
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-stackable molecules assemble into porous crystals displaying concerted cavity-changing motions†
Taewon
Kang‡
 a,
Hongsik
Kim‡
a,
Sungeun
Jeoung‡
b,
Dohyun
Moon
*c,
Hoi Ri
Moon
*b and
Dongwhan
Lee
*a
a,
Hongsik
Kim‡
a,
Sungeun
Jeoung‡
b,
Dohyun
Moon
*c,
Hoi Ri
Moon
*b and
Dongwhan
Lee
*a
aDepartment of Chemistry, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 08826, Korea. E-mail: dongwhan@snu.ac.kr
bUlsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Eonyang-eup, Ulji-gun, Ulsan 44919, Korea
cPohang Accelerator Laboratory, 80 Jigokro-127-beongil, Nam-gu, Pohang 37673, Korea
First published on 1st April 2021
Abstract
With small molecules, it is not easy to create large void spaces. Flat aromatics stack tightly, while flexible chains fold to fill the cavities. As an intuitive design to make open channels inside molecularly constructed solids, we employed propeller-shaped bicyclic triazoles to prepare a series of aromatic-rich three-dimensional (3D) building blocks. This modular approach has no previous example, but is readily applicable to build linear, bent, and branched arrays of non-stackable architectural motifs from existing flat aromatics by single-pot reactions. A letter H-shaped molecule thus prepared self-assembles into porous crystals, the highly unusual stepwise gas sorption behaviour of which prompted in-depth studies. A combination of single-crystal and powder X-ray diffraction analysis revealed multiple polymorphs, and sterically allowed pathways for their reversible interconversions that open and close the pores in response to external stimuli.
Introduction
To construct stress-resistant structures, rigid and shape-persistent building blocks are needed. Otherwise, the assembly would easily collapse. The same principle holds for making non-collapsible open channels by stacking up individual molecules.1–4 With appropriate design, such supramolecular constructs can change the size, shape, and dimension of the pores without compromising their overall architectural integrity, so that small guests can be captured and released by using external stimuli.5–11 From a design perspective, what kind of molecular shape is ideal for such adaptivity? In this paper, we provide a practical answer to this question.Planar aromatic molecules are rigid, but they tend to engage in tight π–π contacts.12–14 As such, they are the least amenable choice of building blocks to make porous materials. How could one spatially organize flat slabs so that they cannot stack well and always leave voids in between? A macroscopic object that satisfies this design requirement is the H-beam used for steel framed buildings (Fig. 1a). Due to the orthogonal relationship between the planes, stacks of letter H-shaped objects should always make edge-to-face contacts, thereby leaving behind a linearly extended empty space, regardless of how they are packed. The size and shape of the resulting channels would change if the H-beams are allowed to slide relative to each other.
