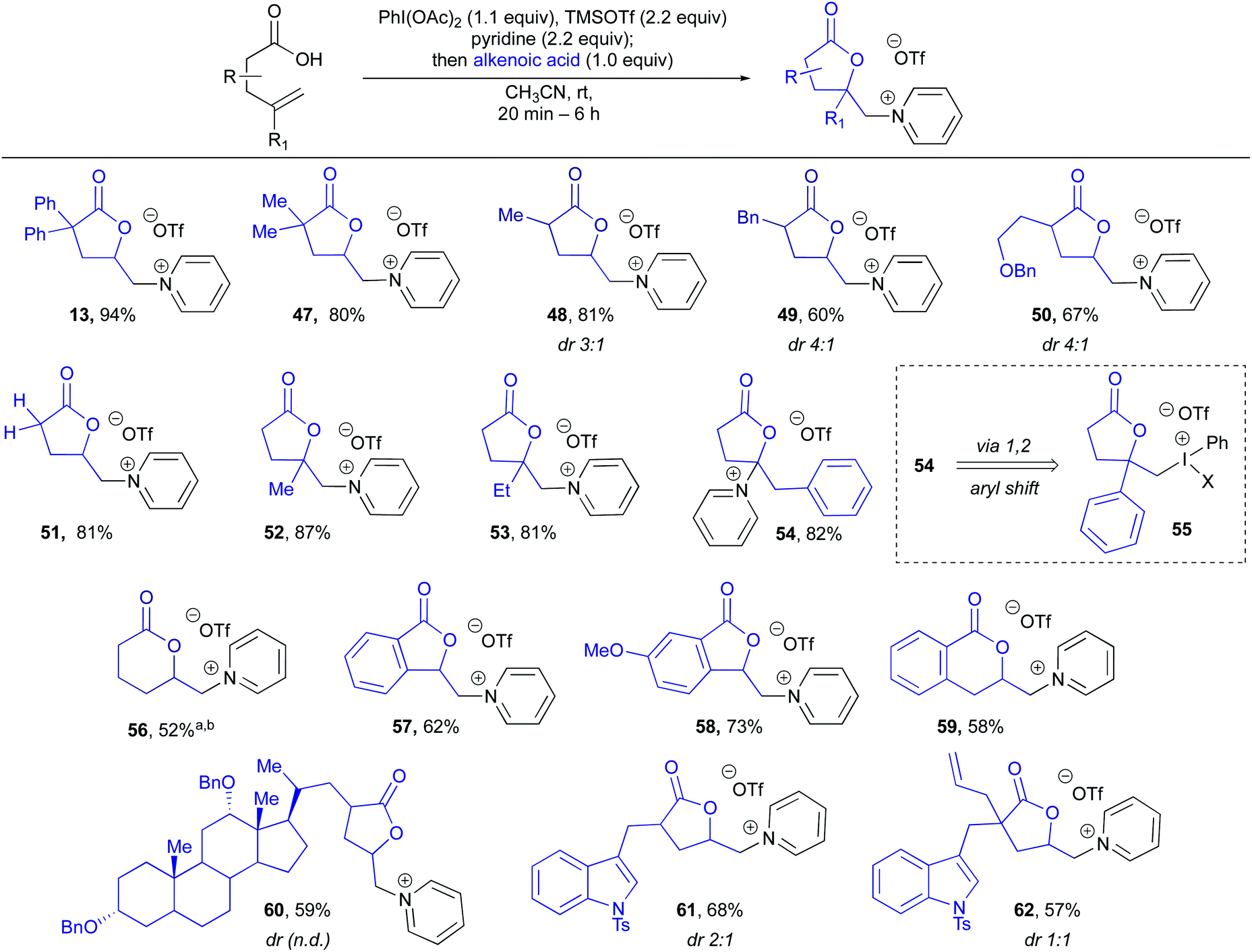
Having established a broad scope with respect to the nitrogen heterocycle, we then turned to diversity at the alkenoic acid (Table 2).43 Beginning with substitution at the α-position, 2,2-dimethyl, 2-Me, 2-Bn, 2-(CH2)2-OBn and α-methylene lactones (47–51) could all be produced in high yields, indicating the Thorpe Ingold effect is not required for efficient cyclization. Substitution on the alkene was then examined, and it was found that 2,2-disubstituted alkenes performed very well, giving 52 and 53 in high yields. Interestingly, a 2-phenyl substituted alkene gave rearranged pyridinium lactone 54, likely through a 1,2-aryl shift via iodonium intermediate 55, which has been previously observed in hypervalent iodine-mediated alkene functionalizations.44 Use of an internal olefin resulted in lactonization, however, unfortunately, no heterocycle incorporation, with the major product arising from elimination to give a terminal alkene (not shown, see ESI† for details). Lactone ring size was not limited to butyrolactones, with 6-membered lactone 56 produced in 52% yield. Vinyl benzoic acid derivatives were then examined and found to give both 5- and 6-membered benzofused pyridinium lactones 57–59 in good yields. Interestingly, 57 and 58 still displayed excellent levels of 5-exo-selectivity, complimentary to the 6-endo-selectivity typically observed for I(III)-mediated lactonizations of vinyl benzoic acids.33 Finally, several more complex substrates, or those containing other reactive functional handles, were examined. Reaction of a protected dehydrocholic acid derivative gave lactone pyridinium salt 60 in 59% yield. Excitingly, even the presence of other reactive alkenes was well tolerated, with protected indole lactones 61 and 62, the latter possessing an additional terminal alkene in a proximal position, being produced in high yields. These examples indicate the potential utility of this method for late stage incorporation of pyridinium salts in synthetic sequences.
|
With regards to the mechanism, our proposed mechanism involved olefin activation (Step 1) followed by 5-exo-trig lactonization to form an intermediate (alkyl)(aryl)iodonium salt (64) (Step 2),45,46 and C–N bond formation would then occur via SN2 displacement with the nitrogen heterocycle (Step 3), (Scheme 5a). Beginning with the substrate activation step, we also considered an alternative pathway involving ligand exchange at iodine by the carboxylic acid to give 65, promoting attack of the olefin on the umpoled oxygen (65, Scheme 5a, inset), more akin to our previous findings on oxygen activation with N-HVIs.26,27 To test this, the reaction was run with methyl ester 68, which would not participate in ligand exchange; this gave near identical yield and reaction rate as the alkenoic acid, lending support to olefin activation being operative (Scheme 5b). Regarding the proposed lactone (alkyl)(aryl)iodonium intermediate 64, unfortunately, all attempts at direct characterization via1H-NMR or X-ray crystallography were unsuccessful; however prior literature lends strong support to formation of such a species via a kinetically favored 5-exo-trig lactonization on a 3-membered iodonium.33,46
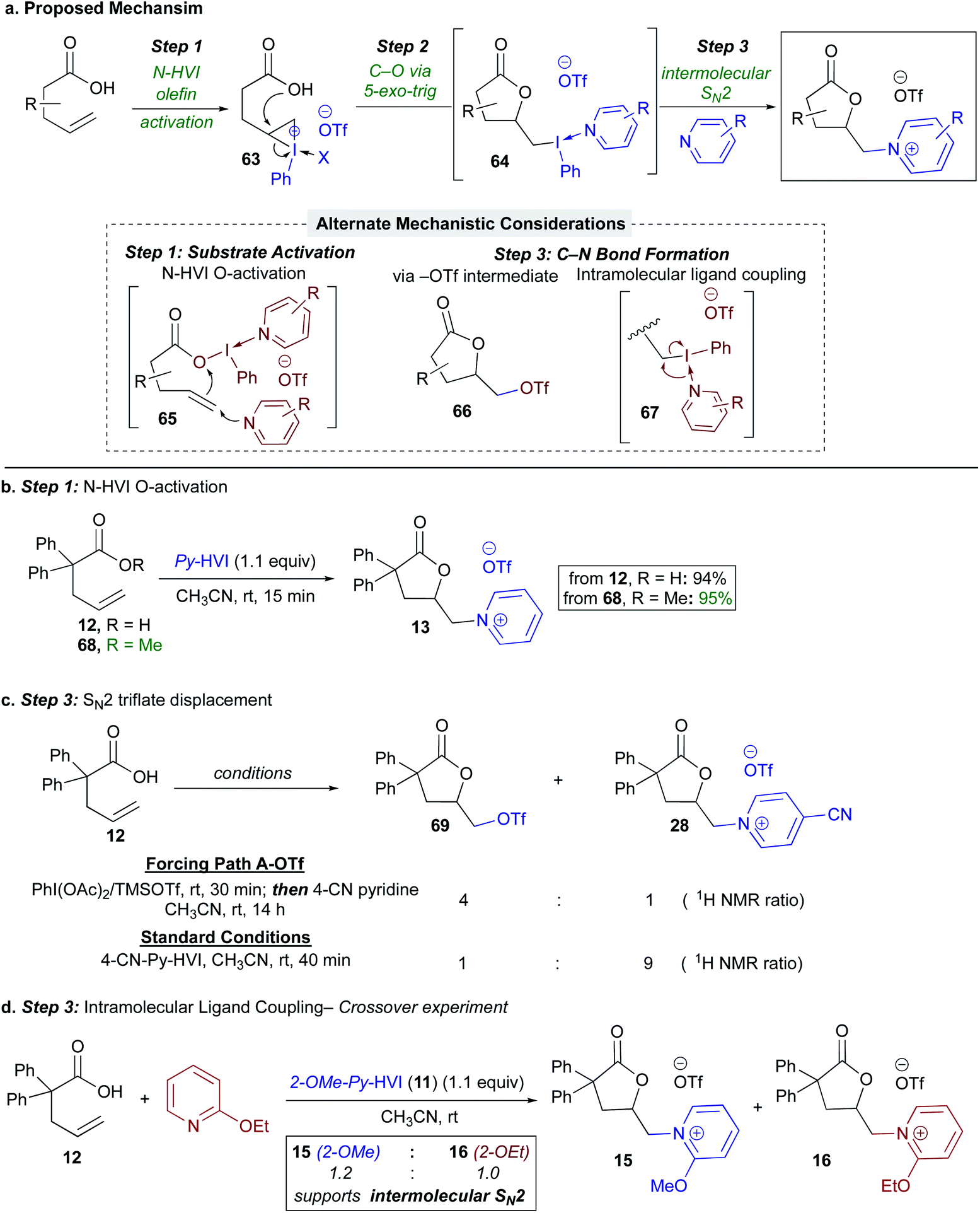 | ||
| Scheme 5 Mechanistic investigation of N-HVI HGT aminolactonization. (a) Proposed mechanism for HGT aminolactonization and considered alternatives (inset). (b) Probing Step 1: Umpolung O-activation. Probing Step 3: (c) Intermolecular –OTf displacement. (d) Inter- vs. intramolecular C–N bonding forming event. | ||
This left us to consider the final C–N bond formation event (Step 3), where we envisioned two alternative pathways: (1) a triflate intermediate such as 66 (Scheme 5a, inset) could form in situ and be competent for product formation, or (2) an intramolecular ligand coupling event from 67.47 To first probe the potential intermediacy of the triflate lactone, 66 was generated in situ via treatment with PhI(OAc)2/TMSOTf,48,49 followed by addition of 4-CN-pyridine (Scheme 5c). While standard conditions for N-HVI HGT gave 90% conversion to the desired salt 28 after 40 minutes by 1H-NMR, 69 gave just 20% product after 14 h. Therefore, while triflate lactone 69 is a viable substrate for heterocycle displacement, it does not appear to be the major operative intermediate. Additionally, this result further emphasizes the unique effectiveness of N-HVIs HGT for (heteroaryl)onium salt synthesis from olefins, as even the highly reactive I(III) species generated from PhI(OAc)2/TMSOTf48,49 was an ineffective activating agent. Finally, a cross over experiment was used to probe for an intramolecular ligand coupling event (67, Scheme 5a, inset), wherein 2-OMe-Py-HVI (11) was used as an activator in the presence of free 2-OEt-pyridine. This experiment showed yielded a 1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0 mixture of lactones 15 and 16, supportive of an intermolecular C–N bond forming event. Taken together, these mechanistic studies support our initially proposed mechanism as shown in Scheme 5a.
:1.0 mixture of lactones 15 and 16, supportive of an intermolecular C–N bond forming event. Taken together, these mechanistic studies support our initially proposed mechanism as shown in Scheme 5a.
Finally, in order to demonstrate the versatility of the resulting (heteroaryl)onium lactones for heterocycle synthesis, we explored a variety of derivatizations to access functionalized and lower oxidation state derivatives (Scheme 6). Full reductions to give saturated piperidines or piperazines (70–74) in excellent yields could be achieved upon hydrogenation with Adam's catalyst (Scheme 6a), providing a means of accessing piperidines with substitution patterns that are either expensive to purchase or challenging to install, such as 3-fluoro- or 3-acyl piperidines (73, 74). Partial hydride reductions led to 3,4-dehydropiperidines 75–78 with complete regioselectivity in all cases, providing vinyl halides or nitriles that serve as functional handles for further diversification (Scheme 6a). Demethylation of 2-OMe pyridinium 15 with NaI gave the corresponding 2-pyridone 79 in 95% yield (Scheme 6b). We then examined the addition of carbon nucleophiles and found addition of a trifluoromethyl group could be achieved with C2-selectivity on 13 to give 80 (Scheme 6c) or that aryl Grignard (Conditions A) or cuprate additions (Conditions B) proceeded with C-2 or C-4 selectivity on 3-Ac- and 3-Bpin-pyridiniums, respectively, to give functionalized 1,2- and 1,4-dihydropyridines (81, 82) (Scheme 6d). In all the above cases, completely selective reaction at the (heteroaryl)onium salt was observed with no competitive reactivity of the lactone moiety, leaving it available for further downstream manipulations.
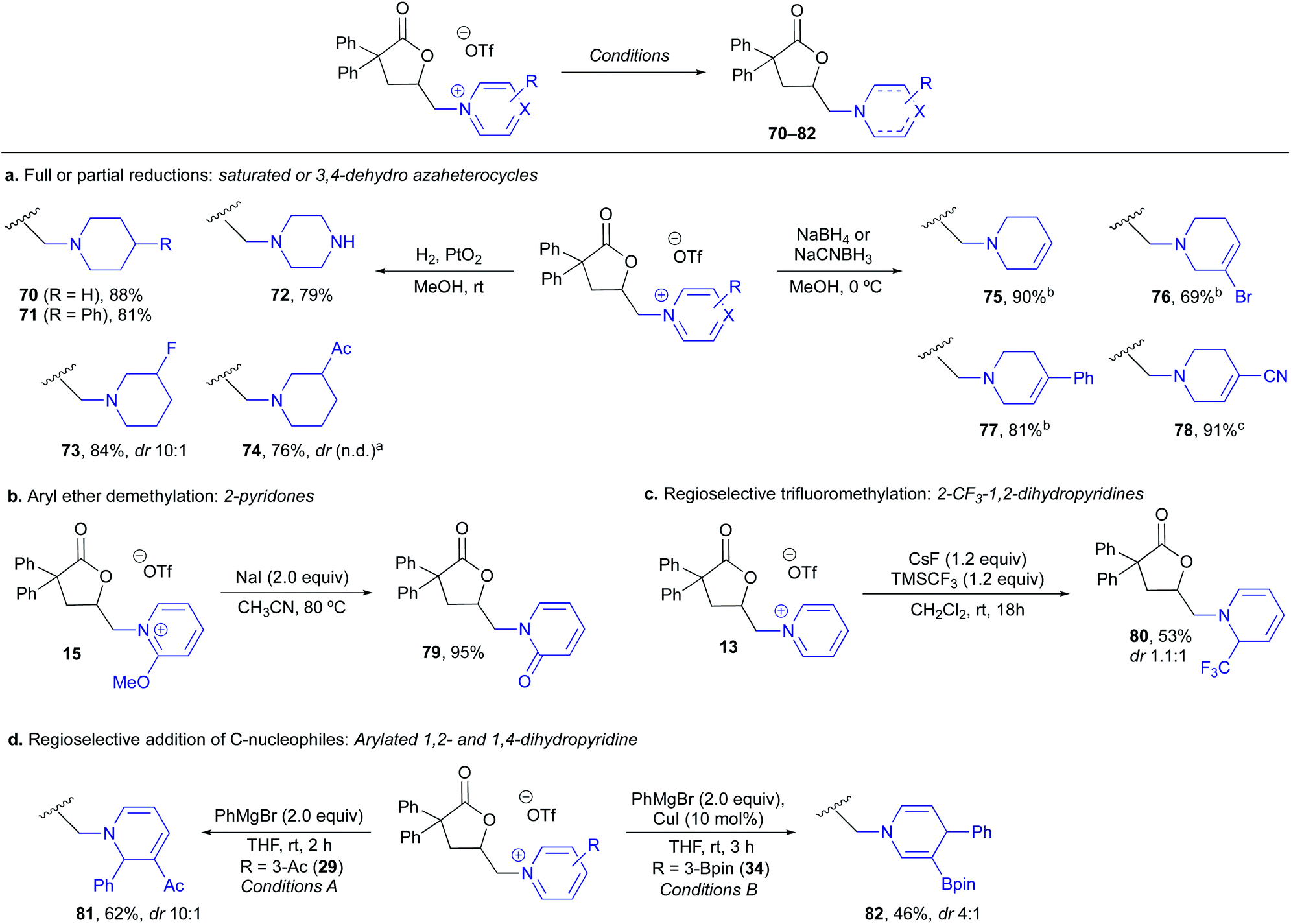 | ||
| Scheme 6 Derivatizations of pyridinium lactones to diverse aminolactonization products. a74 was produced as a ∼1:1 mixture of diastereomers however lack of baseline resolution using several analytical methods prevented definitive determination of ratio. b NaBH4 was used as the reducing agent. c NaCNBH3 was used as the reducing agent. | ||
Conclusions
In conclusion, we report the first example of “heterocyclic group transfer” (HGT) reactions of I(III) N-HVI reagents, providing a new platform for the synthesis of structurally diverse (heteroaryl)onium salts directly from olefins, demonstrated in the aminolactonization of alkenoic acids or esters. The reaction proceeds under remarkably mild conditions, tolerates a broad heterocycle and alkenoic acid scope, can be run without special considerations for air or moisture, and the N-HVIs can be generated in situ, making this strategy both general and practical. For cases involving valuable or sensitive heterocycles, those that are incompatible with N-HVI formation, an enabling “dummy ligand” activation strategy was also developed that allows for their efficient incorporation, further broadening the potential of the methodology. Mechanistic studies indicate that the reaction proceeds via initial olefin activation followed by lactonization and intermolecular SN2 displacement of a (alkyl)(aryl)iodonium salt hypernucleofuge. Representative derivatizations of the resulting (heteroaryl)onium salts demonstrate the power of this platform for broadening the scope of available substitution patterns on the venerable piperidine scaffold for medicinal chemistry. Building on this seminal report, ongoing efforts in our laboratory are working to expand the upon the HGT reactivity of I(III) N-HVIs with the goal of providing a general platform for the incorporation of (heteroaryl)onium salts into organic molecules, fueling the current renaissance of these moieties as functional handles across synthetic chemistry.Author contributions
A. F. T. and S. E. W. conceptualized the work. A. F. T., J. C. W., A. V.-L., X. X., and S. E. W. designed experiments and analyzed data. A. F. T., J. C. W., A. V.-L., and X. X. performed the experiments. S. E. W. wrote the manuscript with editing from J. C. W. and A. F. T.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Institutes of Health (NIH R01 GM123098) and the National Science Foundation (NSF CAREER 1752244) for financial support of this work. We thank Dr Charles DeBrosse (Temple University) for NMR spectroscopic assistance and Dr Charles W. Ross III, Director: Automated Synthesis and Characterization at University of Pennsylvania Chemistry for providing high-resolution mass spectral data.Notes and references
- K. Sepčić, J. Toxicol., 2000, 19, 139–160 Search PubMed.
- P. Madaan and V. K. Tyagi, J. Oleo Sci., 2008, 57, 197–215 CrossRef CAS PubMed.
- B. J. Coe, et al. , Adv. Funct. Mater., 2003, 13, 347–357 CrossRef CAS.
- S. Sowmiah, J. M. S. S. Esperanca, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS PubMed.
- E. V. Dehmlow and S. S. Dehmlow, Phase Transfer Catalysis, Verlag Chemie, Weinheim, Germany, 2nd edn 1983 Search PubMed.
- R. V. Seller, P. V. Reshetov and A. P. Kriven’ko, Chem. Heterocycl. Compd., 2001, 37, 798–821 CrossRef.
- R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141–1156 RSC.
- A. Grozavu, et al. , Nat. Chem., 2019, 11, 242–247 CrossRef CAS PubMed.
- M. Kiamehr, et al. , Tetrahedron, 2012, 68, 9685–9693 CrossRef CAS.
- S. Dong, X. Fu and X. Xu, Asian J. Org. Chem., 2020, 9, 1133–1143 CrossRef CAS.
- J. Zou and P. S. Mariano, Photochem. Photobiol. Sci., 2008, 7, 393–404 CrossRef CAS PubMed.
- J.-N. Derosiers, et al. , Chem. Sci., 2016, 7, 5581–5586 RSC.
- J. A. Bull, J. J. Mousseau, G. Pelletier and A. B. Charette, Chem. Rev., 2012, 112, 2642–2713 CrossRef CAS PubMed.
- J. A. Joule and K. Mills, Heterocyclic Chemistry Ch. 8, John Wiley & Sons Ltd., West Sussex, UK, 2010 Search PubMed.
- Z. S. Ye, M. W. Chen, Q. Chen, L. Shi and Y. Duan, Angew. Chem., Int. Ed., 2012, 51, 10181–10184 CrossRef CAS PubMed.
- D. J. Robinson, S. P. Spurlin, J. D. Gorden and R. R. Karimov, ACS Catal., 2020, 10, 51–55 CrossRef CAS.
- G. Bertuzzi, et al. , ACS Catal., 2016, 6, 6473–6477 CrossRef CAS.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- C. D. Vanderwal, J. Org. Chem., 2011, 76, 9555–9567 CrossRef CAS PubMed.
- F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943–8960 CrossRef CAS.
- S. L. Rössler, et al. , Angew. Chem., Int. Ed., 2020, 59, 9264–9280 CrossRef PubMed.
- For a recent report demonstrating the utility of pyridinium salts other than 2,4,6-triphenylpyridinium in radical cross coupling see: M. Rathnayake and J. D. Weaver, Org. Lett., 2021, 23, 2036–2041 CrossRef CAS PubMed.
- R. Weiss and J. Seubert, Angew. Chem., Int. Ed. Engl., 1994, 33, 891–893 CrossRef.
- R. Corbo and J. L. Dutton, Coord. Chem. Rev., 2018, 375, 69–79 CrossRef CAS.
- B. T. Kelley, J. C. Walters and S. E. Wengryniuk, Org. Lett., 2016, 18, 1896–1899 CrossRef CAS PubMed.
- J. C. Walters, A. F. Tierno, A. H. Dubin and S. E. Wengryniuk, Eur. J. Org. Chem., 2018, 1460–1464 CrossRef CAS PubMed.
- A. De Mico, R. Margarita and G. Piancatelli, Gazz. Chim. Ital., 1995, 215, 325 Search PubMed.
- V. V. Zhdankin, et al. , J. Org. Chem., 2003, 68, 1018–1023 CrossRef CAS PubMed.
- F. Kniep, S. M. Walter, E. Herdtweck and S. M. Huber, Chem.–Eur. J., 2012, 18, 1306–1310 CrossRef CAS PubMed.
- Z. Yuan, R. Cheng, P. Chen, G. Liu and S. H. Liang, Angew. Chem., Int. Ed., 2016, 55, 11882–11886 CrossRef CAS PubMed.
- F. C. S. Sousa, A. F. Tierno and S. E. Wengryniuk, Molecules, 2017, 22, 780 CrossRef PubMed.
- A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed.
- X. Li, P. Chen and G. Liu, Beilstein J. Org. Chem., 2018, 14, 1813–1825 CrossRef CAS PubMed.
- For an example of N-HVIs in pyridination of thio- seleno-, and tellurophenes see: S. Egalahewa, M. Albayer, A. Aprile and J. L. Dutton, Inorg. Chem., 2017, 56(3), 1282–1288 CrossRef CAS PubMed.
- T. J. Donohoe, et al. , Chem.–Eur. J., 2011, 17, 58–76 CrossRef CAS PubMed.
- For an example of the synthesis of vinyl pyridinium salts via olefin C–H pyridination see: L. Liao, R. Guo and X. Zhang, Angew. Chem., Int. Ed., 2017, 56, 3201–3205 CrossRef CAS PubMed.
- V. A. Potapov, R. S. Ishigeev, I. V. Shkurchenko, S. V. Zinchenko and S. V. Amosova, Molecules, 2020, 25, 376–390 CrossRef CAS PubMed.
- For aminolactonization via imidoiodane aziridination see: D. Karila, L. Lemen and R. H. Dodd, Org. Lett., 2011, 13, 5830–5833 CrossRef CAS PubMed.
- For catalytic use of an imidoiodane aziridination for oxyamination see: X.-J. Deng, et al. , J. Org. Chem., 2021, 86, 235–253 CrossRef CAS PubMed.
- For use of a tethered urea in I(III) oxyamination see: B. M. Cochran and F. E. Michael, Org. Lett., 2008, 10, 5039–5042 CrossRef CAS PubMed.
- For a recent report of I(III) oxyamination use a bifunctional carbamate nucleophile see: C. Wata and T. Hashimoto, J. Am. Chem. Soc., 2021, 143, 1745–1751 CrossRef CAS PubMed.
- Studies on N-HVI HGT reactions using other pendant nucleophiles, such as alcohols, amines, and amides, are currently underway in our laboratory and will be reported in due course.
- H. A. Sharma, K. M. Meenie, E. E. Kwan and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 16090–16096 CrossRef CAS PubMed.
- T. Okuyama, T. Takino, T. Sueda and M. Ochiai, J. Am. Chem. Soc., 1995, 117, 3360–3367 CrossRef CAS.
- M. Shah, M. J. Taschner, G. F. Koser and N. L. Rach, Tetrahedron Lett., 1986, 27, 4557–4560 CrossRef CAS.
- T. Okuyama, T. Takino, K. Sato and M. Ochiai, Chem. Lett., 1995, 955–956 Search PubMed.
- Tania, S. D. Houston, L. Sharp-Bucknall, T. B. Poynder, M. Albayer and J. L. Dutton, Chem.–Eur. J., 2020, 26, 15863–15866 CrossRef CAS PubMed.
- T. P. Pell, S. A. Couchman, S. Ibrahim, D. J. D. Wilson, B. J. Smith, P. J. Barnard and J. L. Dutton, Inorg. Chem., 2012, 51, 13034–13040 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00187f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |