
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThickness control of 2D nanosheets assembled from precise side-chain giant molecules†
Fengfeng
Feng
a,
Dong
Guo
e,
Yu
Shao
b,
Xiang
Yan
d,
Kan
Yue
c,
Zhipeng
Pan
a,
Xiangqian
Li
a,
Dongcheng
Xiao
a,
Liang
Jin
a,
Wen-Bin
Zhang
 b and
Hao
Liu
*a
b and
Hao
Liu
*a
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Center for Advanced Low-Dimension Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, P. R. China. E-mail: liuh@dhu.edu.cn
bKey Laboratory of Polymer Chemistry & Physics of Ministry of Education, Center for Soft Matter Science and Engineering, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China
cSouth China Advanced Institute of Soft Matter Science and Technology, School of Molecular Science and Engineering, South China University of Technology, Guangzhou 510640, P. R. China
dSchool of Materials Science and Engineering, Baise University, Baise 533000, P. R. China
eDepartment of Polymer Science, College of Polymer Science and Polymer Engineering, The University of Akron, Akron, Ohio 44325, USA
First published on 26th February 2021
Abstract
The performance of 2D nanomaterials hinges on both the chemical compositions and the morphological structures across different length scales. Among all the three dimensions, thickness is the only one that falls into the nanometer scale and, to some extent, determines the intrinsic properties of 2D nanomaterials. In this study, we report the preparation and precise thickness control of 2D nanosheets assembled from a library of monodispersed amphiphilic giant molecules composed of functional polyhedral oligomeric silsesquioxanes (POSSs) as the side groups. Solution self-assembly of such giant molecules resulted in 2D nanosheets with similar structural configurations, where a bilayer of hydrophobic isobutyl POSS (BPOSS) is sandwiched by two monolayers of hydrophilic POSS bearing carboxylic acid groups (APOSS). The thickness of the obtained nanosheets could be tuned through adjusting the chemical compositions of the pendant POSS cages. Intriguingly, we found that the thickness of the 2D nanosheets was not necessarily proportional to the contour length of the giant molecule nor the total number of POSS cages tethered to the main chain. Indeed, the number ratio of BPOSS to APOSS, rather than the exact number, played a deterministic role in the thickness control. To explain the unusual thickness dependence, we built up a structure model with an in-plane orientation of the giant molecules in the nanosheets, from which a formula was further deduced to semi-quantitatively describe the inverse relationship between the overall thickness and the number ratio of BPOSS to APOSS.
Introduction
Two-dimensional (2D) nanomaterials have received broad attention due to their ultrathin planar geometry and the consequent intrinsic physical properties,1 such as large specific surface area, unique monolayer structure, typical quantum effects and adaptable performance, which jointly boosted their applications in energy storage,2 adsorption,3 catalysis4 and optoelectronics.5 In particular, the pioneering studies on graphene and graphene-derived materials have shown excellent scientific and economic values,6–8 which stimulated the exploration of other 2D materials with specific compositions and structures.9–11 To date, many inorganic 2D nanosheets were prepared by the “top–down” exfoliation method12 that relied on harsh mechanical or chemical conditions to destroy the physical interactions among stacked layers and turn bulk materials into 2D nanosheets. Another important cluster of 2D nanostructures was prepared through the “bottom–up” assembly approach using sub- to several nanometer-size molecular entities as building blocks under relatively mild conditions.13–16 Such an assembly approach has provided a complementary variety of unprecedented 2D nanostructures with intriguing structural and physical properties.17 Since molecular self-assembly depends upon the mutual recognition among collective physical interactions to direct the packing of molecules, the essence of the 2D self-assembly is to locate a particular molecular packing scheme that prefers the formation of 2D over 0D/1D/3D counterparts and meanwhile avoids polymorphic products with different dimensions. Recently, a few prototypes of 2D assemblies have been produced from various building blocks, including small molecules,18 inorganic nanoparticles, block copolymers,19,20 programmed peptides13,21 or peptoids,19,20,22etc. However, there remains a non-trivial challenge to precisely modulate their structural hierarchy in terms of shape, thickness, lateral size and surface functionality. Tackling these problems requires profound understanding of the packing scheme of molecular building blocks.Among all the three dimensions of 2D materials, thickness is the only one that falls into the nanometer scale, which brings in numerous special physical properties that root from their ultrathin planar geometry.23–25 The thickness control of 2D assemblies is of great importance for understanding their self-assembly behaviors and for optimizing their potential properties and functions. To date, the successful thickness modulation of 2D assemblies has only been reported in a few systems. For example, Li26 and Manners27–29 used crystalline polymers as building blocks to fabricate 2D platelets with different topologies and tunable surface functionalities. Since the driving force of the 2D platelet formation is the chain-folding crystallization of polymers, temperature is a critical parameter to tune the thickness. Conticello30,31 and co-workers reported the assembled 2D nanosheets from triple helices of collagen-mimetic peptides that consist of three sequential blocks with positively charged, neutral and negatively charged triads. They managed to increase the thickness of the 2D nanosheets via lengthening the triple helix unit through sequential additions of internal triads. In this case, the nanosheet thickness was positively related to the contour length of the building blocks.
Herein, we report a unique thickness control strategy of 2D nanosheets assembled from side-chain giant molecules32–35 consisting of consecutively connected hydrophobic/hydrophilic polyhedral oligomeric silsesquioxane (BPOSS/APOSS) derivatives as the pendant groups. The selection of POSS-based building blocks is rationalized by their unique preferred arrangement towards 2D aggregates, given a proper driving force.36,37 For instance, Jiang38–40 and co-workers were the first to report the 2D assembly of hyperbranched poly(ether amine) containing anthracene moieties and POSS. A self-seeding technique was applied to control the lateral sizes of the nanosheets. However, the authors did not show thickness control of the 2D nanosheets. In this study, we developed a precise synthetic route based on the thiol-maleimide “click” chemistry41 and “deprotection–addition” cycles42,43 to build a series of side-chain giant molecules with accurate numbers of POSS-based monomers and predetermined sequences. Notably, such chemical precision is a prerequisite to unambiguously deduce the relationship between the molecular parameters of building blocks and the thickness of their 2D assemblies. Intriguingly, we first found that the nanosheet thickness was decreased when only increasing numbers of BPOSS cages. That is to say, the thickness is not necessarily proportional to the contour length of the giant molecule, nor the total number of POSS cages tethered to the main chain. Furthermore, we revealed that the thickness was dictated by the number ratios of BPOSS to APOSS cages, rather than the exact numbers. We also proposed a schematic structure model and formula to semi-quantitatively explain this unusual behavior. We believe that our systematic study on the 2D self-assembly of precisely defined side-chain giant molecules would prompt a deep understanding on the formation rationale and structural modulation of self-assembled 2D nanostructures, and consequently, guiding the precise structural engineering of 2D nanomaterials towards potential functions and applications.44–46
Results and discussion
Synthesis and characterization of the side-chain giant molecules
In the literature, POSS functionalized with various reactive organic groups can be incorporated into virtually any existing polymer system through grafting, copolymerization or blending.47 Nevertheless, in most studies, the backbones of the POSS–polymer composite are synthesized using traditional polymerization techniques (e.g., free radical polymerization and condensation polymerization), and therefore, at least the molecular weights of the resulting composites are not precise. Inspired by the solid-state synthesis of peptides, Cheng was the first to synthesize chain-like giant molecules with precisely defined sequence and composition by interconnecting POSS of versatile functionalities.48–50 However, the reported synthetic route suffers from multi-step reactions, tedious purification and low yield of the target products. Here, we propose a combination of the thiol-maleimide “click” reaction and “deprotection–addition” cycles to advance the precise synthesis of chain-like giant molecules.The general synthetic route and chemical structures of the POSS-based monomers and the resulting giant molecules BnA (n = 1–5) are depicted in Scheme 1. The details are summarized in the ESI.† Here, the highly efficient and selective thiol-maleimide addition reaction, one of the most used “click” reactions, was exploited to compose the backbone of the linear giant molecules. The monomer Mal-XPOSS-ST, where X represents iso-butyl or vinyl groups, was prepared by first coupling a cysteine derivative (Fmoc-Cys-ST, N-(9-fluorenylmethoxycarbonyl)-S-trityl-L-cysteine) with an XPOSS through a routine amidation reaction. After the Fmoc group was carefully deprotected under basic conditions, the maleimide group was then introduced into the amino end. The resulting monomer Mal-XPOSS-ST has a reactive maleimide group (Mal) and a dormant trityl-thiol group (ST). The “deprotection–addition” cycle started from Fmoc-BPOSS-ST as shown in Scheme 1a. First, the trityl group was deprotected under acidic conditions, generating an active thiol group. The following addition reaction between thiol and maleimide groups was catalyzed by triethylamine. In order to avoid tedious separation associated with lots of COOH groups, an intermediate monomer Mal-VPOSS-ST (V denotes vinyl groups) was incorporated into the giant molecules as a precursor of the hydrophilic modification. After completing the synthesis of the target giant molecule, the vinyl groups of VPOSS can be readily transferred into carboxylic acid groups through radical addition by 2-mercaptoacetic acid (see Scheme 1b).