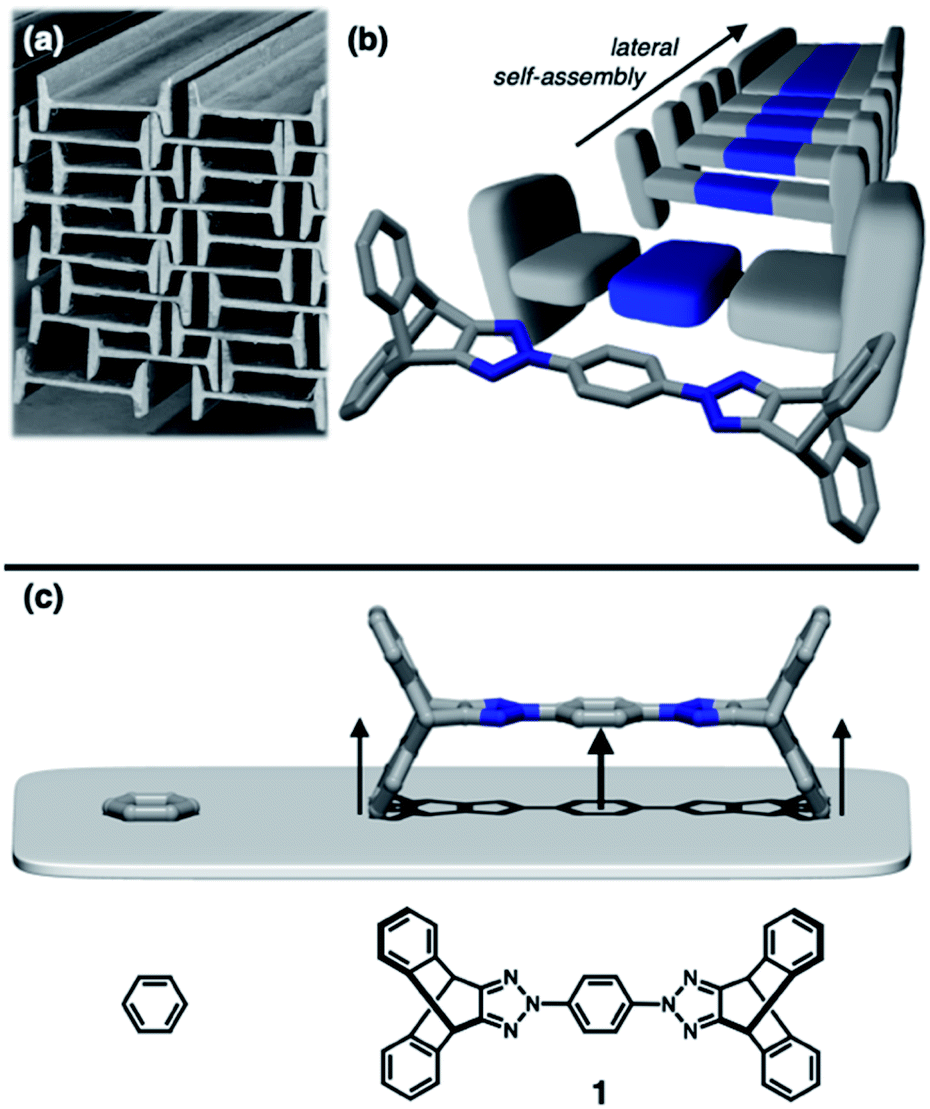 | ||
| Fig. 1 (a) Stacks of steel H-beams leaving non-collapsible open channels, which inspired (b) molecular-level construction of H-beams by lateral self-assembly. (c) Chemical structure of the building block 1, in which two iptycene units are appended to “lift up” the benzene core. In (b), the blue-coloured horizontal slab of the H-shaped 3D model corresponds to the electron-deficient triazole–phenyl–triazole triad within 1, the capped-stick representation of which is overlaid for clarity. | ||
Background and design principles
Translated into molecular language, however, it is impossible to elongate an H-beam to an infinite length by covalent bonds. A synthetically more viable approach would be creating a molecule that resembles a thin slice of H-beam, and laterally connecting each other by non-covalent bonds (Fig. 1b). To turn this idea into reality, we have devised a modular synthetic route for “jacking up” the flat aromatic core by appending propeller-shaped molecular fragments at the periphery (Fig. 1c). This convergent synthesis requires efficient bond-making reactions between structural subcomponents with minimal interference from existing functional groups. For this purpose, we decided to exploit the C–N cross-coupling reactions15 of triazoliptycene (TI)16 and haloarenes (Scheme 1). Unlike other heteroaromatic variants17–19 of the privileged iptycene motif,5,20–23 the triazole-fused TI allows direct attachment of rigid bicyclic scaffolds onto existing aromatic core via single-bonds in the last stage in the synthesis. The highest level of synthetic modularity is achieved this way. A concise point-to-point connection between the vertices of five- and six-membered rings (Fig. 1 and Scheme 1) significantly expands the structural space of “iptycene-capped” aromatics, which are typically accessed by edge-to-edge fusion of two six-membered rings by condensation reactions.20,23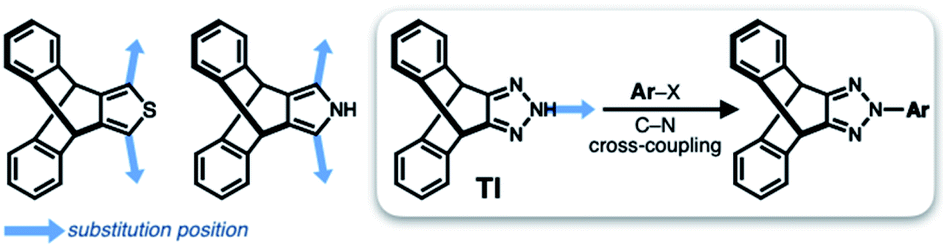 | ||
| Scheme 1 Covalent modification of heteroaromatic-fused iptycenes. | ||
As the newest addition to the heteroaromatic-fused iptycene family, we recently reported the chemistry of TI.16 By transition metal-catalyzed C–N cross-coupling reactions, a wide range of aryl groups can be installed at the N2-position of TI.15,16 As shown in Fig. 2a, one such molecule PhTI forms an antiparallel stacked “dimer” in the solid state to maximize donor–acceptor (D–A) type intermolecular π–π contacts between the electron-rich phenyl ring and the electron-deficient triazole ring.16 A tightly packed array of dimeric (PhTI)2 leaves no void space in the crystal lattice. We postulated that blocking the open end of the N-phenyl group of PhTI by installing additional TI units should effectively suppress the formation of a tightly stacked dimer, thereby opening up void space in the crystal lattice.
