
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Restoration of catalytic activity by the preservation of ligand structure: Cu-catalysed asymmetric conjugate addition with 1,1-diborylmethane†
Changhee
Kim‡
,
Byeongdo
Roh‡
and
Hong Geun
Lee
 *
*
Department of Chemistry, Seoul National University, Seoul 08826, Republic of Korea. E-mail: hgleee@snu.ac.kr
First published on 21st January 2021
Abstract
Reported herein is a novel reaction engineering protocol to enhance the efficiency of a transition metal-catalysed process by strategically preventing ligand degradation. Based on spectroscopic investigations, a decomposition pathway of a chiral phosphoramidite ligand during a Cu-catalysed reaction was identified. The involvement of the destructive process could be minimized under the modified reaction conditions that control the amount of nucleophilic alkoxide base, which is the origin of ligand decomposition. Overall, the strategy has been successfully applied to a new class of asymmetric conjugate addition reactions with bis[(pinacolato)boryl]methane, in which α,β-unsaturated enones are utilised as substrates.
Introduction
The introduction of novel supporting ligands for transition metals has contributed significantly to the development of homogeneous catalysis based on transition metals. The ligands not only ensure the stability of the metal centre, but also contribute to the activity of the system by performing various catalytically relevant functions, such as stereochemical induction or modulation of electronic properties.1 While it is generally accepted that the ligands are structurally invariant species over the course of their catalytic function (Scheme 1a, path a), processes that involve changes in their architecture can take place under the given reaction conditions. For instance, constructive in situ ligand modifications have been reported in the context of cross-coupling2 or addition reactions of organometallic species (Scheme 1a, path b).3 In such cases, the spontaneous conversion of the ligand (L to L′) eventually facilitated the desired reactivity of the catalytic system. If, on the other hand, the ligand originally administered had been converted into an inactive state by structural modifications (L to L′′), the catalytic system would lose its ability to mediate the targeted reaction (Scheme 1a, path c). In these circumstances, it can be difficult to assess the catalyst performance accurately, as either the inherent incompetence of the system or the destruction of the given ligand structure can be the cause of the substandard reactivity. From another point of view, however, this would offer an opportunity for redeeming the reactivity of an unsuccessful system if the origin of malfunction is the latter (Scheme 1b). By strategically preventing the undesirable modification process using suitable reaction conditions, the maximum capacity of the system can be recovered. This could avoid the costly incorporation of a large excess of valuable ligands or tedious re-evaluation of a new ligand system, the success of which is not guaranteed.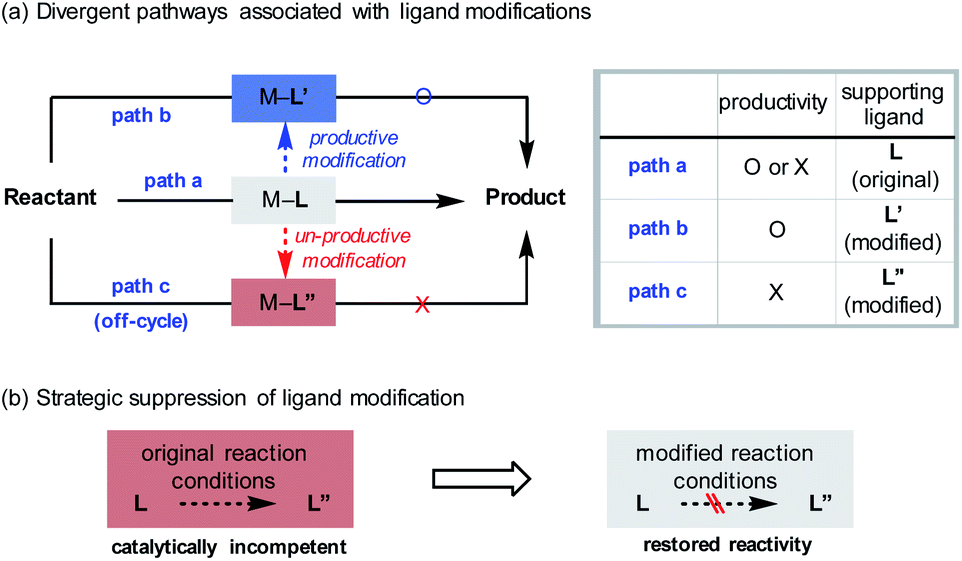 | ||
| Scheme 1 Ligand modifications in transition metal-catalysed reactions. | ||
Here, the possibility of restoring the reactivity of a catalytic system was identified from a transition metal-catalysed stereoselective addition reaction employing 1,1-diborylalkanes (Scheme 2a).4 The synthetically important process was initiated by the activation of a C–B bond through the action of oxygen-based nucleophiles. Subsequent transmetalation to the transition metal catalyst, such as Pd, Cu, or Ir, generates an organometallic species that can react with a variety of electrophilic counterparts to form stereochemically enriched alkylboronate products, a class of compounds that can be diversely functionalized.5 Over the course of the reaction, the involvement of chiral ligands, including bidentate phosphines,5a N-heterocyclic carbenes,5b,c and most commonly phosphoramidites,5d–h plays a critical role in terms of stereochemical induction. In view of the documented susceptibility of phosphoramidite ligands to hydroxide or alkoxide nucleophiles,6 it has been recognized that an undesirable ligand degradation pathway may be involved in reactions with phosphoramidite supporting ligands, especially if the reaction outcome is suboptimal. Accordingly, we evaluated the feasibility of using the ligand preservation approach to promote the insufficient reactivities of reactions with 1,1-diborylalkanes that are based on transition metal-phosphoramidite complexes (Scheme 2b). As a model reaction for investigation, the underdeveloped Cu-catalysed conjugate addition with 1,1-diborylmethane to an α,β-unsaturated ketone substrate was selected.7
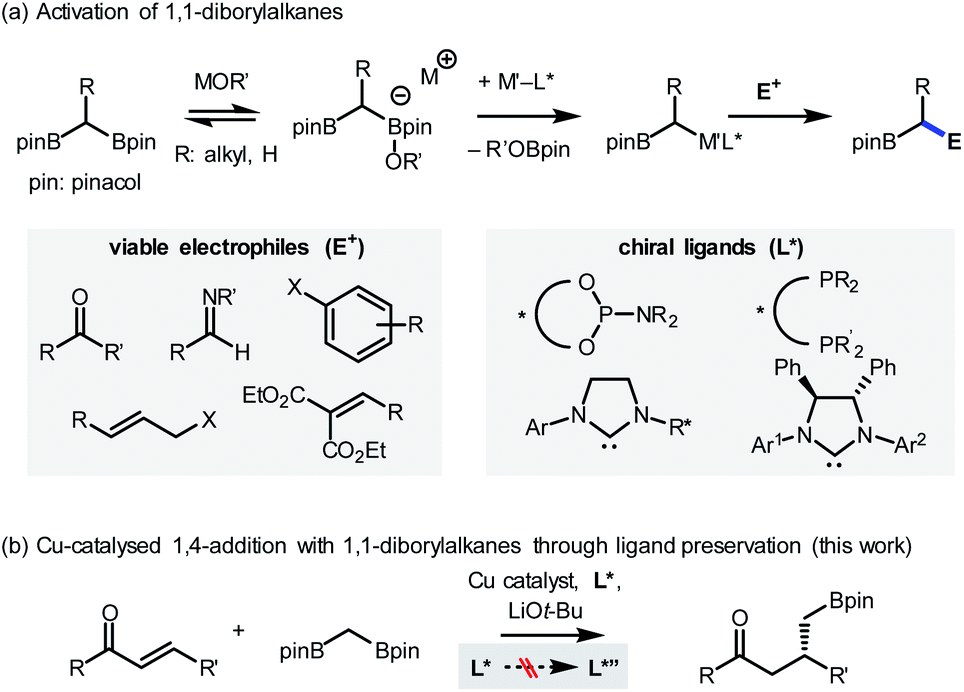 | ||
| Scheme 2 Stereoselective reactions with 1,1-diborylalkanes. | ||
Results and discussion
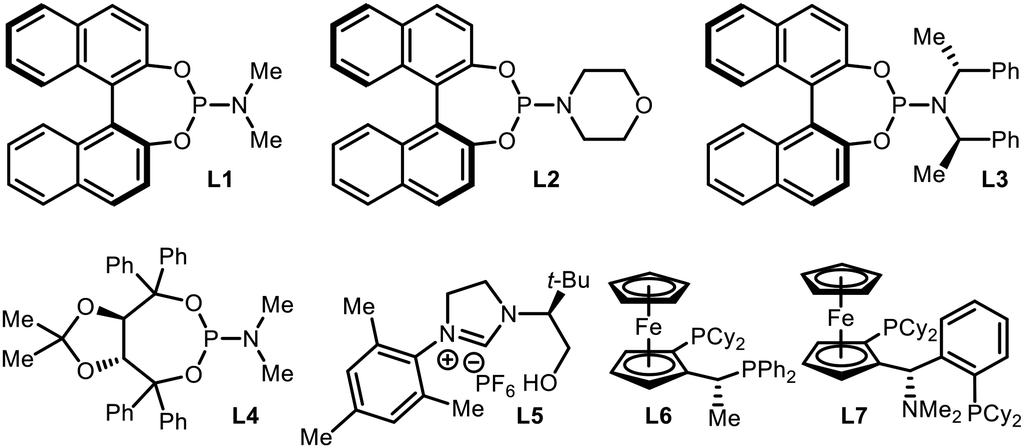
An initial evaluation of the catalyst system with a range of chiral ligands was performed using trans-chalcone as a substrate (Table 1). The stereoselective conjugate addition reaction with bis[(pinacolato)boryl]methane pronucleophile was carried out in the presence of a Cu(I) source, a chiral supporting ligand, and LiOt-Bu, which induces C–B bond cleavage. Ligands with a phosphoramidite backbone exhibited the most pronounced reactivity with concomitant formation of the boron-Wittig product, which accounts for the remainder of the reactivity (entries 2–6).8 Among them, (S)-MonoPhos (L1) demonstrated the best performance in terms of product formation and stereoinduction, although the result was not synthetically ideal (entry 2). Catalyst systems based on other chiral ligands suffered from low levels of conversion (entries 7–9) and/or competitive boron-Wittig reactions. The use of a large excess of ligand, a commonly utilised technique that aids in facilitated ligand ligation, could not promote the reactivity to a practically useful level (entry 3). When attempting to identify conditions with superior reactivity, we found that incorporating Li(acac) into the reaction mixture significantly improved both product yield and enantiomeric excess (entry 10), and additional studies with variations in the catalyst loading showed that 10 mol% catalyst loading was required for the best performance (entries 11 and 12).9 However, the use of other lithium salts, acetylacetonate derivatives, or Lewis acidic entities was not as effective (entries 13–21).10 Eventually, the (S)-MonoPhos-based catalytic system with the Li(acac) additive was chosen as the optimal system.|
|
|||||
|---|---|---|---|---|---|
| Entrya | Ligand | Additive | Conv. (%) | Yieldb (%) | eec (%) |
| a Reaction conditions: CuBr (10 mol%), ligand (12 mol%), LiOt-Bu (1.2 equiv.), additive (1.0 equiv.), 2a (1.5 equiv.), and 1a (0.3 mmol, 0.33 M) in THF (0.9 mL). b Determined by GC analysis with n-dodecane as an internal standard. c Determined by HPLC analysis. d 24 mol% of L1 was used. e 5 mol% of CuBr and 6 mol% of L1 were used. f 2.5 mol% of CuBr and 3 mol% of L1 were used. acac = acetylacetone, TMHD = 2,2,6,6-tetramethyl-3,5-heptanedione, hfacH = hexafluoroacetylacetone. | |||||
| 1 | — | — | >99 | 20 | — |
| 2 | L1 | — | >99 | 63 | 76 |
| 3 | L1 | — | >99 | 68 | 79 |
| 4 | L2 | — | >99 | 40 | 55 |
| 5 | L3 | — | 74 | 19 | 6 |
| 6 | L4 | — | 98 | 40 | 4 |
| 7 | L5 | — | <5 | Trace | — |
| 8 | L6 | — | 28 | Trace | — |
| 9 | L7 | — | 29 | Trace | — |
| 10 | L1 | Li(acac) | >99 | 70 | 92 |
| 11e | L1 | Li(acac) | >99 | 62 | 86 |
| 12f | L1 | Li(acac) | >99 | 35 | 83 |
| 13 | L1 | LiF | 81 | 76 | 77 |
| 14 | L1 | LiCl | 30 | 29 | <5 |
| 15 | L1 | LiBr | 28 | 28 | <5 |
| 16 | L1 | LiClO4 | <5 | Trace | — |
| 17 | L1 | Li(TMHD) | 44 | 35 | <5 |
| 18 | L1 | Na(hfacH) | 32 | 13 | 70 |
| 19 | L1 | TMSCl | 36 | 24 | 7 |
| 20 | L1 | BF3·OEt2 | <5 | Trace | — |
| 21 | L1 | ZnBr2 | <5 | Trace | — |
|
|||||

To better understand the factors that contribute to the successful catalytic system containing Li(acac), catalytically relevant species were prepared and examined using 31P and 11B NMR experiments (Fig. 1). When all the reaction components, including Li(acac), were introduced in the absence of the enone substrate, the active nucleophilic species (2b) was formed. Based on the observed reactivity towards the enone substrates and 31P NMR analysis, the structure of the reactive species was assigned to be the ligand-bound alkyl copper complex (Fig. 1a).11,12 Besides 2b, the uncoordinated ligand L1 was the only significant species that was detected. On the other hand, in the case of an identical mixture that did not contain Li(acac), no free ligand was observed. More importantly, the relative mass balance of 2b diminished significantly. Instead, unidentified chemical species were found (δ 137–147 ppm, box), indicating decomposition of catalytically responsible species based on L1 (vide infra).13
 | ||
| Fig. 1 Spectroscopic analysis of the catalytic system. Reaction conditions: L1 (1.0 equiv., 0.012 mmol), CuBr (0.83 equiv.), 2a (12.5 equiv.), LiOt-Bu (10 equiv.), and Li(acac) (8.3 equiv.) in THF-d8 (0.5 mL). | ||
The origin of the observed catalyst decomposition could be deduced using 11B NMR studies (Fig. 1b). The 11B NMR spectra of the reaction mixture consists of three major signals originating from C-ligated neutral Bpin species, including 2a, 2b, and 2d-Ba (A), heteroatom-bound neutral Bpin species (2c), and anionic complexes, such as 2d-Bb or Li[pinB(Ot-Bu)2] (B).14 Complete quantitative analysis of all relevant species was difficult to perform because of the complexity of the system. It is evident, however, that the application of Li(acac) drove the equilibrium in the direction of increasing the amount of 2c and B. Since the formation of 2c and B stems from the coordination of the alkoxide at the boron centre, the result indicates an escalated level of the entrapped alkoxide nucleophile in the reaction mixture. Eventually, a reduced amount of alkoxide is available for ligand degradation to enhance the catalytic activity of the system.
The impact of added Li(acac) was further investigated in a more controlled experiment in which 2a was independently exposed to the LiOt-Bu nucleophile (Fig. 2). The addition of Li(acac) shifted the equilibrium to the direction of the ate-complex formation (2d). During the reaction, a significant portion of the initially administered LiOt-Bu nucleophiles was consumed: in the case of the Li(acac)-free conditions the effective concentration of LiOt-Bu is 50% higher than that of the conditions with Li(acac).15
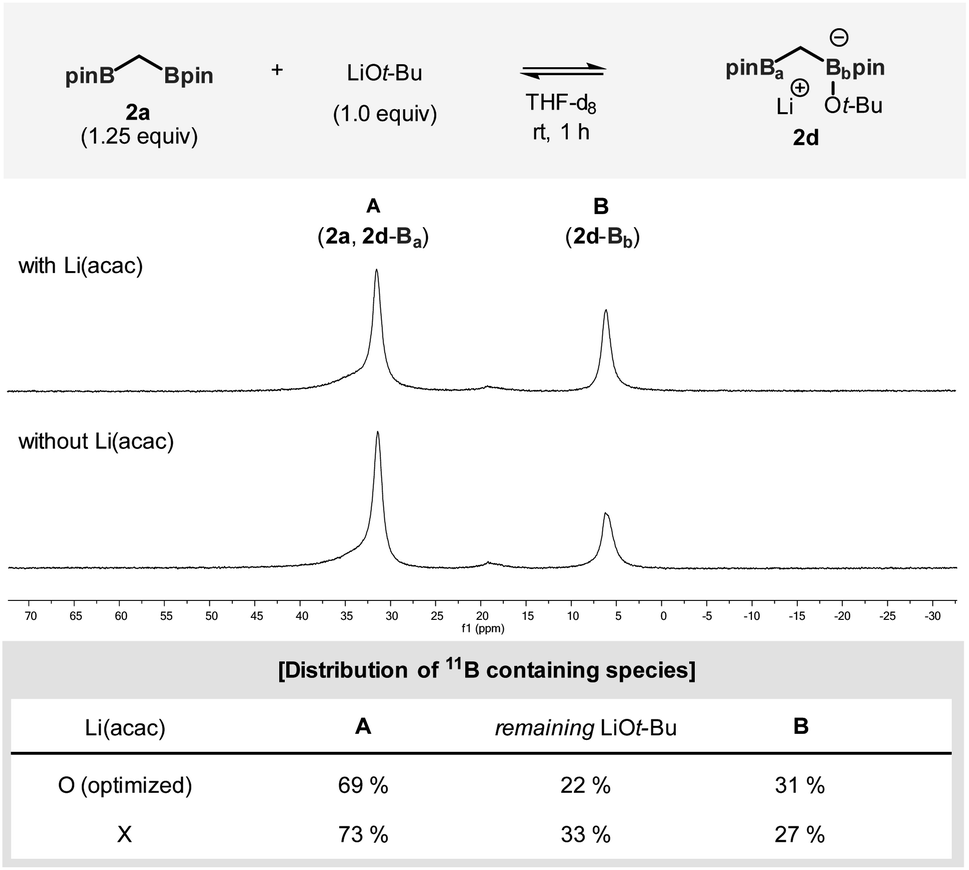 | ||
| Fig. 2 Equilibrium of bis[(pinacolato)boryl]methane with LiOt-Bu monitored by 11B NMR. Reaction conditions: 2a (1.25 equiv., 0.15 mmol), LiOt-Bu (1.0 equiv.), and Li(acac) (0.83 equiv.) in THF-d8 (0.5 mL). | ||
The complete decomposition pathway of the phosphoramidite ligand remains elusive. However, preliminary mass spectroscopic analysis of the reaction mixture suggests the involvement of P–O or P–N bond cleavage in the backbone structure of L1 in the presence of LiOt-Bu.16 It is believed that the initial nucleophilic attack of LiOt-Bu generates an intermediate that leads to other decomposition products.
Based on these observations, a mechanistic scenario that consists of two independent catalytic cycles is presented (Fig. 3). In the productive cycle, the active catalyst I-A, which is generated by the combination of L1, CuBr, and LiOt-Bu, undergoes transmetalation with 2a to generate a copper alkyl species II-A (Fig. 1, 2b). The reactive carbon-based nucleophile should undergo an enantioselective conjugate addition reaction with enone substrate 1 to provide a copper enolate intermediate III-A, an immediate precursor of the desired product with high stereochemical integrity. During the operation of the major catalytic cycle, 2a should be in equilibrium with its ate-complex form 2d, a species responsible for the rate-determining transmetalation, by the involvement of LiOt-Bu.17 Ultimately, the equilibrium controls the amount of the alkoxide base in the reaction mixture that is available to destroy active catalytic species. The process includes the irreversible modification of the phosphoramidite ligand L1, which contributes to furnishing catalytically incompetent species ([Cu]). These species include Cu-complexes ligated with the decomposed ligand (L′′), solvent (S), and/or other heteroatom-containing species present in the reaction mixture. Consequently, an alternative catalytic cycle with lower stereoselectivity operates with the intermediates II-B and III-B. As determined from the NMR experiments, the added Li(acac) controls the extent of the unproductive catalytic cycle by controlling the equilibrium of 2a, LiOt-Bu, and 2d.18
 | ||
| Fig. 3 Plausible mechanistic scenario. | ||
With the optimized conditions in hand, we evaluated the scope of the developed reaction using various α,β-unsaturated enone substrates (Table 2). Reactions with chalcone derivatives bearing electron-donating or electron-withdrawing substituents on the arene ring at the β position of the enones afforded the desired boronic esters with good yield and enantioselectivity (3a–3g), although the placement of an ortho substituent of the arene resulted in substantially diminished yield (3c). It is noteworthy that the efficiency of the reaction could be successfully conserved in a larger-scale reaction, which demonstrates the robustness of the reaction (3a). Variations on the phenyl ring directly attached to the carbonyl group were also tolerated (3h–3j). Importantly, the presence of halogen substituents, which can be used as a handle for further functionalization through cross-coupling reactions, did not affect the outcome of the conjugate addition reaction (3f, 3g, 3i, 3j). Moreover, biologically important heterocycles such as furan or thiofuran could be successfully incorporated into the substrate structure (3k–3n). Additionally, the reactivity could be extended to alkyl-substituted α,β-unsaturated enones. When a diverse array of alkyl groups was introduced at the β-position of the enone substrate, the optimized conditions operated smoothly (3o–3r). Meanwhile, an alkyl group at the carbonyl carbon significantly decreased the efficiency of the product formation due to competitive 1,2-addition reactions. Only the substrate containing a bulky t-butyl group was a viable substrate for the conjugate addition (3s). Finally, an enone with an extended π-system afforded the desired 1,4-addition products (3t). A side product originating from the competing 1,6-addition was not observed.

The synthetic utility of the developed reaction was further demonstrated based on the versatility of the stereochemically enriched γ-ketoboronic ester product (Scheme 3). The Suzuki–Miyaura cross coupling reaction of (S)-3a and bromobenzene afforded the corresponding γ-phenyl ketone product in 72% yield (3ab).19 During the C–C bond-forming reaction, the stereochemical integrity of the stereogenic centre remained unaffected. Moreover, the C–B bond of (S)-3a could be readily oxidized in the presence of NaBO3·4H2O to provide a γ-hydroxy ketone (3ac), which in turn was functionalized into more complex products. A two-step sequence involving Jones oxidation and Fisher esterification furnished the corresponding methyl ester in 51% overall yield (3ad). Alternatively, an enantioselective reduction of the carbonyl group in the presence of the (R)-oxazaborolidine catalyst,20 followed by stereospecific benzylic displacement21 afforded marine natural product calyxolane B in its enantiomeric form (3ae).22
 | ||
| Scheme 3 Transformation of chiral (S)-3a. | ||
Conclusions
In conclusion, a novel reaction engineering strategy has been developed to allow for full utilisation of the catalytic activity of a transition-metal-catalysed reaction. By identifying the ligand decomposition pathway and using an additive-based restoration method, the efficiency of an underdeveloped reaction could be brought to synthetically useful levels. The method has been successfully applied to an enantioselective conjugate addition reaction with 1,1-bis[(pinacolato)boryl]methane catalysed by a Cu-phosphoramidite catalyst system. We believe that the strategy should have a broader impact on reactions catalysed by related metal–ligand complexes. It is anticipated that analogous reaction engineering systems will ultimately be implemented for transition metal catalysis in general.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this research was provided from National Research Foundation of Korea (MSIT, 2018R1C1B6008115). The authors would like to thank Professor Sukbok Chang (Korea Advanced Institute of Science and Technology) for providing chiral ligands.Notes and references
- For selected reviews, see:
(a) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed
; (b) J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486 CrossRef CAS PubMed
-
(a) Q. Shelby, N. Kataoka, G. Mann and J. Hartwig, J. Am. Chem. Soc., 2000, 122, 10718 CrossRef CAS
-
(a) C. Bournaud, C. Falciola, T. Lecourt, S. Rosset, A. Alexakis and L. Micouin, Org. Lett., 2006, 8, 3581 CrossRef CAS PubMed
- For reviews, see:
(a) R. Nallagonda, K. Padala and A. Masarwa, Org. Biomol. Chem., 2018, 16, 1050 RSC
-
(a) C. Sun, B. Potter and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 6534 CrossRef CAS PubMed
-
(a) W. Bannwarth and A. Trzeciak, Helv. Chim. Acta, 1987, 70, 175 CrossRef CAS
- The first asymmetric conjugate addition reaction with 1,1-diborylalkane has recently been reported (Ref. 5c). However, the applicability of the developed reaction has been limited to highly activated substrates containing multiple electron-withdrawing groups. As a result, an extension of the reactivity to an enone substrate was not feasible. For a related conjugate addition based on a different mechanistic pathway, see: W. J. Jang, J. Woo and J. Yun, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202014425
-
(a) D. S. Matteson, R. J. Moody and P. K. Jesthi, J. Am. Chem. Soc., 1975, 97, 5608 CrossRef CAS
- For review of additive effects in asymmetric catalysis, see: L. Hong, W. Sun, D. Yang, G. Li and R. Wang, Chem. Rev., 2016, 116, 4006 CrossRef CAS PubMed
- For additive-promoted advancement of reactivity in conjugate addition, see:
(a) S. Morikawa, S. Yamazaki, M. Tsukada, S. Izuhara, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2007, 72, 6459 CrossRef CAS PubMed
- The assignment is based on the observed reactivity of the mixture to promote the asymmetric conjugate addition upon exposure to the enone substrate. In addition, the independent preparation of related Cu complexes such as L1CuBr (31P: δ 128 ppm) and L1CuOt-Bu (31P: δ 118 ppm) supports this conclusion. See ESI† for details.
- At this time, we cannot rigorously rule out the possibility of 2b existing in its multinuclear form. A related phosphoramidite-based Cu-alkyl species exhibiting such behavior has been prepared and characterized (31P: δ 124 ppm). See: F. von Rekowski, C. Koch and R. M. Gschwind, J. Am. Chem. Soc., 2014, 136, 11389 CrossRef CAS PubMed
- Some of the new species observed in this case could be generated by independently treating L1 with LiOt-Bu. Therefore, it is believed that these species are the decomposition products of L1 and their corresponding Cu complexes. In addition, the new species are being generated over time with concomitant consumption of L1. See ESI† for details.
- For studies associated with the identification of the borate derivatives by 11B NMR, see:
(a) K. Endo, T. Ohkubo and T. Shibata, Org. Lett., 2011, 13, 3368 CrossRef CAS PubMed
- These values were quantitatively determined based on the assumption that all boron species exist as either 2a or 2d. For quantitative analysis based on 11B NMR, see: L. M. Aguilera-Sáez, J. R. Belmonte-Sánchez, R. Romero-González, J. L. Martínez Vidal, F. J. Arrebola, A. Garrido Frenich and I. Fernández, Analyst, 2018, 143, 4707 RSC
- The following species were detected from HRMS analysis (ESI-MS, [M + H]+). See ESI† for details
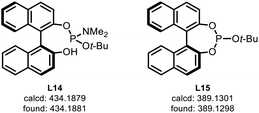 .
. - M. Kim, B. Park, M. Shin, S. Kim, J. Kim, M. H. Baik and S. H. Cho, J. Am. Chem. Soc., 2021, 143, 1069 CrossRef CAS PubMed
- A more active participation of Li(acac) in the catalytic cycle, such as being an activator for 2a or a scavenger for 2c, was disregarded based on control experiments. See ESI† for details.
- C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528 CrossRef CAS PubMed
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS PubMed
- A. Bunrit, P. Srifa, T. Rukkijakan, C. Dahlstrand, G. Huang, S. Biswas, R. A. Watile and J. S. M. Samec, ACS Catal., 2020, 10, 1344 CrossRef CAS
-
(a) G. Hilt, P. Bolze and K. Harms, Chem.–Eur. J., 2007, 13, 4312 CrossRef CAS PubMed

Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data for newly synthesized compounds and other experimental details. See DOI: 10.1039/d0sc06543a |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2021 |