DOI:
10.1039/D1SC00438G
(Edge Article)
Chem. Sci., 2021,
12, 5224-5229
Redox-induced reversible [2 + 2] cycloaddition of an etheno-fused diporphyrin†
Received
24th January 2021
, Accepted 23rd February 2021
First published on 24th February 2021
Abstract
3,5-Ethenoporphyrin is a π-extended porphyrin containing a fused ethene unit between the meso- and β-positions, exhibiting unique contribution of macrocyclic antiaromaticity. We have recently reported that its analogue, etheno-fused diporphyrin, underwent thermal [2 + 2] cycloaddition to furnish X-shaped cyclobutane-linked tetraporphyrins. Here we demonstrate that the cyclobutane-ring formation is dynamically redox-active. Namely, the tetraporphyrin underwent two-step four-electron oxidation to afford two etheno-fused diporphyrin dications. The reduction of the resulting dication regenerated the cyclobutane-linked tetraporphyrin. The dication was sufficiently stable to allow its isolation under ambient conditions. The structure of the dication has been confirmed by 1H NMR spectroscopy and X-ray diffraction analysis. Importantly, the simultaneous double C–C bond cleavage in the cyclopropane ring in the tetraporphyrin is exceptional among dynamic redox (dyrex) systems to achieve large structural changes, thus offering new insights for the design of novel redox-active functional organic materials for electrochromic dyes, organic batteries, and organic memories.
Introduction
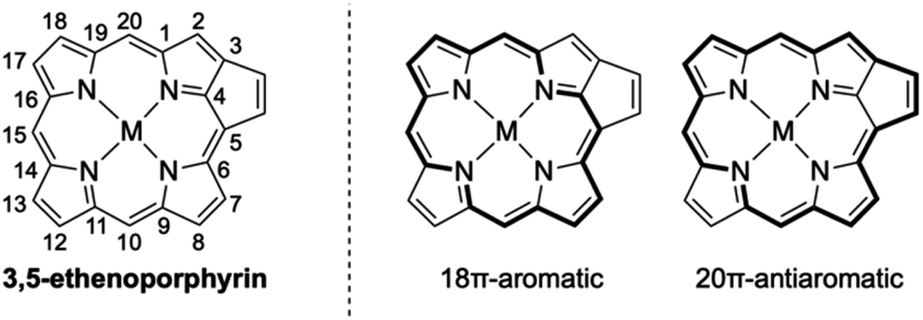
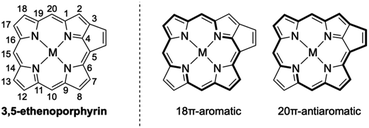
Porphyrins with extended π-conjugation networks exhibit numerous intriguing properties, such as near-infrared absorption, reversible redox activity, characteristic chemical reactivity, and high single-molecule conductance.1 Such porphyrins have attracted considerable attention in various research fields including organic and supramolecular chemistry as well as materials science. 3,5-Ethenoporphyrin is an extraordinary π-extended porphyrin due to the coexistence of 18π-aromaticity and 20π-antiaromaticity in its macrocyclic conjugation (Fig. 1).2,3 Consequently, 3,5-ethenoporphyrin exhibits a narrow HOMO–LUMO gap and high reactivity of the fused C–C double bond.
 |
| | Fig. 1 Aromatic and anti-aromatic conjugation circuits of 3,5-ethenoporphyrin. | |
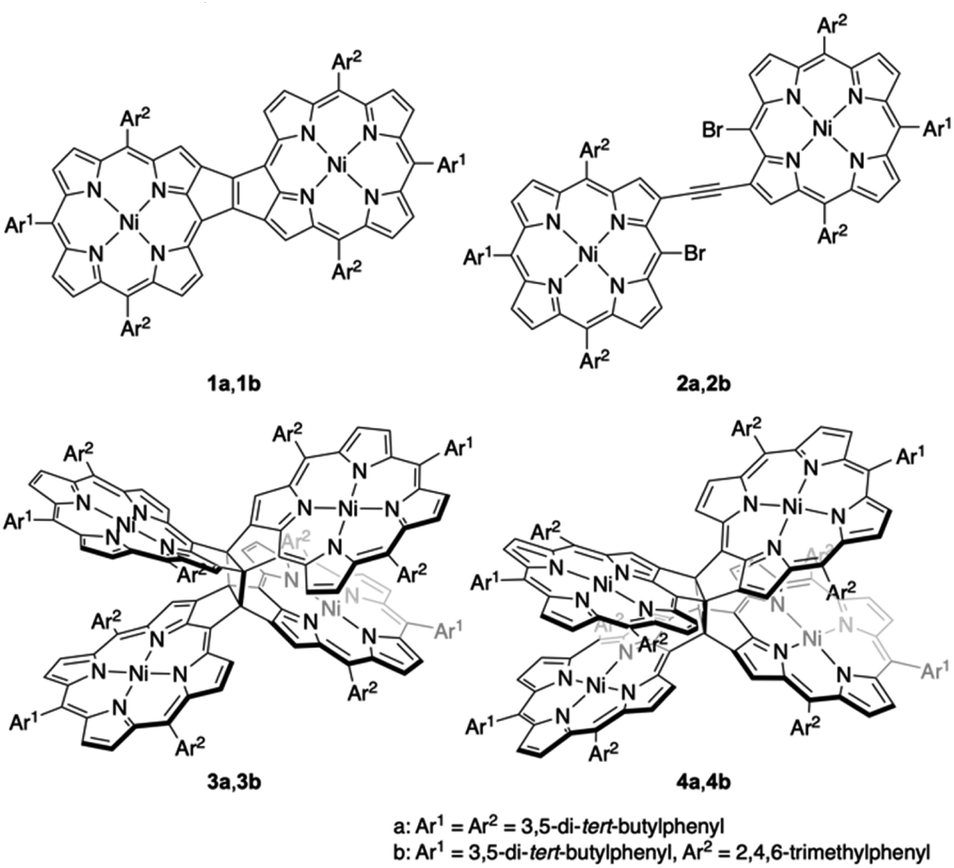
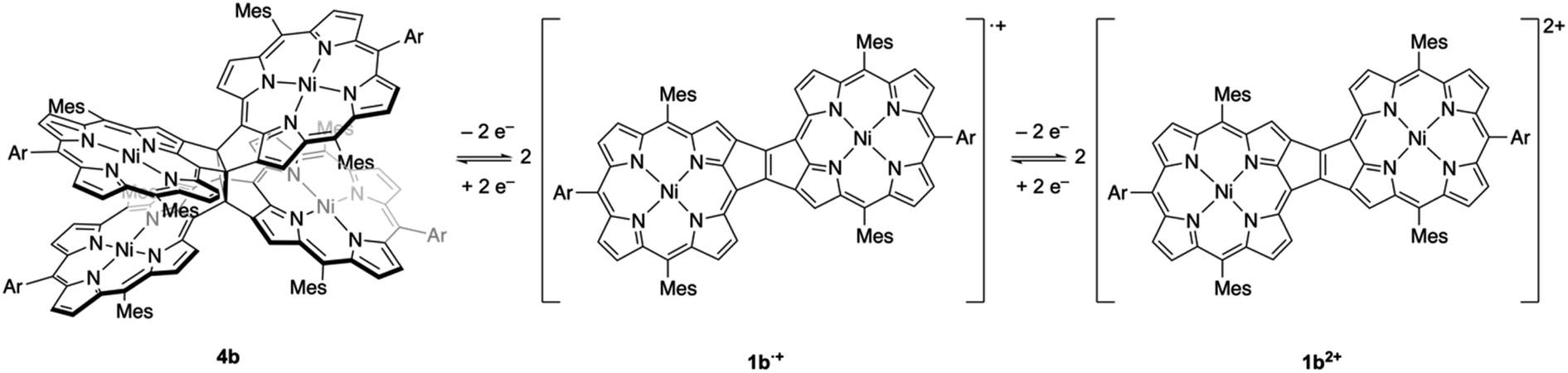
Recently, our research group envisaged the addition of another fused-porphyrin unit to the 3,5-ethenoporphyrin skeleton and attempted the synthesis of etheno-fused diporphyrin 1avia the tandem double-cyclization of β,β-ethynylene-linked dibromodiporphyrin 2a (Fig. 2).4 Unexpectedly, we discovered the formation of cyclobutane-linked tetraporphyrins 3a and 4a. These two tetraporphyrins were formed through the thermal [2 + 2] cycloaddition reaction of in situ-generated 1a. Due to orbital symmetry, the formation of the cyclobutane in 3avia a [2 + 2] cycloaddition is thermally forbidden. Thus, the formation of 3a and 4a suggests the involvement of a thermally activated triplet state of 1a in the thermal [2 + 2] cycloaddition reaction. Indeed, syn-tetramer 4a isomerizes to anti-tetramer 3a upon heating to 160 °C, which implies that the [2 + 2] cycloaddition of 1a is thermally reversible.
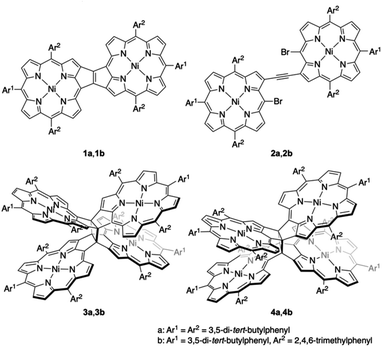 |
| | Fig. 2 Etheno-fused diporphyrin 1, ethynylene-linked dibromodiporphyrin 2, and cyclobutane-linked tetraporphyrins 3 and 4. | |
We then decided to conduct further investigations into these cyclobutane-linked tetraporphyrins with a focus on their redox properties. Here, we disclose a reversible [2 + 2] cycloaddition through electron-transfer-induced cyclobutane ring-opening and -closure. This redox-induced reversible C–C-bond formation constitutes a dynamic redox (dyrex) system; such systems have been actively explored on account of their potential importance as electrochromic dyes, organic batteries, and organic memory devices.5,6 Importantly, the simultaneous double C–C-bond formation/cleavage observed in the tetraporphyrin system is exceptional among reported dyrex systems.
Results and discussion
Synthesis and characterization of cyclobutane-linked tetraporphyrin
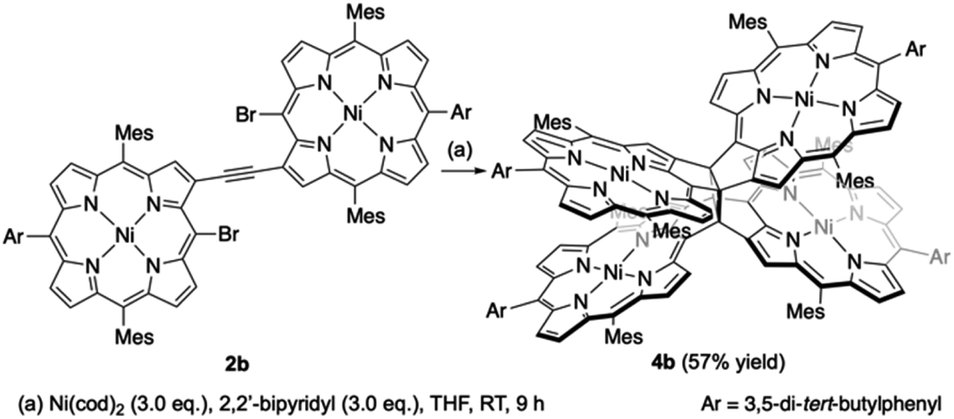
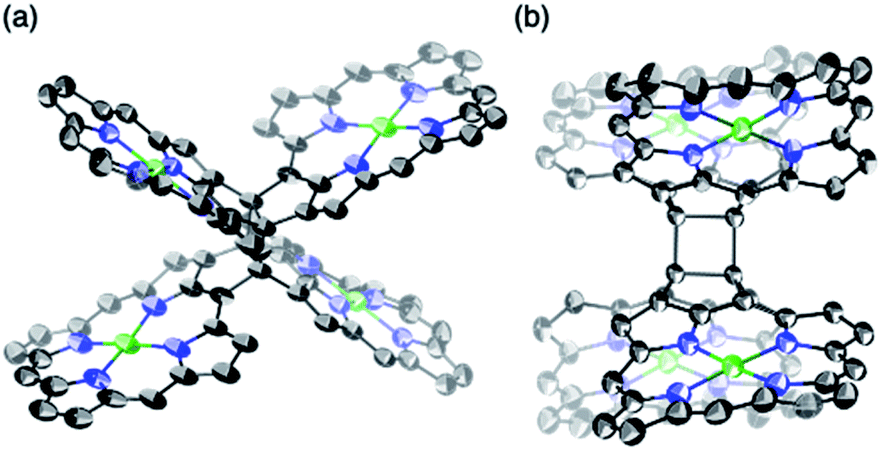
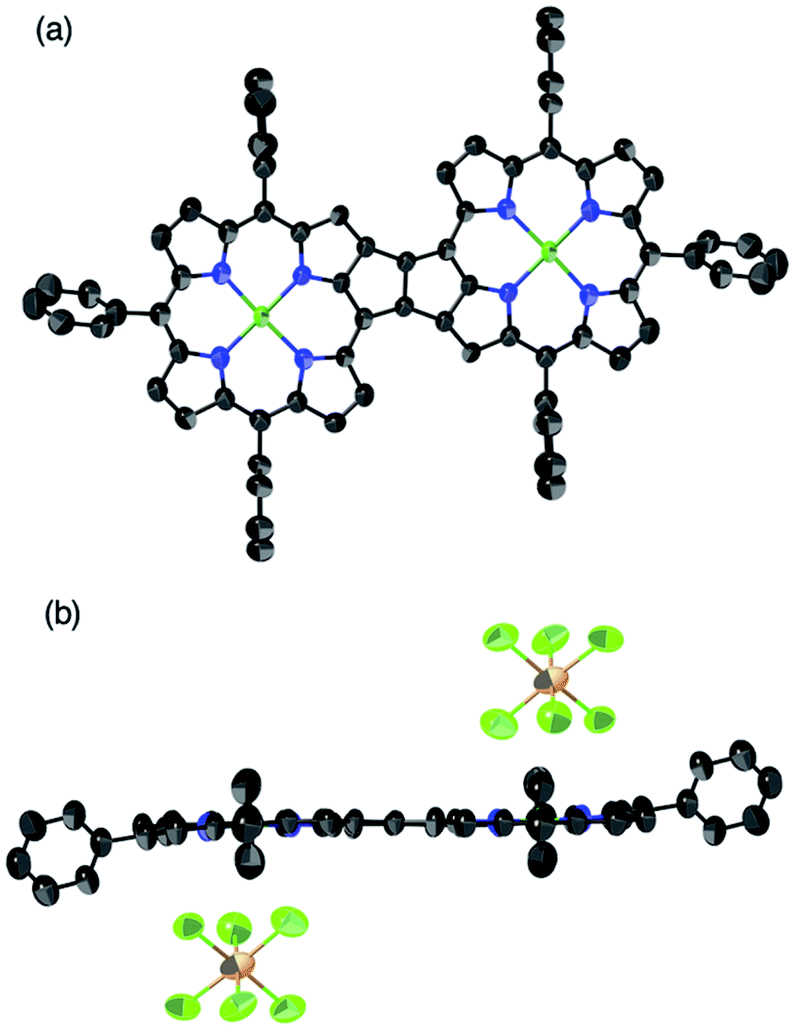
β,β-Ethynylene-linked dibromodiporphyrin 2b was prepared according to a slightly modified literature procedure.4,7 Precursor 2b was subjected to a tandem double cyclization with Ni(cod)2, which afforded syn-tetramer 4b in 57% yield without the formation of anti-tetramer 3b (Scheme 1). anti-Tetramer 3b was not obtained even when the reaction temperature was increased to 60 °C. The selective formation of 4b is due to the steric effect by bulky mesityl groups at the meso-positions. The structure of syn-tetramer 4b was unambiguously determined by single-crystal X-ray diffraction analysis (Fig. 3).
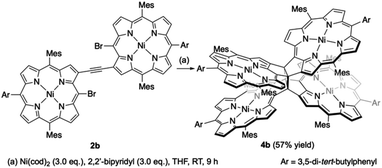 |
| | Scheme 1 Synthesis of syn-tetramer 4b. | |
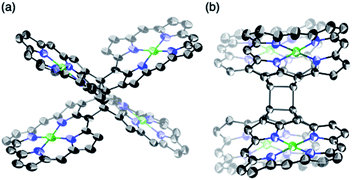 |
| | Fig. 3 Single-crystal X-ray crystal structure of syn-tetramer 4b. (a) General and (b) side view. Thermal ellipsoids are drawn at 50% probability. Solvent molecules, peripheral aryl groups, and all hydrogen atoms are omitted for clarity. | |
Dyrex response of cyclobutane-linked tetraporphyrin
Cyclic voltammogram.
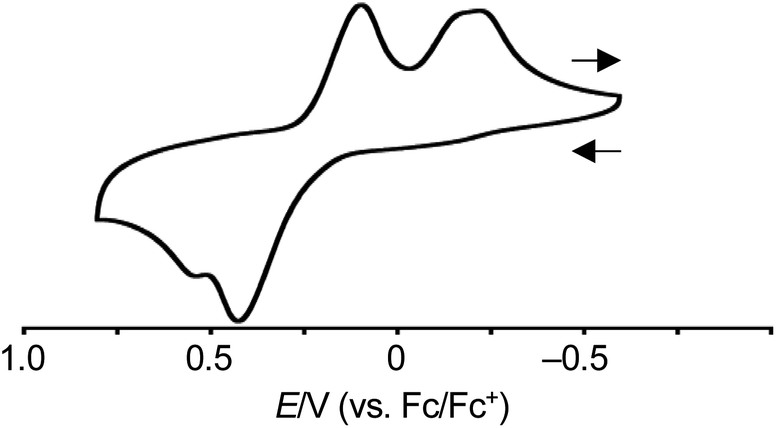
The redox behavior of syn-tetramer 4b was explored. The cyclic voltammogram of 4b was measured in CH2Cl2 with tetrabutylammonium hexafluorophosphate as the supporting electrolyte (Fig. 4 and S20†). The ferrocene/ferrocenium couple (Fc/Fc+) was used as an external reference. In the sweep from −0.60 V to 0.81 V, two peaks were observed at 0.43 and 0.56 V. These values are comparable to the first oxidation peak of porphyrin Ni(II) complexes (ca. 0.58 V).8 In the case of porphyrin, the subsequent back-sweep generates a reduction wave at ca. 0.38 V. In contrast to porphyrin, syn-tetramer 4b displayed reduction peaks at much lower values (0.10 and −0.29 V). The large displacement between the oxidation and reduction peaks implies the occurrence of dynamic structural changes.5,6
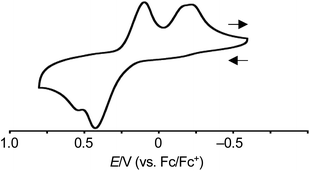 |
| | Fig. 4 Cyclic voltammogram of 4b. Solvent: CH2Cl2; supporting electrolyte: 0.1 M [Bu4N][PF6]; working electrode: glassy carbon; counter electrode: Pt; reference electrode: Ag/AgNO3; scan rate: 0.1 V s−1. | |
Oxidative titration.
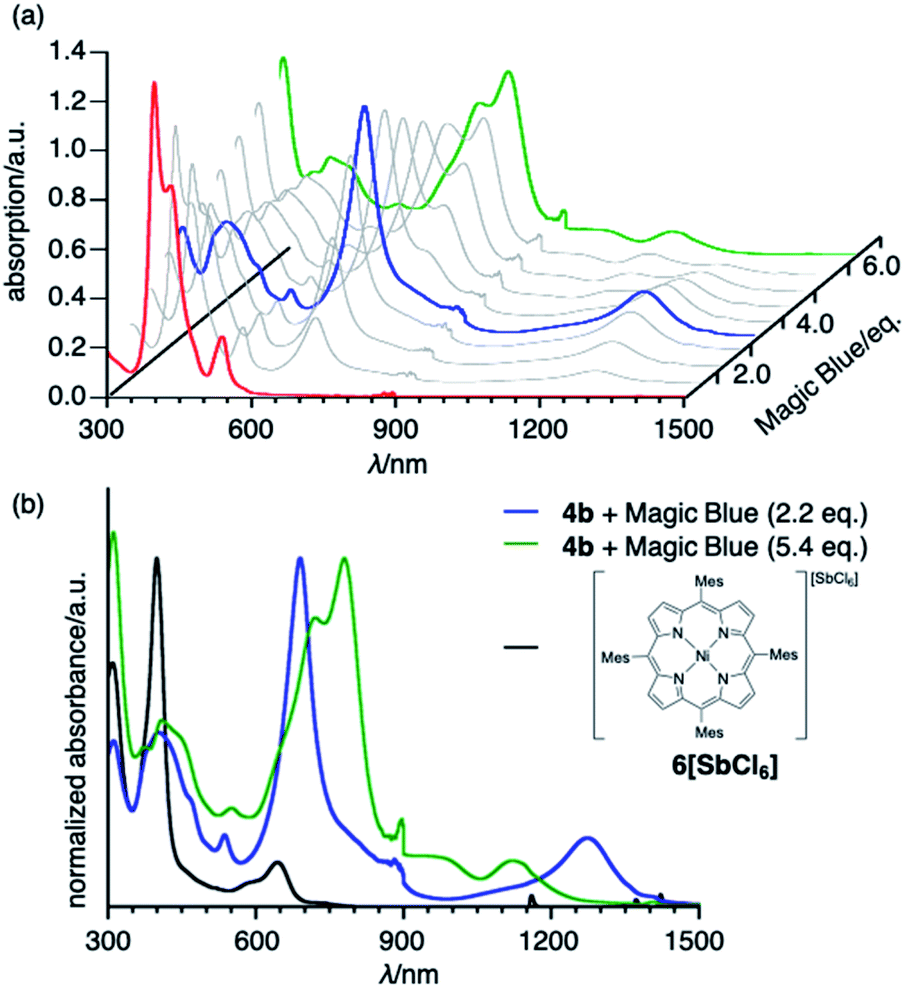
To obtain insight into the unique redox-response of syn-tetramer 4b, we conducted an oxidative titration with tris(4-bromophenyl)aminium hexachloroantimonate (Magic Blue) in CH2Cl2 while measuring its absorption spectra, which demonstrated two-step spectral changes (Fig. 5). Clear isosbestic points were observed in both cases (Fig. S23†). The first change occurred after the consumption of ca. 2 equiv. of Magic Blue, resulting in the appearance of new peaks at 690 and 1274 nm. The further addition of Magic Blue (ca. 2 equiv.) resulted in the second change, which led to a slight blue shift of the absorption tail to ca. 1250 nm. Similar spectral changes were observed during spectroelectrochemical measurements from 0 to 1.2 V (Fig. S21†). Furthermore, the subsequent back-sweep to −0.5 V recovered the absorption peaks of syn-tetramer 4b.
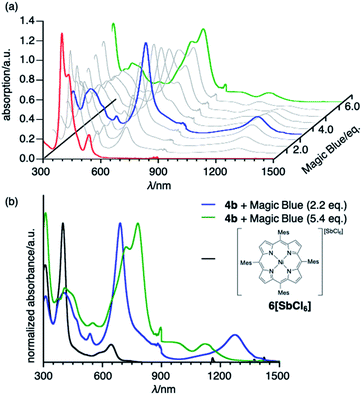 |
| | Fig. 5 (a) Oxidative titration of 4b with Magic Blue. [4b]0 = 3.0 × 10−6 M−1. (b) UV/vis/NIR absorption spectra of 4b with 2.2 equiv. of Magic Blue, 4b with 5.4 equiv. of Magic Blue, and porphyrin radical cation 6[SbCl6]. Solvent: CH2Cl2; λ: wavelength. | |
Isolation of the dication.
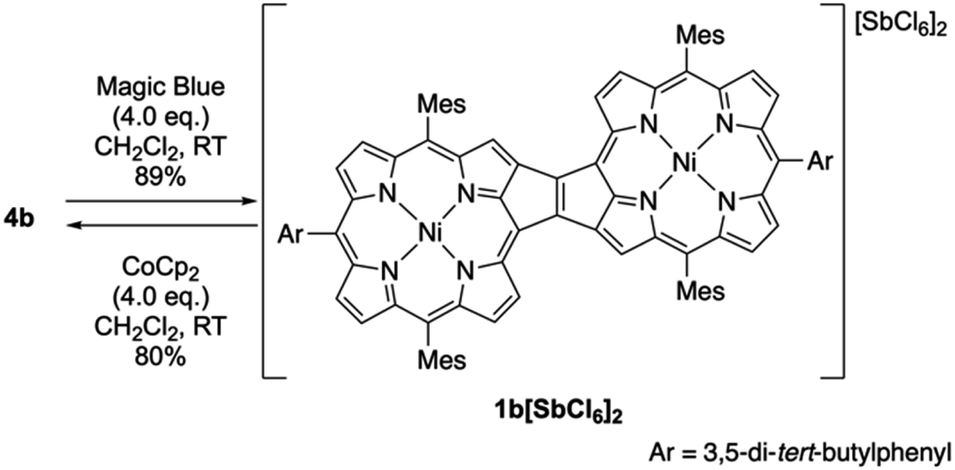
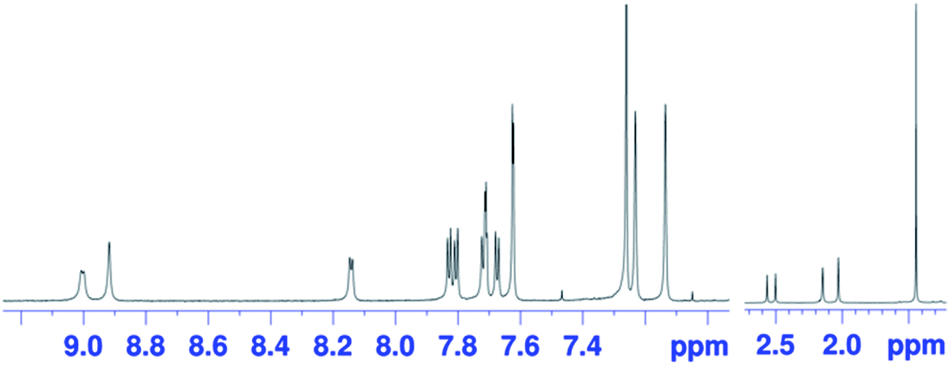
The oxidation of syn-tetramer 4b with 4 equiv. of Magic Blue in CH2Cl2 furnished etheno-fused diporphyrin dication 1b[SbCl6]2 in 89% yield (Scheme 2). The reduction of 1b[SbCl6]2 with an excess of cobaltocene recovered 4b in 80% yield. While dication 1b[SbCl6]2 is sufficiently stable under ambient conditions, repeated recrystallizations were required for its purification, given that 1b[SbCl6]2 decomposes on silica gel. The 1H NMR spectrum of 1b[SbCl6]2 in CDCl3 exhibited one singlet and six doublets (7.67–9.00 ppm) due to the main skeleton (Fig. 6). The calculated nucleus-independent chemical shift (NICS)9 values of 1b[SbCl6]2 nicely coincide with this observation (Fig. S27†). The presence of a distinct diatropic ring current in 1b[SbCl6]2 can be rationalized by the removal of two electrons from the antiaromatic contribution of the etheno-fused diporphyrin core. Importantly, the signals due to the ortho-methyl groups of the mesityl substituents are observed as two singlet signals. Considering that 1b[SbCl6]2 contains two magnetically inequivalent mesityl groups, this result indicates that each set of ortho-methyl groups is magnetically equivalent, supporting that 1b[SbCl6]2 adopts a planar structure. The overall structure of 1b[SbCl6]2 was determined based on single-crystal X-ray diffraction analysis, although the crystal data were not of sufficient quality to allow a detailed structural analysis (Fig. 7). Dication 1b[SbCl6]2 adopts a completely planar structure with a mean plane deviation of 0.07 Å. The UV/vis/NIR absorption spectrum of 1b[SbCl6]2 is in good agreement with that observed after the electrochemical oxidation of syn-tetramer 4b (Fig. S21 and S22†).
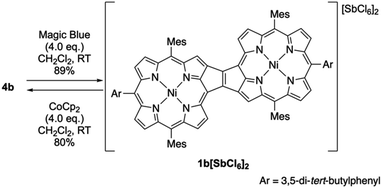 |
| | Scheme 2 Synthesis of etheno-fused diporphyrin dication 1b[SbCl6]2. | |
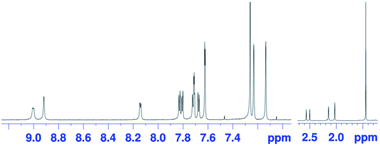 |
| | Fig. 6
1H NMR spectrum of 1b[SbCl6]2 in CDCl3 at 25 °C. | |
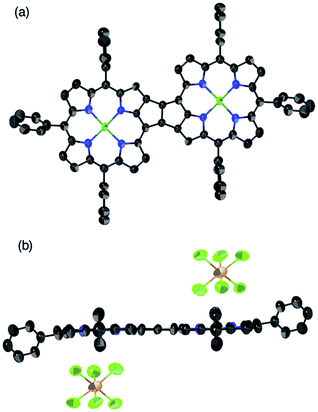 |
| | Fig. 7 Single-crystal X-ray diffraction structure of etheno-fused diporphyrin dication 1b[SbCl6]2. (a) Top and (b) side view. Thermal ellipsoids are drawn at 50% probability. Solvent molecules, methyl groups, tert-butyl groups, and all hydrogen atoms are omitted for clarity. | |
Proposed dyrex-mechanism.
The absorption spectrum after the addition of 2.2 equiv. of Magic Blue is clearly different from that of a porphyrin radical cation (Fig. 5b).10 Considering that the inter-porphyrin interaction in syn-tetramer 4b is essentially negligible due to the non-conjugative nature of the central cyclobutane unit, the initial change in absorption during the titration cannot be explained by the simple oxidation of 4b. Furthermore, this absorption is in good agreement with a theoretical simulation of the radical cation of etheno-fused diporphyrin 1b˙ (Fig. S25†).
Based on the results discussed above, we propose the following redox-response process for syn-tetramer 4b (Scheme 3). The first two-electron oxidation of 4b induces the cleavage of two C–C bonds at the central cyclobutane unit, affording two etheno-fused diporphyrin radical cations 1b˙+. The subsequent oxidation of 1b˙+ furnishes dication 1b2+. The reverse reduction process begins with the one-electron reduction of dication 1b2+. Our previous DFT calculations have predicted that the HOMO level of the etheno-fused diporphyrin is higher than that of a normal porphyrin due to the extended π-system and potential antiaromaticity.4 Hence, the reduction potential of 1b2+ can be expected to be much lower than the oxidation potential of 4b, which would result in a large hysteresis in the cyclic voltammogram. Notably, we have also monitored a similar response during the electrochemical reduction of 4b (Fig. S20†). However, the identification of the reduced species was unsuccessful owing to the instability of these intermediates.
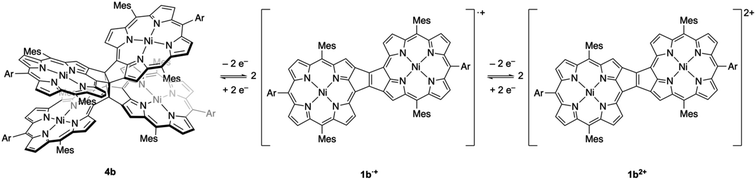 |
| | Scheme 3 Dyrex response of syn-tetramer 4b. | |
Importantly, the redox response of 4b, namely the reversible cycloreversion involving the cleavage of two C–C bonds, is exceptional among dyrex systems.6,7 Quadricyclane, anthracene-dimer, and acridizinium dimer undergo double C–C bond cleavage upon electron-transfer, providing norbornadiene, anthracene, and acridizinium, respectively.11–13 In these cases, however, the reverse C–C bond formation requires light-irradiation. The unique reactivity of the etheno-fused diporphyrin is attributed to the contribution of antiaromaticity in its macrocyclic conjugation. We believe that the current study offers a general insight that antiaromatic molecules15 are a promising candidate for the design of novel redox-active functional organic materials including electrochromic dyes, organic batteries, and organic memory devices.
Thermal and photo-induced cycloreversion of cyclobutane-linked tetraporphyrin
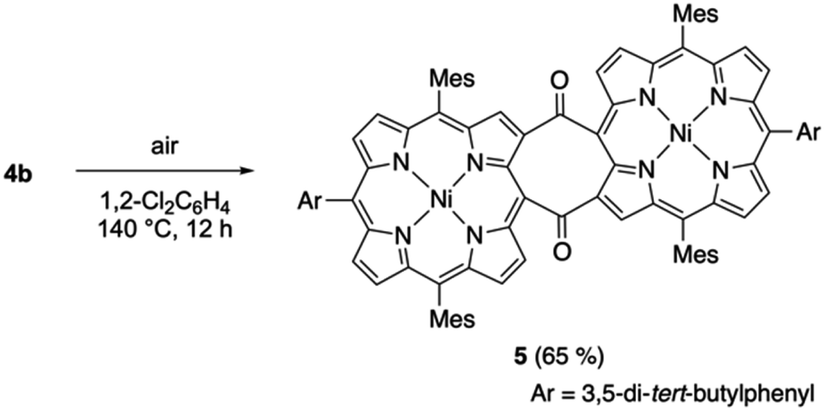
We also examined the thermal cycloreversion of syn-tetramer 4b, which was monitored using variable-temperature NMR and UV/vis absorption spectroscopy techniques. The 1H NMR spectrum of 4b in 1,2-dichlorobenzene-d4 showed slight changes (up to 0.4 ppm) upon increasing the temperature from 20 °C to 120 °C (Fig. S18†). However, the absorption spectrum of 4b in 1,2-dichlorobenzene displayed negligible changes upon heating (Fig. S19†). Consequently, the temperature-dependent change of the 1H NMR chemical shifts can be attributed to the dynamic motion of the meso-aryl groups. Notably, heating the dichlorobenzene solution of 4b to 140 °C afforded diketodiporphyrin 5 in 65% yield (Scheme 4). This result suggests the transient generation of etheno-fused diporphyrin 1b, which is instantly oxidized to diketone 5. A similar diketodiporphyrin was formed in our previous study with the corresponding zinc(II) complexes.4 The excited state of nickel(II) porphyrins generally undergoes a rapid decay through the metal (d,d) state.14 Consequently, the formation of diketodiporphyrin 5 implies that the thermally activated triplet state of in situ-generated etheno-fused diporphyrin 1b reacted with triplet oxygen.
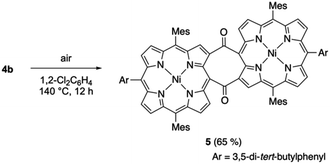 |
| | Scheme 4 Thermal conversion of syn-tetramer 4b to 5 under aerobic conditions. | |
We also examined the effect of photo-irradiation on cycloreversion. A CH2Cl2 solution of 4b was irradiated by a high-pressure mercury lamp equipped with a sharp cut filter (λ > 380 nm) (Fig. S24†). However, no detectable change was observed.
Conclusions
We have prepared X-shaped cyclobutane-linked tetraporphyrin 4b and examined the thermal and redox-mediated cycloreversion of its cyclobutane-ring. Heating 4b in 1,2-dichlorobenzene resulted in negligible changes in the 1H NMR and UV/vis absorption spectra. Instead, syn-tetramer 4b undergoes a two-step four-electron oxidation to afford etheno-fused diporphyrin dication 1b2+. This redox-mediated cyclobutane-ring cycloreversion proceeds in a reversible manner and exhibits a large hysteresis in the cyclic voltammogram. Importantly, this process is accompanied by the cleavage of two C–C bonds, which is exceptional among dyrex systems. The current research highlights the unique reactivity of antiaromatic molecules and offers fundamental insights for the design of novel redox-active functional organic materials including electrochromic dyes, organic batteries, and organic memory devices.
Author contributions
The manuscript was written through contributions of all authors. All authors have approved the final version of the manuscript. H. S. supervised the project and contributed to conceptualization, project administration, and writing (review & editing) the manuscript. K. M. carried out the synthesis and characterization. I. H. collected the X-ray data of 4b. N. F. wrote the original draft.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI grant JP17H01190 and JP20H05863. H. S. gratefully acknowledges financial support from the Sumitomo Foundation. X-ray diffraction data of 4b were collected at the BL40XU beamline of SPring-8 with approval of JASRI (proposal 2019B1134). The authors also thank Dr Nobuhiro Yasuda at SPring-8 (JASRI) for the collection of the X-ray diffraction data.
Notes and references
-
(a) H. L. Anderson, Chem. Commun., 1999, 2323 RSC;
(b) M. G. H. Vicente, L. Jaquinod and K. M. Smith, Chem. Commun., 1999, 1771 RSC;
(c) J. R. Reimers, N. S. Hush and M. J. Crossley, J. Porphyrins Phthalocyanines, 2002, 6, 795 CrossRef CAS;
(d) S. Fox and R. W. Boyle, Tetrahedron, 2006, 62, 10039 CrossRef CAS;
(e) N. Aratani, D. Kim and A. Osuka, Chem.–Asian J., 2009, 4, 1172 CrossRef CAS PubMed;
(f) C. Jiao and J. Wu, Synlett, 2012, 171 CAS;
(g) J. P. Lewtak and D. T. Gryko, Chem. Commun., 2012, 48, 10069 RSC;
(h) A. M. V. M. Pereira, S. Richeter, C. Jeandon, J.-P. Gisselbrecht, J. Wytko and R. Ruppert, J. Porphyrins Phthalocyanines, 2012, 16, 464 CrossRef CAS;
(i) H. Mori, T. Tanaka and A. Osuka, J. Mater. Chem. C, 2013, 1, 2500 RSC;
(j) M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900 CrossRef CAS PubMed;
(k) T. Tanaka and A. Osuka, Chem. Soc. Rev., 2015, 44, 943 RSC.
-
(a) A. Nakano, N. Aratani, H. Furuta and A. Osuka, Chem. Commun., 2001, 1920 RSC;
(b) A. K. Sahoo, S. Mori, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 7972 CrossRef CAS PubMed;
(c) N. Fukui, H. Yorimitsu, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2014, 53, 4395 CrossRef CAS PubMed;
(d) N. Fukui, S. Arai, H. Shinokubo, H. Yorimitsu and A. Osuka, Heterocycles, 2015, 90, 252 CrossRef CAS.
-
(a) S. Fox and R. W. Boyle, Chem. Commun., 2004, 1322 RSC;
(b) D.-M. Shen, C. Liu and Q.-Y. Chen, Chem. Commun., 2005, 4982 RSC;
(c) D.-M. Shen, C. Liu and Q.-Y. Chen, J. Org. Chem., 2006, 71, 6508 CrossRef CAS PubMed;
(d) S. Hayashi, Y. Matsubara, S. Eu, H. Hayashi, T. Umeyama, Y. Matano and H. Imahori, Chem. Lett., 2008, 37, 846 CrossRef CAS;
(e) G. Bringmann, D. C. G. Götz, T. A. M. Gulder, T. H. Gehrke, T. Bruhn, T. Kupfer, K. Radacki, H. Braunschweig, A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2008, 130, 17812 CrossRef CAS PubMed;
(f) T. D. Lash, B. E. Smith, M. J. Melquist and B. A. Godfrey, J. Org. Chem., 2011, 76, 5335 CrossRef CAS PubMed;
(g) A. M. V. M. Pereira, M. G. P. M. S. Neves, J. A. S. Cavaleiro, C. Jeandon, J.-P. Gisselbrecht, S. Choua and R. Ruppert, Org. Lett., 2011, 13, 4742 CrossRef CAS PubMed;
(h) T. Ishizuka, Y. Saegusa, Y. Shiota, K. Ohtake, K. Yoshizawa and T. Kojima, Chem. Commun., 2013, 49, 5939 RSC;
(i) N. Fukui, W.-Y. Cha, S. Lee, S. Tokuji, D. Kim, H. Yorimitsu and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 9728 CrossRef CAS PubMed;
(j) K. Ota, T. Tanaka and A. Osuka, Org. Lett., 2014, 16, 2974 CrossRef CAS PubMed;
(k) N. Fukui, S.-K. Lee, K. Kato, D. Shimidzu, T. Tanaka, S. Lee, H. Yorimitsu, D. Kim and A. Osuka, Chem. Sci., 2016, 7, 4059 RSC.
- T. Nagai, A. Takiguchi, M. Ueda, K. Oda, S. Hiroto and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 8392 CrossRef CAS PubMed.
-
T. Suzuki, H. Tamaoki, J.-i. Nishida, H. Higuchi, T. Iwai, Y. Ishigaki, K. Hanada, R. Katoono, H. Kawai, K. Fujiwara and T. Fukushima, in Organic Redox Systems: Synthesis, Properties, and Applications, ed. T. Nishinaga, Wiley, Hoboken, 2015, ch. 2, pp. 13–37 Search PubMed.
-
(a) H. Horner and S. Hünig, J. Am. Chem. Soc., 1977, 99, 6120 CrossRef;
(b) J. Jubb, C. Floriani, A. Chiesi-Villa and C. Rizzoli, J. Am. Chem. Soc., 1992, 114, 6571 CrossRef CAS;
(c) T. Suzuki, J.-i. Nishida and T. Tsuji, Angew. Chem., Int. Ed. Engl., 1997, 36, 1329 CrossRef CAS;
(d) T. Muramatsu, A. Toyota, M. Kudou, Y. Ikegami and M. Watanabe, J. Org. Chem., 1999, 64, 7249 CrossRef CAS;
(e) S. Hünig, C. A. Briehn, P. Bäuerle and A. Emge, Chem.–Eur. J., 2001, 7, 2745 CrossRef;
(f) S. Iwashita, E. Ohta, H. Higuchi, H. Kawai, K. Fujiwara, K. Ono, M. Takenaka and T. Suzuki, Chem. Commun., 2004, 2076 RSC;
(g) J.-i. Nishida, T. Miyagawa and Y. Yamashita, Org. Lett., 2004, 6, 2523 CrossRef CAS PubMed;
(h) J. K. Mahoney, V. Regnier, E. A. Romero, F. Molton, G. Royal, R. Jazzar, D. Martin and G. Bertrand, Org. Chem. Front., 2018, 5, 2073 RSC;
(i) A. Gosset, L. Wilbraham, Š. N. Lachmanová, R. Sokolová, G. Dupeyre, F. Tuyèras, P. Ochsenbein, C. Perruchot, H.-P. J. de Rouville, H. Randriamahazaka, L. Pospíšil, I. Ciofini, M. Hromadová and P. P. Lainé, J. Am. Chem. Soc., 2020, 142, 5162 CrossRef CAS PubMed.
- K. Fujimoto and A. Osuka, Chem.–Eur. J., 2018, 24, 6530 CrossRef CAS PubMed.
- K. Fujimoto, H. Yorimitsu and A. Osuka, Chem.–Eur. J., 2015, 21, 11311 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
-
(a) A. Wolberg and J. Manassen, J. Am. Chem. Soc., 1970, 92, 2982 CrossRef CAS PubMed;
(b) D. Dolphin, T. Niem, R. H. Felton and I. Fujita, J. Am. Chem. Soc., 1975, 97, 5288 CrossRef CAS PubMed.
-
(a) P. G. Gassman, R. Yamaguchi and G. F. Koser, J. Org. Chem., 1978, 43, 4392 CrossRef CAS;
(b) O. Brummel, D. Besold, T. Döpper, Y. Wu, S. Bochmann, F. Lazzari, F. Waidhas, U. Bauer, P. Bachmann, C. Papp, H.-P. Steinrück, A. Görling, J. Libuda and J. Bachmann, ChemSusChem, 2016, 9, 1424 CrossRef CAS PubMed.
-
(a) R. A. Barber, P. de Mayo, K. Okada and S. K. Wong, J. Am. Chem. Soc., 1982, 104, 4995 CrossRef CAS;
(b) J. M. Masnovi and J. K. Kochi, J. Am. Chem. Soc., 1985, 107, 6781 CrossRef CAS.
- H. Ihmels and J. Luo, J. Photochem. Photobiol., A, 2008, 200, 3 CrossRef CAS.
- C. M. Drain, C. Kirmaier, C. J. Medforth, D. J. Nurco, K. M. Smith and D. Holten, J. Phys. Chem., 1996, 100, 11984 CrossRef CAS.
-
(a) K. B. Wiberg, Chem. Rev., 2001, 101, 1317 CrossRef CAS PubMed;
(b) T. Nishinaga, T. Ohmae and M. Iyoda, Symmetry, 2010, 2, 76 CrossRef CAS;
(c) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903 RSC;
(d) H. Hopf, Angew. Chem., Int. Ed., 2013, 52, 12224 CrossRef CAS PubMed;
(e) M. Rosenberg, C. Dahlstrand, K. Kilsa and H. Ottosson, Chem. Rev., 2014, 114, 5379 CrossRef CAS PubMed;
(f) C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977 CrossRef CAS PubMed;
(g) Y. M. Sung, J. Oh, W.-Y. Cha, W. Kim, J. M. Lim, M.-C. Yoon and D. Kim, Chem. Rev., 2017, 117, 2257 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2055688 and 2055689. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc00438g |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Norihito
Fukui
a and
Hiroshi
Shinokubo
b,
Norihito
Fukui
a and
Hiroshi
Shinokubo