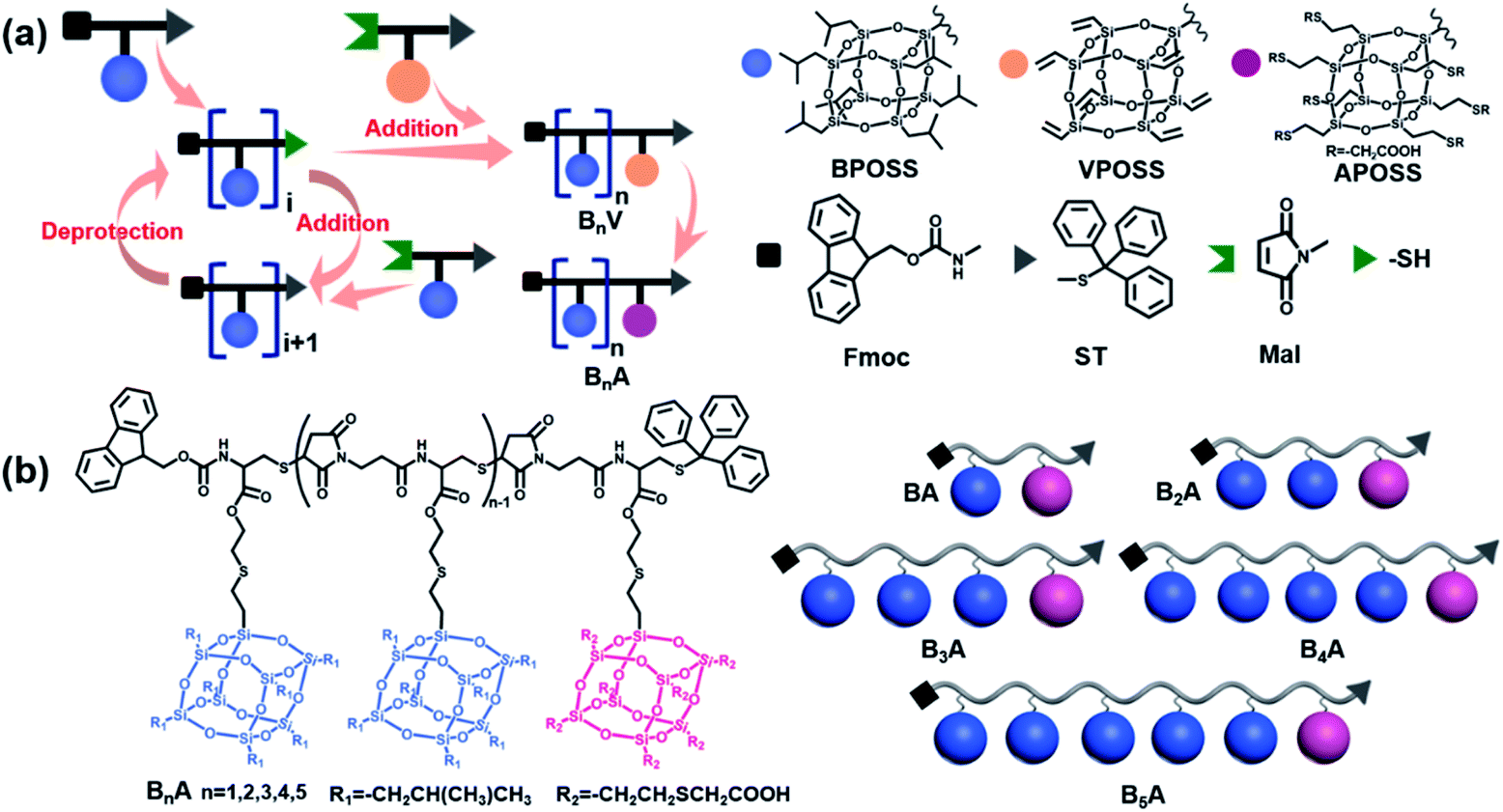 | ||
| Scheme 1 Synthetic routes and chemical structures of side-chain giant molecules. (a) Illustration of the general “deprotection–addition” cycle to prepare the giant molecules BnA (n = 1–5). The different colors and shapes represent specific POSS species or functional groups. (b) Chemical structures and cartoon illustrations of side-chain giant molecules BnA (n = 1–5). The blue spheres and pink spheres represent BPOSS and APOSS cages, respectively. | ||
Taking advantage of the “deprotection–addition” cycle, a series of linear giant molecules were feasibly prepared with specific numbers and sequences of BPOSS and APOSS cages. Fig. 1a summarizes the proton nuclear magnetic resonance (1H NMR) spectra from the monomer Mal-BPOSS-ST to the intermediate product B5V (see peak assignments in Scheme S1†). Four more intermediate products, BV–B4V, were accordingly synthesized and characterized by gel permeation chromatography (GPC) and matrix assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectra to confirm the structural precision. All GPC traces (Fig. 1b) of the intermediate BnV molecules were symmetric and narrowly distributed, showing the expected gradual shift towards low retention time with increased numbers of POSS cages. Additionally, in the MALDI-TOF mass spectra (Fig. 1c), five single lines exhibiting the measured molecular weights of the corresponding BnV molecules matched well with the calculated values, providing even starker proof for the successful synthesis and purity of products. Notably, the incorporation of VPOSS at the end of the giant molecule gives rise to the 1H NMR peaks at around δ 6.2–5.8 ppm. Those peaks totally disappear after all the vinyl groups react with 2-mercaptoacetic acid that transfers B5V to B5A. The detailed analysis of the 1H NMR spectra of other final products (BA–B4A) can be found in Fig. S3.†
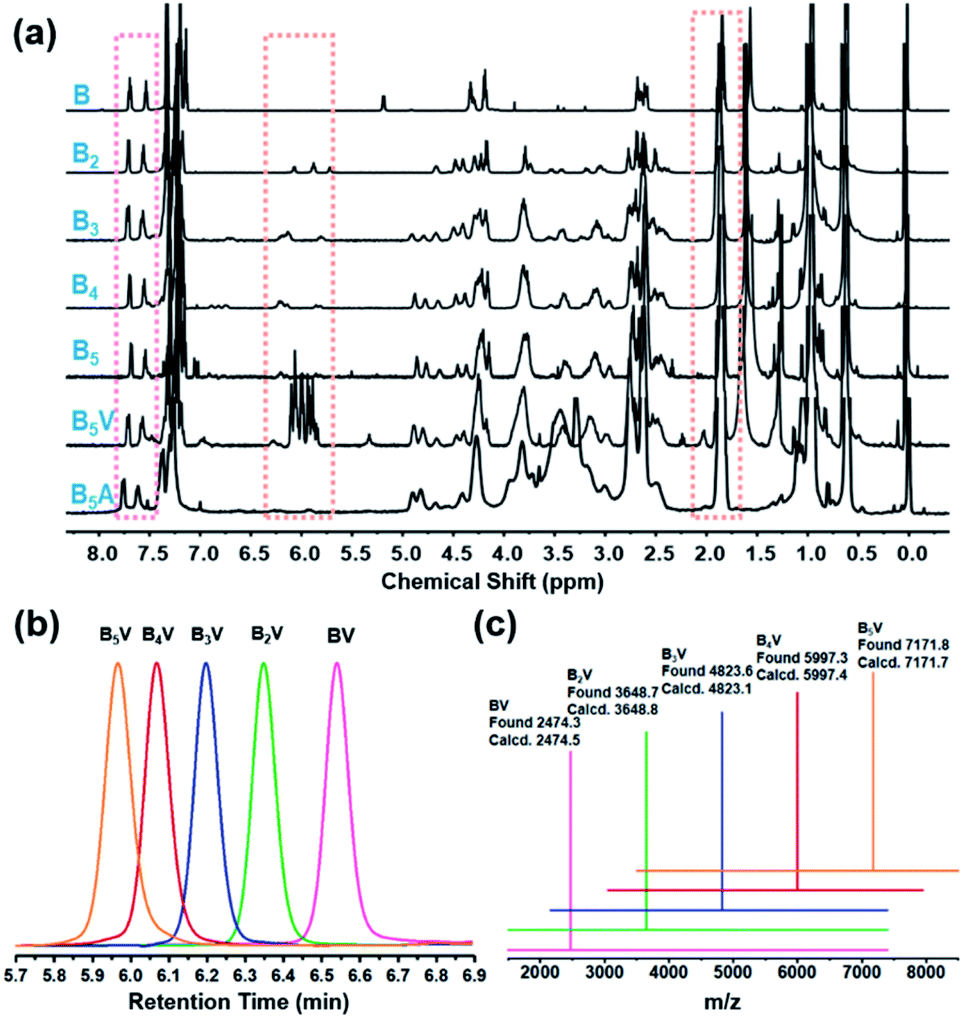 | ||
| Fig. 1 Molecular characterization of the giant molecules and the related intermediate compounds. (a) 1H NMR spectra of molecules from Fmoc-BPOSS-ST (denoted as “B” in the spectra) to one final product B5A. (b) GPC traces and (c) MALDI-TOF MS spectra of BnV (n = 1–5). All the samples in (c) show peaks of [M·Na]+ corresponding to the calculated molecular weights. | ||
Self-assembled 2D nanosheets of the BnA molecules
To facilitate the solution self-assembly process, a mixture of toluene and N,N-dimethylformamide (DMF) was applied to dissolve the BnA samples, since toluene and DMF are good solvents for BPOSS and APOSS, respectively. 2D nanosheets of BnA (n = 1–5) were readily obtained by placing a small droplet of the sample solution (∼0.2 mg mL−1) on a flat substrate and letting it slowly evaporate at ambient temperature. Considering the solubility difference of BnA (n = 1–5), optimization of the initial concentration and toluene/DMF ratio was individually performed (see details in the ESI†). Previously, we reported that the use of a base could deprotonate the carboxylic acid groups on the APOSS cage and result in the formation of 2D nanosheets.34 Thanks to the lower boiling point and faster evaporation rate of toluene than DMF, the BPOSS cages started to crystallize when the volume fraction of toluene decreased to a critical point. In this study, however, we found that no extra basic species was needed. One possible reason is that the carboxylic acid groups could be partially disassociated in DMF, providing electrostatic repulsion to inhibit the growth of the crystalline nanosheets along the normal direction.51,52 It is also reasonable that the solvation effect between DMF and APOSS cages helps stabilize the 2D nanosheets from physical stacking or aggregation.The bright-field (BF) transmission electron microscope (TEM) images of the 2D nanosheets assembled from BnA (n = 1–5) are displayed in Fig. 2a. Samples were prepared on carbon-coated mica substrates and then transported to copper grids after the carbon films were floated on a water surface. Obviously, all the samples were able to form discrete 2D nanosheets with the regular parallelogram shape. The lateral size of the nanosheets was around several micrometers. Moreover, atomic force microscopy (AFM) analysis of the nanosheets showed an average thickness of 6.2 ± 0.3 nm, 5.1 ± 0.2 nm, 4.6 ± 0.2 nm, 3.9 ± 0.2 nm, and 3.8 ± 0.2 nm for BA–B5A, respectively (Fig. 2b). Very interestingly, the observed thickness of the nanosheets gradually decreased as the number of BPOSS cages increased. The change in average thickness from this series of samples was also confirmed using X-ray scattering techniques. From the grazing incidence wide-angle X-ray diffraction (GIWAXD) data (Fig. 2c), strong diffractions along the substrate normal direction could be observed, which originated from those physically stacked nanosheets. The first order diffraction peaks suggested d-spacing values of 6.40 nm, 4.69 nm, 4.24 nm, 4.07 nm, and 3.95 nm for BA–B5A, respectively (Fig. 2d). Other diffraction arches were attributed to the inner crystalline BPOSS layers of the 2D nanosheets. The small angle X-ray scattering (SAXS) profiles collected from powder BnA samples also confirmed the d-spacing values. Although not exactly the same with the thickness values from AFM analysis due to the different measuring principles, the same trend where the thickness of the self-assembled nanosheets decreased with increasing BPOSS numbers could be clearly validated (Fig. 2e).
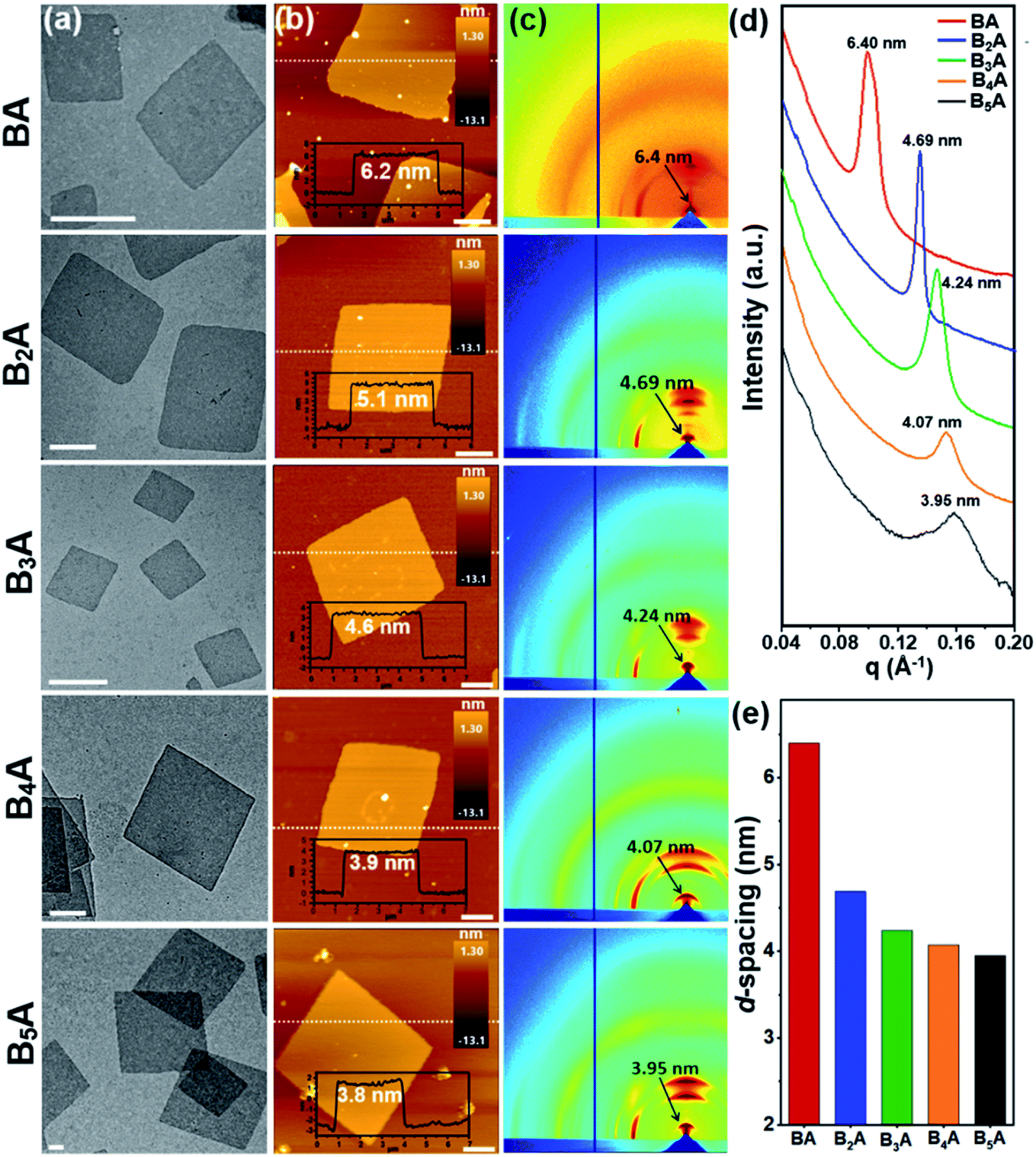 | ||
| Fig. 2 Characterization of 2D nanosheets assembled from BnA (n = 1–5). (a) TEM BF images of the 2D nanosheets (scale bar = 500 nm). (b) AFM images and height traces of the dashed lines shown as the insets (scale bar = 1 μm). (c) 2D GIWAXD patterns with the d-spacing values assigned to the first order diffraction arches. (d) SAXS profiles collected from powder samples in transmission mode. Intensity is vertically offset for clarity. (e) Histograms of the 2D nanosheet thickness based on the SAXS data in (d). | ||
Structure model construction and thickness formula deduction
From these results and our previous study,36,37 we hypothesized that such nanosheets were composed of a sandwich-like structure: two layers of crystalline BPOSS covered by two layers of amorphous APOSS/backbone (see the WAXD of APOSS in Fig. S6†). To prove this, selected area electron diffraction (SAED) was utilized to investigate the crystalline structure of the nanosheets. Fortunately, the 3D crystal structure of octaisobutylsilsesquioxane (T8 BPOSS) has already been determined to possess a triclinic unit cell53 with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (a = 1.00 nm, b = 1.08 nm, c = 1.10 nm, α = 96.54°, β = 91.27°, γ = 99.44°). We further hypothesized that the two BPOSS layers within the nanosheets can be regarded as two crystal planes dragged out from T8 BPOSS's 3D crystals (Table 1).
space group (a = 1.00 nm, b = 1.08 nm, c = 1.10 nm, α = 96.54°, β = 91.27°, γ = 99.44°). We further hypothesized that the two BPOSS layers within the nanosheets can be regarded as two crystal planes dragged out from T8 BPOSS's 3D crystals (Table 1).
| Sample | M total | M b , | N BPOSS | N APOSS | r | h AFM | d-Spacingd | Φ BPOSS , | Φ APOSS , | h calc. , |
|---|---|---|---|---|---|---|---|---|---|---|
| a Exact molecular weight (Da). b The averaged molecular weight of the backbone monomers calculated from total molecular weight of backbone divided by the sum of BPOSS and APOSS numbers. c The averaged value after measuring 10 individual nanosheets at different locations. d Calculated from the SAXS patterns using d = 2π/q. e Calculated volume fractions of BPOSS and APOSS. f Calculation carried out by approximating the densities of BPOSS, APOSS and backbone to be 1.25 g cm−3, 1.25 g cm−3 and 1.3 g cm−3, respectively. g Calculated 2D nanosheet thickness. | ||||||||||
| BA | 3098.6 | 455.6 | 1 | 1 | 1 | 6.2 | 6.40 | 29% | 43% | 6.99 |
| B2A | 4275 | 403.8 | 2 | 1 | 2 | 5.1 | 4.69 | 41% | 31% | 4.83 |
| B3A | 5451.2 | 378.3 | 3 | 1 | 3 | 4.6 | 4.24 | 49% | 24% | 4.11 |
| B4A | 6627.4 | 362.7 | 4 | 1 | 4 | 3.9 | 4.07 | 53% | 20% | 3.75 |
| B5A | 7803.7 | 352.2 | 5 | 1 | 5 | 3.8 | 3.95 | 57% | 17% | 3.53 |
| B2A2 | 5885.8 | 378.3 | 2 | 2 | 1 | 6.1 | 6.08 | 30% | 45% | 6.65 |
| B3A2 | 7061.7 | 362.7 | 3 | 2 | 1.5 | 4.9 | 4.89 | 38% | 37% | 5.32 |
| B4A2 | 8237.6 | 352.2 | 4 | 2 | 2 | 4.1 | 4.57 | 43% | 32% | 4.65 |
Computer simulation was then carried out with Accelrys Cerius2 software to calculate electron diffraction (ED) patterns of several possible crystal zones. Fig. 3a and S7† list the crystal packing of T8 BPOSS along the [001], [010] and [100] zones with their corresponding simulated ED patterns (Fig. 3b). Fig. 3c and d display the experimental SAED data collected from B3A, B4A and B5A nanosheets, respectively. It is quite clear that BnA (n = 1–5) shares the same SAED patterns that better match the simulated [001] zone ED pattern after we carefully compare the relative intensities of each diffraction point and reciprocal axis angles (see Fig. S8† for the SAED patterns of BA and B2A). Furthermore, wide angle X-ray diffraction (WAXD) was exploited to confirm the crystal structure of inner BPOSS layers of BA–B5A (Fig. S9†). The very similar WAXD patterns with tiny discrepancies, associated with the SAED analysis, indicate that the BPOSS cages in BA–B5A nanosheets take almost the same molecular packing scheme. After clarifying this point, we can go back to explaining the thickness decreasing tendency with increasing BPOSS numbers.
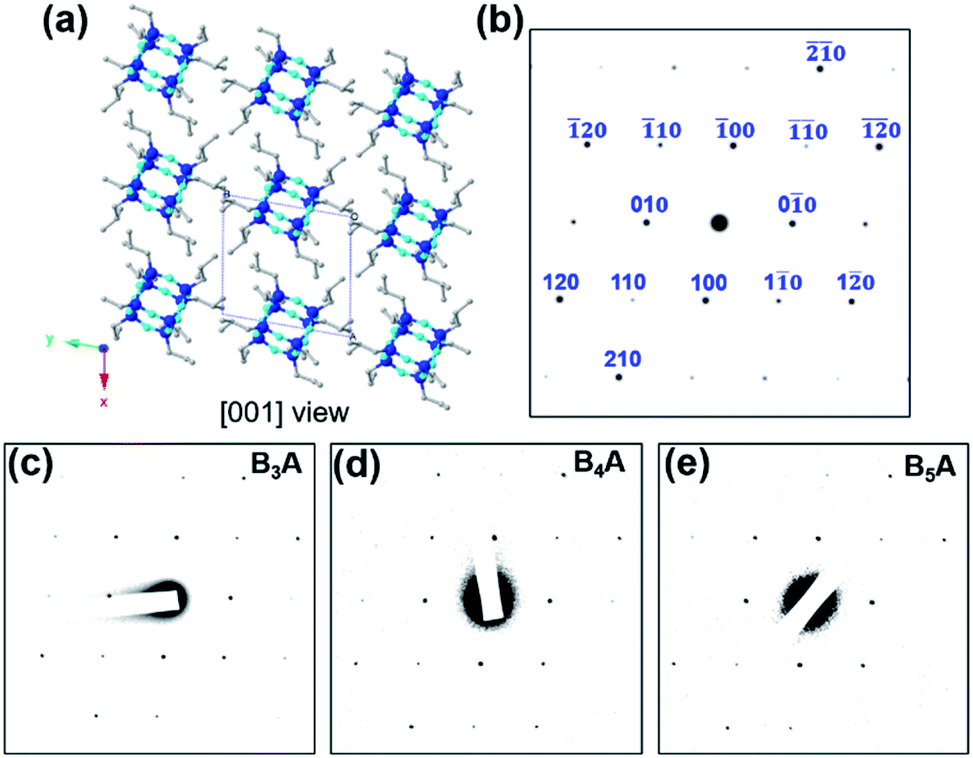 | ||
| Fig. 3 Structure identification of BPOSS bilayers in the 2D nanosheets. (a) [001] zone view of the crystal lattice model of T8 BPOSS, and (b) the corresponding simulated ED pattern with indices. (c), (d) and (e) are experimental SAED patterns of 2D nanosheets assembled from B3A, B4A and B5A. Color inversion was conducted on the original SAED data for a better contrast. | ||
In Fig. 4, we schematically depict the overall 2D nanosheet structures of three samples. It should be noted that the APOSS cages and backbones have no long-range translational or rotational order. Apparently, the total thickness originates from three parts: the crystalline BPOSS bilayers, amorphous backbones and amorphous APOSS layers. As mentioned above, all the nanosheets possess the same crystalline BPOSS bilayers which contribute around 2.0 nm to the total thickness based on its unit cell size. The rest contributions are from the amorphous backbone and APOSS cages. Accounting for the geometry of the nanosheets, a simple calculation gives the following formula of the total thickness h (see detailed deductions in the ESI†):
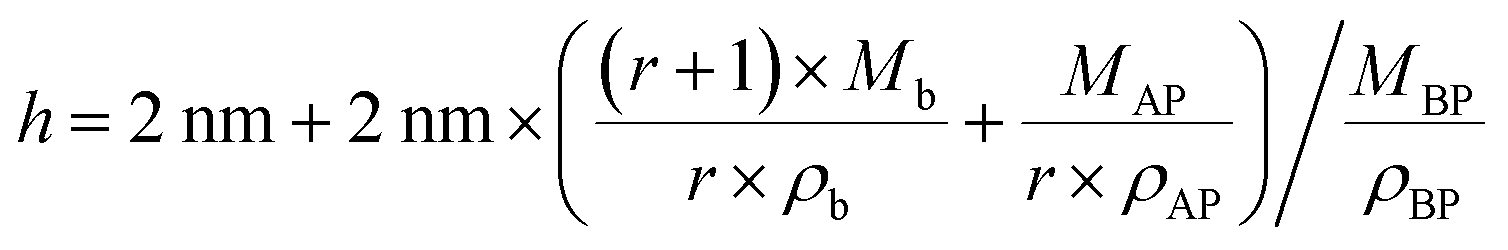 | (1) |
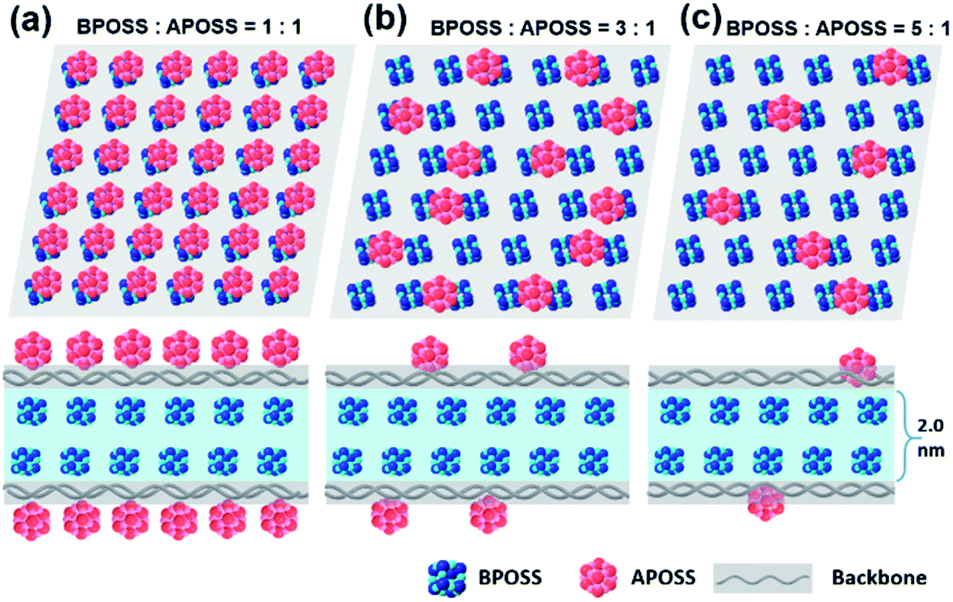 | ||
| Fig. 4 Representative illustration of the thickness declining tendency with increased number ratios of BPOSS to APOSS. (a), (b) and (c) are top and side views of the proposed structure configurations of BA, B3A, and B5A, respectively. Some of the chemical structure details are omitted for clarity. BPOSS layers are crystalline, but APOSS and backbone layers are not supposed to possess any long-range order. | ||
Validation of the structure model by the BnA2 molecules
The models shown in Fig. 4 associated with eqn (1) can reasonably explain our experimental observations on the self-assembly behaviors of the BnA molecules. As mentioned in our previous publications,36,37 the prerequisite of nanosheet formation is that the number of BPOSS in the giant molecule would not be smaller than that of the APOSS. Otherwise, the BPOSS cages cannot crystallize to form the inner bilayers due to the steric hindrance. To prove it, we synthesized BA2 and no nanosheets were obtained from this sample (see details in the ESI and Fig. S10†). Therefore, eqn (1) is only valid when r ≥ 1. From eqn (1), it can be further concluded that at a fixed number of BPOSS, increasing the number of APOSS will enlarge the total thickness of 2D nanosheets as long as r ≥ 1 is fulfilled. To test this deduction, three more samples B2A2, B3A2 and B4A2 were intentionally synthesized (Fig. 5a) and characterized (see details in the ESI†). Their r values are 1, 1.5 and 2, respectively.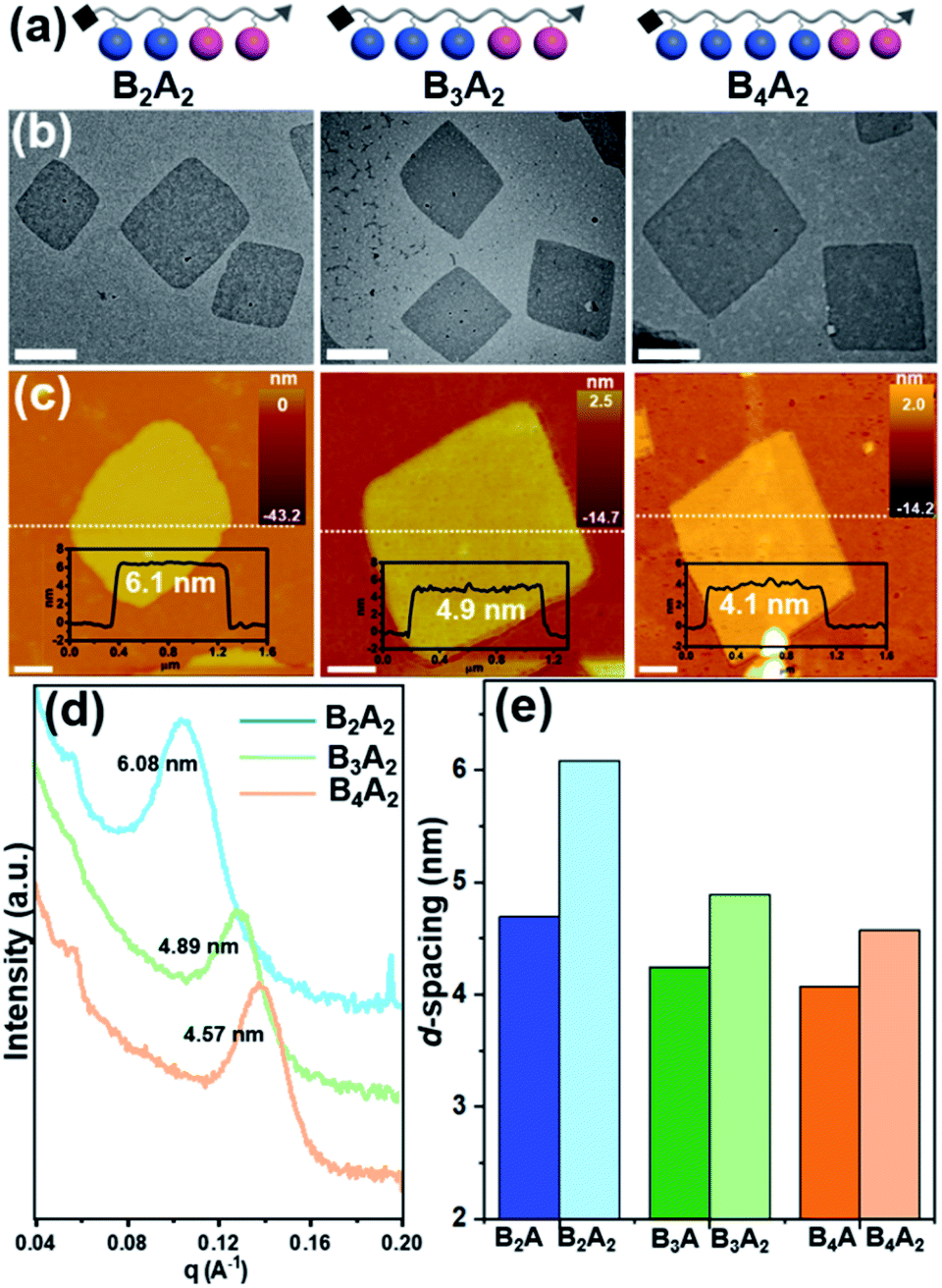 | ||
| Fig. 5 Characterization of 2D nanosheets assembled from BnA2 (n = 2–4). (a) Cartoon illustrations of side-chain giant molecules BnA2 (n = 2–4). (b) TEM BF images of the 2D nanosheets (scale bar = 500 nm). (c) AFM images and height traces of the dashed lines shown as the insets (scale bar = 1 μm). (d) SAXS profiles collected from powder samples in transmission mode. Intensity is vertically offset for clarity. (e) Histograms of the 2D nanosheet thickness based on the SAXS data in (c) and Fig. 2d. | ||
The 2D nanosheets for microscopy characterization were accordingly prepared by the abovementioned methods. BF TEM images in Fig. 5b suggest all the three nanosheet samples hold the same parallelogram shapes with lateral size around several micrometers (see SAED patterns in Fig. S8†). AFM measurements in Fig. 5c give the averaged thickness of 6.1 ± 0.2 nm, 4.9 ± 0.2 nm and 4.4 ± 0.2 nm for B2A2, B3A2 and B4A2, respectively. The corresponding d-spacing values obtained from SAXS are 6.08 nm, 4.89 nm and 4.57 nm (Fig. 5d), matching well with the AFM results. On the other hand, the d-spacing values of B2A, B3A and B4A are 4.82 nm, 4.23 nm and 3.89 nm (Fig. 2d). A comparative histogram in Fig. 5e directly reflects the enlarged thickness with increasing APOSS numbers at fixed BPOSS numbers (4.82 nm → 6.08 nm, 4.23 nm → 4.89 nm and 3.89 nm → 4.57 nm). Finally, even at the same r value, the thickness of 2D nanosheets is still affected by the absolute number of backbone monomers. Such an impact can be revealed by comparing the d-spacing values of BA/B2A2 (6.40 nm/6.08 nm) and B2A/B4A2 (4.82 nm/4.57 nm). Although the density of the amorphous component is identical with the same r, the averaged Mb values are different for BA/B2A2 (456 g mol−1/362 g mol−1) and B2A/B2A2 (378 g mol−1/362 g mol−1). Therefore, the total thickness is also influenced by the two bulky end groups. In the future study, we plan to design sequential or regio-isomers54,55 to gain a deeper understanding on the counterintuitive molecular packing of these 2D materials.
Conclusions
In summary, a family of precisely defined side-chain giant molecules with different contour lengths and compositions (BnAi, n = 1–5, i = 1–2) were modularly synthesized via the “deprotection–addition” cycle. Under optimized conditions, such giant molecules can self-assemble into 2D nanosheets through a simple solvent evaporation process. Those nanosheets have a sandwich-like structure configuration. We further observed that the thickness of obtained nanosheets was very sensitive to the structural parameters of the side-chain giant molecules, specifically the number ratio of BPOSS to APOSS. For BnA (n = 1–5, r = 1–5) with one APOSS located at the chain end, the nanosheet thickness gradually decreases with increasing numbers of BPOSS, as evidenced by AFM, GIWAXD and SAXS measurements. A simple structure model was proposed to explain the variation in thickness, from which a formula was deduced to describe the decreasing tendency of nanosheet thickness with increasing r values. Importantly, this model was reaffirmed by the BnA2 (n = 2–4) samples. This study has clarified the relationship between the chemical structure of giant molecule chains and the thickness of their 2D assemblies. We believe that this study represents a profound understanding towards the fabrication and structural control of 2D nanostructures via the self-assembly strategy and would also expand our toolbox for structure engineering of 2D functional nanomaterials.Author contributions
F. F., H. L. and K. Y. designed the studies and prepared the manuscript. D. G. and X. Y. carried out the preliminary synthesis of the monomers. F. F. and Z. P. modified the premature synthetic routes and finished the synthesis/characterization of all samples. F. F. carried out all the AFM tests. TEM characterization, including bright field images and SAED patterns, was conducted by X. L. and D. X. GIWAXD experiments were carried out by D. G. SAXS characterization was finished by Y. S. and W.-B. Z. All GPC experiments were conducted by L. J. Computer simulation was carried out by H. L. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22003008) and the Science and Technology Commission of Shanghai Municipality (No. 19YF1400400). The authors also thank Prof. Jinlin He and Prof. Zhengbiao Zhang at Soochow University for assistance with the MALDI-TOF mass spectrometry. We had very constructive suggestions from Prof. Bernard Lotz at Université de Strasbourg for the structure model building and formula deduction. We thank the onsite staff of Beamline BL16B1 at the Shanghai Synchrotron Radiation Facility (SSRF) for assistance with the SAXS experiments and staff of Sector 8-ID-E at the Advanced Photon Source of Argonne National Laboratory for assistance with the GIWAXD measurements.References
- X. Zhang and Y. Xie, Chem. Soc. Rev., 2013, 42, 8187–8199 RSC.
- P. Zhang, F. Wang, M. Yu, X. Zhuang and X. Feng, Chem. Soc. Rev., 2018, 47, 7426–7451 RSC.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS.
- A. Splendiani, L. Sun, Y. Zhang, T. Li, J. Kim, C. Y. Chim, G. Galli and F. Wang, Nano Lett., 2010, 10, 1271–1275 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2009, 110, 132–145 CrossRef PubMed.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano, V. R. Cooper, L. Liang, S. G. Louie, E. Ringe, W. Zhou, S. S. Kim, R. R. Naik, B. G. Sumpter, H. Terrones, F. Xia, Y. Wang, J. Zhu, D. Akinwande, N. Alem, J. A. Schuller, R. E. Schaak, M. Terrones and J. A. Robinson, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS.
- X. Zhang, L. Hou, A. Ciesielski and P. Samorì, Adv. Energy Mater., 2016, 6, 1600671 CrossRef.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS PubMed.
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568–571 CrossRef CAS PubMed.
- L. Liu, L. H. Klausen and M. Dong, Nano Today, 2018, 23, 40–58 CrossRef CAS.
- K. T. Nam, S. A. Shelby, P. H. Choi, A. B. Marciel, R. Chen, L. Tan, T. K. Chu, R. A. Mesch, B. C. Lee, M. D. Connolly, C. Kisielowski and R. N. Zuckermann, Nat. Mater., 2010, 9, 454–460 CrossRef CAS PubMed.
- Y. Lin, M. R. Thomas, A. Gelmi, V. Leonardo, E. T. Pashuck, S. A. Maynard, Y. Wang and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 13592–13595 CrossRef CAS PubMed.
- W. Bai, Z. Jiang, A. E. Ribbe and S. Thayumanavan, Angew. Chem., Int. Ed., 2016, 55, 10707–10711 CrossRef CAS PubMed.
- C. E. Boott, A. Nazemi and I. Manners, Angew. Chem., Int. Ed., 2015, 54, 13876–13894 CrossRef CAS PubMed.
- Y. Kim, S. Shin, T. Kim, D. Lee, C. Seok and M. Lee, Angew. Chem., Int. Ed., 2013, 52, 6426–6429 CrossRef CAS PubMed.
- B. Li and C. Y. Li, J. Am. Chem. Soc., 2006, 129, 12–13 CrossRef PubMed.
- M.-S. Hsiao, W. Y. Chen, J. X. Zheng, R. M. Van Horn, R. P. Quirk, D. A. Ivanov, E. L. Thomas, B. Lotz and S. Z. D. Cheng, Macromolecules, 2008, 41, 4794–4801 CrossRef CAS.
- Y. Lin, M. R. Thomas, A. Gelmi, V. Leonardo, E. T. Pashuck, S. A. Maynard, Y. Wang and M. M. Stevens, J. Am. Chem. Soc., 2017, 139, 13592–13595 CrossRef CAS PubMed.
- E. J. Robertson, A. Battigelli, C. Proulx, R. V. Mannige, T. K. Haxton, L. Yun, S. Whitelam and R. N. Zuckermann, Acc. Chem. Res., 2016, 49, 379–389 CrossRef CAS PubMed.
- L. Dou, A. B. Wong, Y. Yu, M. Lai, N. Kornienko, S. W. Eaton, A. Fu, C. G. Bischak, J. Ma, T. Ding, N. S. Ginsberg, L.-W. Wang, A. P. Alivisatos and P. Yang, Science, 2015, 349, 1518 CrossRef CAS PubMed.
- A. Ciarrocchi, A. Avsar, D. Ovchinnikov and A. Kis, Nat. Commun., 2018, 9, 919 CrossRef PubMed.
- C. Tan, X. Cao, X.-J. Wu, Q. He, J. Yang, X. Zhang, J. Chen, W. Zhao, S. Han, G.-H. Nam, M. Sindoro and H. Zhang, Chem. Rev., 2017, 117, 6225–6331 CrossRef CAS PubMed.
- B. Li, C. Ni and C. Y. Li, Macromolecules, 2007, 41, 149–155 CrossRef.
- X. He, Y. He, M.-S. Hsiao, R. L. Harniman, S. Pearce, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2017, 139, 9221–9228 CrossRef CAS PubMed.
- X. He, M.-S. Hsiao, C. E. Boott, R. L. Harniman, A. Nazemi, X. Li, M. A. Winnik and I. Manners, Nat. Mater., 2017, 16, 481–488 CrossRef CAS PubMed.
- Z. M. Hudson, C. E. Boott, M. E. Robinson, P. A. Rupar, M. A. Winnik and I. Manners, Nat. Chem., 2014, 6, 893–898 CrossRef CAS PubMed.
- A. D. Merg, G. Touponse, E. van Genderen, X. Zuo, A. Bazrafshan, T. Blum, S. Hughes, K. Salaita, J. P. Abrahams and V. P. Conticello, Angew. Chem., Int. Ed., 2019, 131, 13641–13646 CrossRef.
- A. D. Merg, E. van Genderen, A. Bazrafshan, H. Su, X. Zuo, G. Touponse, T. B. Blum, K. Salaita, J. P. Abrahams and V. P. Conticello, J. Am. Chem. Soc., 2019, 141, 20107–20117 CrossRef CAS PubMed.
- W. B. Zhang, X. F. Yu, C. L. Wang, H. J. Sun, I. F. Hsieh, Y. W. Li, X. H. Dong, K. Yue, R. Van Horn and S. Z. D. Cheng, Macromolecules, 2014, 47, 1221–1239 CrossRef CAS.
- W.-B. Zhang and S. Z. D. Cheng, Giant, 2020, 1, 100011 CrossRef.
- Y. Liu, T. Liu, X.-y. Yan, Q.-Y. Guo, J. Wang, R. Zhang, S. Zhang, Z. Su, J. Huang, G.-X. Liu, W. Zhang, W. Zhang, T. Aida, K. Yue, M. Huang and S. Z. D. Cheng, Giant, 2020, 4, 100031 CrossRef.
- G. Li, Z. Gan, Y. Liu, S. Wang, Q.-Y. Guo, Z. Liu, R. Tan, D. Zhou, D. Kong, T. Wen and X.-H. Dong, ACS Nano, 2020, 14, 13816–13823 CrossRef CAS PubMed.
- H. Liu, C.-H. Hsu, Z. Lin, W. Shan, J. Wang, J. Jiang, M. Huang, B. Lotz, X. Yu, W.-B. Zhang, K. Yue and S. Z. D. Cheng, J. Am. Chem. Soc., 2014, 136, 10691–10699 CrossRef CAS.
- H. Liu, J. Luo, W. Shan, D. Guo, J. Wang, C.-H. Hsu, M. Huang, W. Zhang, B. Lotz, W.-B. Zhang, T. Liu, K. Yue and S. Z. D. Cheng, ACS Nano, 2016, 10, 6585–6596 CrossRef CAS.
- B. Yu, X. Jiang and J. Yin, Macromolecules, 2014, 47, 4761–4768 CrossRef CAS.
- B. Yu, X. Jiang and J. Yin, Chem. Commun., 2013, 49, 603–605 RSC.
- B. Yu, X. Jiang and J. Yin, Macromolecules, 2012, 45, 7135–7142 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- Z. Huang, Q. Shi, J. Guo, F. Meng, Y. Zhang, Y. Lu, Z. Qian, X. Li, N. Zhou, Z. Zhang and X. Zhu, Nat. Commun., 2019, 10, 1918 CrossRef PubMed.
- Z. Liu, Z. Yang, X. Chen, R. Tan, G. Li, Z. Gan, Y. Shao, J. He, Z. Zhang, W. Li, W.-B. Zhang and X.-H. Dong, JACS Au, 2021, 1, 79–86 CrossRef CAS.
- Z. Li, H. Li, J. Zhang, X. Liu, Z. Gu and Y. Li, Chin. J. Polym. Sci., 2020, 38, 1149–1156 CrossRef CAS.
- Z. Li, J. Hu, L. Yang, X. Zhang, X. Liu, Z. Wang and Y. Li, Nanoscale, 2020, 12, 11395–11415 RSC.
- Z. Li, J. Zhang, Y. Fu, L. Yang, F. Zhu, X. Liu, Z. Gu and Y. Li, J. Mater. Chem. B, 2020, 8, 7018–7023 RSC.
- S.-W. Kuo and F.-C. Chang, Prog. Polym. Sci., 2011, 36, 1649–1696 CrossRef CAS.
- W. Zhang, M. Huang, H. Su, S. Zhang, K. Yue, X.-H. Dong, X. Li, H. Liu, S. Zhang, C. Wesdemiotis, B. Lotz, W.-B. Zhang, Y. Li and S. Z. D. Cheng, ACS Cent. Sci., 2016, 2, 48–54 CrossRef CAS PubMed.
- W. Zhang, S. Zhang, Q. Guo, X. Lu, Y. Liu, J. Mao, C. Wesdemiotis, T. Li, Y. Li and S. Z. D. Cheng, ACS Macro Lett., 2018, 7, 635–640 CrossRef CAS.
- W. Zhang, X. Lu, J. Mao, C.-H. Hsu, G. Mu, M. Huang, Q. Guo, H. Liu, C. Wesdemiotis, T. Li, W.-B. Zhang, Y. Li and S. Z. D. Cheng, Angew. Chem., Int. Ed., 2017, 56, 15014–15019 CrossRef CAS PubMed.
- X. Yu, W.-B. Zhang, K. Yue, X. Li, H. Liu, Y. Xin, C.-L. Wang, C. Wesdemiotis and S. Z. D. Cheng, J. Am. Chem. Soc., 2012, 134, 7780–7787 CrossRef CAS PubMed.
- Y. A. Fadeeva and L. P. Safonova, J. Solution Chem., 2011, 40, 980–988 CrossRef CAS.
- A. R. Bassindale, Z. Liu, I. A. MacKinnon, P. G. Taylor, Y. Yang, M. E. Light, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2003, 14, 2945–2949 RSC.
- Y. Shao, S. Yang and W.-B. Zhang, Chem.–Eur. J., 2020, 26, 2985–2992 CrossRef CAS PubMed.
- X.-M. Wang, Y. Shao, P.-F. Jin, W. Jiang, W. Hu, S. Yang, W. Li, J. He, P. Ni and W.-B. Zhang, Macromolecules, 2018, 51, 1110–1119 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The details of synthesis, chemical structure characterization, additional results and discussions. See DOI: 10.1039/d1sc00021g |
| This journal is © The Royal Society of Chemistry 2021 |