
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic probes for detecting deacylase and demethylase activity towards post-translationally-modified lysine residues†
Yuichiro
Hori
 ab,
Miyako
Nishiura
a,
Tomomi
Tao
a,
Reisuke
Baba
a,
Steven D.
Bull
c and
Kazuya
Kikuchi
*abd
ab,
Miyako
Nishiura
a,
Tomomi
Tao
a,
Reisuke
Baba
a,
Steven D.
Bull
c and
Kazuya
Kikuchi
*abd
aGraduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: kkikuchi@mls.eng.osaka-u.ac.jp
bIFReC, Osaka University, Suita, Osaka 565-0871, Japan
cDepartment of Chemistry, University of Bath, Bath, BA27AY, UK
dQuantum Information and Quantum Biology Division, Osaka University, Suita, Osaka 565-0871, Japan
First published on 7th January 2021
Abstract
Reversible enzymatic post-translational modification of the ε-amino groups of lysine residues (e.g. N-acylation reactions) plays an important role in regulating the cellular activities of numerous proteins. This study describes how enzyme catalyzed N-deprotection of lysine residues of non-fluorescent peptide-coumarin probes can be used to generate N-deprotected peptides that undergo spontaneous O- to N-ester transfer reactions (uncatalyzed) to generate a highly fluorescent N-carbamoyl peptide. This enables detection of enzyme catalyzed N-deacetylation, N-demalonylation, N-desuccinylation and N-demethylation reactions activities towards the N-modified lysine residues of these probes using simple ‘turn on’ fluorescent assays.
Introduction
Posttranslational modification (PTM) of the ε-amino groups of lysine residues in proteins is an important epigenetic mechanism that is used to control a wide range of biological phenomena.1–3 For example, enzyme catalyzed acetylation and methylation reactions of lysine groups of histones are known to be important for the regulation of gene expression and maintaining cell homeostasis.1 Covalent modification of selected lysine side chains in histones leads to chromatin being remodeled, resulting in transcription levels being activated and associated gene expression levels being modulated.1 The ε-amino groups of protein lysine residues are modified using other types of acyl donor, including their conversion into N-succinyl, N-malonyl-, N-glutaryl-, N-crotonyl-, N-propionyl-, N-butyryl- and N-2-hydroxyisobutyryl-lysine derivatives.2,3 These PTMs are not only used to reversibly functionalize the lysine residues of histone proteins, but are also present in cytosolic and mitochondrial proteins that play important roles in controlling gene regulation and metabolic pathways.2,3 However, the mechanisms and specificity profiles of many of the lysine N-modification enzymes responsible for these type of PTMs remain unexplored in many cases.4 Similarly, the effect of many of these lysine N-modification reactions on protein structure and function and their biological significance still needs to be more fully explored.4–7The availability of simple analytical methods to detect enzyme catalyzed N-functionalization and/or N-deprotection reactions of lysine residues in proteins would be very useful to explore how these type of PTMs affect cellular processes. Previous studies have explored the activity of lysine N-modification enzymes using radioisotope,8 high-performance liquid chromatography (HPLC),8 mass spectrometry,8,9 enzyme-coupled assays8–12 and antibody-based methods.9,13 However, these approaches often employ complicated multistep procedures, require multiple enzymatic reactions, are time-consuming or lack high-throughput capabilities. Therefore, the availability of simple turn ‘on’ fluorescent assays that could be used to detect different types of enzyme catalyzed lysine N-modification activities would be very useful for exploring the biological consequences of lysine PTM.
We have previously described the development of fluorogenic probes to detect lysine deacetylase (Sirt1) activity using a simple one-pot additive-free fluorescence turn ‘on’ protocol.14–16 Non-fluorescent probe P-N(H)Ac [previously reported as LN1(Ac)],16 that contains a histone H3 (8–13) recognition peptide domain, an N-acetyl-lysine residue and an N-terminus linked coumarin fragment was prepared as a histone deacetylase substrate (Fig. 1). The 7-hydroxy group of the coumarin fragment of P-N(H)Ac is protected as a carbonate ester which results in its fluorescence being ‘turned off’. Histone deacetylase catalyzed hydrolysis of P-N(H)Ac results in N-deacetylation to afford the deprotected lysine residue of P-NH2. The free ε-amino group of P-NH2 then undergoes spontaneous intramolecular attack at the carbonyl group of its carbonic ester moiety,16 which results in an intramolecular O- to N-ester transfer reaction. The resultant transfer peptide P-N(H)CO2Bu contains a more stable N-carbamoyl-lysine residue and a highly fluorescent 7-hydroxycoumarin fragment. Therefore, histone deacetylase activity towards P-N(H)Ac probe can be visualized using a simple ‘turn-on’ fluorescence assay that detects the appearance of the fluorescent transfer peptide P-N(H)CO2Bu as a rearranged product of the enzyme catalyzed hydrolysis reaction of P-N(H)Ac.
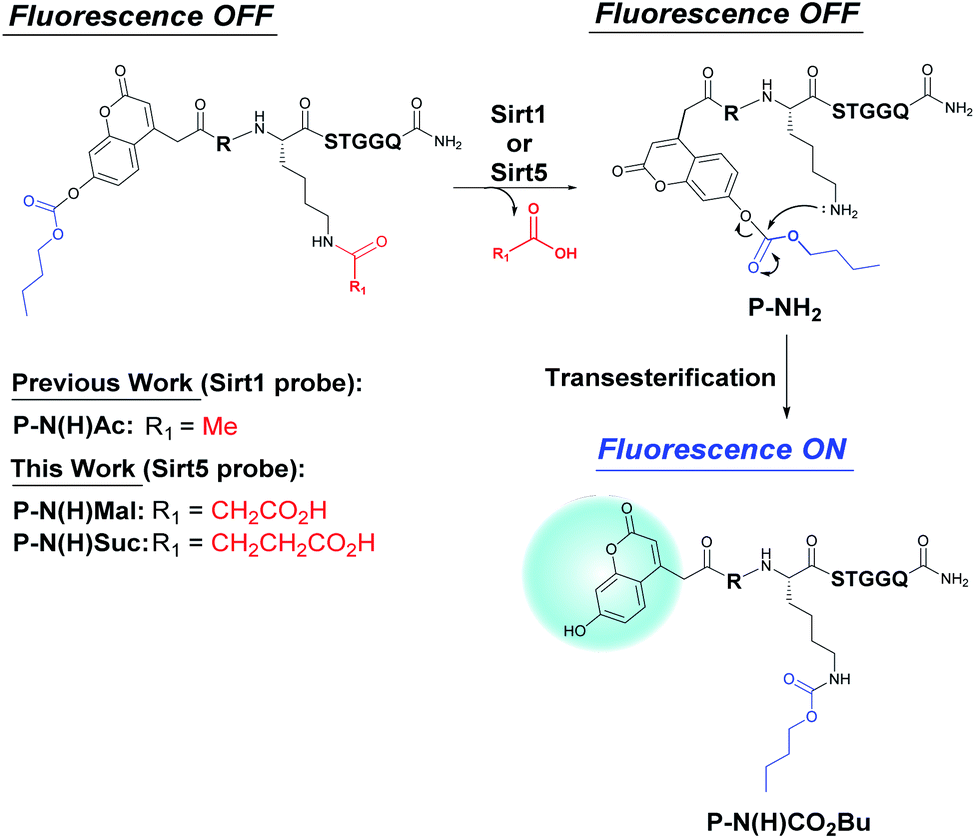 | ||
| Fig. 1 Enzyme mediated deacetylation of P-N(H)Ac, demalonylation of P-N(H)Mal and desuccinylation of P-N(H)Suc affords P-NH2 that undergoes an intramolecular O- to N-ester transfer reaction to afford the fluorescent N-carbamoyl transfer peptide P-N(H)CO2Bu. | ||
Other groups subsequently reported different probes for the fluorescent detection of lysine deacetylase activity using reactions that are based on imine formation,17,18 nucleophilic aromatic substitution reactions to generate changes in a probe fluorescence response,19 or self-immolative intramolecular cyclization reactions.20 Förster resonance energy transfer (FRET) based probes that contain quencher and fluorophore fragments attached to the ε-lysine side chain of non-natural substrates have also been described. These probes rely on enzymatic cleavage of either their quencher or fluorophore fragments to produce a fluorescence reponse.21,22 For example, the N-decrotonylase activity of HDAC3 has been detected using a chemical probe containing an N-crotonyl-lysine group, however these activity levels were 16 times lower than HDAC3 mediated N-deacetylation activity levels towards an N-acetyl-lysine probe analogue.18 However, no ‘turn on’ fluorescent probes for detecting enzymatic N-deprotection reactions (e.g. N-demethylation) towards other types of N-derivatized lysine residues (e.g. ε-N-dimethylated-lysine residues) have been reported to date. Consequently, we now describe how the fluorescent assay that we previously developed to detect lysine deacetylase activity has been adapted to develop new 2nd-generation probes for the fluorescent “turn-on” detection of enzyme catalyzed N-desuccinylase, N-demalonylase and N-demethylase activities towards N-modified lysine residues that are present in peptide substrates.
Results and discussion
Enzyme activatable fluorogenic probes for detecting lysine N-demalonylase and N-desuccinylase activities
Sirt5 is a lysine deacylase that employs NAD+ as a cofactor to catalyze the reversible malonylation/succinylation of the ε-amino groups of selected lysine residues in proteins.23,24 Sirt5 has previously been shown to demonstrate good activity for deacylation of N-malonyl and N-succinyl lysine residues of proteins that are known to regulate metabolism in mitochondria,25,26 as well as for deacylating proteins involved in neurodegeneration pathways.27 Therefore, it was decided that development of a fluorogenic assay to detect Sirt5 activity would be a good demonstration of the potential of using our enzyme catalyzed N-deprotection/transesterification fluorescence strategy to develop new probes to image other types of deacylase activity.Target peptides P-N(H)Mal and P-N(H)Suc were designed as potential enzyme activatable fluorogenic probes for detecting Sirt5 mediated demalonylase and desuccinylase activities, respectively (Fig. 1). It was reasoned that enzymatic hydrolysis of the N-acyl fragments of both probes would result in formation of P-NH2 whose free amino group would then undergo intramolecular rearrangement to afford P-N(H)CO2Bu, thus resulting in a ‘turn on’ of fluorescence. The non-fluorogenic coumarin–peptides P-N(H)Mal and P-N(H)Suc were prepared using standard solid-phase synthesis employing N-Fmoc protected α-amino acids and standard peptide coupling agents (see ESI “Materials and methods”, Scheme S1†). High-performance liquid chromatography (HPLC) analyses revealed that incubation of Sirt5 with P-N(H)Mal and P-N(H)Suc in HEPES buffer (pH 8.0) resulted in enzyme catalyzed hydrolysis of their N-acyl groups to afford P-NH2 (non-fluorescent), with intramolecular transesterification then affording the rearranged fluorescent transfer peptide P-N(H)CO2Bu. Therefore, incubation of P-N(H)Mal (peak at ∼15 min) with Sirt5 resulted in its complete consumption after 15 min to afford a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 mixture of P-NH2 (peak at ∼9.5 min) and the rearranged N-carbamoyl peptide P-N(H)CO2Bu (peak at ∼12 min), with all of the P-NH2 fully rearranged after 30 min (Fig. 2a). Sirt5 hydrolyzed P-N(H)Suc (peak at ∼14 min) even more quickly, with all the probe consumed after 5 min and P-NH2 fully rearranged into P-N(H)CO2Bu after 20 min (Fig. 2b). Switching from UV to fluorescence detection confirmed that the peak for the rearranged peptide P-N(H)CO2Bu at ∼12 min was highly fluorescent (Fig. 2c and d). The identity of P-NH2 and P-N(H)CO2Bu were confirmed through ESI-MS analysis which gave accurate mass values for the HPLC peak fractions eluting at ∼9.5 and ∼12 min, respectively (Fig. S1†). MALDI-TOF/TOF-MS analysis of the peak at ∼12 min also confirmed that O- to N-ester transfer from the coumarin fragment of P-NH2 to the lysine residue of P-N(H)CO2Bu had occurred (Fig. S2†).
:7 mixture of P-NH2 (peak at ∼9.5 min) and the rearranged N-carbamoyl peptide P-N(H)CO2Bu (peak at ∼12 min), with all of the P-NH2 fully rearranged after 30 min (Fig. 2a). Sirt5 hydrolyzed P-N(H)Suc (peak at ∼14 min) even more quickly, with all the probe consumed after 5 min and P-NH2 fully rearranged into P-N(H)CO2Bu after 20 min (Fig. 2b). Switching from UV to fluorescence detection confirmed that the peak for the rearranged peptide P-N(H)CO2Bu at ∼12 min was highly fluorescent (Fig. 2c and d). The identity of P-NH2 and P-N(H)CO2Bu were confirmed through ESI-MS analysis which gave accurate mass values for the HPLC peak fractions eluting at ∼9.5 and ∼12 min, respectively (Fig. S1†). MALDI-TOF/TOF-MS analysis of the peak at ∼12 min also confirmed that O- to N-ester transfer from the coumarin fragment of P-NH2 to the lysine residue of P-N(H)CO2Bu had occurred (Fig. S2†).
 | ||
| Fig. 2 HPLC analyses of P-N(H)Mal and P-N(H)Suc reacted with Sirt5 (100 nM) in HEPES buffer (pH 8.0) over time. UV absorption HPLC traces of: (a) P-N(H)Mal (5 μM); and (b) P-N(H)Suc (5 μM). Fluorescence HPLC traces of: (c) P-N(H)Mal (5 μM); and (d) P-N(H)Suc (5 μM). The peaks labelled with an asterisk in HPLC traces (a and b) are from DTT present in the reaction buffer. | ||
Fluorescence measurements were then carried out to monitor Sirt5 activity towards the P-N(H)Mal and P-N(H)Suc probes over time (Fig. 3). Both probes showed a large increase in fluorescence intensity when they were incubated with Sirt5, with negligible increases in fluorescence intensity observed when heat denatured Sirt5 or no Sirt5 was present. Kinetic parameters for Sirt5 towards both probes were determined using a modified Michaelis–Menten equation that allowed for the fact that fluorescence is generated via a two-step process that proceeds via an enzyme catalyzed deacylation step followed by an uncatalyzed rearrangement reaction (Fig. S3, S4, Tables S1 and S2†). Sirt5 exhibited a kcat/Km value of 1.3 × 105 M−1 s−1 towards P-N(H)Suc and a kcat/Km value of 6.5 × 104 M−1 s−1 towards P-N(H)Mal, meaning that the hydrolytic activity of Sirt5 towards P-N(H)Suc is 2-fold greater than for P-N(H)Mal. Nevertheless, both of these new probes were sufficiently active to enable rapid fluorescent detection of Sirt5 demalonylase and desuccinylase activities using a fluorogenic assay that takes <30 min to complete.
 | ||
| Fig. 3 Fluorescence detection of Sirt5 (100 nM) activity towards P-N(H)Mal and P-N(H)Suc in HEPES buffer (pH 8.0). Fluorescense intensities of (a) P-N(H)Mal (5 μM) and (b) P-N(H)Suc (5 μM) were recorded in the presence of Sirt5; heat-denatured Sirt5 (100 nM); and no Sirt5. Excitation and emission wavelengths were 371 and 466 nm, respectively. | ||
The cross reactivities of the P-N(H)Mal and P-N(H)Suc probes towards other deacylases were investigated by reacting them with Sirtuins (Sirt1, Sirt3 and Sirt6) and Zn(II)-dependent deacetylases (HDAC1, HDAC2, HDAC3, HDAC6 and HDAC11) (Fig. S5†). These results revealed that only Sirt5 enhanced the fluorescence intensities of these probes, thus demonstrating that they are highly selective for Sirt5. The probes contain carbonate ester groups that could potentially be hydrolyzed by cellular esterase activities, leading to undesirable increases in background fluorescence intensity levels. However, we found that their fluorescence intensities did not increase in the presence of carboxylesterases 1 or 2 (CES1 or CES2) that is expressed in human cells,28 thus demonstrating that their carbonate ester bonds are resistant to CES mediated hydrolysis (Fig. S6†). Incubation of the P-N(H)Mal and P-N(H)Suc probes with cell lysates from HeLa cells containing exogenously added Sirt5 resulted in a 2-fold increase in fluorescence intensity when compared to probe containing cell lysates without Sirt5. This demonstrates that the probes can be used for the fluorescence detection of Sirt5 deacylase activities in complex cellular mixtures using a simple assay that can potentially be adapted for high-throughput analysis (Fig. S7†).
An enzyme activatable fluorogenic probe for detecting lysine N-demethylase activity
Development of fluorescent assays to detect demethylase activities towards N-dimethylated lysine residues (R–N+(H)Me2 → RN+(H2)Me) are challenging, because the ε-amino group remains positively charged throughout the course of the N-demethylase reaction. N-methyl groups are also less sterically demanding than N-acyl groups and so it is more difficult to selectively recognize a dimethylated amino group. Furthermore, lysine N-demethylases are known to exhibit much stricter specificity profiles for N-methyl-lysine containing peptide sequences that they recognise,29–32 as compared to lysine deacetylases that generally exhibiting a more relaxed specificity profile.33–35 A promising fluorescence displacement assay has previously been developed to detect demethylase activity towards peptide probes that contain trimethylated lysine residues (R–N+(Me)3).36 However, this N-demethylase sensor is not particularly sensitive, because it employs a fluorescence ‘turn off’ mechanism that only produces a relatively small reduction in fluorescent intensity (∼20%).Jumonji domain-containing 2 (JMJD2) demethylase was chosen as a target enzyme to develop a fluorescent ‘turn on’ probe to detect lysine demethylase activity. This enzyme employs a Fe(II)- and 2-oxoglutarate (2-OG)-dependent deoxygenation mechanism to catalyze N-demethylation of the tertiary amine (H3K9-NMe2) and quaternary ammonium (H3K9-N+Me3) residues of histone H3 lysine 9, respectively.37 This type of JMJD2 demethylase is known to be involved in controlling cellular differentiation and cancer development and so is a potentially attractive target for developing anticancer therapies.38,39
A non-fluorescent di-N-methylated peptide probe (P-NMe2) was designed that contained a peptide sequence H3(7–16) known to be recognized by JMJD2E demethylase,40 a lysine-9 residue containing an ε-NMe2 group and a C-terminal linked O-butyroyl protected 7-hydroxycoumarin fragment (Fig. 4). It was anticipated that JMJD2E demethylase would oxidatively deprotect one of the N-methyl groups of P-NMe2 (non-fluorescent) to produce the secondary amine group of P-NHMe. The secondary amino group of P-NHMe (non-fluorescent) would then undergo a spontaneous O → N intramolecular transesterification reaction to afford P-N(Me)CO2Bu, thus resulting in the fluorescence of its deprotected coumarin fragment being ‘turned on’.
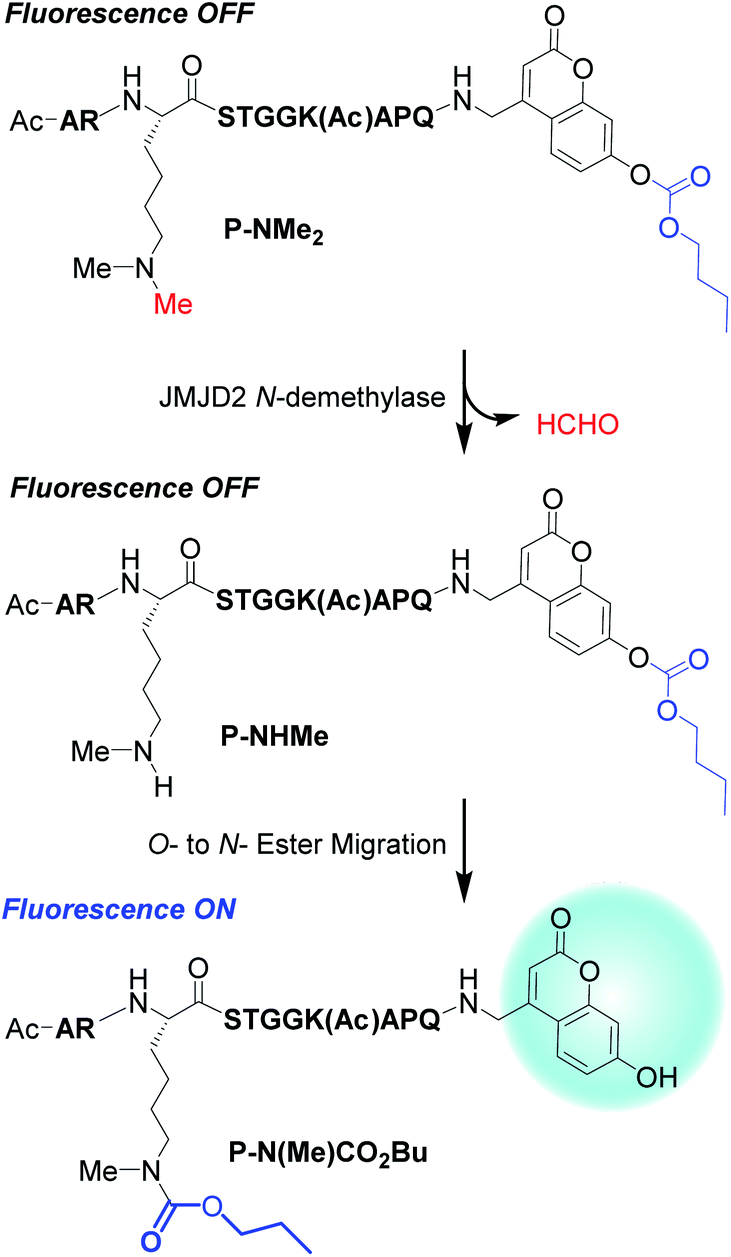 | ||
| Fig. 4 JMJD2 mediated demethylation of P-NMe2 affords P-NHMe that undergoes an intramolecular O- to N-ester transfer reaction to afford the fluorescent N-carbamoyl transfer peptide P-(N(Me)CO2Bu. | ||
The peptides P-NHMe and P-NMe2 were prepared using Fmoc solid-phase synthesis, with both of the peptides containing a glutamine residue at their C-termini for ease of synthesis and N-acetyl-Lys14 residues to prevent any competing intramolecular transesterification reactions from occurring (Scheme S2†). The ability of P-NHMe to undergo the desired spontaneous O- to N-transesterification reaction was investigated by dissolving it in aqueous HEPES buffer (pH 8.0) (no demethylase) and analyzing the resultant solution using HPLC (Fig. 5) and mass spectrometry. HPLC analysis revealed that the peak for P-NHMe at ∼23 min was consumed after 3 h, with the new fluorescent peak that appeared at ∼26 min corresponding to the O → N rearranged P-N(Me)CO2Bu confirmed by ESI-MS and MALDI-TOF/TOF-MS analysis (Fig. S8 and S9†). Incubation of P-NMe2 under the same conditions did not produce a fluorescent peak for P-N(Me)CO2Bu, however a small fluorescent peak was detected at ∼14 min, with an accurate mass value corresponding to a coumarin containing peptide fragment whose carbonate ester had been hydrolyzed (Fig. S10†).
 | ||
| Fig. 5 HPLC analyses of the transesterification reaction of P-NHMe in HEPES buffer (pH 8.0) using: (a) UV detection; and (b) fluorescence detection. The peak labelled by an asterisk in the HPLC traces originates from DTT that is present in the buffer. | ||
Incubation of the non-fluorescent probe P-NMe2 (peak at ∼23 min) with JMJD2E demethylase (5 μM) in HEPES buffer resulted in its complete oxidative N-demethylation to afford P-N(Me)CO2Bu after 3 h (Fig. 6). ESI-MS analysis of the peak fraction eluting at ∼23 min after 15 min revealed that it was a coeluting mixture of P-NMe2 and P-NHMe (Fig. S11†). ESI and MALDI-TOF/TOF-MS analyses of the fluorescent peak that appeared at ∼26 min revealed an accurate mass value corresponding to P-N(Me)CO2Bu, thus indicating that transesterification of the monomethylated lysine had occurred (Fig. S12 and S13†). A relatively high concentration of JMJD2E demethylase (5 μM) was required to catalyze the N-demethylation reaction of P-NMe2, which is consistent with previous reports that elevated levels of this demethylase are necessary to effectively catalyze N-demethylation reactions of lysine residues.41 Fluorescence measurements were then carried out to monitor JMJD2E demethylase activity towards the P-NMe2 probe over time (Fig. 7). Unfortunately, the carbonate ester bond of the P-NMe2 probe proved to be more labile than for the corresponding N-acyl probes, which produces free 7-hydroxycoumarin that results in a greater increase in uncatalyzed background fluorescence intensity over time. However, exposure of the P-NMe2 probe to JMJD2E demethylase (10 μM) resulted in a significant increase in the rate of fluorescence enhancement, with an 9.4:1 ratio of fluorescent intensities in the presence/absence of demethylase observed after 5 minutes (Fig. 7). This result demonstrates that the rate of increase in fluorescence intensity when the P-NMe2 probe is incubated with JMJD2E is sufficient to enable the N-demethylase activity to be rapidly detected using this fluorescent assay system.
 | ||
| Fig. 6 HPLC analyses of JMJD2E demethylase (5 μM) catalyzed N-demethylation reaction of P-NMe2 (5 μM) in HEPES buffer (pH 8.0) using: (a) UV detection; (b) fluorescence detection. The peak labelled as *1 is a coumarin containing peptide formed from background hydrolysis of the carbonate fragment of P-NMe2. The non-fluorescent peak labelled as *2 is DTT that is present as a component of the reaction buffer. | ||
 | ||
| Fig. 7 Fluorescence detection of JMJD2E demethylase activity using P-NMe2 as a probe. Graphs show fluorescence increases produced when P-NMe2 (5 μM) is incubated with (a) JMJD2E demethylase (10 μM); (b) heat-denatured JMJD2E (5 μM); or (c) no enzyme in HEPES buffer (pH 8.0). | ||
The lower demethylase activity of JMJD2E towards P-NMe2 resulted in initial attempts to use this probe to fluorescently detect JMJD2E activity in cell lysates proving unsuccessful. This was because addition of P-NMe2 to cell lysate (no JMJDE2) produced a significant background fluorescence over the 1 h time period required to carry out the JMJD2E assay (Fig. S14†). We reasoned that this increase in background fluorescence could have been being caused by the action of cellular esterases catalyzing unwanted hydrolysis of the probe's carbonate ester bond to produce a fluorescent cleavage product. Indeed, although the fluorescence intensity of P-NMe2 did not increase when incubated with CES1, a 2.8-fold increase in fluorescence intensity was observed when it was treated with CES2 for 1 h (Fig. S15†). Alternatively, we also considered that the background cellular fluorescence might be caused by biological nucleophiles (e.g. glutathione) reacting with the carbonate ester bond of P-NMe2 to release a fluorescent cleavage product. Consequently, N-ethylmaleimide (NEM) was incorporated into the cell lysate assays as a sacrificial conjugate acceptor that would react rapidly with any endogenous thiol nucleophiles that were present. This resulted in a 75% suppression of the background fluorescence of cell lysate containing P-NMe2 probe (no JMJD2E) after 1 h incubation. This meant that exogenous addition of JMJD2E to cell lysate containing P-NMe2 and NEM now resulted in a 2.4-fold increase in fluorescence intensity. Therefore, modifying the cell lysate assay to include NEM as a thiol scavenger enables P-NMe2 to be used to fluorescently detect JMJD2E demethylase activity in complex cellular extracts for the first time.
Conclusions
This study describes the development of fluorogenic probes for detection of enzymatic demalonylation, desuccinylation and demethylation activities towards peptide probes that contain modified N-acyl or N-dimethylated lysine residues, respectively. Enzymatic N-deprotection of lysine residues of these non-fluorescent probes produce peptides whose ε-amino groups undergo O- to N-ester migration reactions to produce peptides containing fluorescent 7-hydroxycoumarin fragments. This study demonstrates that this intramolecular transesterification strategy is potentially applicable for the fluorescent detection of enzyme activities towards different types of N-modified lysine groups present in different PTM peptide sequences. We believe that the probes and principle developed in this study will promote future researches regarding posttranslational lysine modification of proteins. Importantly, this study demonstrates that this detection strategy will be highly versatile and applicable to different peptide probes with other lysine modification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI (Grant Numbers JP19K22255, JP18H03935, JP17H06409 “Frontier Research on Chemical Communications” to K. K. and JP17H02210, JP18K19402, JP20H02879 to Y. H.), JSPS A3 Foresight Program, JSPS Asian CORE Program, “Asian Chemical Biology Initiative”, Japan (JSPS)-UK (RSC) Research Cooperative Program (JPJSBP120195705 to K. K.), Toray Science Foundation (19-6008) and Royal Society International Exchange (IEC\R3\183068 to S. D. Bull).References
- R. DesJarlais and P. J. Tummino, Biochemistry, 2016, 55, 1584–1599 CrossRef CAS.
- B. R. Sabari, D. Zhang, C. D. Allis and Y. Zhao, Nat. Rev. Mol. Cell Biol., 2017, 18, 90–101 CrossRef CAS.
- C. Choudhary, B. T. Weinert, Y. Nishida, E. Verdin and M. Mann, Nat. Rev. Mol. Cell Biol., 2014, 15, 536–550 CrossRef CAS.
- Z. A. Wang and P. A. Cole, Cell Chem. Biol., 2020, 27, 953–969 CrossRef CAS.
- A. Sreedhar, E. K. Wiese and T. Hitosugi, Genes Dis, 2019, 7, 166–171 CrossRef.
- J. Wan, H. Liu, J. Chu and H. J. Zhang, Cell. Mol. Med., 2019, 23, 7163–7169 CrossRef.
- Z. Wu, J. Connolly and K. K. Biggar, FEBS J., 2017, 284, 2732–2744 CrossRef CAS.
- Y. Li, T. Liu, S. Liao, Y. Lan, A. Wang, Y. Wang and B. He, Biochem. Biophys. Res. Commun., 2015, 467, 459–466 CrossRef CAS.
- Y. Shi, F. Lan, C. Matson, P. Mulligan, J. R. Whetstine, P. A. Cole and R. A. Casero, Cell, 2004, 119, 941–953 CrossRef CAS.
- D. Wegener, F. Wirsching, D. Riester and A. Schwienhorst, Chem. Biol., 2003, 10, 61–68 CrossRef CAS.
- C. Roessler, C. Tuting, M. Meleshin, C. Steegborn and M. Schutkowski, J. Med. Chem., 2015, 58, 7217–7223 CrossRef CAS.
- P. A. Cloos, J. Christensen, K. Agger, A. Maiolica, J. Rappsilber, T. Antal, K. H. Hansen and K. Helin, Nature, 2006, 442, 307–311 CrossRef CAS.
- D. Herman, K. Jenssen, R. Burnett, E. Soragni, S. L. Perlman and J. M. Gottesfeld, Nat. Chem. Biol., 2006, 2, 551–558 CrossRef CAS.
- R. Baba, Y. Hori, S. Mizukami and K. Kikuchi, J. Am. Chem. Soc., 2012, 134, 14310–14313 CrossRef CAS.
- K. Dhara, Y. Hori, R. Baba and K. Kikuchi, Chem. Commun., 2012, 48, 11534–11536 RSC.
- R. Baba, Y. Hori and K. Kikuchi, Chem.–Eur. J., 2015, 21, 4695–4702 CrossRef CAS.
- D. R. Rooker and D. Buccella, Chem. Sci., 2015, 6, 6456–6461 RSC.
- D. R. Rooker, Y. Klyubka, R. Gautam, E. Tomat and D. Buccella, Chembiochem, 2018, 19, 496–504 CrossRef CAS.
- Y. Xie, J. Ge, H. Lei, B. Peng, H. Zhang, D. Wang, S. Pan, G. Chen, L. Chen, Y. Wang, Q. Hao, S. Q. Yao and H. Sun, J. Am. Chem. Soc., 2016, 138, 15596–15604 CrossRef CAS.
- X. Liu, M. Xiang, Z. Tong, F. Luo, W. Chen, F. Liu, F. Wang, R. Q. Yu and J. H. Jiang, Anal. Chem., 2018, 90, 5534–5539 CrossRef CAS.
- S. Schuster, C. Roessler, M. Meleshin, P. Zimmermann, Z. Simic, C. Kambach, C. Schiene-Fischer, C. Steegborn, M. O. Hottiger and M. Schutkowski, Sci. Rep., 2016, 6, 22643 CrossRef CAS.
- M. Kawaguchi, S. Ikegawa, N. Ieda and H. Nakagawa, Chembiochem, 2016, 17, 1961–1967 CrossRef CAS.
- J. Du, Y. Zhou, X. Su, J. J. Yu, S. Khan, H. Jiang, J. Kim, J. Woo, J. H. Kim, B. H. Choi, B. He, W. Chen, S. Zhang, R. A. Cerione, J. Auwerx, Q. Hao and H. Lin, Science, 2011, 334, 806–809 CrossRef CAS.
- J. L. Feldman, K. E. Dittenhafer-Reed and J. M. Denu, J. Biol. Chem., 2012, 287, 42419–42427 CrossRef CAS.
- M. J. Rardin, W. He, Y. Nishida, J. C. Newman, C. Carrico, S. R. Danielson, A. Guo, P. Gut, A. K. Sahu, B. Li, R. Uppala, M. Fitch, T. Riiff, L. Zhu, J. Zhou, D. Mulhern, R. D. Stevens, O. R. Ilkayeva, C. B. Newgard, M. P. Jacobson, M. Hellerstein, E. S. Goetzman, B. W. Gibson and E. Verdin, Cell Metab., 2013, 18, 920–933 CrossRef CAS.
- Y. Nishida, M. J. Rardin, C. Carrico, W. He, A. K. Sahu, P. Gut, R. Najjar, M. Fitch, M. Hellerstein, B. W. Gibson and E. Verdin, Mol. Cell, 2015, 59, 321–332 CrossRef CAS.
- F. Li and L. Liu, Front. Cell. Neurosci., 2016, 10, 171 Search PubMed.
- M. J. Hatfield, R. A. Umans, J. L. Hyatt, C. C. Edwards, M. Wierdl, L. Tsurkan, M. R. Taylor and P. M. Potter, Chem. Biol. Interact., 2016, 259, 327–331 CrossRef CAS.
- F. Forneris, C. Binda, M. A. Vanoni, E. Battaglioli and A. Mattevi, J. Biol. Chem., 2005, 280, 41360–41365 CrossRef CAS.
- F. Forneris, C. Binda, A. Dall'Aglio, M. W. Fraaije, E. Battaglioli and A. Mattevi, J. Biol. Chem., 2006, 281, 35289–35295 CrossRef CAS.
- S. S. Ng, K. L. Kavanagh, M. A. McDonough, D. Butler, E. S. Pilka, B. M. Lienard, J. E. Bray, P. Savitsky, O. Gileadi, F. von Delft, N. R. Rose, J. Offer, J. C. Scheinost, T. Borowski, M. Sundstrom, C. J. Schofield and U. Oppermann, Nature, 2007, 448, 87–91 CrossRef CAS.
- J. R. Horton, A. K. Upadhyay, H. H. Qi, X. Zhang, Y. Shi and X. Cheng, Nat. Struct. Mol. Biol., 2010, 17, 38–43 CrossRef CAS.
- G. Blander, J. Olejnik, E. Krzymanska-Olejnik, T. McDonagh, M. Haigis, M. B. Yaffe and L. Guarente, J. Biol. Chem., 2005, 280, 9780–9785 CrossRef CAS.
- A. L. Garske and J. M. Denu, Biochemistry, 2006, 45, 94–101 CrossRef CAS.
- B. C. Smith, B. Settles, W. C. Hallows, M. W. Craven and J. M. Denu, ACS Chem. Biol., 2011, 6, 146–157 CrossRef CAS.
- Y. Liu, L. Perez, A. D. Gill, M. Mettry, L. Li, Y. Wang, R. J. Hooley and W. Zhong, J. Am. Chem. Soc., 2017, 139, 10964–10967 CrossRef CAS.
- R. J. Klose, E. M. Kallin and Y. Zhang, Nat. Rev. Genet., 2006, 7, 715–727 CrossRef CAS.
- P. A. Cloos, J. Christensen, K. Agger and K. Helin, Genes Dev., 2008, 22, 1115–1140 CrossRef CAS.
- W. L. Berry and R. Janknecht, Cancer Res., 2013, 73, 2936–2942 CrossRef CAS.
- L. Hillringhaus, W. W. Yue, N. R. Rose, S. S. Ng, C. Gileadi, C. Loenarz, S. H. Bello, J. E. Bray, C. J. Schofield and U. Oppermann, J. Biol. Chem., 2011, 286, 41616–41625 CrossRef CAS.
- N. R. Rose, S. S. Ng, J. Mecinović, B. M. Liénard, S. H. Bello, Z. Sun, M. A. McDonough, U. Oppermann and C. J. Schofield, J. Med. Chem., 2008, 51, 7053–7056 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, synthetic schemes, supplementary tables, and supplementary figures. See DOI: 10.1039/d0sc06551j |
| This journal is © The Royal Society of Chemistry 2021 |