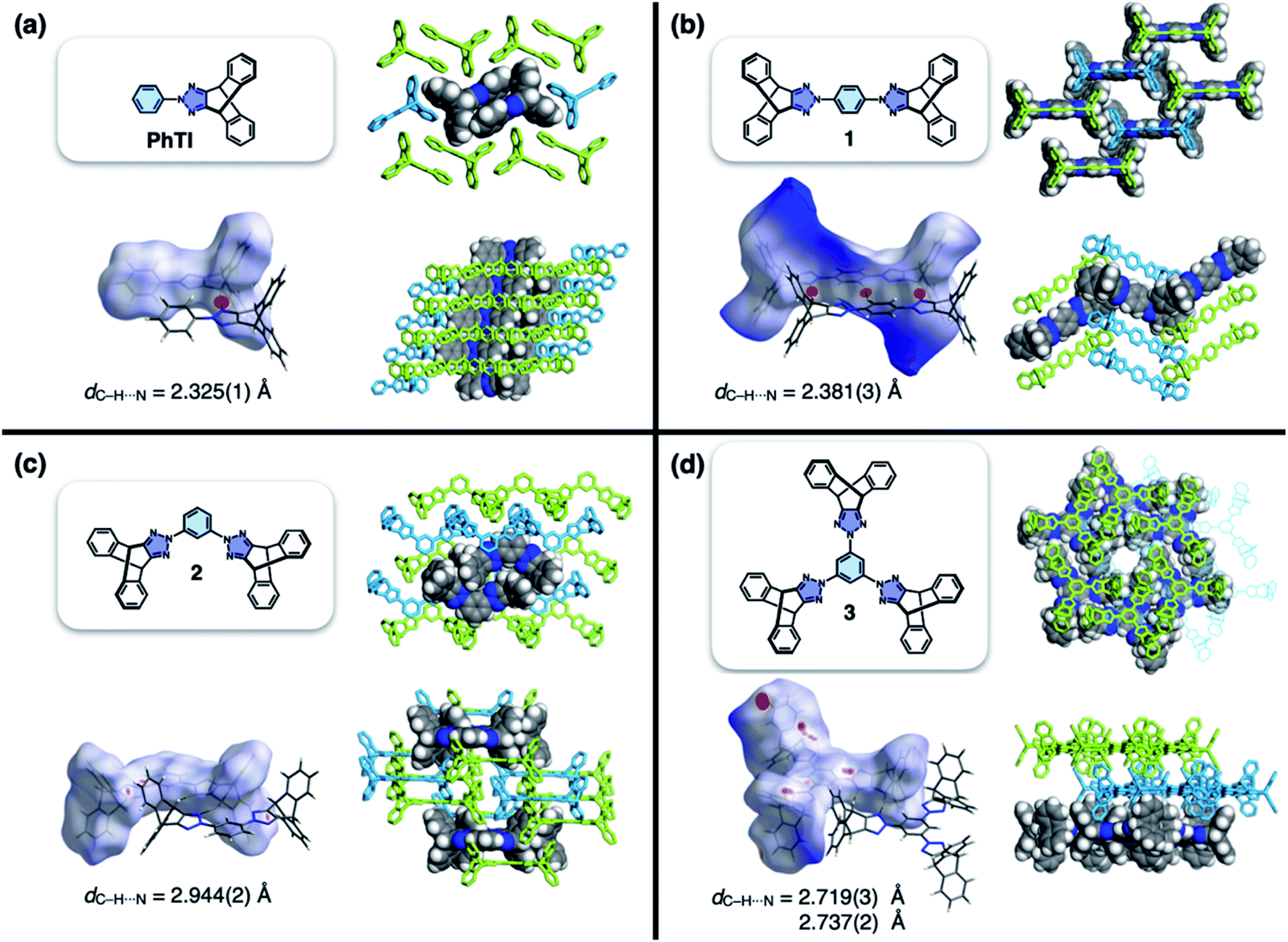 | ||
| Fig. 2 Molecular structures, Hirshfeld surfaces24 mapped with dnorm values, and single-crystal packing diagrams of (a) PhTI, (b) 1 (1A phase), (c) 2, and (d) 3, constructed with crystallographically determined atomic coordinates. Disordered solvent molecules are omitted for clarity. For each structure, the hydrogen bond length (dC–H⋯N) denotes the interatomic distance between hydrogen and nitrogen atoms. | ||
Results and discussion
Non-stackable 3D aromatics: modular synthesis and solid-state assembly
To test the validity of this intuitive steric model, TI was subjected to palladium-catalyzed C–N cross-coupling reactions with di- or tribromobenzene to produce 1–3 in high yields (up to 95%, Scheme 2).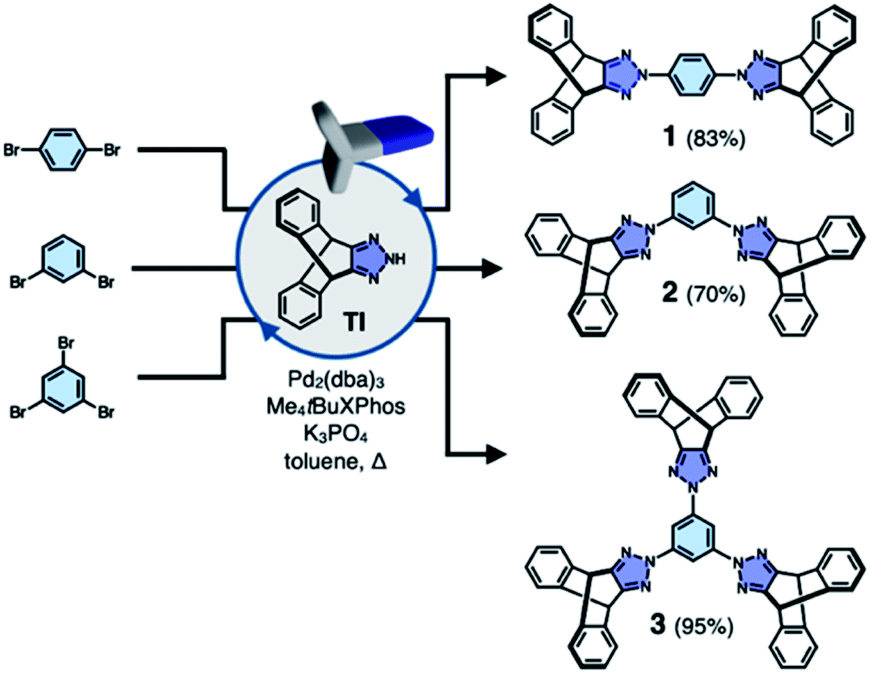 | ||
| Scheme 2 Modular construction by one-pot C–N cross-coupling reactions. | ||
Single-crystal X-ray diffraction (SC-XRD) studies revealed that 1 indeed affords a porous structure having open channels (Fig. 2b), whereas its regioisomer 2 assembles into a non-porous structure with isolated voids failing to define continuous channels (Fig. 2c). A close inspection of the crystal structure showed that the crescent-shaped 2 makes tight van der Waals (vdW) contacts between the concave and convex faces of neighboring molecules (Fig. 2c). This shape complementary vdW interaction is reinforced by the C–H⋯N hydrogen bonds (dC–H⋯N = 2.944(2) Å, Fig. 2c), which is also observed for 3 having three TI units surrounding the central benzene ring (Fig. 2d). The molecule crystallizes to form a hexagonal packing held by C–H⋯N hydrogen bonds (dC–H⋯N = 2.719(3), 2.737(2) Å, Fig. 2d) but without open channels.
Comparative structure analysis of 1–3 (Fig. 2) points toward the importance of the H-shaped molecular geometry. Unlike 2 or 3, molecule 1 assembles into a porous structure since it cannot find shape-complementary pairs from any orientations. The intermolecular interaction thus extends in the lateral direction to maximize the number of C–H⋯N hydrogen bonds (dC–H⋯N = 2.381(3) Å, Fig. 2b). By design, close π–π contacts are not allowed for the resulting H-beam like assemblies (Fig. 1); they instead utilize weak C–H⋯π interactions to surround open channels (Fig. 2b).
Open channels displaying stepwise gas sorption behaviour
To investigate the functional relevance of the open channels found in crystalline 1, sorption isotherms were obtained for N2, H2, and CO2 gases (Fig. S1a–c†). Prior to measurements, solid samples were activated to remove solvent molecules. Interestingly, 1 showed guest-dependent gas sorption behaviour. Double-step adsorption isotherm and desorption hysteresis were observed for N2 (T = 77 K, Fig. S1a†), although 1 does not seem to have specific binding sites for N2. Deviation from type I isotherm25 is one of the prominent characteristics of structural dynamics during the sorption process.26–33 In stark contrast, only desorption hysteresis was observed for H2 gas (Fig. S1b†); simple type I adsorption occurred for CO2 gas (Fig. S1c†).Our attempts to understand this intriguing gas sorption behaviour were initially met with insistent and uninterpretable experimental results. For example, powder X-ray diffraction (PXRD) analysis on the thermally activated 1 produced what appears to be a hopelessly complicated intensity pattern that could not be modeled with the initially obtained SC-XRD structure of 1 (Fig. S1d†). More problematic was the batch-to-batch irreproducibility of the PXRD data of 1, which suggested the presence of multiple crystalline domains within the bulk material. Apparently, the relative proportions of these putative polymorphs vary from batch to batch, depending on how the microcrystallites of 1 were prepared before sorption measurements.
Metastable porosity of interconverting polymorphs
To identify the polymorph that is responsible for the double-step N2 sorption (Fig. S1a†), we first needed to decipher the complicated PXRD pattern. An exhaustive empirical screening was thus carried out by changing the conditions for (i) precipitating microcrystallites of 1 from the solution, and (ii) removing entrapped solvent molecules from the harvested material. During this investigation, a total of four polymorphs of 1 were identified, including the initially determined structure 1A (Fig. 2b). All these polymorphs, 1A, 1B, 1C, and 1Z, were characterized by SC-XRD. The results are summarized in Fig. 3 and S2.†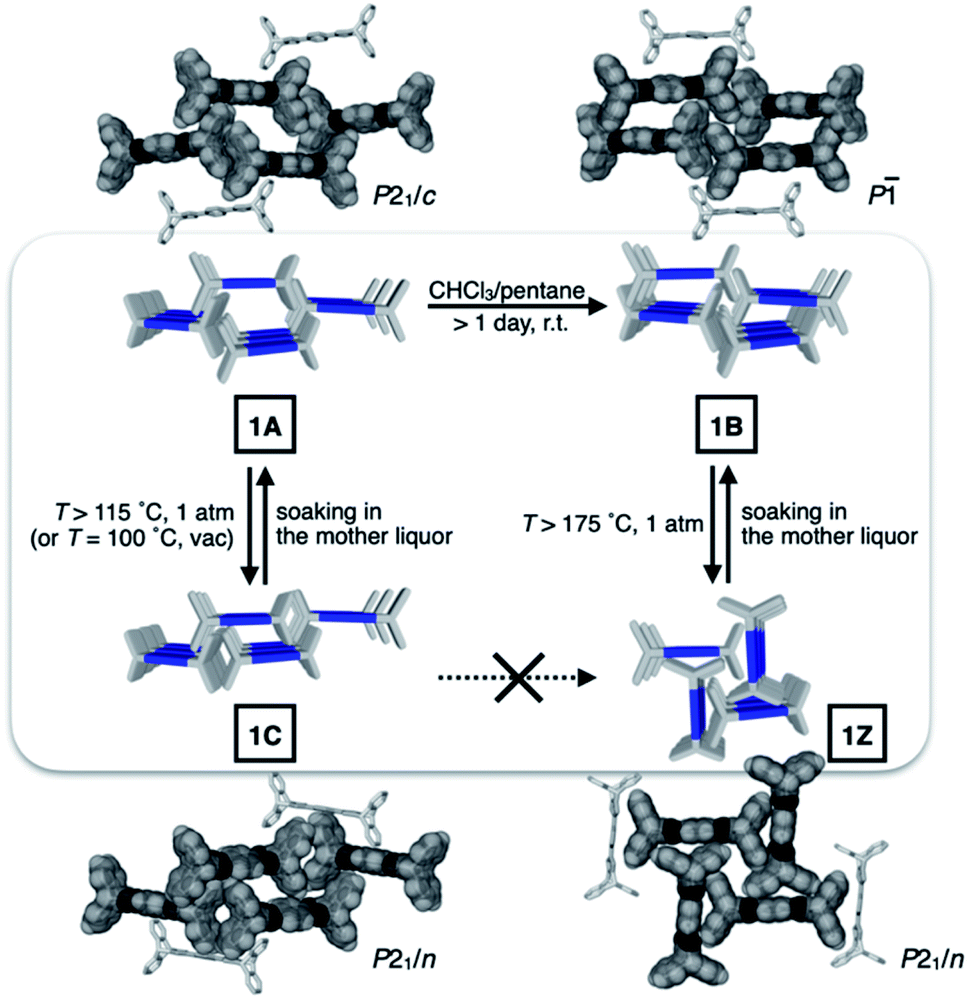 | ||
| Fig. 3 Schematic representation of the structural interconversions between the four polymorphs. The capped-stick packing diagrams are constructed with crystallographically determined atomic coordinates, and overlaid with space-filling models to show intermolecular contacts and pores. | ||
We found that the addition of pentane into a chloroform solution sample immediately produced 1A, whereas aging of the crystalline material in the mother liquor transformed it into 1B (Fig. 3 and S3a†). Removal of entrapped solvents from 1A and 1B produced 1C and 1Z, respectively (Fig. 3, S3b and d†). These processes could be reversed, as evidenced by the PXRD analysis on the bulk sample prior to and after solvent back-filling (Fig. S3c and e†). The three polymorphs, 1A, 1B, and 1C, have open channels as potential conduits for exogenous guests (Fig. 3). Even for the most tightly packed non-porous polymorph 1Z, no close π–π contact could be established due to the geometrical mismatch between adjacent H-shaped building blocks (Fig. 3).
Structural dynamics captured during gas uptake and release
With the SC-XRD and PXRD data of the four polymorphs in hand, we proceeded to standardize the sample preparation and activation protocols to obtain reproducible gas sorption data. Specifically, samples of 1B needed to be activated in a vacuum at T = 100 °C to achieve the prominent double-step adsorption isotherm and desorption hysteresis (Fig. 4a). Increasing the activation temperature induced 1B-to-1Z conversion; the sample is fully transformed into non-porous 1Z at T = 175 °C (Fig. 3 and S3d†). From the PXRD analysis of the bulk material used for gas sorption, we found that activated 1B contains a small amount of 1C phase (red asterisks; Fig. 4b and c). In addition, the entire structure was slightly perturbed, as reflected on the splitting of the (021) peak (green and blue asterisks; Fig. 4b and c), which is related to the plane bisecting the pores of 1B phase (Fig. S4†). Except for this minor variation, other PXRD peaks of 1B are fully maintained. Presumably, evacuation and heating in the activation process could shift the pores as the entrapped solvent molecules escape, but without significantly altering the cell parameters.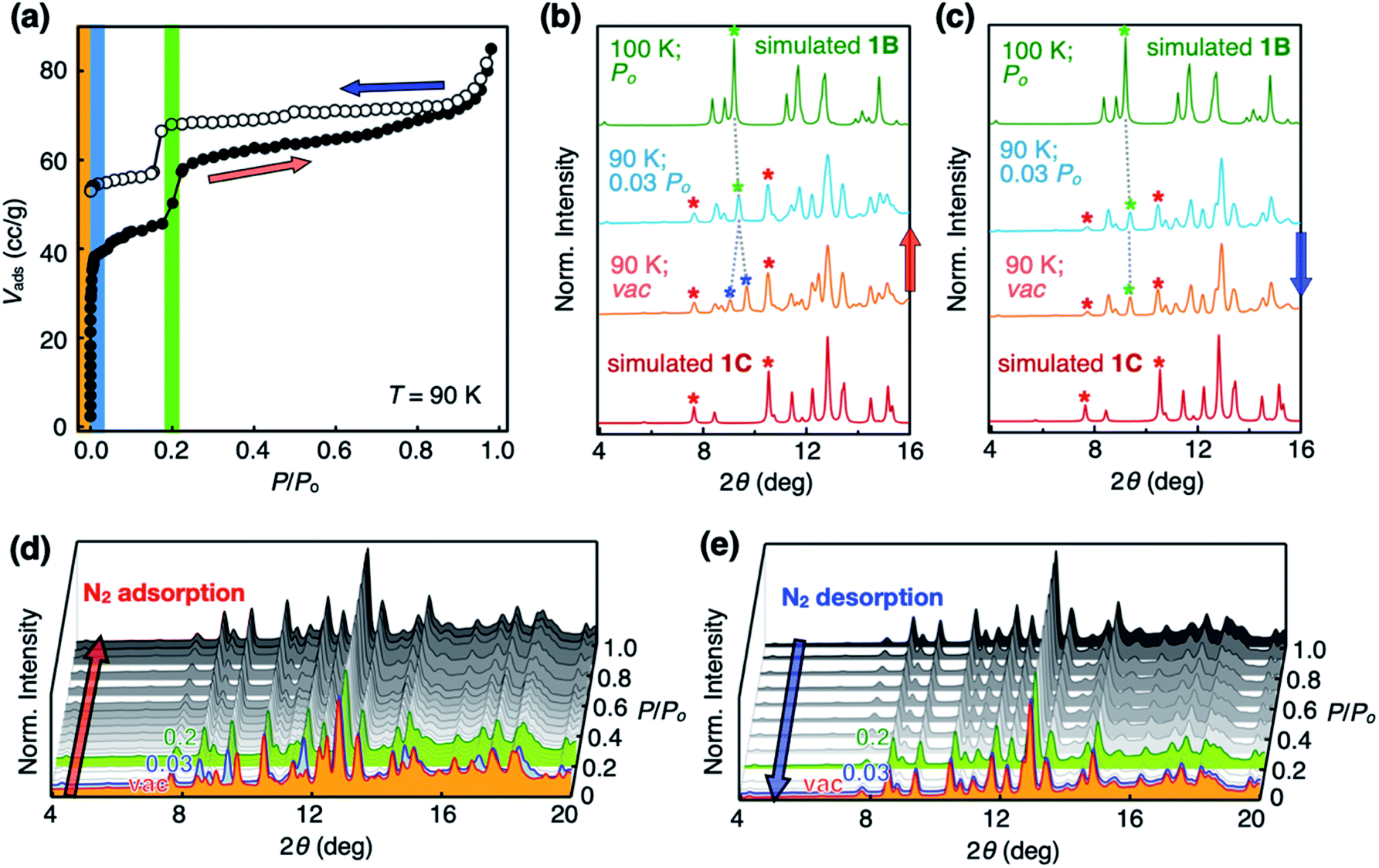 | ||
| Fig. 4 N2 sorption isotherms and in situ PXRD pattern changes recorded during the sorption process. (a) N2 adsorption (filled circles; red arrow) and desorption (empty circles; blue arrow) isotherms of activated 1B at T = 90 K showing double-step adsorption isotherm and desorption hysteresis. (b) Structural changes at the initial stage of N2 uptake, as reflected on the two split (021) peaks (denoted by blue asterisks) merging into a single peak (denoted by a green asterisk), which indicates the recovery of the 1B phase by pore-opening by N2 molecules. Red asterisks denote small amount of 1C phase formed during the activation process. (c) PXRD pattern at the final stage of N2 desorption showing no noticeable changes of the (021) peak, which is consistent with the N2 desorption hysteresis. PXRD pattern changes were recorded during the entire (d) adsorption, and (e) desorption processes. | ||
Changes in the PXRD patterns recorded under gas sorption conditions indicated the recovery of pure 1B phase immediately after N2 gas filled the vacuum (Fig. 4b and d). This phase transition is best interpreted as the pore-opening process that is responsible for the sharp increase in the Vadsvs. P/Po curve at the very initial step (P/Po < 0.03; Fig. 4a). At the second step (P/Po ≈ 0.2) where a sudden rise in gas uptake occurred again, however, the PXRD pattern does not show noticeable differences (Fig. 4d). In other words, this second structural transition involves only minor changes in the cell parameters yet a significant increase in the adsorption area. One possible explanation is an effective pore expansion by freezing out the bond-rotating motions34–36 of the phenylene spacer group of 1, which would have a negligible impact on the lattice constants.35
In the desorption process, no changes were observed in the PXRD pattern (Fig. 4c and e) at P/Po < 0.03, indicating that gas sorption-induced structural transformation at low pressure is irreversible. This experimental observation is consistent with the hysteresis in the desorption process (Fig. 4a). Meanwhile, independently prepared crystals of 1C displayed simple type I isotherm (Fig. S5†), thus ruling out its participation in the double-step sorption process.
Molecular origin of cavity-changing motions
To gain a detailed molecular-level understanding of the pore-changing motions observed in gas sorption, we revisited the solid-state intermolecular interactions captured by SC-XRD on the different phases of 1. As summarized in Fig. 5a, all four phases share a common hierarchical assembly pattern involving three types of intermolecular forces: hydrogen bonding, electrostatic contact, and vdW interaction. Conceptually, the solid-state packing commences with lateral self-assembly of 1 by C–H⋯N hydrogen bonds (dC–H⋯N = 2.372(3)–2.472(2) Å, Tables S1–S6 and Fig. S6†) to form a 1-D array that resembles an H-beam (Fig. 1b and 5a). In the crystalline lattice, these molecular H-beams buttress each other by C–H⋯π contacts. For either phenyl⋯triazole or phenyl⋯phenyl pair, the electropositive end of the C–H dipole and electronegative π-clouds of the aromatic rings establish weak C–H⋯π contacts (dC–H⋯π = 3.404(3)–3.485(4) Å for phenyl⋯triazole; 3.517(4)–3.942(3) Å for phenyl⋯phenyl; Table S7†).37 As such, the C–H dipole ends can slide freely on the π-surfaces, until the steric clash with the vertical iptycene unit stops the movement. This degree of freedom allows 1 to sample multiple packing patterns without significant energy barriers (Fig. 5b). A conceptual analogy could be drawn to a sliding door that can stop anywhere along the door track.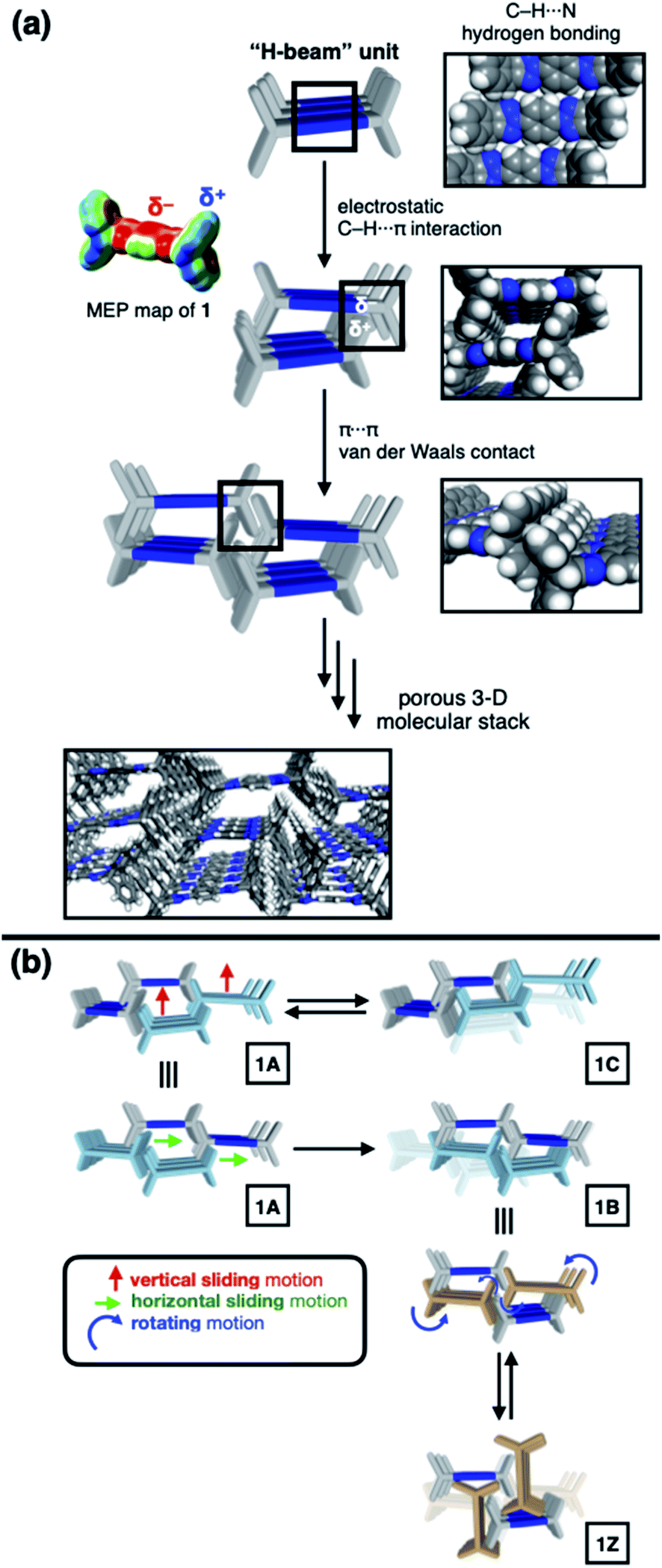 | ||
| Fig. 5 (a) Schematic representation of lateral association of 1 to form H-beam units, and their organization into the 3-D lattice. At each stage of this conceptual reconstruction, space-filling models based on the X-ray structures of 1B are shown next. The molecular electrostatic potential (MEP) map illustrates charge distribution that is responsible for intermolecular C–H⋯π interactions. (b) Synchronized sliding (either horizontal for 1A-to-1B, or vertical for 1A-to-1C) and rotating (for 1B-to-1Z) motions of the H-beam units in the solid state to drive phase transitions. | ||
Depending on how these H-beam substructures are arranged in space, different polymorphs of 1 are produced (Fig. 3). The four polymorphs of 1 are structurally similar, thus making it possible for us to draw intuitive correlations and deduce the mechanism of their interconversion by molecular motions within the confines of the crystal lattice. Taking the principle of least action, the 1A-to-1B transition could best be explained by horizontal sliding of the H-beam units along the central triazole–phenylene–triazole triad (Fig. 5b). On the other hand, the interconversion between 1A and 1C entails vertical sliding of the H-beam units along the peripheral benzene rings (Fig. 5b). More drastic rotating motions are required to interconvert porous 1B and non-porous 1Z (Fig. 5b). These processes are accompanied by changes in the cavity dimensions (Table S8†), which follow the order 1A > 1B ≫ 1C > 1Z.
Conclusions
Imparting dynamic properties is a powerful strategy to design stimuli-responsive porous materials.1–11 The non-planar architecture of 1 effectively prevents close vdW contacts, thereby creating open channels. The pseudo-orthogonal arrangement of the π-faces constituting the H-shaped molecular scaffold functions as steric barriers for the sliding and rotating motions in the solid state. Such physical boundary conditions limit the number of possible intermolecular arrangements, thereby simplifying the reaction coordinates of their interconversion in response to external stimuli. Efforts are currently underway in our laboratory to expand this design concept, and find practical applications in host–guest chemistry.Author contributions
T. Kang and H. Kim: conceptualization, formal analysis, investigation, methodology, visualization, writing-original draft, writing-review & editing. S. Jeoung: conceptualization, formal analysis, investigation, methodology, visualization. D. Moon: conceptualization, formal analysis, funding acquisition, methodology, supervision, validation. H. R. Moon: conceptualization, funding acquisition, supervision, validation. D. Lee: conceptualization, funding acquisition, project administration, supervision, validation, visualization, writing-original draft, writing-review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Whanchul Shin for helpful discussions. This work was supported by the Samsung Science and Technology Foundation (SSTF-BA1701-10680). The work at UNIST was supported by the National Research Foundation (NRF) of Korea (2020R1A2C3008908) funded by the Ministry of Science and ICT (MSIT). The X-ray crystallography experiments at PLS-II BL2D-SMC beamline were supported in part by MSIT and POSTECH. T. K. thanks the NRF-2016-Global PhD Fellowship Program (2016H1A2A1906550) through the NRF.Notes and references
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef CAS PubMed
.
- R.-B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC
- J. R. Holst, A. Trewin and A. I. Cooper, Nat. Chem., 2010, 2, 915–920 CrossRef CAS PubMed
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed
- J. H. Chong, S. J. Ardakani, K. J. Smith and M. J. MacLachlan, Chem.–Eur. J., 2009, 15, 11824–11828 CrossRef CAS PubMed
- M. I. Hashim, H. T. M. Le, T.-H. Chen, Y.-S. Chen, O. Daugulis, C.-W. Hsu, A. J. Jacobson, W. Kaveevivitchai, X. Liang, T. Makarenko, O. Š. Miljanić, I. Popovs, H. V. Tran, X. Wang, C.-H. Wu and J. I. Wu, J. Am. Chem. Soc., 2018, 140, 6014–6026 CrossRef CAS PubMed
- T.-H. Chen, W. Kaveevivitchai, A. J. Jacobson and O. Š. Miljanić, Chem. Commun., 2015, 51, 14096–14098 RSC
- H. Wang, B. Li, H. Wu, T.-L. Hu, Z. Yao, W. Zhou, S. Xiang and B. Chen, J. Am. Chem. Soc., 2015, 137, 9963–9970 CrossRef CAS
- H. Yamagishi, H. Sato, A. Hori, Y. Sato, R. Matsuda, K. Kato and T. Aida, Science, 2018, 361, 1242–1246 CrossRef CAS PubMed
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed
- M. J. Bojdys, M. E. Briggs, J. T. A. Jones, D. J. Adams, S. Y. Chong, M. Schmidtmann and A. I. Cooper, J. Am. Chem. Soc., 2011, 133, 16566–16571 CrossRef CAS PubMed
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed
- S. E. Wheeler, Acc. Chem. Res., 2013, 46, 1029–1038 CrossRef CAS
- H. Choi, S. Ogi, N. Ando and S. Yamaguchi, J. Am. Chem. Soc., 2021, 143, 2953–2961 CrossRef CAS PubMed
- S. Ueda, M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8944–8947 CrossRef CAS PubMed
- T. Kang, H. Kim and D. Lee, Org. Lett., 2017, 19, 6380–6383 CrossRef CAS PubMed
- V. E. Williams and T. M. Swager, Macromolecules, 2000, 33, 4069–4073 CrossRef CAS
- Y. Ramondenc, R. Schwenninger, T. Phan, K. Gruber, C. Kratky and B. Kräutler, Angew. Chem., Int. Ed. Engl., 1994, 33, 889–891 CrossRef
-
R. R. Gupta, M. Kumar and V. Gupta, Heterocyclic Chemistry Volume II: Five-Membered Heterocycles, Springer-Verlag, Berlin Heidelberg, 1998 Search PubMed
-
C.-F. Chen and Y.-X. Ma, Iptycenes Chemistry: From Synthesis to Applications, Springer, Berlin, 2013 Search PubMed
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC
-
(a) L. Ueberricke and M. Mastalerz, Chem. Rec., 2021, 21, 558–573 CrossRef CAS PubMed
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC
- M. D. Donohue and G. L. Aranovich, Adv. Colloid Interface Sci., 1998, 76–77, 137–152 CrossRef CAS
- S. Kitagawa and R. Matsuda, Coord. Chem. Rev., 2007, 251, 2490–2509 CrossRef CAS
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CrossRef CAS
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357–361 CrossRef CAS PubMed
- M. K. Taylor, T. Runčevski, J. Oktawiec, M. I. Gonzalez, R. L. Siegelman, J. A. Mason, J. Ye, C. M. Brown and J. R. Long, J. Am. Chem. Soc., 2016, 138, 15019–15026 CrossRef CAS PubMed
- S. Jeoung, S. Lee, J. H. Lee, S. Lee, W. Choe, D. Moon and H. R. Moon, Chem. Commun., 2019, 55, 8832–8835 RSC
- S.-m. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920–1925 CrossRef CAS PubMed
- A. Comotti, S. Bracco and P. Sozzani, Acc. Chem. Res., 2016, 49, 1701–1710 CrossRef CAS PubMed
- Q. Huang, W. Li, Z. Mao, L. Qu, Y. Li, H. Zhang, T. Yu, Z. Yang, J. Zhao, Y. Zhang, M. P. Aldred and Z. Chi, Nat. Commun., 2019, 10, 3074 CrossRef PubMed
- S. Nandi, P. De Luna, R. Maity, D. Chakraborty, T. Daff, T. Burns, T. K. Woo and R. Vaidhyanathan, Mater. Horiz., 2019, 6, 1883–1891 RSC
- The dC–H⋯π values were determined by measuring the distance between the aromatic ring root-mean-square plane and the closest carbon atom of the adjacent aromatic ring in the X-ray structure. Please see Table S7† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data (1H and 13C NMR spectra, FT-IR spectra, HR-MS) of the newly synthesized compounds. CCDC 1980363–1980368, 2048700, and 2048701. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc01163d |
| ‡ T. Kang, H. Kim, and S. Jeoung contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |